
Fe3N/Fe3O4 hetero-nanocrystals embedded in porous carbon fibers for enhanced lithium storage†
Yuanxiao
Ma
a,
Chen
Hu
a,
Miaobing
Ruan
a,
Yigang
Li
a,
Xuefan
Wang
a,
Zepeng
Chen
a,
Ze-Xing
Cai
 a,
Yan
Han
b,
Shenghong
Liu
*a and
Haibin
Sun
*a
a,
Yan
Han
b,
Shenghong
Liu
*a and
Haibin
Sun
*a
aKey Laboratory of Microelectronics and Energy of Henan Province, Department of Physics and Electronic Engineering, Xinyang Normal University, Xinyang, 464000, P. R. China. E-mail: liush@xynu.edu.cn; sunhaibin@xynu.edu.cn
bSchool of Physics and Electronic Information, Nanchang Normal University, Nanchang, 330032, P. R. China
First published on 12th December 2024
Abstract
Energy storage devices have applications in large-scale portable and smart devices due to their high energy density and long lifespan, but the limited theoretical capacity of the graphite anode in lithium-ion batteries has slowed the development of portable electronic devices. Herein, we prepared porous fibers with heterogeneous Fe3N/Fe3O4 nanocrystals wrapped by a carbon layer. A series of measurements, such as TEM images, Raman spectra, XRD pattern and XPS analysis, were used to unveil the formation of Fe3N/Fe3O4 nanocrystals. Due to the synergistic effect of the large specific surface area originating from the porous structure and the heterogeneous nanocrystals, the porous Fe3N/Fe3O4@C fibers exhibit a good electron/ion transmission route and rich active sites. As anode materials for lithium-ion batteries (LIBs), porous Fe3N/Fe3O4@C fibers delivered a reversible capacity of 964 mA h g−1 after 200 cycles at 2 A g−1 and long-term cycling stability (282 mA h g−1 after 2000 cycles at 5 A g−1). This work provides a method to regulate biphasic anode materials with desirable structures to enhance the reversibility of LIBs.
Introduction
Graphite-based lithium-ion batteries (LIBs) are widely used in common energy storage devices due to their safety, reversibility, and economical production.1 Nevertheless, the disadvantages of LIBs during the charge/discharge process include a low theoretical capacity (372 mA h g−1), unsatisfactory cycling stability, and serious volume expansion.2 Recently, anode materials in LIBs have included nickel-based sulfides (NiS, NiS2, Ni3S2, and Ni3S4) and iron-based carbides/nitrides (Fe3C, FeN, Fe2N, and Fe3N), which have achieved better electrochemical performance because of their high theoretical capacities (450–880 mA h g−1).3–8 However, their advantages were hampered by substantial volume fluctuations and nanoparticle pulverization during repeated lithiation/delithiation.9 Therefore, the development of alternative anode materials for LIBs is a key challenge to achieve a high capacity, excellent cycling stability, and superior rate performance.Phase engineering can greatly improve the adsorption and transportation of Li+ by an internal electric field. For example, Liu et al. reported a biphasic Ni3Se4/NiSe2 heterostructure with excellent Li storage capacity and a long cycle life.10 Sun et al. synthesized LIBs with a high rate capacity facilitated by Ru doped Ni3Se4/NiSe@NC nanotubes.11 Hu et al. optimized heterogeneous SnO2−x/Fe2O3−y nanocrystals for high-power LIBs because of rich oxygen vacancies and the self-catalysis of Fe2O3.12 Their superior electrochemical performance was attributed to the formation of heteroatomic structures that enhanced the conductivity, accelerated the transport of electrons and ions, and boosted the number of active sites.
To avoid the agglomeration/pulverization of the biphasic hetero-nanocrystals during the long-term cycling process, carbon (C) frameworks were frequently used to provide sufficient volume expansion for outstanding electrochemical properties. For example, Jin et al. constructed a MOF-derived N-doped porous carbon to encapsulate Co3O4/Fe2O3 nanoparticles for superior stability (621 mA h g−1 at 1 A g−1 after 1000 cycles).13 Zhang et al. prepared small Co/Co3O4 nanoparticles embedded in N-doped porous carbon to deliver excellent cycling stability and high-rate capability (1313 mA h g−1 after 500 cycles at 2 A g−1).14 Li et al. designed a hollow Ni/NiO@N-doped porous carbon to improve the cycling stability and rate performance (695.1 mA h g−1 after 200 cycles at 0.1 A g−1).15 Wang et al. used a pomegranate-like porous carbon to coat CuxSny/Sn/SnO2 submicrospheres for use as anode materials for LIBs, exhibiting a capacity of 604 mA h g−1 after 150 cycles at 200 mA g−1.16 Those excellent electrochemical capacities can be attributed to the synergistic effect that stemmed from the two components, i.e., heterogeneously structured biphasic materials and the in situ formed porous carbon support, can lead to excellent lithium storage performance.
In this work, we designed and prepared highly dispersed Fe3N/Fe3O4 heterogeneous nanocrystals embedded in porous electrospinning carbon fibers by consecutive carbonization and nitridation treatment. Observations by SEM, TEM, Raman spectroscopy, XRD, and XPS demonstrate the formation of heteroatomic Fe–O and Fe–N bonds. Consequently, the porous Fe3N/Fe3O4@C fibers delivered excellent lithium storage capacity due to the enhanced electrochemical reaction kinetics and the greater number of active sites.
Experimental section
Synthesis of Fe3N/Fe3O4@C
Material characterization
The crystalline structures of as-prepared samples were identified by X-ray diffraction (XRD) using a Cu Kα radiation source and Raman spectroscopy (Renishaw inVia confocal Raman spectrometer). The morphologies were characterized by cold-field-emission scanning electron microscopy (SEM, Hitachi S-4800), transmission electron microscopy (TEM, JEOL 2100F at 200 kV), and energy-dispersive X-ray spectrometry (EDX). Thermogravimetric analysis (TGA, TA Q600, Waters) was employed to characterize the carbon content in the temperature range of 40–800 °C with a heating rate of 10 °C min−1. The nitrogen sorption isotherms were measured at 77 K using an ASAP 2460 system (Micromeritics Instrument Corp.). The specific surface area and the pore size distribution were analyzed using the Brunauer–Emmett–Teller (BET) method. X-ray photoelectron spectroscopy (XPS) was performed with monochromatic Mg Kα (1253.6 eV) radiation. The magnetic properties were measured using a comprehensive physical property testing system (DyaCool-9T, Quantum Design) in the magnetic field range of −3–3 kOe at 300 K.Electrochemical measurements
Coin-type (CR2032) cells were assembled inside an Ar-filled glovebox. Fe3N/Fe3O4@C served as the working electrode, which was mixed with 80 wt% active material, 10 wt% Kochen black, 10 wt% polyvinylidene difluoride (PVDF), and N-methyl-2-pyrrolidone (NMP) to form a slurry. Then, the slurry was coated onto a copper sheet and dried under vacuum at 80 °C for 12 h. The mass loading of active materials was about ∼0.5 mg cm−2. Lithium metal was used as the counter electrode and a glass fiber membrane as the separator. The electrolyte solution was 1 M LiPF6 dissolved in a mixed solvent of ethylene carbonate, dimethyl carbonate (DMC), and ethyl methyl carbonate (EMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1). Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed using a CHI 660D workstation. The galvanostatic charge–discharge (GCD) tests were performed and rate performances were measured using a NEWARE battery testing system (BTS-610) with cut-off voltages of 0.01 V and 3.0 V (vs. Li/Li+).
:1:1). Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed using a CHI 660D workstation. The galvanostatic charge–discharge (GCD) tests were performed and rate performances were measured using a NEWARE battery testing system (BTS-610) with cut-off voltages of 0.01 V and 3.0 V (vs. Li/Li+).
Results and discussion
As shown in Fig. 1a, Fe3N/Fe3O4@C fibers were synthesised by the electrospinning method with consecutive carbonization and nitridation treatment. The biphasic heterostructure of Fe3N/Fe3O4 is depicted in Fig. 1b. Fe and N atoms constitute a structure, while Fe atoms are bonded to O atoms, reflecting the top and side views of Fe3N/Fe3O4 hetero-nanocrystal structures. The morphology and microstructures of Fe3N/Fe3O4@C samples were observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM images in Fig. 2a and b show that the carbon fibers were cross-connected to form a freestanding membrane. Abundant pores were formed on the carbon fibers, as shown in the zoomed-in SEM image in Fig. 2c. As a comparison, Fig. S1† shows that many carbon fibers did not have pores due to the absence of carbonization treatment under an Ar atmosphere. The TEM image in Fig. 2d shows that Fe3N/Fe3O4@C particles were uniformly embedded in the carbon nanofibers. The high-resolution TEM image in Fig. 2e exhibits clear lattice fringes related to the (100) plane of Fe3N, the (400) plane of Fe3O4, and the (101) plane of C. The corresponding elemental maps in Fig. 2f confirm the existence of Fe, N, O, and C elements, revealing the successful construction of Fe3N/Fe3O4 hetero-nanocrystals. | ||
| Fig. 1 (a) Illustration of the synthesis route of porous Fe3N/Fe3O4@C fibers. (b) Top and side views of Fe3N/Fe3O4 hetero-nanocrystal structures. | ||
 | ||
| Fig. 2 Morphologies of porous Fe3N/Fe3O4@C fibers. (a–c) SEM images, (d–f) TEM images, and the corresponding elemental maps. | ||
The Raman spectrum in Fig. 3a exhibits two characteristic peaks at 1342 and 1588 cm−1, which were attributed to the D band for defect-induced disordered carbon and the G band for sp2 hybridized graphitic carbon.17 The intensity ratio of the two peaks (ID/IG ≈ 1.17) illustrates the presence of a large amount of defective carbon and active sites in porous Fe3N/Fe3O4@C fibers.18 Additionally, the three main peaks at 214, 272, and 381 cm−1 confirmed the existence of Fe3N.7 The XRD patterns in Fig. 3b show the presence of characteristic peaks of Fe3N, which closely match its crystal phases (PDF# 49-1662). The other diffraction peaks were indexed to Fe3O4 (PDF#19-0629), which is in good agreement with the TEM results.
 | ||
| Fig. 3 Crystal structure of porous Fe3N/Fe3O4@C fibers. (a) Raman spectrum, (b) XRD patterns, (c) TGA curve, (d) and pore size distribution. | ||
From the thermogravimetric analysis (TGA) curve seen in Fig. 3c, a slight weight loss below 400 °C can be observed by the continuous evaporation of the adsorbed water. A sharp weight loss appeared between 400 °C and 500 °C, which can be ascribed to the oxidation of carbon (C + O2 → CO2), the subsequent oxidation of Fe3N(Fe3N + O2 → Fe2O3 + NOx), and the further oxidation of Fe3O4 (Fe3O4 + O2 → 2Fe2O3). The weight loss after 500 °C was estimated to account for about 73.1% of Fe3N/Fe3O4@C fibers. This might be used to verify the importance of carbonization treatment in achieving porous fibers. The porous microstructure was analyzed using the Brunauer–Emmett–Teller (BET) method. As shown in Fig. 3d and Fig. S2,† the specific surface area of porous Fe3N/Fe3O4@C fibers was estimated to be 328.9 m2 g−1 larger than that of the nonporous structure (18.8 m2 g−1, as shown in Fig. S3†). The corresponding pore size distribution revealed the presence of microporous structures, in accordance with a type-IV adsorption/desorption isotherm.19 Thus, the highly porous structure provided abundant space and active sites that could accommodate the transmission of ions, which should accelerate the reaction kinetics to enhance lithium-ion storage.20
X-ray photoelectron spectroscopy (XPS) was used to investigate the elemental valence states and chemical bonds, as shown in Fig. S4.† The high-resolution Fe 2p spectrum in Fig. 4a shows two major peaks at 712.3 eV and 724.8 eV, respectively, which corresponded to the Fe 2p3/2 and Fe 2p1/2 spin–orbit doublets of Fe in Fe3O4.6 They could be divided into several peaks at 711.0 eV and 719.2 eV for Fe2+, and at 713.3 eV and 724.8 eV for Fe3+.21 The satellite peak at 707.4 eV indicates the presence of Fe–N bonds. The high-resolution N 1s spectrum in Fig. 4b was split into five peaks at 398.2, 399.7, 401.0, 401.6, and 402.7 eV, which belonged to pyridinic N, Fe–N, pyrrolic N, graphitic N, and oxidized N.22 The high-resolution O 1s spectrum is shown in Fig. 4c, which demonstrates the presence of Fe–O bonds in Fe3N/Fe3O4@C fibers. Additionally, the presence of C–N bonds in the high-resolution C 1s spectrum (Fig. 4d) may improve the conductivity and provide additional active sites for lithium storage.23
 | ||
| Fig. 4 High-resolution XPS (a) Fe 2p, (b) N 1s, (c) O 1s, and (d) C 1s spectra of porous Fe3N/Fe3O4@C fibers. | ||
The electrochemical performance of the porous Fe3N/Fe3O4@C fibers was examined in half-cells using metal lithium as counter electrodes. Fig. 5a shows the first three cyclic voltammograms (CVs) achieved at a scan rate of 0.2 m V s−1 within 0.01–3.0 V. During the first discharge process, reduction peaks were observed at 1.54 and 0.67 V, indicating the gradual conversion of Fe3N and Fe3O4 into Li3N and Li2O, as well as the generation of a solid electrolyte interface (SEI) layer, respectively.7,12 Subsequently, the reduction peak at 0.67 V disappeared from the CV profiles in the second and third discharge processes, indicating the formation of a stable SEI layer on the electrode surface. During the subsequent charge process, the corresponding oxidation peaks appeared at 1.04 V and 1.60 V, indicating that the transformation of Fe3N and Fe3O4 was reversible. The subsequent CV profiles nearly overlapped, indicating excellent electrochemical reversibility due to the synergistic effects between the porous structure and biphasic heterostructure. Fig. 5b shows the first three galvanostatic charge/discharge (GCD) profiles at a current density of 0.1 A g−1. The potential plateaus closely matched the peaks in CV curves. The subsequent cycles nearly overlapped, indicating that the GCD profiles remained stable, with a close to 100% coulombic efficiency. Compared with the nonporous Fe3N/Fe3O4@C fibers shown in Fig. 5c, the porous Fe3N/Fe3O4@C fibers showed a large specific capacity (1984 mA h g−1 at 0.1 A g−1) and coulombic efficiency (62.7%) in the initial GCD profiles.
 | ||
| Fig. 5 (a) First three CV profiles at 0.2 mV s−1, (b) GCD curves of porous Fe3N/Fe3O4@C fibers at 0.1 A g−1. Comparison with the nonporous Fe3N/Fe3O4@C fibers: (c) first GCD profiles at 0.1 A g−1, (d) magnetic hysteresis profiles at 300 K, (e) rate capabilities, (f) comparison with previous reports, and (g) cycling performance at 2 A g−1. (h) Long-term cycling performance of porous Fe3N/Fe3O4@C fibers at 5 A g−1. | ||
The spin polarization of Fe atoms was explored by performing magnetic measurements after the first discharge process. The magnetic hysteresis (MH) profiles in Fig. 5d show S-like loops, suggesting the ferromagnetic properties of the samples.24 The porous Fe3N/Fe3O4@C fibers exhibited a much larger saturation magnetization (MS) of 11.6225 emu g−1 than nonporous Fe3N/Fe3O4@C fibers (9.0075 emu g−1). The remanent magnetization (MR) and coercive force (HC) of the former were lower than those of the latter, indicating the presence of many more reduced Fe atoms in porous Fe3N/Fe3O4@C fibers than in nonporous Fe3N/Fe3O4@C fibers.25 The magnetization of porous Fe3N/Fe3O4@C fibers was measured with zero-field-cooled (ZFC) and field-cooled (FC) temperature at 10 Oe and shown in Fig. S5.† The electrode showed a ferromagnetic signature with a blocking temperature of around 297 K, in good agreement with the MH measurement. The resultant high spin polarization of porous Fe3N/Fe3O4@C fibers enhanced the bonding of N atoms with Li atoms to provide superior lithium-ion storage.
Fig. 5e shows their GCD profiles at current densities in the range of 0.1–10 A g−1. The sample delivered the highest reversible capacity of 1194.9 mA h g−1 at 0.1 A g−1, while it was only 492.4 mA h g−1 at 5 A g−1. When the current density returned to 0.1 A g−1, the capacity reached 905.8 mA h g−1. The rate capacity of porous Fe3N/Fe3O4@C fibers, as shown in Fig. 5f, surpassed that of the nonporous samples and many previous results7,26–29 due to the enhanced active sites and increased Li+ adsorption. The cycling test results in Fig. 5g show that porous Fe3N/Fe3O4@C fibers delivered a specific capacity of 964 mA h g−1 after 200 cycles at 2 A g−1, while this was only 318 mA h g−1 for nonporous Fe3N/Fe3O4@C fibers. Additionally, a notable increase was observed in porous Fe3N/Fe3O4@C fibers, suggesting the high activation of active materials during the first charge/discharge cycle. The coulombic efficiency was nearly 100%. As shown in Fig. 5h, the specific capacity of porous Fe3N/Fe3O4@C fibers exhibited an increasing trend during the high current density of 5 A g−1, reaching up to the maximum specific capacity of 640 mA h g−1 after 844 cycles. This case can be ascribed to the rich microporous/mesoporous structure to promote quick electron/ion transport and the additional electrochemical activation sites. In the subsequent cycles, the specific capacity faded to 282 mA h g−1 after 2000 cycles. The capacity loss was due to the irreversible formation of a SEI film to suppress the electron/ion transport between the electrode materials and the electrolyte. Additionally, the capacity retention was about 69% over 2000 cycles at 5 A g−1. This excellent cycling stability indicated synergistic effects between the porous architecture and the hetero-nanocrystals of Fe3N/Fe3O4 to promote charge transport and structural durability.
The Li+ storage process of porous Fe3N/Fe3O4@C fibers was investigated using CV measurements at scan rates from 0.2 to 1.2 mV s−1 (Fig. 6a). The CV profiles produced similar redox peaks during the discharge–charge process, indicating superior reversibility. The electrochemical kinetics were explored using the relationship between the peak current (i) and scan rate (v), which was expressed using the following formulas:30
| i = avb | (1) |
| log(i) = blog(v) + log(a) | (2) |
| i = k1v + k2v1/2 | (3) |
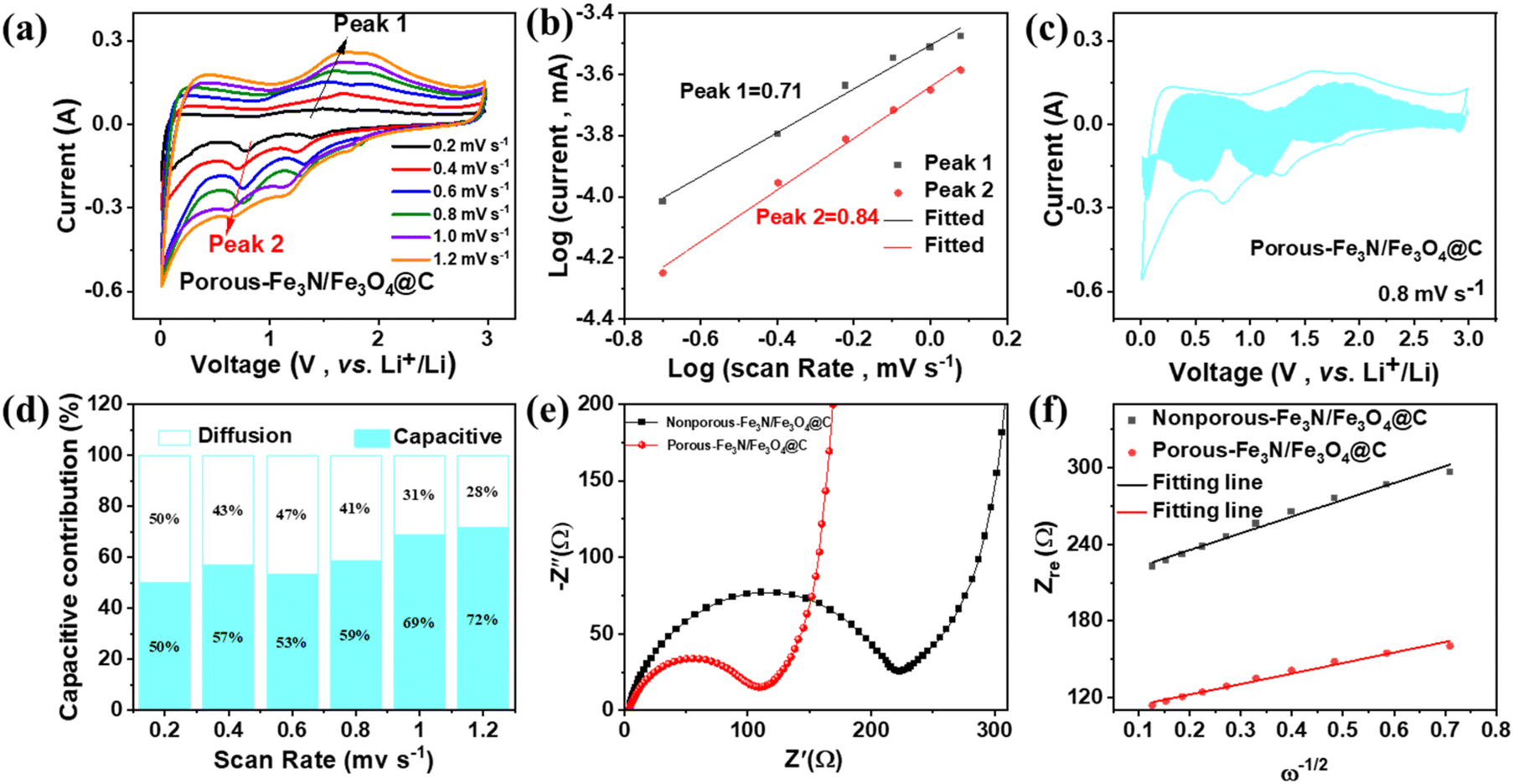 | ||
| Fig. 6 (a) CV curves at scan rates from 0.2 to 1.2 mV s−1, (b) Log (i) versus log (v) at different scan rates, (c) capacitive (reseda) and diffusion (white) contributions at 0.8 mV s−1, and (d) contribution ratio of the capacitive behavior of Ni3Se4/NiSe@NC at different scan rates. Comparison with the nonporous Fe3N/Fe3O4@C fibers: (e) EIS spectra before cycling, and (f) the relationship between Zre and ω−1/2 at a low frequency before cycling. | ||
Fig. 6e shows the electrochemical impedance spectroscopy (EIS) spectra of the initial two electrodes. The Nyquist plots were composed of a semicircle in the high-frequency region and a straight line in the low-frequency region. The charge transfer resistance (Rct) value of porous Fe3N/Fe3O4@C fibers (110.8 Ω) was smaller than that of nonporous Fe3N/Fe3O4@C fibers (221.7 Ω). The Li+ diffusion coefficient (DLi+) was explored using the following formulas:34
| ω = 2πf | (4) |
| Zre = R + σω−1/2 | (5) |
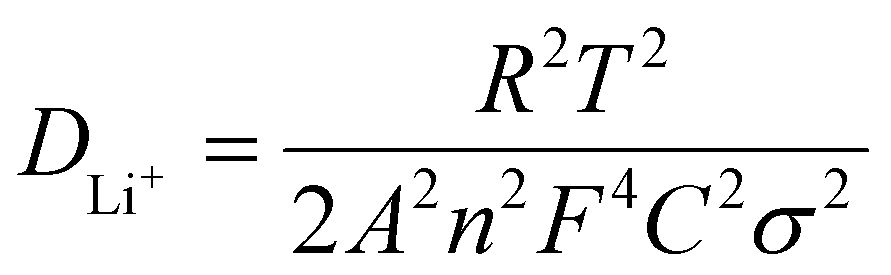 | (6) |
500 C mol−1), respectively. Thus, the slope (σ) can be estimated by the function relation of the Warburg diffusion process (Zre) and ω−1/2, as shown in Fig. 6f. The porous Fe3N/Fe3O4@C fibers exhibited a lower σ value (82.4) than the nonporous Fe3N/Fe3O4@C fibers (131.1), once again confirming the superior Li+ diffusion coefficient and stronger electrochemical dynamics.35
Conclusions
Fe3N/Fe3O4@C porous fibers were prepared by electrospinning, and consecutive carbonization and nitridation. XPS analysis unveiled the formation of Fe–N and Fe–O bonds, which further confirmed the formation of heterogeneous nanocrystals by TEM and XRD. The porous Fe3N/Fe3O4@C fibers delivered a capacity of 1984 mA h g−1 at 0.1 A g−1, a rate capacity of 407.8 mA h g−1 at 10 A g−1, and a specific capacity of 282 mA h g−1 after 2000 cycles at 5 A g−1. Overall, this biphasic porous heterostructure with a high specific surface area and wrapped by a carbon layer greatly improved the electrochemical performance and structural stability of LIBs.Author contributions
Yuanxiao Ma: data curation and writing – original draft. Chen Hu: investigation. Miaobing Ruan: methodology and formal analysis. Yigang Li: magnetization measurement. Xuefan Wang: XPS measurement and analysis. Zepeng Chen: sample preparation. Ze-Xing Cai: visualization. Yan Han: review & editing. Shenghong Liu: review & editing. Haibin Sun: conceptualization, supervision, and funding acquisition.Data availability
All data on the measured ecosystem variables indicating ecosystem functions that support the findings of this study are included within this paper and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (11874317 and 22305207), the Key Scientific Research Projects of Colleges and Universities in Henan Province (22A140009), and the Program for Young Scholars of Xinyang Normal University. This work was supported by the Analysis and Testing Center of XYNU.References
- X. Yu, J. Xiang, Q. Shi, L. Li, J. Wang, X. Liu, C. Zhang, Z. Wang, J. Zhang, H. Hu, A. Bachmatiuk, B. Trzebicka, J. Chen, T. Guo, Y. Shen, J. Choi, C. Huang and M. H. Rümmel, Small, 2024, 12, 2406309 CrossRef.
- Q. Liu, X. Su, D. Lei, Y. Qin, J. Wen, F. Guo, Y. A. Wu, Y. Rong, R. Kou and X. Xiao, Nat. Energy, 2018, 3, 936 CrossRef.
- V. Mullaivananathan, N. K. Shakkeel and N. Kalaiselv, Energy Fuels, 2021, 35, 8991–9000 CrossRef.
- D. Zhou, X. Guo, Q. Zhang, Y. Shi, H. Zhang, C. Yu and H. Pang, Adv. Funct. Mater., 2022, 32, 2107928 CrossRef.
- X. Qian, J. Cheng, Y. Wang, L. Jin, J. Chen, Q. Hao and K. Zhang, Phys. Chem. Chem. Phys., 2023, 25, 5559–5568 RSC.
- J. He, A. Bhargav, H. Sul and A. Manthiram, Angew. Chem., Int. Ed., 2023, 62, e202216267 CrossRef PubMed.
- S. Liu, W. Zheng, W. Xie, H. Cui, Y. Li, C. Zhang, Z. Ji, F. Liu, R. Chen, H. Sun and J. Xu, Carbon, 2022, 192, 162–169 CrossRef.
- M. Idrees, A. S. Haidyrah, A. Rehman, Q. Zhang, X. Li and S. M. Abbas, J. Alloys Compd., 2021, 883, 160824 CrossRef.
- H. Cheng, J. G. Shapter, Y. Li and G. Gao, J. Energy Chem., 2021, 57, 451–468 CrossRef.
- Z. Liu, D. Wang, Z. Liu, W. Li, R. Zhang, L. Wu, H. Mu, Y. Hou, Q. Gao, L. Feng and G. Wen, J. Colloid Interface Sci., 2022, 627, 716–729 CrossRef.
- H. Sun, C. Liu, Y. Liang, S. Liu, Y. Qiao, C. Li, W. Xie, G. Ge and Z.-X. Cai, ACS Appl. Nano Mater., 2024, 7, 13017–13026 CrossRef.
- C. Hu, L. Chen, Y. Hu, A. Chen, L. Chen, H. Jiang and C. Li, Small, 2021, 17, 2103532 CrossRef PubMed.
- M. Jin, G. Sun, J. Yuan, Y. Wang, J. Zhou, J. Li, X. Ni, X. Pan and E. Xie, J. Alloys Compd., 2022, 922, 166231 CrossRef.
- C. Zhang, Y. Song, L. Xu and F. Yin, Chem. Eng. J., 2020, 380, 122545 CrossRef.
- Y. Li, H. Yu, L. Miao, L. Wang and Y. Song, J. Alloys Compd., 2024, 1005, 176080 CrossRef.
- H. Wang, X. Du, X. Jiang, Y. Chai, X. Yang and R. Yuan, Chem. Eng. J., 2017, 313, 535–543 CrossRef.
- H. Sun, J. Xu, C. Wang, G. Ge, Y. Jia, J. Liu, F. Song and J. Wan, Carbon, 2016, 108, 356–362 CrossRef.
- H. Sun, X. Kong, H. Park, F. Liu, Z. H. Lee and F. Ding, Adv. Mater., 2022, 9, 2107587 CrossRef.
- W. Xie, Q. Wang, J. Xu, Y. Yu, R. Zhao, N. Li, M. Li, Y. Du, S. Peng and G. Cao, J. Mater. Chem. A, 2019, 7, 10523–10533 RSC.
- S. Liu, W. Zheng, C. Hu, Y. Li, H. Cui, X. Chu, X. Li, Y. Xue, W. Xie, F. Liu, H. Sun and J. Xu, J. Power Sources, 2024, 605, 234536 CrossRef.
- J. Zhao, Y. Weng, S. Xu, A. Sheb, X. Wen and G. Yang, J. Power Sources, 2020, 464, 228246 CrossRef.
- X. Zhang, F. Ma, K. Srinivas, B. Yu, X. Chen, B. Wang, X. Wang, D. Liu, Z. Zhang, J. He and Y. Chen, Energy Storage Mater., 2022, 45, 656–666 CrossRef.
- H. Huang, S. Gao, A.-M. Wu, K. Cheng, X.-N. Li, X.-X. Gao, J.-J. Zhao, X.-L. Dong and G.-Z. Cao, Nano Energy, 2017, 31, 74–83 CrossRef.
- S. Liang, C. Liu, H. Sun, C. Li, M. Feng, S. Gao, S. Liu, H. Pan and G. Ge, J. Energy Storage, 2023, 59, 106540 CrossRef.
- Z. Li, Y. Zhang, X. Li, F. Gu, L. Zhang, H. Liu, Q. Xia, Q. Li, W. Ye, C. Ge, H. Li, H. Hu, S. Li, Y.-Z. Long, S. Yan, G.-X. Miao and Q. Li, J. Am. Chem. Soc., 2021, 143, 12800–12808 CrossRef PubMed.
- Y. Zhang, H. Xu, P. Li, W. Li, H. Yue, H. Li, L. Wu, W. Fa, Q. Yu and Q. Guo, Ionics, 2024, 30, 1329–1337 CrossRef.
- L. Tian, Y. Xie, J. Lu, T. Liu, Q. Hu, Y. Xiao, X. Zhu and X. Su, J. Alloys Compd., 2022, 922, 166208 CrossRef.
- S. Liu, W. Zheng, M. Huang, Y. Xu, W. Xie, H. Sun and Y. Zhao, Nanotechnology, 2022, 33, 135401 CrossRef PubMed.
- D. Zhang, C. Zhang, X. Shi, H. Xu, S. Shi, Y. Li, B. Luo, G. Liu, X. Liu, C. Yu and X. Li, J. Power Sources, 2023, 579, 233288 CrossRef.
- W. Xie, W. Wang, L. Duan, W. Zheng, S. Liang, S. Liu, F. Liu, X. Wang, H. Sun and X. Sun, J. Alloys Compd., 2022, 918, 165687 CrossRef.
- D. Wang, Y. Chao, K. Guo, Z. Wang, M. Yang, J. Zhu, X. Cui and Q. Xu, Adv. Funct. Mater., 2024, 2405642 CrossRef.
- Q. Wei, X. Chang, D. Butts, R. DeBlock, K. Lan, J. Li, D. Chao, D.-L. Peng and B. Dunn, Nat. Commun., 2023, 14, 7 CrossRef PubMed.
- H. Sun, W. Wang, L. Zeng, C. Liu, S. Liang, W. Xie, S. Gao, S. Liu and X. Wang, Dalton Trans., 2022, 51, 12071 RSC.
- J. Liu, X. Xu, R. Hu, L. Yang and M. Zhu, Adv. Energy Mater., 2016, 6, 1600256 CrossRef.
- T.-F. Yi, T.-T. Wei, Y. Li, Y.-B. He and Z.-B. Wang, Energy Storage Mater., 2020, 26, 165–197 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt02999b |
| This journal is © The Royal Society of Chemistry 2025 |