
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and evaluation of dansyl-derived chemosensors for disulfide-cleavage-triggered detection: photophysical, metal sensing, and thermometric applications†
Igor
Lourenço‡
a,
Frederico
Duarte‡
a,
Georgi M.
Dobrikov
bc,
Atanas
Kurutos
*bd,
Ivaylo
Slavchev
b,
José Luis
Capelo-Martínez
ae,
Hugo M.
Santos
ae and
Carlos
Lodeiro
 *ae
*ae
aBIOSCOPE Research Group, LAQV-REQUIMTE, Chemistry Department, NOVA School of Science and Technology, FCT NOVA, Universidade NOVA de Lisboa, 2829-516 Caparica, Portugal. E-mail: cle@fct.unl.pt
bInstitute of Organic Chemistry with Centre of Phytochemistry, Bulgarian Academy of Sciences, Acad. G. Bonchev str., bl. 9, 1113 Sofia, Bulgaria. E-mail: Atanas.Kurutos@orgchm.bas.bg
cNational Centre of Excellence Mechatronics and Clean Technologies, Sofia, Bulgaria
dUniversity of Chemical Technology and Metallurgy, 8 St. Kliment Ohridski blvd, 1756 Sofia, Bulgaria
ePROTEOMASS Scientific Society, 2825-466 Costa de Caparica, Portugal
First published on 10th February 2025
Abstract
In this work, two new dansyl-derived chemosensors have been designed with the aim of developing a disulfide-cleavage-triggered probe. These stable new ligands, containing a thioether (L1) and a disulfide bridge (L2), have been synthesized and evaluated for their photophysical properties in both solution and the solid state. Different fluorescence emission responses have been observed for both compounds due to the self-quenching nature of the disulfide bond in L2 compared to L1, with the latter reaching quantum yields close to 50%. Efforts to characterize solute–solvent interactions through Kamlet–Taft studies have revealed a positive solvatofluorochromic response for both compounds. Furthermore, the aggregation-induced emission phenomenon was demonstrated at increasing water fractions. The presence of free thiol groups enhances the complexation ability, making L1 particularly beneficial for sensing Cu2+ and Hg2+ ions, achieving detection limits as low as 2 μM in the case of mercury. Finally, incorporating L1 into polymer-doped films demonstrated promising results as molecular thermometers, while the response of L2 to sodium dithionite underscores its potential for disulfide-cleavage-triggered applications.
1. Introduction
The design of new sensors for metals and anions has been a topical area of chemistry in recent years. Contamination of water (including water in biological objects) with heavy metals is continuously increasing. Nonetheless, universally applicable and entirely effective sensors for their detection and removal are not available. It is well known that most heavy metals (particularly Hg, Cu, Pb and Cd) possess high affinity to organic molecules containing sulfur, due to the formation of a strong donor–acceptor bond with this nonmetal element. Furthermore, the design of organosulfur fluorescent compounds may provide unique chemical properties that enable the interaction of particular substrates in biological systems. Some examples of biologically active substances include the aforementioned metal ions, reactive sulfur, oxygen and nitrogen species (RSS, ROS, and RNS) and biomacromolecules.1 Among the common classes of organosulfur compounds, thioether and disulfide bond-containing probes may possess a complementary framework since efforts have been made to develop disulfide-cleavage-activated probes. This comes from the fact that disulfide bonds present strong reactivity or biological activity through the reductive disulfide cleavage reaction producing two thiols, or the photocleavage reaction via a radical mechanism.2 Several studies have been published on the basis of the presence of this bond for the sensing of biologically relevant molecules and metal ions due to the often observed self-quenching effects through fluorescence resonance energy transfer or photoinduced electron transfer prior cleavage.3–9Over the last few decades, few studies have explored sulfur containing ligands, possessing a dansyl moiety as a source of fluorescence. Many of them were used for different applications such as protein labeling,10,11 immobilization onto polymers,12 enzyme detection,13 cell imaging14 and metal ion detection.15,16 In addition, the presence of a sulfonamide functionality can significantly improve the characteristics of such organic ligands17 (especially their ability for complexation).
Our previous expertise in the synthesis of sulfur containing dansyl ligands for the detection of heavy metals reveals that the incorporation of sulfur into aromatic rings leads to less efficient ligands, whereas the best ligands contain sulfur, bound to aliphatic carbon chains.18,19 Conversely, the existence of free thiol groups in the ligands provides optimal complexation capability; nevertheless, such compounds are often unstable and very susceptible to oxidation, even from atmospheric exposure.
In this work, we provide two novel stable ligands (Scheme 1, L1 and L2), including thio- and dithio- bridges connecting two identical dansyl-substituted aromatic groups. The chemical structures of these ligands were designed in order to ensure strong chelation of heavy metals by the introduction of sulfur atoms and sulfonamide functionalities. Furthermore, efforts have been made to highlight the photophysical contrast of both compounds towards the sensing of sodium dithionite or sodium hydrosulfite, Na2S2O4, which is a strong reductant and broadly used in several industries as a bleaching agent, however classified as a highly toxic agent.20–25 To the best of our knowledge, very few studies have been published using dansyl moieties for the sensing of sodium dithionite with early reports from Nicholas Rattray et al. where a sterically constrained bisdansyl chromophore would undergo fluorescence changes after reduction of the azo-bond locking group, and the detection of azo-reductase expressing bacteria was also evaluated.26 In contrast, Xingxing Cao et al. were able to sense sodium dithionite by simultaneous immobilization in silicon nanowires of a dansyl group and dabsyl group, the latter would act as a quencher and recognizing group due the strong absorption at 470 nm overlapping with the dansyl group's emission. This system was found to exhibit high selectivity and good linearity between emission intensities and dithionite concentrations.27
 | ||
| Scheme 1 Synthetic approach to the preparation of dansyl derivatives L1 and L2. | ||
Finally, the aggregation-induced emission (AIE) behaviour with increasing water fractions and the incorporation of L1 into polymeric thin films towards temperature-dependent emission studies have been elucidated.
2. Results and discussion
2.1. Synthesis
As in our recent study,18 the target compounds L1 and L2 were synthesised in dry pyridine via the reaction of dansyl chloride (1) with commercially available amines 2 and 3, respectively (Scheme 1). Since 3 is presumably sensitive to oxidation, synthesis of L2 was carried out at room temperature. Both L1 and L2 were obtained in pure form and good yields after column chromatography. They were unambiguously characterized by using different 1D and 2D NMR experimental techniques, melting point temperatures and HRMS.2.2. Photophysical characterization
The bisdansylated compounds L1 and L2 bearing single sulfur and disulfide bonds were studied according to their photophysical properties both in solution and in the solid state. Fig. 1 displays the chemical structure with the photoluminescence data in acetonitrile with the remaining solvents shown in Fig. S3 and S4 (ESI†) while a summary of all the data is present in Table 1.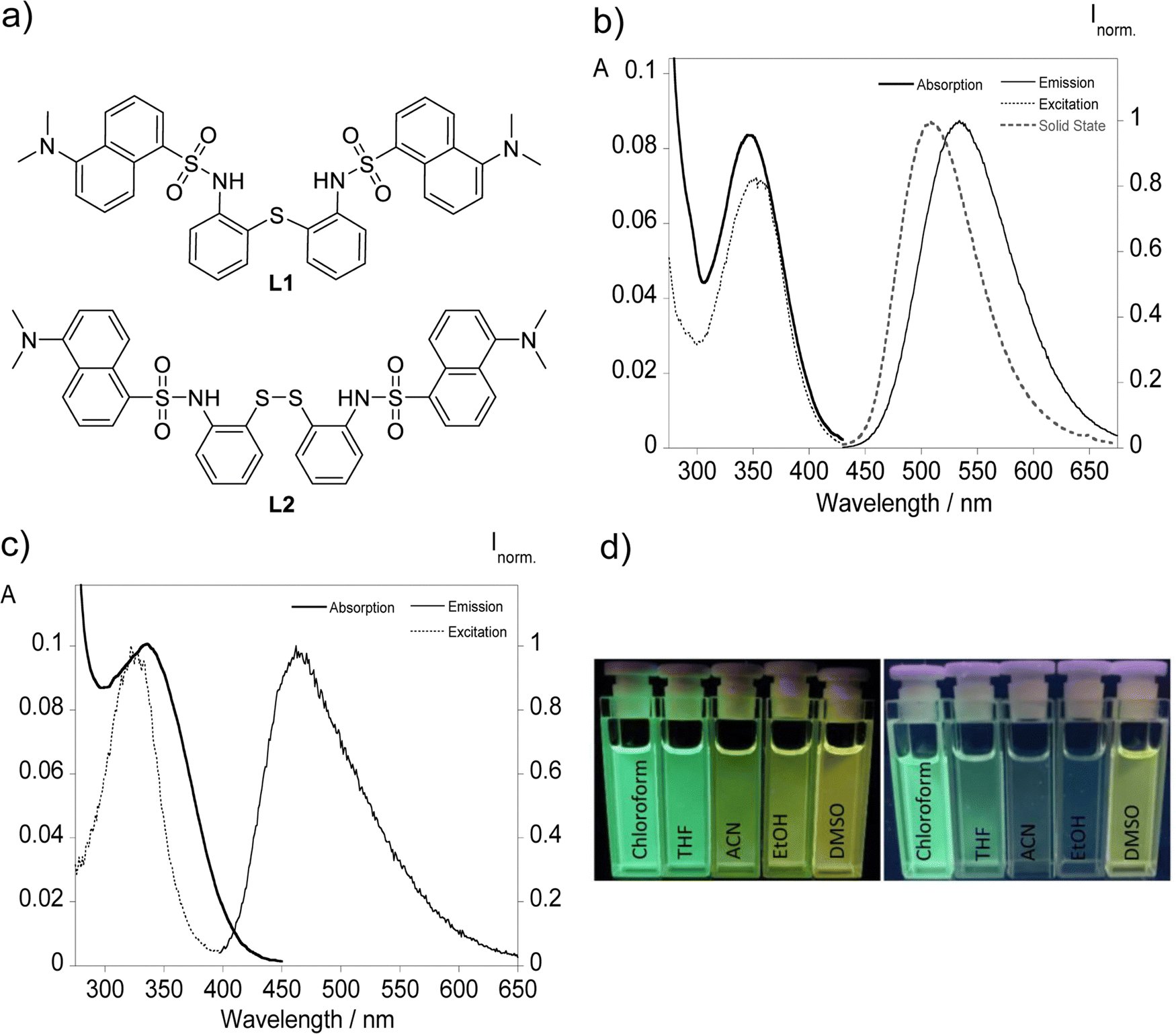 | ||
| Fig. 1 (a) Molecular structure of the dansyl derived compounds L1 and L2 bearing single sulfur and disulfide bridges. Photophysical characterization of derivatives L1 (b) and L2 (c) in acetonitrile ([L1] = [L2] = 10 μM). Images under UV light (365 nm) of (d) L1 (left) and L2 (right) in different solvents with varying polarity. | ||
| Cpd. | Solv. | λ abs [nm] | λ em [nm] | ε (104) [cm−1 M−1] | Stokes shift [cm−1] | Φ (%) | λ em Solid [nm] | τ [ns] |
|---|---|---|---|---|---|---|---|---|
| L1 | DMSO | 345 | 545 | 0.76 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 637 637 |
17.7 | 508 | 12.6 |
| EtOH | 340 | 525 | 0.81 | 10364 |
21.2 | 11.4 | ||
| CH3CN | 345 | 535 | 0.86 | 10294 |
18.8 | 11.7 | ||
| THF | 343 | 511 | 0.88 | 9804 | 41.1 | 13.7 | ||
| CHCl3 | 350 | 510 | 1.01 | 8964 | 48.3 | 15.5 | ||
| L2 | DMSO | 340 | 529 | 0.91 | 10508 |
4.6 | — | 4.8 |
| EtOH | 335 | 508 | 0.98 | 10166 |
2.9 | 7.3 | ||
| CH3CN | 340 | 464 | 0.98 | 8299 | 2.5 | 10.6 | ||
| THF | 330 | 500 | 1.24 | 10303 |
3.5 | 12.5 | ||
| CHCl3 | 333 | 487 | 1.21 | 9496 | 3.6 | 12.9 |
The UV-vis spectral data of both compounds reveal maxima centered at ca. 345 and 335 nm corresponding to the π–π* transitions of the dansyl chromophore supporting the observation of colorless solutions with the naked eye. Clear differences rise upon proper excitation of the samples with the visualization of a greenish light with the emission maximum centered at 535 for L1 and a faint bluish green emission characterized by a maximum around 464 nm observed for L2. This behaviour is present across all solvents used, and it can be visually identified in Fig. 1d with the reason behind it being the presence of each type of sulfur bond that links both chromophores. Disulfide bonds when incorporated in fluorescent molecules are often associated with the quenching of the emission by promoting photoinduced electron transfer (PeT) or Förster resonance energy transfer (FRET).3–9 This divergence observed between both ligands presents a key advantage that will be explored further ahead. On a similar subject, solid state emission is only present for L1 with a maximum around 508 nm that is blue-shifted compared with in solution resembling the emission profile of chloroform and THF.
The fluorescence quantum yields of the two compounds were also evaluated, using dansylamide as a standard for compounds L1 and L2, with compound L1 reaching a quantum yield of 0.48 in chloroform, and for compound L2 the expected lower values were obtained across all solvents with fluorescence efficiency observed below 5%. Time-correlated single photon counting suggests double digit nanosecond times among all solvents between 11.4 and 15.5 ns for L1 while apolar solvents provide higher fluorescence lifetimes.
The presence of a solvent environment notably influences the stabilization of the ligand's excited state, primarily due to changes in the fluorophore's dipole moment. Considering the data gathered in Table 1, a correlation between the increase in solvent polarity and the energy gap, translating to a greater Stokes shift, ultimately resulting in a bathochromic shift can be inferred. This shift can be attributed to a positive solvatofluorochromic effect, where the environment-sensitive dansyl derivatives adjust their fluorescence maximum wavelengths in response to solvent polarity, a behavior clearly illustrated in Fig. 2.
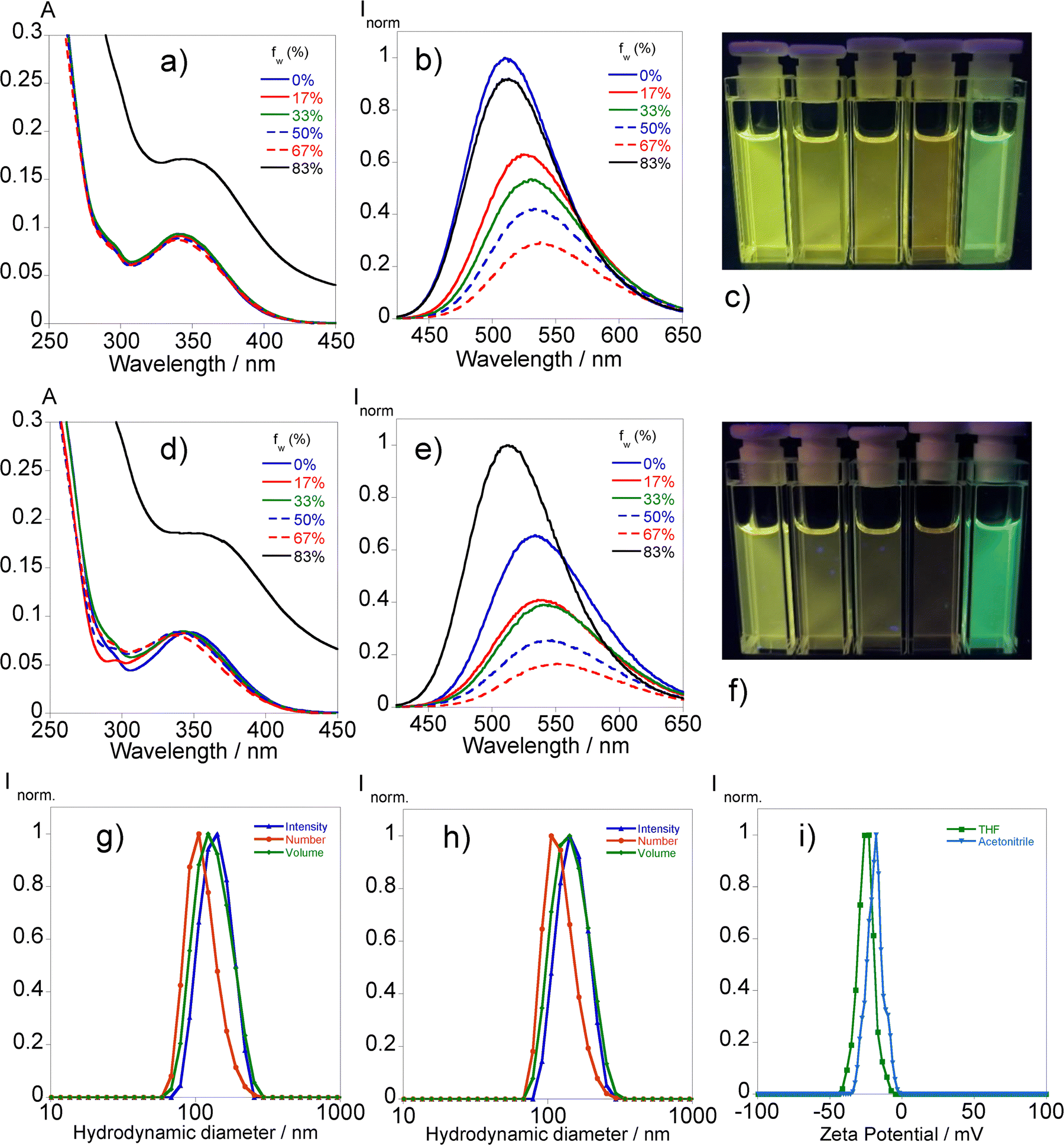 | ||
| Fig. 2 Absorption ((a) and (d)) and emission ((b) and (e)) spectra of L1 in THF ((a) and (b)) and in acetonitrile ((d) and (e)) with different water fractions fw (0%, 17%, 33%, 55%, 67%, and 83%), [L1]= 10 μM. Images of L1 in (c) THF and (f) acetonitrile with different water fractions under UV light (365 nm). The hydrodynamic diameter of L1 aggregates in THF (g) and acetonitrile (h) according to the number, volume, and intensity distributions and zeta potential (i) of the solutions of 83% water fractions. | ||
In response to these results, significant efforts have been focused on thoroughly describing the interactions between the solvent and the molecules.
To this end, three solute-dependent parameters (υ0, a, b, and p) have been determined via multiparametric fitting using the Kamlet–Taft equation (eqn (1)).
| υ = υ0 + aα + bβ + pπ* | (1) |
Table 2 summarizes the reference wavenumber value υ0, the fitted parameters (a, b and p) with the slopes and correlation coefficients attained through the fitting of a linear plot of υexp.versus υcalc, by applying the Kamler–Taft model excluding acetonitrile.
| υ 0 | a | b | p | Slope | R 2 | |
|---|---|---|---|---|---|---|
| L1 | 21492 |
−509 | −911 | −2451 | 1.00 | 1 |
| L2 | 21943 |
−65 | −1652 | −1784 | 1.00 | 1 |
Key distinctions can be elucidated regarding the predisposition of each compound towards the characteristics of different solvents. The fitted parameters reveal low hydrogen bond donor sensitivity with extended negative values for L1, while L2 is more prone to stabilization by solvents with more hydrogen bond acceptor character due to the presence of an extra electronegative atom that forms a disulfide bridge. Additionally, highly polarizable solvents can stabilize the excited state of L1 more effectively, as the more negative p value suggests higher polarizability, leading to stronger solvation effects.
2.3. Aggregation-induced emission behaviour in water
The assessment of the photophysical properties of L1 with different ratios of water in THF and acetonitrile mixtures has been conducted via absorption and fluorescence spectroscopy.Fig. 2 illustrates that the augmentation of the water fraction results in a reduction in fluorescence in both solvents, accompanied by a little red shift in emission until reaching a 67% water content.
The absorption profile changes significantly in the mixture of 83% water content with the rise of the baseline indicating the presence of aggregates. This phenomenon translates into substantial changes in the emission profile by the sudden increase in the fluorescence intensity followed by a hypsochromic shift suggesting an aggregation-induced emission (AIE) effect. In the case of THF, the emission intensity surpasses the initial one while in acetonitrile it is equalized with the associated fluorescence quantum yields of 12% and 8%, respectively. The heterogeneity observed in the absorbance measurements led us to further characterize the presence of aggregates through dynamic light scattering analysis. The results, presented in Fig. 2, suggest hydrodynamic sizes of 142.5 ± 7.9 nm (THF/water) and 146.0 ± 2.5 nm (acetonitrile/water) with associated polydispersity index values of 0.037 ± 0.07 and 0.045 ± 0.01, respectively.
2.4. Metal ion sensing
Dansyl derivatives have shown promise as efficient probes for the detection of metal ions, specifically evaluating the efficacy of compound L1 was carried out, since L2 showed negligible emission from the start. For this reason, several metal ions including Co2+, Cu+, Cu2+, Zn2+, Ag+, Cd2+, Hg+, and Hg2+ in acetonitrile and Pb2+ in water were tested. Fig. 3 shows a consolidation of the changes in the emission intensity with the addition of 0.5, 1, and 5 equivalents of the metal ions. Notably, the absorption and emission spectra showed significant variations exclusively in the presence of mercury(II) and copper(II) ions. The introduction of a single equivalent of each metal ion significantly diminished the emission signal. Furthermore, the fluorescence intensity was completely quenched upon the addition of 5 equivalents. | ||
| Fig. 3 Maximum emission intensities of L1 (a) after the addition of 0.5, 1, and 5 equivalents of Co2+, Cu+, Cu2+, Zn2+, Ag+, Cd2+, Hg+, Hg2+ and Pb2+ metal ions in acetonitrile. Spectrophotometric and spectrofluorometric titrations of compound L1 with increased additions of Cu2+ ([L1] = 16.8 μM) (b) and Hg2+ ([L1] = 20 μM) (c) in acetonitrile. The insets present the emission as a function of [Cu2+]/[L1] at 534 nm (b) and [Hg2+]/[L1] at 534 nm (c). | ||
This signifies that both compounds exhibit high selectivity for these particular metal ions.
The efficacy of compound L1 in detecting Cu2+ and Hg2+ metal ions was promising, prompting titrations with these metals in acetonitrile. Fig. 3 shows the spectrophotometric and spectrofluorometric titrations towards both metals exhibiting similar absorption spectral behaviour with a decrease in absorption at 250 nm and 347 nm and an increase in the absorbance at 289 nm. As for the emission spectra, a suppression of the emission intensity was observed at 534 nm. Fig. 3 also shows the fluctuation of the emission intensity maxima of compound L1 with the increase of the metal concentration in solution. The suppression of the emission caused by Hg2+ comes from the fact that, as a heavy atom, it has photophysical properties such as spin–orbit coupling, making non-radiative pathways compete with fluorescence and therefore having a mechanism for quenching the emission after chelation (CHEQ). The Cu2+ ion is also commonly detected by CHEQ due to its paramagnetic nature by having unpaired electrons in the d-layer, which occurs through either electron transfer or energy transfer mechanisms. However, once filled Cu+ becomes a CHEF system, which is why Cu+ does not quench the emission intensity of compound L1. This system can recognize either Cu2+ from Cu+ or Hg2+ from Hg+, being a good example for metal speciation.
As a favorable characteristic of a molecular probe, the binding reversibility of L1 with Hg2+ and Cu2+ was evaluated against a competitive ligand, i.e., Na2EDTA. As shown in Fig. S8 (ESI†), the addition of 2.2 equivalents of Cu2+ leads to fluorescence quenching of L1 that showed no significant recovery of the emission intensity following the addition of equimolar Na2EDTA, indicating that the complex formed is not reversible in the presence of this competitive ligand. In contrast, the addition of 1.0 equivalents of Hg2+ also causes fluorescence quenching; however, the emission intensity is recovered with equimolar Na2EDTA in solution, suggesting the regeneration of free L1. These reversibility studies were performed 5 times (Fig. S6, ESI†), providing the results to support the capacity for L1 to be a reversible fluorescent probe for the detection of Hg2+ but not for Cu2+. Furthermore, the formation of the complexes has been followed by ESI-HRMS suggesting the stoichiometric proportion of 1:1 as it can be seen in Fig. S7 and S8 (ESI†). (L1 + Cu2+): [M + H]+ for C36H34CuN4O4S3 = 746.1111 m/z (−0.01 ppm). Calculated [M + H]+ for C36H34CuN4O4S3 = 746.111093 m/z. (L1 + Hg2+): [M + H]+ for C36H34HgN4O4S3 = 885.1519 m/z (−0.2 ppm). Calculated [M + H]+ for C36H34HgN4O4S3 = 885.152138 m/z.
To thoroughly evaluate the sensing capabilities for Hg2+ and Cu2+ ions, the interaction constants of all complexes were calculated using HypSpec software.30 Additionally, the detection and quantification limit parameters for both compounds in relation to Hg2+ and Cu2+ were determined. The association constants, along with the LOD (limit of detection) and LOQ (limit of quantification), are provided in Table 3.
| Compounds | Metal (M) | Association constants (logKass), L:M |
LOD (μM) | LOQ (μM) |
|---|---|---|---|---|
| L1 | Cu2+ | 5.73 ± 0.01 | 4 | 8 |
| Hg2+ | 5.80 ± 0.02 | 2 | 4 |
The stability constant values for L1 towards both metal ions evidence similar values in a stoichiometric proportion of 1:1 ligand to metal with a slightly higher value in the case of Hg2+ (logKass. = 5.80 ± 0.02).
The detection and quantification limits have also been determined, reaching values as low as 2 and 4 μM for Hg2+, respectively. These values indicate that L1 could be highly useful in applications such as environmental monitoring, where it is crucial to detect and measure Hg2+ at low concentrations.
2.5. Sensing of sodium dithionite
The potential use of L2 in the induced turn-on sensing of sodium dithionite by disulfide bond cleavage has been evaluated in light of the duality observed in the photophysical profile of both compounds, which is a result of the self-quenching characteristic of disulfide systems. Fig. 4 shows the UV-vis and the fluorescence response in acetonitrile upon consecutive additions of an aqueous solution of sodium dithionite up to 9 equivalents alongside the visual representation of the turn-on effect after the addition of 5 equivalents, while the batch-based sensing can be seen in Fig. S9 (ESI†).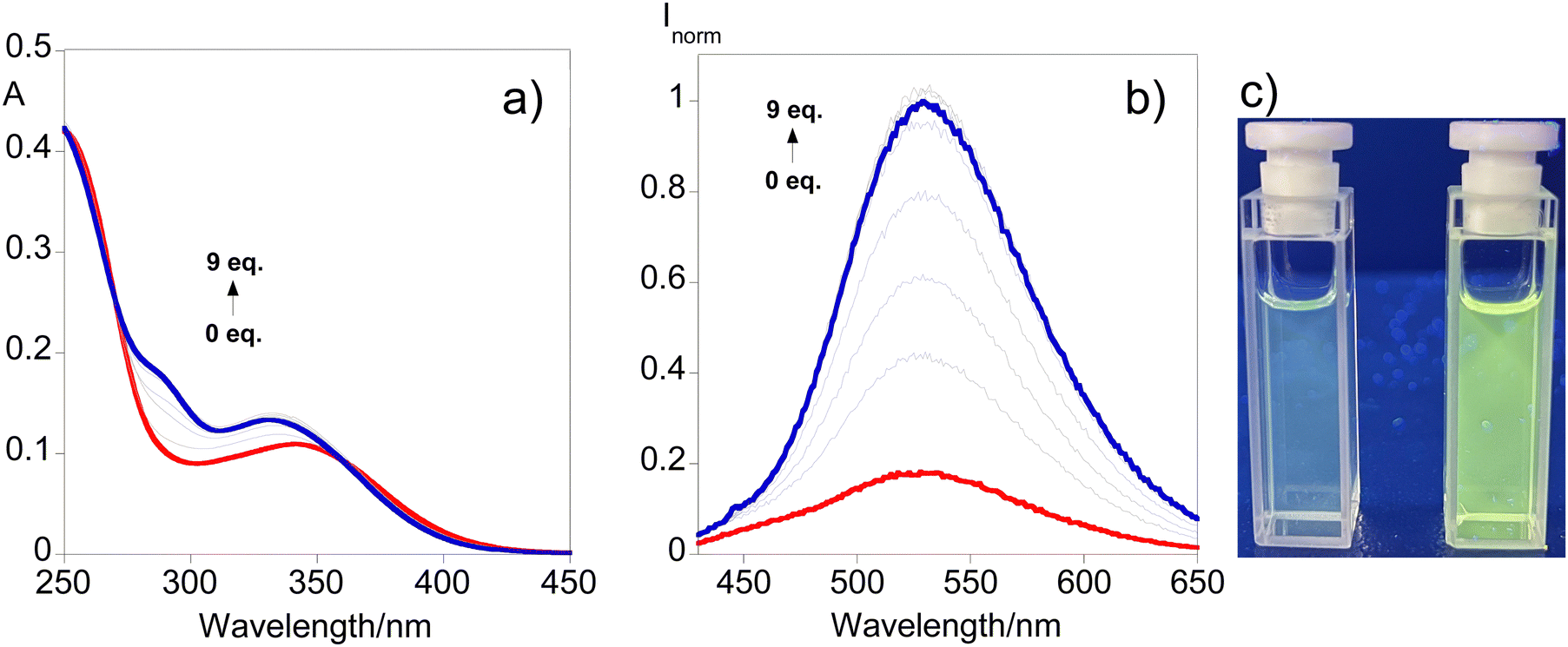 | ||
| Fig. 4 UV-vis (a) and emission (b) spectral responses after the addition of up to 9 equivalents of sodium dithionite to L2 solution in acetonitrile. The image under UV-light (365 nm) (c) of the initial solution (left) and after the addition of 5 equivalents (right). | ||
Fig. 4 reveals that as the concentration of dithionite increases, a progressive blue shift of the absorption profile is noticed with the emergence of a shoulder around 290 nm. Regarding the fluorescence response, the results show that the emission band observed initially successively increases with the addition of dithionite indicating the cleavage of the disulfide bond. Also, ESI-HRMS adds support to this suggestion due to the formation of the monomer moiety depicted in Fig. S10 (ESI†). ([M + H]+ for C18H18N2O2S2 = 359.0882 m/z (−0.1 ppm) and calculated [M + H]+ for C18H18N2O2S2 = 359.088246 m/z.) This behavior can be perceived through Fig. 4c where a turn-on sensing mechanism is observed highlighting the usefulness of compound L2 for disulfide-cleavage-activated applications.
Although a clear contribution of compound L2 towards the development of new disulfide-based chemosensors has been established, some considerations might become useful for the improvement of the future design of these types of compounds. We propose that improving the selectivity of the sensor molecule must be considered since these compounds may interact with multiple analytes. Some examples include the soft character of sulfur atoms that can be easily combined with Hg2+ as an example of a soft metal according to the hard–soft–acid–base theory or the implications of the reducing environment when applying for a specific reactive species in a biological system. This can be avoided through the design of a multiple lock design method to counteract false positives. Considerations about water solubility should be fulfilled alongside biocompatibility by implementing changes in their molecular structure. Finally, a switch-on or clear-cut ratiometric fluorescence mechanism may provide a comfortable level of certainty regarding the physical property changes in these systems.1,2
2.6. NMR titrations of the free ligands
The full structural elucidation of the free ligands L1 and L2 was achieved with the help of 1D and 2D NMR experiments carried out in various solvents including CDCl3 (Fig. SX1–SX20 for L1 and Fig. SX59–SX82 for L2, ESI†), CD3CN (Fig. SX21–SX40 for L1 and Fig. SX83–SX98 for L2, ESI†), and DMSO-d6 (Fig. SX41–SX58 for L1 and Fig. SX99–SX115 for L2, ESI†). In addition to acetonitrile-d3, deuterated DMSO was also employed as an alternative bearing in mind the relatively poor solubility of the salts (mercury(II) acetate, copper(II) triflate, and sodium dithionite) in CD3CN. For all experiments we used 1 equiv. of each salt as the maximum amount in order to obtain clear NMR working solutions of suitable concentration.The experiment of L1 with mercury(II) acetate in acetonitrile-d3 (Fig. SX145–SX171, ESI†) revealed almost no visible change with respect to the chemical shifts in the 1H- and the 13C-NMR spectra. The only notable difference involves broadening of the NH signal (Fig. SX164 and SX165, ESI†), which nevertheless retained the integral for a total of 2 protons. Changing the media to DMSO-d6, the 1H-NMR spectra of L1 with Hg2+ resulted in rather noisy spectra (including a broad baseline) with side bands around the main signals, the appearance of two new multiplets, namely a triplet a doublet of doublets (with a small JH–H coupling constant) at 6.30 ppm, along with a new doublet at about 8.69 ppm (Fig. SX192 and SX193, ESI†). The full identification of the mixture is hampered taking into consideration the broad signals and baseline, which remain broad and noisy even after 29 days. Despite the non-emissive nature of L2 preventing the evaluation of the sensing capabilities through fluorescence techniques, attempts to record spectra of L2 in deuterated acetonitrile after the addition of 1 equiv. of Hg2+only showed very broad signals along the whole spectral range once again (Fig. SX200, ESI†). The shape remained unaltered over several days. To shed more light, we attempted to perform the same measurement in DMSO-d6. In the case of ligand L2, changing the solvent revealed very pronounced spectral changes, pointing out to the spontaneous formation of a new product (Fig. SX225–SX246, ESI†). Intriguingly, the presence of mercury(II) acetate exhibits more pronounced spectral changes in DMSO-d6 for both L1 and L2. The addition of Hg2+ to L2 resulted in the complete disappearance of the NH proton. The new spectra showed no sign of either an NH, or an SH proton signal. With respect to the 13C-NMR spectra, the most pronounced changes were found for the 2 carbon atoms on the benzene ring, which are attached to the nitrogen and sulfur heteroatoms (Fig. SX245, ESI†). We assume that the S–S bond is most likely cleaved while Hg is allocated between the two heteroatoms.
While keeping DMSO-d6 as the working media, the addition of 1 equiv. of copper(II) triflate to ligand L2 (Fig. SX205–SX224, ESI†) showed no difference in comparison to the pure ligand as seen from the 1H-NMR spectra, where only a minor shift of the majority of the signals is present in the carbon spectra. In the 13C-NMR spectra, the triflate quartet with a JC–F coupling constant of around 320 Hz (123.87 ppm, 120.95 ppm, 117.75 ppm, and 114.20 ppm) was also reported (Fig. SX209, ESI†). The shape of the 1H-NMR spectrum remained the same after 17 days. Therefore, we repeated the same experiment in acetonitrile-d3 (Fig. SX194–SX199, ESI†). Likewise to the data obtained in DMSO-d6, the 13C-NMR in acetonitrile-d3 comprises the triflate quartet with a JC–F coupling constant of around 320 Hz (126.74 ppm, 123.55 ppm, 120.36 ppm, and 117.19 ppm as). The addition of 1 equiv. of copper(II) triflate to ligand L1 (Fig. SX188–SX191, ESI†) leads to only negligible changes (of about 0.02–0.03 ppm towards a stronger field) as seen from the 1H-NMR spectra in DMSO-d6 (Fig. SX188 and 189, ESI†). By analogy, the 13C-NMR exhibits a shift of around 0.6 ppm for all signals (Fig. SX190 and 191, ESI†). Both spectra of the free ligand and after the addition of copper(II) triflate appear to be highly similar accounting for the same number of protons and carbons in the chemical structure of the fluorophore. In turn, working with acetonitrile-d3 for L2 showed somehow more pronounced shifts for both the 1H- and the 13C-NMR spectra (Fig. SX194–SX199, ESI†). In particular several shifts are observed for the whole dansyl moiety (including the dimethylamino group and the aromatic protons), whereas the o-substituted benzene ring remains unaltered. A shift of the signals was present for the N(CH3)2 - from 2.81 ppm to 3.04 ppm when copper(II) triflate is added (Fig. SX194, ESI†). The 13C-spectra also show distinctive changes, even to the carbon atom directly linked to the dimethylamino group, since an approximately 5 ppm shift is evident (Fig. SX190, ESI†). L2 also exhibits the characteristic triflate quartet with a JC–F coupling constant of around 320 Hz (126.74 ppm, 123.56 ppm, 120.37 ppm, and 117.19 ppm). Ligand L2 exhibits very distinctive shifts in acetonitrile-d3 – mainly relative to the dansyl moiety (up to 0.5 ppm). In particular, the singlet corresponding to the methyl protons of N(CH3)2 shifts from 2.78 ppm to 3.41 ppm (Fig. SX194, ESI†). Notable changes are also evident in the 13C-NMR spectra, where the carbon attached to the dimethylamino shifts from 153.09 ppm to 139.51 ppm (Fig. SX197, ESI†). Other dansyl carbon signals also move by about 4–5 ppm. On the other hand, only minor changes were visible for the benzene ring containing the two ortho S and N heteroatoms (Fig. SX198, ESI†).
The addition of 1 equiv. of sodium dithionite to L2 in acetonitrile-d3 (Fig. SX201–SX204, ESI†) furnished the appearance of new proton signals allocated at 9.82 (s), 8.20 (dd), 7.53 (m), 7.44 (dd), 7.38 (dd), 7.29 (m), and 7.05 (td) and a negligible change of the NH shift from 7.82 (s) to 7.86 (s). The kinetics of this experiment are very slow, since even after 30 days we observe a mixture of the free ligand (about 57%) and an additional product (about 43%) – the ratio was determined based on the integration of the 1H-NMR. Due to the low concentration and the very poor solubility of sodium dithionite in acetonitrile, clear HMBC cross peaks of the XH signals were not observed. Despite the relatively low concentration, the NOESY experiment also did not yield any further information. This in turn hampered the full assignment of the newly developed product in deuterated acetonitrile. Next, we attempted to repeat the experiment in deuterated DMSO (Fig. SX247–SX254, ESI†). Despite changing the media, the solubility of the dithionite salt was not notably enhanced. Adding 1 equiv. of sodium dithionite to ligand L2 in DMSO-d6 yielded the broadening of the NH proton at 10.22 ppm (no disappearance – integral is retained), while a new sharp signal was developed around 9.86 ppm (Fig. SX248–SX250, ESI†). Several new signals were developed over time of which the more evident were found centered at 8.24 ppm (dt), 8.18 ppm (dd), 7.60 ppm (m), 7.42 ppm (dt), and 7.40 ppm (t). In parallel, the intensity of several multiplets was diminished, while others were enhanced accompanied by the appearance of overlapping multiplets. Accounting for the relatively clear region around the dimethylamino protons, we accounted for a mixture of at least two main components (ligand + newly formed product) in almost equal quantities (Fig. SX250, ESI†). This slow transformation to about 50% of a new product takes more than 24 days under these experimental conditions. Bearing in mind the strong overlap of the majority of the aromatic protons, full identification of the mixture by means of NMR is currently limited and beyond the scope of our current investigation.
2.7. Temperature-dependent emission of polymer doped films
Since compound L1 has manifested solvatochromism aligned with the fact that it is the only compound exhibiting emission in the solid state, efforts have been put forth for the design of solid supports that would act as molecular thermometers. Bearing this in mind, doped polymer films have been produced following the procedure depicted in the experimental section using PMMA and TPU as polymer matrixes. Fig. 5 shows the emission spectra of L1 in the solid state and doped in the polymer materials together with the images under natural and UV light.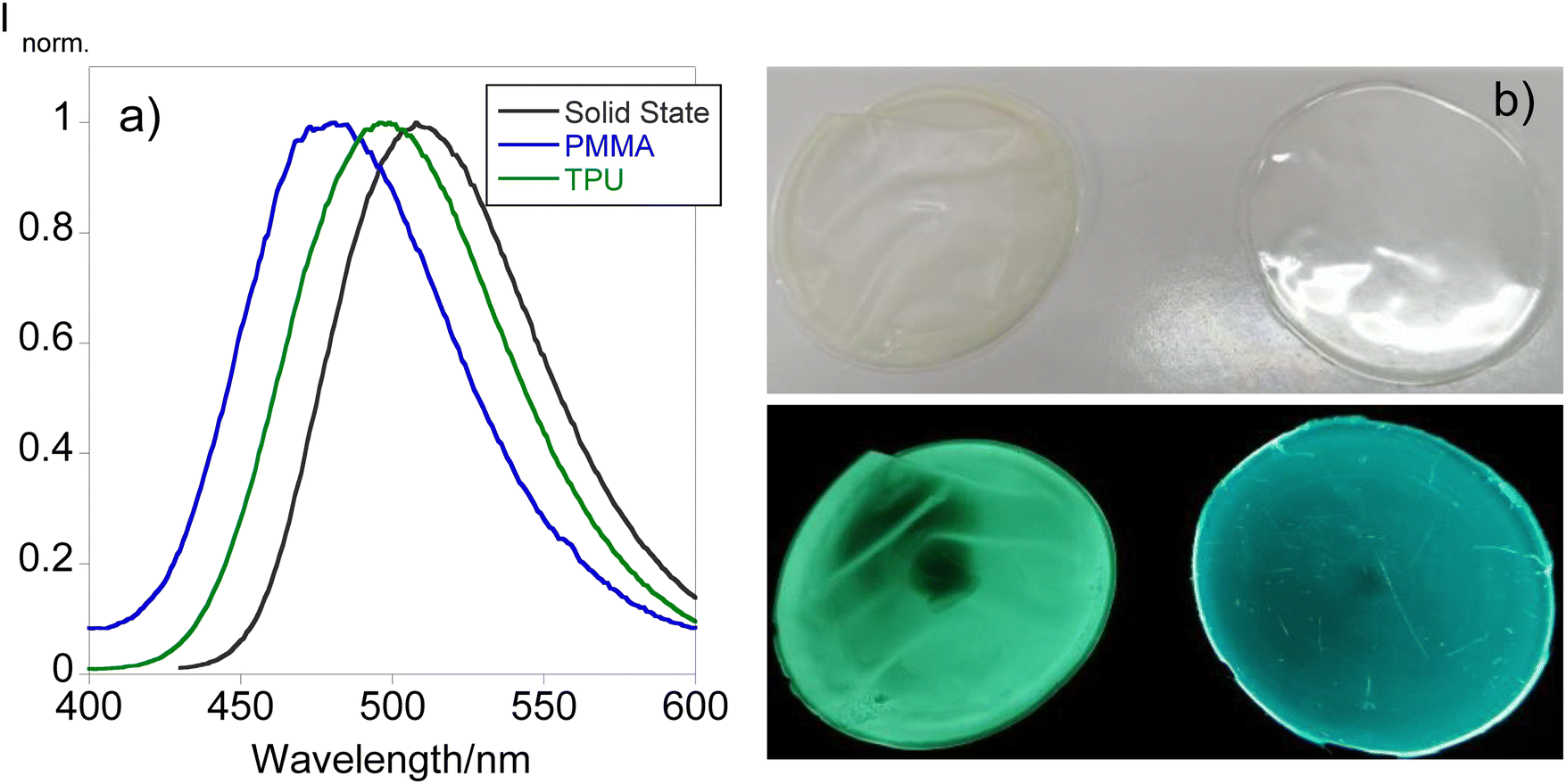 | ||
| Fig. 5 Comparison between the emission spectra of the different polymer thin films doped with (a) L1 and the (b) images under natural (up) and UV light (down) of TPU (left) and PMMA (right). | ||
From Fig. 5, it can be inferred that both polymer matrixes are able to shift the emission of the final material causing a blue shift from 508 in the solid state to 496 and 481 nm in TPU and PMMA, respectively. This phenomenon is also tangible in the images of the polymers found in Fig. 5 since TPU shows a greenish emission while PMMA's emission resembles a more bluish color. These observations have been described in previous studies demonstrating the potential of the use of dansyl compounds towards the development of tunable solid materials for diverse applications.18 Considering these findings, the fabricated polymer doped materials have been tested to be used as molecular thermometers by recording the emission at different temperatures. Fig. 6 presents the temperature-dependent emission spectra of both doped materials.
 | ||
| Fig. 6 Temperature-dependent emission spectra of L1 doped in (a) TPU during the heating cycle [inset: Inorm. vs. T plot at 496 nm upon heating: 32 to 149 °C (Y = 1.1685 − 0.0050481x), yielding R = 0.9902]; (b) TPU during the cooling cycle [inset: Inorm. vs. T plot at 496 nm upon cooling: 30 to 182 °C (Y = 0.10949 − 0.0050184x), yielding R = 0.9933]; (c) PMMA during the heating cycle [inset: Inorm. vs. T plot at 481 nm upon heating: 100 to 220 °C (Y = 1.481 − 0.0062098x), yielding R = 0.9963]; (d) PMMA during the cooling cycle [inset: Inorm. vs. T plot at 481 nm upon cooling: 51 to 227 °C (Y = 1.1278 − 0.0044644x), yielding R = 0.9925]. | ||
A reduction in the fluorescence signal is seen in both polymeric materials as the temperature increases, reaching values of up to 227 °C. Regarding the TPU-doped material, a clear total quenching of the fluorescence signal was achieved at 200 °C. Throughout the increases in temperature over the course of the cycle, linearity has been found between 32 and 149 °C; however, only 10% of the initial emission is restored after cooling suggesting that further increases in temperature may cause physical damage to the polymer preventing reusability of the material.
In the case of PMMA doped with L1, higher temperatures could be achieved in this solid substrate along with a linearity range at higher temperatures between 100 and 200 °C shifting its potential towards industrial applications. During the cooling cycle, linearity was retained as the temperature decreased and in contrast with the TPU substrate, the physical changes enabled the recovery of the emission past the initial one due to the increased rigidity observed.
Thus, these materials provide insights into the design of solid materials that can be tuned in terms of their emission depending on the polymer matrix used, and the linearity of the fluorescence signal sustains the use of these doped polymers as thermometers.
3. Conclusions
Two new dansyl compounds bearing thio- and dithio- functionalities were synthesized and their photophysical characteristics were investigated. Using the Kamlet–Taft equation, the solute–solvent interactions were analyzed in detail, revealing a positive solvatofluorochromic behavior for both compounds. Additionally, the compounds showed enhanced emission in the presence of water, exhibiting aggregation-induced emission (AIE) at a high water content. This AIE effect was further confirmed by dynamic light scattering experiments, which revealed hydrodynamic sizes of 142.5 ± 7.9 nm (THF/water) and 146.0 ± 2.5 nm (acetonitrile/water) for L1. Taking an environmental remediation standpoint, L1 was found sensitive to Hg2+ and Cu2+ ions by modulating their emission profile with the highest association constant towards Hg2+ (logKass. = 5.80) in tandem with the lowest LOD and LOQ values of 2 and 4 μM, respectively. Given that the key photophysical differences were attributed to the presence of disulfide bridges in L2 promoting self-quenching behavior, a proof of concept for sodium dithionite sensing was put forth to highlight the potential use of this compound towards disulfide-cleavage-triggered sensing. Finally, the PMMA and TPU polymer-doped films were fabricated in order to demonstrate the possibility of modulating the emission maximum wavelength in the solid state, and the use of these materials as molecular thermometers has been elucidated.
Author contributions
Igor Lourenço: formal analysis, investigation, writing – original draft, review & editing, and visualization. Frederico Duarte: formal analysis, investigation, writing – original draft, review & editing, and visualization. Georgi Dobrikov: methodology, validation, formal analysis, investigation, writing – original draft, review & editing, resources, and visualization. Atanas Kurutos: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, review & editing, resources, visualization, supervision, and funding acquisition. Ivaylo Slavchev: investigation. Jose Luis Capelo-Martinez: resources, writing – review & editing, and funding acquisition. Hugo M. Santos: investigation, resources, validation, and writing – review & editing. Carlos Lodeiro: conceptualization, methodology, validation, formal analysis, investigation, resources, writing – review & editing, visualization, supervision, funding acquisition, and project administration.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work received support from PT national funds (FCT/MCTES, Fundação para a Ciência e Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) through the projects UIDB/50006/2020 and UIDP/50006/2020. This work received support from the PROTEOMASS Scientific Society through the General Funding Grant 2022-2023, and the projects #PM001/2019 and #PM003/2016. The financial support from the Bulgarian National Science Fund (BNSF) under grant – “Novel styryl and polymethine fluorophores as potential theranostic agents” contract No KP-06-M59/1 from 15.11.2021 is gratefully acknowledged by A. K. This work is also developed and acknowledged by A. K. as part of contract No: BG-RRP-2.004-0002-C01, Laboratory of Organic Functional Materials (Project BiOrgaMCT), Procedure BG-RRP-2.004 “Establishing of a network of research higher education institutions in Bulgaria”, funded by the BULGARIAN NATIONAL RECOVERY AND RESILIENCE PLAN. This work was supported by the Associate Laboratory for Green Chemistry LAQV that is financed by the national funds from Fundação para a Ciência e Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior (FCT/MCTES) through the projects UIDB/50006/2020 and UIDP/50006/2020. The PROTEOMASS Scientific Society (Portugal) is acknowledged for the funding provided through the General Funding Grant 2023-2024 and for the funding provided to the Laboratory for Biological Mass Spectrometry Isabel Moura (#PM001/2019 and #PM003/2016). F. D. thanks the FCT/MCTES (Portugal) for his doctoral grant 2021.05161.BD. H. M. S. acknowledges the Associate Laboratory for Green Chemistry LAQV (LA/P/0008/2020) funded by the FCT/MCTES for his research contract. The financial support by the Bulgarian National Science Fund (BNSF) under grant – “Novel styryl and polymethine fluorophores as potential theranostic agents” contract No KΠ-06-M59/1 from 15.11.2021 is gratefully acknowledged by A. K. This work is also developed and acknowledged by A. K. as part of contract No: BG-RRP-2.004-0002-C01, Laboratory of Organic Functional Materials (Project BiOrgaMCT), Procedure BG-RRP-2.004 “Establishing of a network of research higher education institutions in Bulgaria”, funded by the BULGARIAN NATIONAL RECOVERY AND RESILIENCE PLAN. G. D. acknowledges that this work was supported by the European Regional Development Fund under the “Research Innovation and Digitization for Smart Transformation” program 2021-2027 under the Project BG16RFPR002-1.014-0006 “National Centre of Excellence Mechatronics and Clean Technologies”. Research equipment of this project was used for experimental work.References
- G. Hu, H. Dong Xu and J. Fang, Sulfur-based fluorescent probes for biological analysis: A review, Talanta, 2024, 279, 126515, DOI:10.1016/j.talanta.2024.126515.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Disulfide-Cleavage-Triggered Chemosensors and Their Biological Applications, Chem. Rev., 2013, 113, 5071–5109, DOI:10.1021/cr300358b.
- Y. Zhang, S. Xia, S. Wan, T. E. Steenwinkel, T. Vohs, R. L. Luck, T. Werner and H. Liu, Ratiometric Detection of Glutathione Based on Disulfide Linkage Rupture between a FRET Coumarin Donor and a Rhodamine Acceptor, ChemBioChem, 2021, 22, 2282–2291, DOI:10.1002/cbic.202100108.
- Z. Zheng, Y. Huyan, H. Li, S. Sun and Y. Xu, A lysosome-targetable near infrared fluorescent probe for glutathione sensing and live-cell imaging, Sens. Actuators, B, 2019, 309, 127065, DOI:10.1016/j.snb.2019.127065.
- Z. Wang, R. Jing, Y. Li, D. Song, Y. Wan, N. Fukui, H. Shinokubo, Z. Kuang and A. Xia, Intrinsic Photostability in Dithiolonaphthalimide Achieved by Disulfide Bond-Induced Excited-State Quenching, J. Phys. Chem. Lett., 2023, 14, 8485–8492, DOI:10.1021/acs.jpclett.3c02260.
- G. J. S. Cheng, J. M. Qin, X. Li and Q. Y. Cao, A naphthalimide-based fluorescent probe with mitochondria targeting for GSH sensing and cancer cell recognition, Dyes Pigm., 2023, 211, 111089, DOI:10.1016/j.dyepig.2023.111089.
- J. Mishra, R. Kaur, A. K. Ganguli and N. Kaur, Urea/thiourea based dipodal nanoreceptors: Aqueous medium fluorescent chemosensor for Zn(II) and Hg(II) ions by photoinduced electron transfer, Microchem. J., 2024, 206, 111501, DOI:10.1016/j.microc.2024.111501.
- J. Qiang, Y. Wang, Y. Li, Z. Guo, L. Jiang, F. Wang, S. Lu and X. Chen, A dual-locked near-infrared fluorescent probe based on ESIPT and FRET for improved discrimination between normal and cancer cells, Sens. Actuators, B, 2024, 405, 135344, DOI:10.1016/j.snb.2024.135344.
- A. K. Ghosh, A. H. Khan and P. K. Das, Naphthalimide-Based AIEgens for Sensing Protein Disulfide Isomerase through Thiol−Disulfide Redox Exchange, Anal. Chem., 2023, 95, 13638–13648, DOI:10.1021/acs.analchem.3c02442.
- J. Torres-Kolbus, C. Chou, J. Liu and A. Deiters, Synthesis of Non-linear Protein Dimers through a Genetically Encoded Thiol-ene Reaction, PLoS One, 2014, 9(9), e105467, DOI:10.1371/journal.pone.0105467.
- Y. Li, M. Yang, Y. Huang, X. Song, L. Liu and P. R. Chen, Genetically encoded alkenyl–pyrrolysine analogues for thiol–ene reaction mediated site-specific protein labeling, Chem. Sci., 2012, 3, 2766–2770, 10.1039/C2SC20433A.
- J. Sánchez-Bodón, M. Diaz-Galbarriatu, L. Pérez-Álvarez, J. L. Vilas-Vilela and I. Moreno-Benítez, Expanding (Bio)Conjugation Strategies: Metal-Free Thiol-Yne Photo-Click Reaction for Immobilization onto PLLA Surfaces, Coatings, 2024, 14, 839, DOI:10.3390/coatings14070839.
- H. Kurosaki, H. Yasuzawa, Y. Yamaguchi, W. Jin, Y. Arakawa and M. Goto, Detection of a metallo-lactamase (IMP-1) by fluorescent probes having dansyl and thiol groups, Org. Biomol. Chem., 2003, 1, 17–20, 10.1039/b209086d.
- J. Youziel, A. R. Akhbar, Q. Aziz, M. E. B. Smith, S. Caddick, A. Tinker and J. R. Baker, Bromo- and thiomaleimides as a new class of thiol-mediated fluorescence ‘turn-on’ reagents, Org. Biomol. Chem., 2014, 12, 557–560, 10.1039/c3ob42141d.
- V. Tharmaraj and K. Pitchumani, A highly selective ratiometric fluorescent chemosensor for Cu(II) based on dansyl-functionalized thiol stabilized silver nanoparticles, J. Mater. Chem. B, 2013, 1, 1962–1967, 10.1039/c3tb00534h.
- A. Jose, R. Sahadevan, M. Vijay, S. Sadhukhan and M. Porel, Dansyl-appended sequence-defined oligomers for selective ultrasensitive detection of Hg2+ in water, paper strips, living cells and its efficient removal, Sens. Actuators, B, 2023, 380, 133335, DOI:10.1016/j.snb.2023.133335.
- M. Batool, Z. Afzal, H. M. Junaid, A. R. Solangi and A. Hassan, Sulfonamides as Optical Chemosensors, Crit. Rev. Anal. Chem., 2024, 54(5), 954–981, DOI:10.1080/10408347.2022.2105135.
- F. Duarte, G. Dobrikov, A. Kurutos, J. L. Capelo-Martinez, H. M. Santos, E. Oliveira and C. Lodeiro, Development of fluorochromic polymer doped materials as platforms for temperature sensing using three dansyl derivatives bearing a sulfur bridge, J. Photochem. Photobiol., A, 2023, 445, 115033, DOI:10.1016/j.jphotochem.2023.115033.
- G. Pedro, F. Duarte, G. M. Dobrikov, A. Kurutos, H. M. Santos, J. L. Capelo-Martínez, E. Oliveira and C. Lodeiro, Optical evaluation of dansyl derivatives and their implementation in low-cost and flexible dye-doped PMMA platforms for efficient detection of hazardous chemical vapours, Dyes Pigm., 2024, 224, 112042, DOI:10.1016/j.dyepig.2024.112042.
- Y. Zha, R. Xin, M. Zhang, X. Cui and N. Li, Stimuli-responsive azobenzene-quantum dots for multi-sensing of dithionite, hypochlorite, and azoreductase, Microchim. Acta, 2020, 187, 481, DOI:10.1007/s00604-020-04455-9.
- J. Li, X. Yu, D. Shu, H. Liu, M. Gu, K. Zhang, G. Mao, S. Yang and R. Yang, Accelerated Activity-Based Sensing by Fluorogenic Reporter Engineering Enables to Rapidly Determine Unstable Analyte, Anal. Chem., 2024, 96, 7723–7729, DOI:10.1021/acs.analchem.4c00945.
- B. Dong, W. Song, X. Kong, N. Zhang and W. Lin, Visualizing cellular sodium hydrosulfite (Na2S2O4) using azo-based fluorescent probes with a high signal-to-noise ratio, J. Mater. Chem. B, 2019, 7, 730–733, 10.1039/c8tb02487a.
- A. Chevalier, C. Mercier, L. Saurel, S. Orenga, P. Y. Renard and A. Romieu, The first latent green fluorophores for the detection of azoreductase activity in bacterial cultures, Chem. Commun., 2013, 49, 8815–8817, 10.1039/c3cc44798g.
- X. Yu, L. Xiang, S. Yang, S. Qu, X. Zeng, Y. Zhou and R. Yang, A near-infrared fluorogenic probe with fast response for detecting sodium dithionite in living cells, Spectrochim. Acta, Part A, 2021, 245, 118887, DOI:10.1016/j.saa.2020.118887.
- S. Wang, W. Wu, J. Lv, Q. Qi and W. Huang, Fast detection of sodium dithionite in sugar using a xanthylium-based f luorescent probe, Food Chem., 2024, 452, 139547, DOI:10.1016/j.foodchem.2024.139547.
- N. J. W. Rattray, W. A. Zalloum, D. Mansell, J. Latimer, M. Jaffar, E. V. Bichenkova and S. Freeman, Chemical and bacterial reduction of azo-probes: monitoring a conformational change using fluorescence spectroscopy, Tetrahedron, 2013, 69, 2758–2766, DOI:10.1016/j.tet.2013.01.086.
- X. Cao, L. Mu, M. Chen and G. She, A facile fluorescent sensor based on silicon nanowires for dithionite, Appl. Surf. Sci., 2018, 441, 388–393, DOI:10.1016/j.apsusc.2018.01.313.
- S. Mocanu, G. Ionita and I. Matei, Solvatochromic characteristics of dansyl molecular probes bearing alkyl diamine chains, Spectrochim. Acta, Part A, 2020, 237, 118413, DOI:10.1016/j.saa.2020.118413.
- E. Oliveira, R. M. F. Baptista, S. P. G. Costa, M. M. M. Raposo and C. Lodeiro, Solvatochromic Effects of bis(indolyl)thienylaril derivatives as new Colored Materials, Photochem. Photobiol. Sci., 2014, 13, 492–498, 10.1039/C3PP50352F.
- P. Gans, A. Sabatini and A. Vacca, Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs, Talanta, 1996, 43, 1739–1753, DOI:10.1016/0039-9140(96)01958-3.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc04750h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |