
Advancing cancer therapy with a heptamethine carbocyanine dye-conjugated radionuclide drug†
Mingxing
Huang‡
a,
Weichen
Wang‡
a,
Yingwei
Wang
 b,
Rang
Wang
a,
Lili
Pan
b,
Xin
Li
a,
Bo
Li
c,
Jingsong
Zhang
c,
Ruoxiang
Wang
d,
Yi
Zhang
*e and
Rong
Tian
*a
b,
Rang
Wang
a,
Lili
Pan
b,
Xin
Li
a,
Bo
Li
c,
Jingsong
Zhang
c,
Ruoxiang
Wang
d,
Yi
Zhang
*e and
Rong
Tian
*a
aDepartment of Nuclear Medicine, West China Hospital, Sichuan University, Chengdu, China. E-mail: rongtiannuclear@126.com
bDepartment of Nuclear Medicine and Clinical Nuclear Medicine Research Lab, West China Hospital, Sichuan University, Chengdu, China
cNuclear Power Institute of China, National Engineering Research Center of Isotope and Medicine, and Radioisotope Engineering Technology Research Center of Sichuan, Chengdu, China
dDepartment of Medicine, Cedars-Sinai Medical Center, Los Angeles, California 90048, USA
eDepartment of Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles, California 90048, USA
First published on 1st November 2024
Abstract
Malignant tumors pose substantial treatment challenges due to their heterogeneity, metastatic potential, and therapeutic resistance, underscoring the urgent need for more effective treatment options. In this study, a novel radionuclide drug conjugate (RDC) was developed and characterized employing a tumor-targeting heptamethine carbocyanine dye (DZ) for its high specificity and favourable safety profile. The RDC, DOTA-DZ-HX, was synthesized by conjugating DZ with dodecane tetraacetic acid (DOTA) through a dipeptide linker. The anti-tumor efficacy and biodistribution of [177Lu]Lu-DOTA-DZ-HX were assessed in H1975 and HeLa xenograft models. [177Lu]Lu-DOTA-DZ-HX showed high stability, specific tumor accumulation, and substantial radioactivity retention at the tumor site for seven days. Biodistribution analysis revealed that the drug exhibited prolonged blood circulation with gradual clearance. Additionally, [177Lu]Lu-DOTA-DZ-HX could effectively and selectively inhibit tumor growth, as evidenced by a significant reduction in tumor volume. These findings suggest that heptamethine carbocyanine dye-directed radionuclide targeting could provide an effective treatment for cancer and potentially other malignancies, representing a significant advancement in cancer therapy.
Introduction
Malignant tumors pose significant health challenges. Although oncological treatments have progressed over the past decade, tumor cell heterogeneity and therapeutic resistance frequently lead to clinical failure. The use of chemotherapy is often limited by its adverse effects,1 while immunotherapy and existent targeted therapy are effective only for certain tumor types.2,3 There is an urgent need for the development of broad-spectrum and tumor-specific anti-tumor strategies.Recent advancements in radionuclide therapy, particularly with the introduction of radionuclide drug conjugates (RDCs), have established effective and safe alternatives for treating various cancer types.4 RDCs use specific targeting mechanisms to deliver radionuclides directly to cancer cells, concentrating the radioactive energy on tumor tissues while sparing healthy cells. Precision targeting also reduces the required molecular dosage compared to traditional chemotherapy, typically to the sub-nanomolar level, further minimizing side effects.4,5 Moreover, RDCs may provide dual functionality, serving both imaging and therapeutic purposes, which exemplifies the concept of theranostics.6
The delivery of radionuclides by RDCs currently involves small molecules,7 peptides,8 and antibodies9 that are specific to certain types of cancers. Their versatility in treating a wide range of cancer types remains restricted.10 Additionally, significant challenges, such as crossing physiological barriers like the blood–brain barrier (BBB) and the blood–tumor barrier (BTB), as well as issues related to biocompatibility and stability, pose considerable obstacles to their successful application.
In 2010, Chung and colleagues discovered that heptamethine carbocyanine dyes (HMCDs), such as IR-783 and MHI-148, specifically accumulate in the mitochondria and lysosomes of tumor cells, but not in normal cells.11 These dyes could be retained in xenograft or spontaneous tumors for up to 15 days.12 Further studies revealed that HMCD's tumor targeting is mediated mainly by proteins in the family of organic anion-transporting polypeptides (OATPs), which are over-expressed in cancer cells, and their expression is further enhanced under intra-tumoral hypoxic conditions through hypoxia-inducible factor 1α (HIF-1α).12 Emitting near infrared (NIR) fluorescence, HMCDs represent a group of rare small molecules whose tumor-cell specificity could be exploited for tumor imaging and targeting.
Extensive investigations have been conducted into the potential applications of various HMCDs for the diagnosis and treatment of cancers.13–17 Even at doses exceeding the imaging dose by 100 times, HMCDs do not exhibit systemic toxicity.11,18,19 HMCD–drug conjugates were identified with significant specificity and prolonged retention in various malignancies, including prostate, lung, breast, liver, gastric, kidney, pancreatic, and gliomas.20–24 Compared with other delivery vehicles, HMCDs offer several advantages: (1) they exhibit excellent in vivo stability and a well-tolerated safety profile; (2) they are preferentially taken up by various cancer cells over normal ones; and (3) they target both localized and metastatic tumors and can cross the BBB and BTB. HMCDs are recognized as broad-spectrum tumor-targeting agents with favourable safety profiles and high biocompatibility.25
A HMCD known as DZ-1 has been selected as our tumor-targeting vehicle due to its high tumor specificity and ease of chemical modification.26–29 In this study, we report the characterization of an innovative broad-spectrum tumor-targeting RDC, [177Lu]Lu-DOTA-DZ-HX. This complex is designed with the targeting vehicle DZ-1 linked by a dipeptide to the tetraxetan (DOTA), which chelates with 177Lu. The report focuses on the tumor-killing efficacy and biosafety of this complex in both in vitro and in vivo settings.
Methods
Materials
DOTA-tris(t-Bu ester) was purchased from Confluore Biotech (Xi'an, China). Aspartic acid (Asp) and lysine (Lys) were obtained from Bide Pharmatech Ltd (Shanghai, China). All other reagents and chemicals for synthesis were commercially obtained from Aladdin Reagent (Shanghai, China) or Sigma-Aldrich (St. Louis, MO, United States) and used without further purification unless otherwise stated. Carrier-free [177Lu]Cl3 in 0.04 M HCl solution (specific activity: ≥ 1480 GBq mg−1) was purchased from Syncor Pharmaceuticals (Chengdu, China). Solutions were prepared using deionized ultrapure water with a resistivity of 18.2 MΩ using a Milli-Q Direct Ultrapure Water System from Millipore (Billerica, MA, USA). All synthesized compounds were characterized by 1H nuclear magnetic resonance (NMR) spectroscopy using a 400 MHz Bruker Avance II spectrometer (Bruker, Billerica, MA). Liquid chromatography–mass spectrometry (LC–MS) was conducted using an Infinity Lab mass spectrometer connected to an Agilent 1260 series instrument with an Eclipse Plus C18 column (4.6 mm × 100 mm, 3.5 μm) and a UV wavelength of 254 nm. The radiochemical purity was recorded by radio-thin-layer chromatography (rTLC) and reversed-phase high-performance liquid chromatography (RP-HPLC) using a Kinetex C18 column (250 mm × 4.6 mm, 5 μm) with an additional γ-detector (Beijing PET Technology, Beijing). The gradient and flow rate of the water/(0.1%TFA)/acetonitrile mobile phase were modified for the individual products. The synthesis and radiolabeling methods for [177Lu]Lu-DOTA-DZ-HX are shown in Scheme 1. | ||
| Scheme 1 Reagents and conditions: (i) (1) Ethyl chloroformate, Et3N, 1 h; (2) L-aspartate-4-tert-butyl ester; (ii) (1) HATU, Et3N, Nα-Fmoc-L-lysine tert-butyl ester; (2) piperidine; (iii) (1) EDC, HOBt; (2) TFA; (iv) [177Lu]Cl3. | ||
Synthesis of DOTA-DZ-HX
Radiolabeling
For radiolabeling with [177Lu]LuCl3, a solution containing 37 MBq–74 MBq of [177Lu]LuCl3 was mixed with 0.2 mL of 1 M sodium acetate (NaOAc) buffer, adjusted to pH 5.0–5.5. This mixture was then incubated with DOTA-DZ-HX (50 μg) at 90 °C for 20 minutes. The radiochemical purity of [177Lu]Lu-DOTA-DZ-HX was evaluated using paper chromatography and radio-HPLC. For paper chromatography, 0.1 M citric acid was used as the mobile phase. For radio-HPLC, the separation was carried out with a gradient system consisting of solvent A (acetonitrile, ACN) and solvent B (0.1% formic acid, HCOOH in water). The gradient was programmed as follows: from 0 to 15 minutes: 60% solvent A and 40% solvent B; from 15 to 20 minutes: 90% solvent A and 10% solvent B; from 20 to 25 minutes: 10% solvent A and 90% solvent B; the flow rate was maintained at 1.0 mL min−1.Stability study
To evaluate the in vitro stability of [177Lu]Lu-DOTA-DZ-HX, 3.7 MBq of the radiolabeled compound (100 μL) was incubated with an equal volume of either phosphate-buffered saline (PBS, pH 7.4) or bovine serum at 37 °C. The radiochemical purity was assessed by rTLC at multiple time points, such as 0, 4, 24, 48, 120, and 168 hours. All experiments were conducted in triplicate to ensure reproducibility.Cell culture and tumor models
Human lung adenocarcinoma H1975 and Human cervical cancer HeLa cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Beijing, China). The cells were cultured in RPMI-1640 and the DMEM (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Waltham, MA, USA) and 1% penicillin–streptomycin (100 μg mL−1) and streptomycin (100 μg mL−1). Cultures were maintained in a 5% CO2 humidified atmosphere at 37 °C. All animal studies were approved by the Animal Care and Use Committee of Sichuan University (approval ID: ID20230707002) and were conducted in compliance with national laws governing animal experimentation. For the establishment of animal models, female BALB/c nude mice (5 weeks old) were purchased from Charles River Laboratories (Beijing, China). To induce tumor formation, 5 × 106 cells were subcutaneously inoculated into the right upper limbs of the mice. When the tumor volume reached approximately 50–100 mm3, the mice were randomized into 4–5 groups and subjected to fluorescence imaging studies and radioligand therapeutic assays.Cytotoxicity assays
For the in vitro radioisotope therapy experiment, H1975 and HeLa cells were seeded in 96-well plates at a density of 5 × 103 cells per well and incubated overnight at 37 °C to allow for cell attachment. The next day, varying amounts of [177Lu]LuCl3 and [177Lu]Lu-DOTA-DZ-HX (0.000, 0.462, 0.925, 1.850, 2.775, and 3.700 MBq) were added to the wells. The cells were then incubated with the radioactive compounds for 48 hours. Cell viability was assessed using the CCK-8 assay, following the manufacturer's protocol (Dojindo Molecular Technologies, USA).Blood clearance
For the blood clearance study of [177Lu]Lu-DOTA-DZ-HX and [177Lu]Lu-DOTA, eight normal ICR mice (6 weeks old) (Charles River Laboratories, Beijing, China) were used as a single experimental group. Each mouse received an intravenous injection of 0.74 MBq of radiolabelled compounds. Blood samples (0.01 mL) were collected from the tail vein at 2, 5, 10, 25, 30, 60, 120, 240, 480, 1440, 2880, and 4320 minutes post-injection. The collected blood was weighed, and the radioactivity was measured using an automatic γ-counter (PerkinElmer Wizard, Shelton, CT, USA). The percentage of tracer uptake was calculated as the percentage of the injected dose per gram of tissue (% ID per g). This value was decay-corrected to the start time of counting, reflecting the activity associated with the tissue per gram of organ weight relative to the actual injected dose.Whole-body NIR fluorescence imaging
To monitor the distribution and accumulation of the radiolabeled compound, tumor-bearing mice were anesthetized using isoflurane. Each mouse received an i.v injection of DOTA-DZ-HX (∼100 μg per mouse, n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 3). NIR fluorescence images were captured at various time points post-injection with an IVIS imaging spectrum system combined with IVIS Living Image 3.0 software (PerkinElmer, USA). Imaging parameters were set as follows: excitation wavelength (λex) = 745 nm, emission wavelength (λem) = 820 nm, binning = 1, and exposure time = 0.1 seconds.
= 3). NIR fluorescence images were captured at various time points post-injection with an IVIS imaging spectrum system combined with IVIS Living Image 3.0 software (PerkinElmer, USA). Imaging parameters were set as follows: excitation wavelength (λex) = 745 nm, emission wavelength (λem) = 820 nm, binning = 1, and exposure time = 0.1 seconds.
Micro-SPECT/CT imaging
Tumor-bearing mice (n = 3) were investigated using a small animal single photon emission computed tomography (SPECT)/computed tomography (CT) scanner (Novel Medical, Beijing, China) when the tumor size reached 150 to 200 mm3. SPECT was performed under isoflurane anaesthesia at 24, 48, 72, 120, and 168 hours after a single i.v injection of [177Lu]Lu-DOTA-DZ-HX (37 MBq) in saline. Simultaneously, [177Lu]Lu-DOTA was used as a negative control group for imaging. The acquisition time for each scan was approximately 25 min. The detailed information on image acquisition and processing data is listed in Tables S1 and S2 (ESI†).
Biodistribution
H1975 and HeLa tumor-bearing mice were randomly assigned into five or six groups (n = 4). Each mouse received an intravenous injection of 0.2 mL of [177Lu]Lu-DOTA-DZ-HX (1.85 MBq). Time-dependent biodistribution studies were conducted by sacrificing the mice at 1, 4, 24, 48, 72, and 96 hours post-injection. The tumor, together with blood, brain, heart, liver, spleen, lung, kidney, stomach, intestine, muscle, and bone tissues, was harvested and weighed, and the radioactivity was measured using an automatic γ-counter (PerkinElmer Wizard, Shelton, CT, USA). The organ uptake was calculated as the percentage of injected dose per gram of wet tissue mass (% ID per g).Radiotherapy study
Following a xenograft implant, studies were initiated when the average tumor volume reached approximately 50–100 mm3 in H1975 and HeLa tumor-bearing mice. The mice were randomly divided into four groups (n = 5), each receiving solvent control (PBS), [177Lu]Lu-DOTA (37 MBq), [177Lu]Lu-DOTA-DZ-HX (37 MBq, 18.5 MBq), and [177Lu]Lu-DOTA-DZ-HX (11.1 MBq, 7.4 MBq) in 100 μL of saline. These were administered intravenously (i.v.) once. The individual tumor size (mm3) was calculated using the formula: 1/2 × length × width2. Tumor volumes and body weights of all mice were monitored every two days. Mice were euthanized if they experienced a weight loss of 40% or if the tumor volume exceeded 1500 mm3. Potential adverse events were assessed based on body mass and histological analysis.Statistical analysis
Statistical analysis was performed with GraphPad Prism software version 10.0 (Bethesda, MD, USA) or with IBM SPSS version 23.0 (IBM, Armonk, NY, USA). The reported values were presented as mean ± standard deviation. In the unpaired student t-test, p-values < 0.05 were considered statistically significant.Results
Synthesis and characterization of DOTA-DZ-HX
The cancer-targeting vehicle DZ-1 was synthesized according to established protocols.30 As illustrated in Scheme 1, the synthesis of DOTA-DZ-HX began with the modification of DZ-1. Initially, the side chain terminal carboxylic group of DZ-1 was activated using ethyl chloroformate and triethylamine in anhydrous dichloromethane for one hour. This activated compound then reacted with L-aspartate-4-tert-butyl ester to form DZ-asp tert-butyl ester. Subsequently, DOTA-tris (t-Bu ester) was conjugated with Nα-Fmoc-L-lysine tert-butyl ester using HATU and TEA as coupling agents, yielding DOTA-lys tert-butyl ester. This intermediate was then reacted with DZ-asp tert-butyl ester using EDCI and HOBt as coupling agents. The final compound, DOTA-DZ-HX, was obtained after deprotection with TFA. The product, DOTA-DZ-HX, was purified by reverse-phase semi-preparative RP-HPLC and characterized by 1H NMR (Fig. 1A) and high-resolution mass spectrometry (HR-MS) (Fig. 1B). | ||
| Fig. 1 Characterization and stability analysis of [177Lu]Lu-DOTA-DZ-HX. (A) 1H NMR spectrum of DOTA-DZ-HX (400 MHz, MeOD). (B) High-resolution mass spectrum of DOTA-DZ-HX. (C) Radio-HPLC chromatogram of [177Lu]Lu-DOTA-DZ-HX detected using a gamma detector. (D) Stability of [177Lu]Lu-DOTA-DZ-HX in PBS and FBS. | ||
Radiolabeling and stability of the [177Lu]Lu-DOTA-DZ-HX conjugate
[177Lu]Lu-DOTA-DZ-HX was generated with a radiochemical purity of 95.7 ± 0.32% under heating at 90 °C for 20 minutes and used without further purification. The specific activity and the molar activity of the radiolabeled complex were 0.74 MBq μg−1 and 0.25 MBq μmol−1, respectively. As displayed in Fig. 1C, the retention time of [177Lu]Lu-DOTA-DZ-HX on radio-HPLC was 9.1 minutes. [177Lu]Lu-DOTA-DZ-HX demonstrated stability, maintaining radiochemical purities of over 95% in PBS and over 93% in FBS after 168 hours of incubation, with almost no degradation observed (Fig. 1D).In vitro efficacy of [177Lu]Lu-DOTA-DZ-HX
The results demonstrated a dose-dependent cytotoxic effect of [177Lu]Lu-DOTA-DZ-HX on tumor cells, with increased dosage correlating with enhanced cytotoxicity. In the experimental group treated with [177Lu]Lu-DOTA-DZ-HX, the cell survival rates at 3.7 MBq dose treatment were 41.81% ± 5.67 for H1975 cells and 26.51% ± 2.38 for HeLa cells, compared to 68.54% ± 5.47 for the control group (Fig. 2A). This enhanced effectiveness is attributed to the compound's ability to directly target subcellular organelles.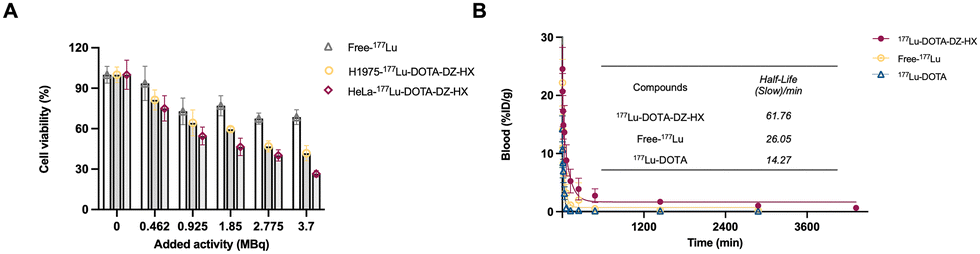 | ||
| Fig. 2 Tumor-cell killing activity and blood clearance of [177Lu]Lu-DOTA-DZ-HX. (A) Tumor-cell killing activity was evaluated by treating H1975 lung cancer cells and HeLa cervical cancer cells with serially diluted conjugates for 48 hours. (B) Blood clearance profiles of [177Lu]Lu-DOTA-DZ-HX, [177Lu]Lu, and [177Lu]Lu-DOTA. | ||
Blood clearance studies revealed that [177Lu]Lu-DOTA-DZ-HX is eliminated from the bloodstream in a biphasic manner. Initially, it displayed a rapid clearance with a half-life of 3.52 minutes, followed by a much slower secondary phase with a half-life of 61.76 minutes. In contrast, both [177Lu]LuCl3 and [177Lu]Lu-DOTA exhibited rapid metabolism in the bloodstream, with a half-life of 26.05 minutes and 14.27 minutes, respectively (Fig. 2B).
Fluorescence imaging
The tumor specificity of the non-radiolabeled precursor DOTA-DZ-HX was validated using a mouse xenograft model by in vivo fluorescence imaging. Fluorescence imaging revealed that the precursor DOTA-DZ-HX exhibited exceptional targeting capabilities in the H1975 xenograft tumors. The pharmacokinetics of the drug indicated a slow metabolism in vivo, with an initial systemic distribution followed by progressive accumulation at the tumor site. The peak tumor uptake was observed 24 hours post-administration (1.83 × 109 photons per s per cm2 per steradian), with the highest tumor-to-background ratio recorded at 108.15 ± 10.75 (Fig. 3 and Table S3, ESI†).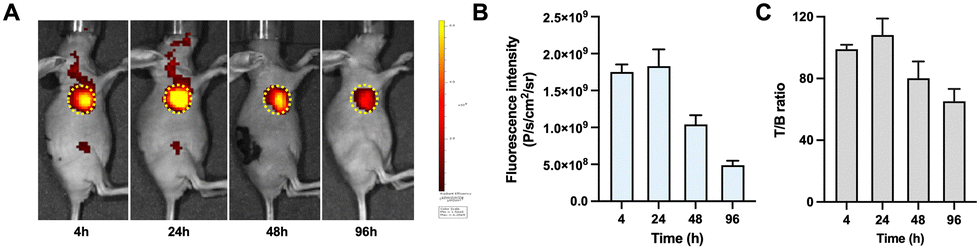 | ||
| Fig. 3 Fluorescence imaging of the non-radiolabeled precursor DOTA-DZ-HX. (A) Fluorescence imaging at different time points after drug injection into H1975 xenograft models (n = 3 per group). (B) and (C) Semi-quantitative delineation of tumor and tumor-to-background fluorescence intensity. The dashed circle indicates the tumor. | ||
Micro-SPECT imaging and distribution of [177Lu]Lu-DOTA-DZ-HX
Longitudinal SPECT/CT images are presented in Fig. 4. Following the administration of [177Lu]Lu-DOTA-DZ-HX, the tumor was distinctly visible at 24 hours and remained detectable for up to 7 days. For the H1975 xenograft models (Fig. 4A and B), the tumor uptake of the radioligands reached its peak between 24 and 48 hours post-injection, with values recorded at 14.71 ± 0.65% ID per g at 24 hours, 15.23 ± 0.37% ID per g at 48 hours, 11.76 ± 1.41% ID per g at 72 hours, and 7.32 ± 0.89% ID per g at 120 hours. For the HeLa xenograft models (Fig. 4C and D), the tumor uptake of the radioligands peaked at 48 hours post-injection (17.16 ± 4.91% ID per g), which was higher than the H1975 xenograft tumor. The uptake remained relatively sustained over time. A higher background uptake was noted at earlier time points, with radioligands predominantly excreted via the renal pathway. In contrast, [177Lu]Lu-DOTA cleared from the body within 24 hours, underscoring its lack of sustained tumor specificity (Fig. S1, ESI†).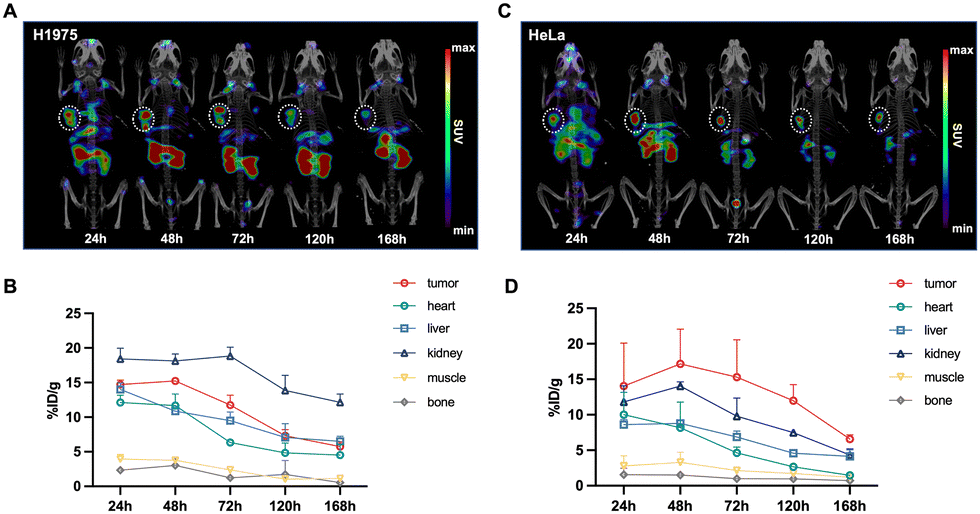 | ||
| Fig. 4 The in vivo targeting properties of [177Lu]Lu-DOTA-DZ-HX. (A) and (B) SPECT imaging of [177Lu]Lu-DOTA-DZ-HX and semi-quantitative delineation of % ID per g for major organs on the H1975 xenograft models (n = 3 per group). (C) and (D) SPECT imaging of [177Lu]Lu-DOTA-DZ-HX and semi-quantitative delineation of % ID per g for major organs on the HeLa xenograft models (n = 3 per group). The dashed circle indicates the tumor. | ||
Fig. 5 shows the organ uptake characteristics of [177Lu]Lu-DOTA-DZ-HX and tumor-to-blood (T/B), tumor-to-muscle (T/M), tumor-to-kidney (T/K), and tumor-to-liver (T/L) ratios at the indicated time points. Detailed information on the biodistribution of H1975 and HeLa xenograft tumors is shown in Tables S4 and S5 (ESI†). [177Lu]Lu-DOTA-DZ-HX exhibited prolonged blood circulation with gradual clearance. For the H1975 xenograft models (Fig. 5A), the uptake value of blood was 36.72% ID per g at 1 h, decreasing to 7.83% ID per g at 48 h and 0.69% ID per g at 96 h. The notable uptake of [177Lu]Lu-DOTA-DZ-HX at an earlier time (4 h and 24 h) was observed for kidneys, lungs, liver, and spleen. The tumor uptake continued to increase over time (5.75 ± 0.30% ID per g at 1 h), reaching a maximum of 13.50 ± 1.40% ID per g at 24 h. [177Lu]Lu-DOTA-DZ-HX exhibited a similar specificity property to HeLa xenograft tumors (Fig. 5C). The tumor uptake continued to increase over time and was significantly higher than the H1975 tumor at 96 h (11.94% ID per g vs. 2.76% ID per g). The observed difference in uptake between the two models may be attributed to the higher expression of HIF-1α and OATP in HeLa, which are associated with dye-seeking in tumors (Fig. 8). Additionally, the T/B and T/M ratios of the drug progressively increased, reaching maximum values of 4.14 and 11.79 for the H1975 tumors (Fig. 5B) and 2.67 and 11.08 for the HeLa tumors (Fig. 5D).
 | ||
| Fig. 5 The in vivo targeting properties of [177Lu]Lu-DOTA-DZ-HX. (A) and (B) Decay-corrected biodistribution data and tumor-to-normal tissue ratios in H1975 xenograft models (n = 4 per group). (C) and (D) Decay-corrected biodistribution data and tumor-to-normal tissue ratio in HeLa xenograft models (n = 4 per group). | ||
In vivo efficacy of [177Lu]Lu-DOTA-DZ-HX
Treatment with different doses of [177Lu]Lu-DOTA-DZ-HX could inhibit H1975 and HeLa xenograft tumor growth (Fig. 6 and 7). Additionally, a decrease in body weight was observed in the high-dose group (37 MBq), which experienced an 18% reduction (Fig. 6B). However, reducing the dose could achieve the same therapeutic effect without causing weight loss (Fig. 6E).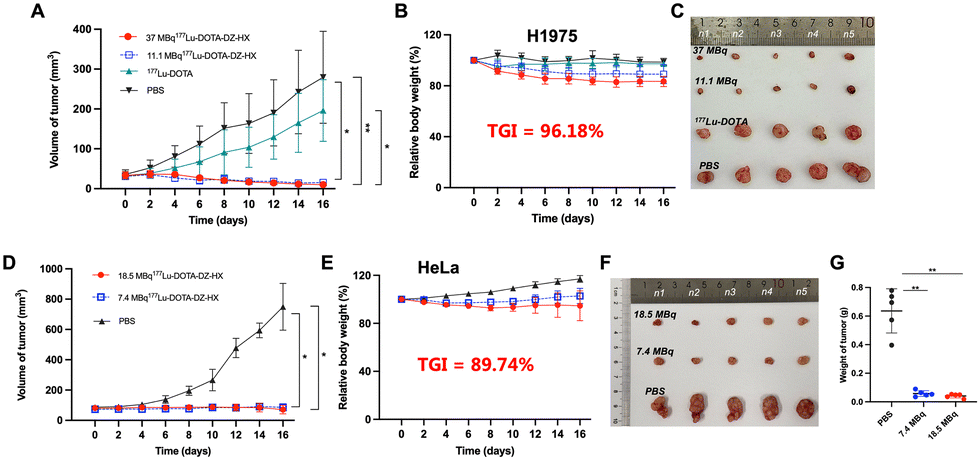 | ||
| Fig. 6 Therapeutic effects of [177Lu]Lu-DOTA-DZ-HX on H1975 lung cancer and HeLa cervical cancer in tumor-bearing mice. (A)–(C) Tumor volume growth curves, relative body weight changes, and representative images of tumor tissues from H1975 xenograft-bearing mice, recorded over a 16-day treatment period (n = 5 per group). (D)–(G) Tumor volume growth curves, relative body weight changes, representative tumor images, and tumor weights from HeLa xenograft-bearing mice, recorded over a 16-day treatment period (n = 5 per group). A statistical comparison between groups was made with the Kruskal–Wallis test, and data are presented as mean ± SD. Only significant differences are presented in the figure. *p < 0.05, **p < 0.01. | ||
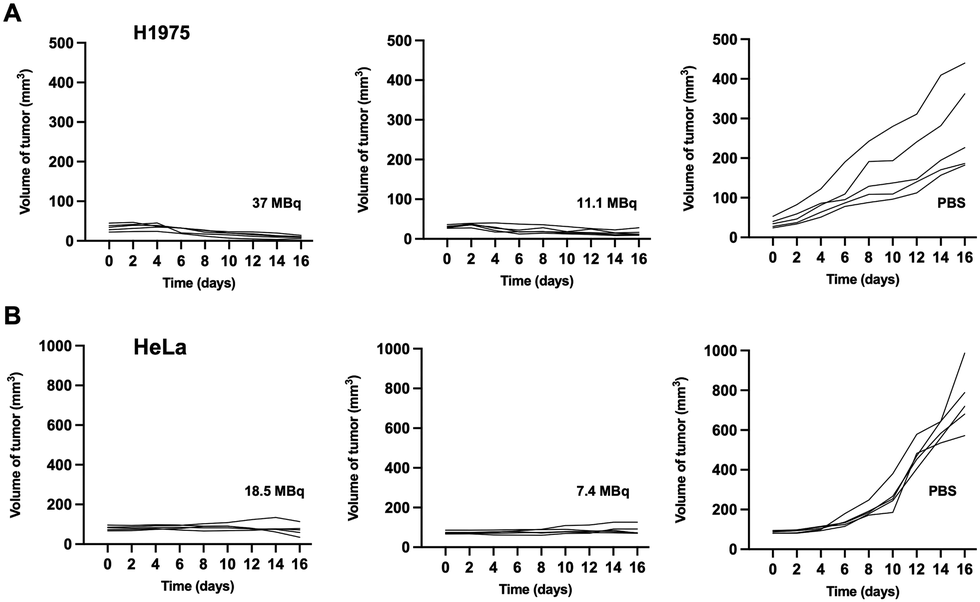 | ||
| Fig. 7 Individual tumor volume at the indicated time point after injecting [177Lu]Lu-DOTA-DZ-HX in two types of xenograft models. (A) The H1975 xenograft tumor volume curves. (B) The HeLa xenograft tumor volume curves (n = 5 per group). | ||
Representative photographs visually showed that the growth of H1975 and HeLa tumors was inhibited across different dose ranges (7.4 MBq to 37 MBq) of [177Lu]Lu-DOTA-DZ-HX, exhibiting comparable levels of suppression (Fig. 6C and F). For the H1975 tumor volume, 37 MBq [177Lu]Lu-DOTA-DZ-HX showed the most potent tumor inhibition at 16 days, which was significantly smaller than the PBS group (10.01 ± 2.75 mm3vs. 279.37 ± 115.53 mm3, P < 0.01), achieving a relative tumor inhibition (TGI) rate of 96.18% (Fig. 6A). 11.1 MBq [177Lu]Lu-DOTA-DZ-HX also showed significant tumor suppression, with a tumor volume of 15.48 ± 7.51 mm3 (Table S6, ESI†). For the more aggressive HeLa tumors, both 7.4 MBq and 18.5 MBq of the [177Lu]Lu-DOTA-DZ-HX group demonstrated marked reductions in the tumor volume and tumor weight, with measurements of 71.52 ± 28.86 mm3 and 0.04 g for the high-dose group, compared to 749.67 ± 154.30 mm3 and 0.64 g in the control group (Fig. 6D and G). Notably, the 18.5 MBq dose of [177Lu]Lu-DOTA-DZ-HX achieved a substantial relative tumor inhibition rate of 89.74% (Table S7, ESI†). In contrast, neither the [177Lu]Lu-DOTA nor the PBS group inhibited tumor growth. Fig. 7 further substantiates these effects through individual tumor volume tracking, revealing consistent growth inhibition across varying doses in both models.
Immunohistochemistry revealed that H1975 and HeLa tumor tissues express high levels of HIF-1α (38.86% in H1975 and 58.87% in HeLa) and OATP2B1 (32.15% in H1975 and 79.38% in HeLa), which are crucial for DZ-mediated tumor targeting. Further analysis using the terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labelling (TUNEL) assay revealed notable differences in apoptosis rates between the experimental and control groups. In the H1975 tumor model, the high-dose group exhibited an apoptosis rate of 60.92%, compared to 18.57% in the low-dose group and 2.02% in the control group. Similarly, in the HeLa tumor model, the high-dose group showed an apoptosis rate of 27.30%, while the low-dose group displayed a rate of 13.28%, with no apoptosis observed in the control group (Fig. 8 and Table S8, ESI†).
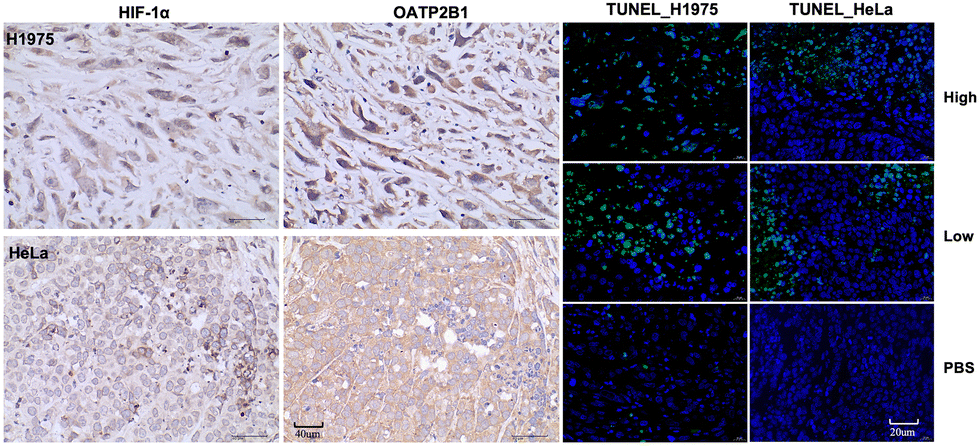 | ||
| Fig. 8 Immunohistochemical analysis for markers HIF-1α, OATP2B1, and TUNEL on H1975 and HeLa tumors. Scale bars: 40 μm for HIF-1α and OATP2B1; 20 μm for TUNEL. | ||
Safety evaluation
The body weight was monitored for all groups in H1975 and HeLa models to evaluate the health conditions of mice (Fig. 6B and E). A gradual drop in the body weight was observed in [177Lu]Lu-DOTA-DZ-HX groups, especially 37 MBq groups, mainly due to the relatively high radiation dose received. The weight loss in the 11.1 and 18.5 MBq [177Lu]Lu-DOTA-DZ-HX groups was slightly better compared to that in 37 MBq [177Lu]Lu-DOTA-DZ-HX (5%, 10% vs. 18%). No weight loss was observed in the 7.4 MBq [177Lu]Lu-DOTA-DZ-HX group, [177Lu]Lu-DOTA only group and the PBS group. Moreover, H&E staining results showed no obvious pathological change in the liver, spleen, and kidneys, and there was no significant difference between the Lutetium treatment groups and control groups (Fig. S2, ESI†). These results indicated that the application of [177Lu]Lu-DOTA-DZ-HX in our study was safe in lung cancer and cervical cancer models.Discussion
Resistance to chemotherapy and targeted therapy in tumor cells is a significant barrier to effective cancer treatment. Current anti-tumor therapies often have limited efficacy and are associated with severe adverse effects. DZ-1, identified as a novel type of HMCD, has emerged as a versatile delivery vehicle for a broad range of anti-tumor therapeutics. Further research has shown that DZ-1 is enriched and retained within subcellular organelles such as lysosomes and mitochondria, enhancing its therapeutic potential. DZ-1-mediated subcellular targeted therapy has effectively inhibited tumor growth and circumvented therapeutic resistance in tumor cells. Our research team has, for the first time, synthesized an HMCD-based RDC to deliver the therapeutic radionuclide 177Lu to tumor cells. This study employs an image-guided platform to conduct a preclinical investigation of the RDC, focusing on its specificity in targeting tumor cells and its effectiveness in inhibiting tumor growth, which have been validated in the H1975 lung and HeLa cervical mouse models.Initial studies showed that the 177Lu-DZ-1 complex, formed through a DOTA chelator and linked via a lysine linker, [177Lu]Lu-DOTA-DZ-Lys, exhibited significant instability issues, impeding its potential therapeutic application. To overcome this issue, we modified the linker by incorporating an aspartic acid residue between DZ and lysine, resulting in the creation of a derivative named DOTA-DZ-HX. This modified compound was subsequently complexed with 177Lu to form [177Lu]Lu-DOTA-DZ-HX.
While DZ-1 conjugated chemotherapy drugs have demonstrated significant anti-tumor efficacy, the high molecular dose required for the chemotherapy agents necessitates a substantial amount of the DZ-1 dye, which introduces a degree of toxicity. For diagnostic and therapeutic applications involving radioactive nuclides and their carriers, the required molecular dose of DZ-1 is minimal, resulting in negligible side effects. Given that DZ-1 conjugated nuclides accumulate in the mitochondria and lysosomes of tumor cells, the radiation energy emitted by the nuclides more effectively damages the organelles of cancer cells, thereby eliminating drug-resistant tumors. Hence, DZ-1 conjugated radioactive nuclides demonstrate significantly improved potential for clinical applications.
Currently, there are no reports on DZ-1 or other HMCD conjugated nuclides being used for tumor treatment. Our results demonstrate that the [177Lu]Lu-DOTA-DZ-HX complex exhibits exceptional stability in serum, effectively enhancing the biostability of the DZ conjugate. The in vitro cancer cell-killing efficacy of [177Lu]Lu-DOTA-DZ-HX in targeting and eradicating H1975 and HeLa cells surpasses that of free [177Lu]LuCl3. SPECT/CT imaging further confirmed the tumor-targeting capabilities of the radiolabeling conjugate. In mice injected with [177Lu]Lu-DOTA-DZ-HX, tumors were distinctly visible, and the radiolabeled compound remained localized within the tumors for up to seven days. Therapeutic evaluations have demonstrated that [177Lu]Lu-DOTA-DZ-HX effectively and selectively eradicates tumor cells in the H1975 and HeLa xenograft tumors. This treatment significantly inhibited tumor growth and induced tumor regression.
The observation that different doses (7.4 MBq to 37 MBq) led to comparable tumor inhibition could be due to several factors. First, [177Lu]Lu-DOTA-DZ-HX may have a sufficiently broad therapeutic window, enabling effective tumor suppression at lower doses. This is consistent with previous studies,31 which have shown that radionuclide-labelled compounds can exert antitumor effects across a range of doses, potentially due to their extended circulation and retention within tumor tissues. Additionally, at the higher dose (37 MBq), we observed an increase in weight loss among treated animals, indicating a degree of dose-dependent toxicity. This toxicity might have moderated some of the therapeutic effects, which could explain the limited additional benefit at higher doses. Lastly, the prolonged tumor retention of DZ-HX may have supported sustained antitumor activity at lower doses, allowing for effective treatment while reducing the risk of toxicity.
These findings underscore the potential of [177Lu]Lu-DOTA-DZ-HX as a potent and selective therapeutic candidate, capable of inducing substantial tumor regression while sparing vital organs from significant damage.
Conclusions
For the first time, we synthesized and characterized the [177Lu]Lu-DOTA-DZ-HX complex, a HMCD-based radionuclide complex. This study demonstrates the complex's high stability and specificity towards H1975 and HeLa cancer cells. The conjugate effectively eliminates cancer cells and significantly suppresses tumor growth. Specifically, the highest dose achieved the greatest tumor suppression, while lower doses maintained comparable efficacy without inducing adverse effects. This research establishes a foundational framework for developing HMCD-based radionuclide complexes aimed at targeted therapy for lung cancer, cervical cancer, and other aggressive malignancies.Author contributions
Mingxing Huang: investigation, methodology, formal analysis, and original draft writing. Weichen Wang: investigation, methodology, formal analysis, and original draft writing. Yingwei Wang: methodology (optimized synthesis of DZ-HX), formal analysis, and original draft editing. Rang Wang: investigation, formal analysis (radiolabeling), and original draft editing. Lili Pan: investigation, methodology (optimized synthesis of DZ-HX), formal analysis, and original draft editing. Xin Li: investigation, methodology (fluorescence imaging), formal analysis, and original draft editing. Bo Li: funding acquisition, resources, and original draft editing. Jingsong Zhang: investigation, resources, and original draft editing. Ruoxiang Wang: investigation, methodology, formal analysis, and original draft writing. Yi Zhang: conceptualization, methodology, supervision, writing and editing the original draft, and original draft – review and editing. Rong Tian: conceptualization, resources, methodology, funding acquisition, supervision, writing and editing the original draft, and original draft – review and editing.Data availability
The data supporting the findings of this study, including experimental details, biodistribution data, and tumor volume measurements, are provided in the ESI.† This study does not involve data from human participants or the use of crystallographic data or analysis code.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 82472022).Notes and references
- K. Nurgali, R. T. Jagoe and R. Abalo, Front. Pharmacol., 2018, 9, 245 CrossRef.
- S. H. Vu, P. Vetrivel, J. Kim and M. S. Lee, Int. J. Mol. Sci., 2022, 23, 18 Search PubMed.
- S. H. Shin, A. M. Bode and Z. Dong, npj Precis. Oncol., 2017, 1, 28 CrossRef PubMed.
- G. Sgouros, L. Bodei, M. R. McDevitt and J. R. Nedrow, Nat. Rev. Drug Discovery, 2020, 19, 589–608 CrossRef CAS PubMed.
- G. Sgouros, Cancer Res., 2019, 79, 5479–5481 CrossRef CAS PubMed.
- N. Pirooznia, K. Abdi, D. Beiki, F. Emami, S. S. Arab, O. Sabzevari and S. Soltani-Gooshkhaneh, Bioorg. Chem., 2020, 102, 104100 CrossRef CAS PubMed.
- S. Pratihar, K. K. Bhagavath and T. Govindaraju, RSC Chem. Biol., 2023, 4, 826–849 RSC.
- J. Lindberg, J. Nilvebrant, P. A. Nygren and F. Lehmann, Molecules, 2021, 26, 19 Search PubMed.
- S. Parakh, S. T. Lee, H. K. Gan and A. M. Scott, Cancers, 2022, 14, 6 CrossRef PubMed.
- K. Zakeri, D. Narayanan, G. Evans, P. Prasanna, J. C. Buchsbaum, B. Vikram and J. Capala, J. Nucl. Med., 2019, 60, 41–49 CrossRef CAS.
- X. Yang, C. Shi, R. Tong, W. Qian, H. E. Zhau, R. Wang, G. Zhu, J. Cheng, V. W. Yang, T. Cheng, M. Henary, L. Strekowski and L. W. K. Chung, Clin. Cancer Res., 2010, 16, 2833–2844 CrossRef CAS PubMed.
- C. Shi, J. B. Wu, G. C. Chu, Q. Li, R. Wang, C. Zhang, Y. Zhang, H. L. Kim, J. Wang, H. E. Zhau, D. Pan and L. W. Chung, Oncotarget, 2014, 5, 10114–10126 CrossRef PubMed.
- X. Yang, C. Shi, R. Tong, W. Qian, H. E. Zhau, R. Wang, G. Zhu, J. Cheng, V. W. Yang, T. Cheng, M. Henary, L. Strekowski and L. W. Chung, Clin. Cancer Res., 2010, 16, 2833–2844 CrossRef CAS PubMed.
- J. Liu, J. Zhang, Y. Zhang, W. Wei, M. Zhan, Z. Zhang, B. Liu, X. Hu and W. He, Biomater. Sci., 2024, 12, 2614–2625 RSC.
- Y. Park, M. H. Park and H. Hyun, Int. J. Mol. Sci., 2024, 25, 10 Search PubMed.
- S. Wangngae, S. Siriwibool, K. Chansaenpak, S. Wet-Osot, R. Y. Lai and A. Kamkaew, ChemMedChem, 2022, 17, e202100780 CrossRef CAS PubMed.
- C. Sun, W. Du, B. Wang, B. Dong and B. Wang, BMC Chem., 2020, 14, 21 CrossRef CAS PubMed.
- A. Raveendran, S. Poilil Surendran, J. Ser, K. Alam, H. Cho and Y. Y. Jeong, Int. J. Nanomed., 2021, 16, 7169–7180 CrossRef CAS.
- C. Zhang, T. Liu, Y. Su, S. Luo, Y. Zhu, X. Tan, S. Fan, L. Zhang, Y. Zhou, T. Cheng and C. Shi, Biomaterials, 2010, 31, 6612–6617 CrossRef CAS PubMed.
- Q. Lv, X. Yang, M. Wang, J. Yang, Z. Qin, Q. Kan, H. Zhang, Y. Wang, D. Wang and Z. He, J. Controlled Release, 2018, 279, 234–242 CrossRef CAS PubMed.
- K. M. Pflug, D. W. Lee, A. Tripathi, V. A. Bankaitis, K. Burgess and R. Sitcheran, Mol. Pharmaceutics, 2023, 20, 6140–6150 CrossRef CAS PubMed.
- E. Cooper, P. J. Choi, W. A. Denny, J. Jose, M. Dragunow and T. I. Park, Front. Oncol., 2021, 11, 654921 CrossRef CAS PubMed.
- X. Yang, C. Shao, R. Wang, C. Y. Chu, P. Hu, V. Master, A. O. Osunkoya, H. L. Kim, H. E. Zhau and L. W. K. Chung, J. Urol., 2013, 189, 702–710 CrossRef CAS PubMed.
- N. Zhao, C. Zhang, Y. Zhao, B. Bai, J. An, H. Zhang, J. B. Wu and C. Shi, Oncotarget, 2016, 7, 57277–57289 CrossRef PubMed.
- P. J. Choi, T. I. H. Park, E. Cooper, M. Dragunow, W. A. Denny and J. Jose, Bioconjugate Chem., 2020, 31, 1724–1739 CrossRef CAS PubMed.
- L. Xiao, Y. Zhang, W. Yue, X. Xie, J.-P. Wang, M. D. Chordia, L. W. K. Chung and D. Pan, Nucl. Med. Biol., 2013, 40, 351–360 CrossRef CAS.
- J. Yuan, X. Yi, F. Yan, F. Wang, W. Qin, G. Wu, X. Yang, C. Shao and L. W. K. Chung, Mol. Med. Rep., 2015, 11, 821–828 CrossRef CAS PubMed.
- C. Zhang, Y. Zhao, H. Zhang, X. Chen, N. Zhao, D. Tan, H. Zhang and C. Shi, Int. J. Mol. Sci., 2017, 18, 6 Search PubMed.
- C. Zhang, Y. Zhao, N. Zhao, D. Tan, H. Zhang, X. Chen, H. Zhang, J. An, C. Shi and M. Li, Contrast Media Mol. Imaging, 2018, 2018, 4979746 Search PubMed.
- Y. Guan, Y. Zhang, L. Xiao, J. Li, J. P. Wang, M. D. Chordia, Z. Q. Liu, L. W. Chung, W. Yue and D. Pan, Mol. Pharmaceutics, 2017, 14, 1–13 CrossRef CAS PubMed.
- M. Verhoeven, J. Haeck, E. de Blois, F. Orlandi, D. Barbato, M. Tedesco, M. Konijnenberg and S. U. Dalm, Mol. Imaging Biol., 2024, 26, 114–123 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01798f |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |