DOI:
10.1039/D5DT00135H
(Paper)
Dalton Trans., 2025,
54, 5286-5292
C–C and C–O bond formation reactivity of nickel complexes supported by the pyridinophane MeN3C ligand†
Received
17th January 2025
, Accepted 25th February 2025
First published on 26th February 2025
Abstract
The pyridinophane ligands RN3CX (X = H, Br) are well-established scaffolds that facilitate and stabilize nickel oxidative addition complexes to the proximal C(aryl)–X bond. In this study, we report the synthesis, detailed characterization, and reactivity of a series of NiII and NiIII complexes supported by the MeN3CX ligand. Our findings demonstrate that NiII complexes can be oxidized to readily yield well-defined NiIII species. Excitingly, the Ni-disolvento complexes exhibit catalytic trifluoroethoxylation to generate the C–O coupled product. In addition, the NiIII-halide complex undergoes transmetallation with a Grignard reagent and subsequent C–C reductive elimination, while the β-hydride elimination side reaction is suppressed, outperforming its NiII analogue.
Introduction
High-valent organometallic nickel complexes have emerged as fascinating subjects of study due to their unique properties and potential applications in catalysis to forge new chemical bonds.1–25 Among the various ligands employed to stabilize NiIII centers, the tetradentate pyridinophane ligands, RN3CX, have supported high-valent Ni species with remarkable stability.1 Understanding the stability of these complexes is crucial for reactivity study, and in particular, the role of substituent effects has been a topic of interest.26–29 In this regard, replacing the tert-butyl (tBu) N-substituents with the less bulky neopentyl (Np) groups (yielding the NpN3C system) has been found to reduce stability and enhance the reactivity of the high valent nickel species compared to the tBuN3CX system.28–30 Since the introduction of these systems, numerous metalloorganic systems based on this framework and incorporating first- and second-row transition metals such as Mn, Fe, Co, and Pd have been utilized to investigate their reactivity and catalytic processes.31–40 Building upon the remarkable reactivity enhancement observed with Np N-substituents,28,29 our current investigation explores the potential of the less bulky N-methyl analogues in stabilizing NiIII intermediates.31 In this case, the use of these methyl-substituted ligands aimed to elucidate the influence of steric effects on the reactivity of the nickel complexes. To generate the NiIII centers, we employed both mild oxidants, allowing us to compare the reactivity and stability of the resulting complexes. The investigation aimed to shed light on the factors influencing the strength and significance of the agostic interactions between the Ni center and the ipso C(aryl)–H bond. Finally, in addition to stabilizing NiIII intermediates and studying agostic interactions, we examined the oxidative addition of low-valent nickel systems into C–H bonds. This step allowed us to investigate the feasibility of C–H bond activation and its potential as a key step in various catalytic transformations.
Results and discussion
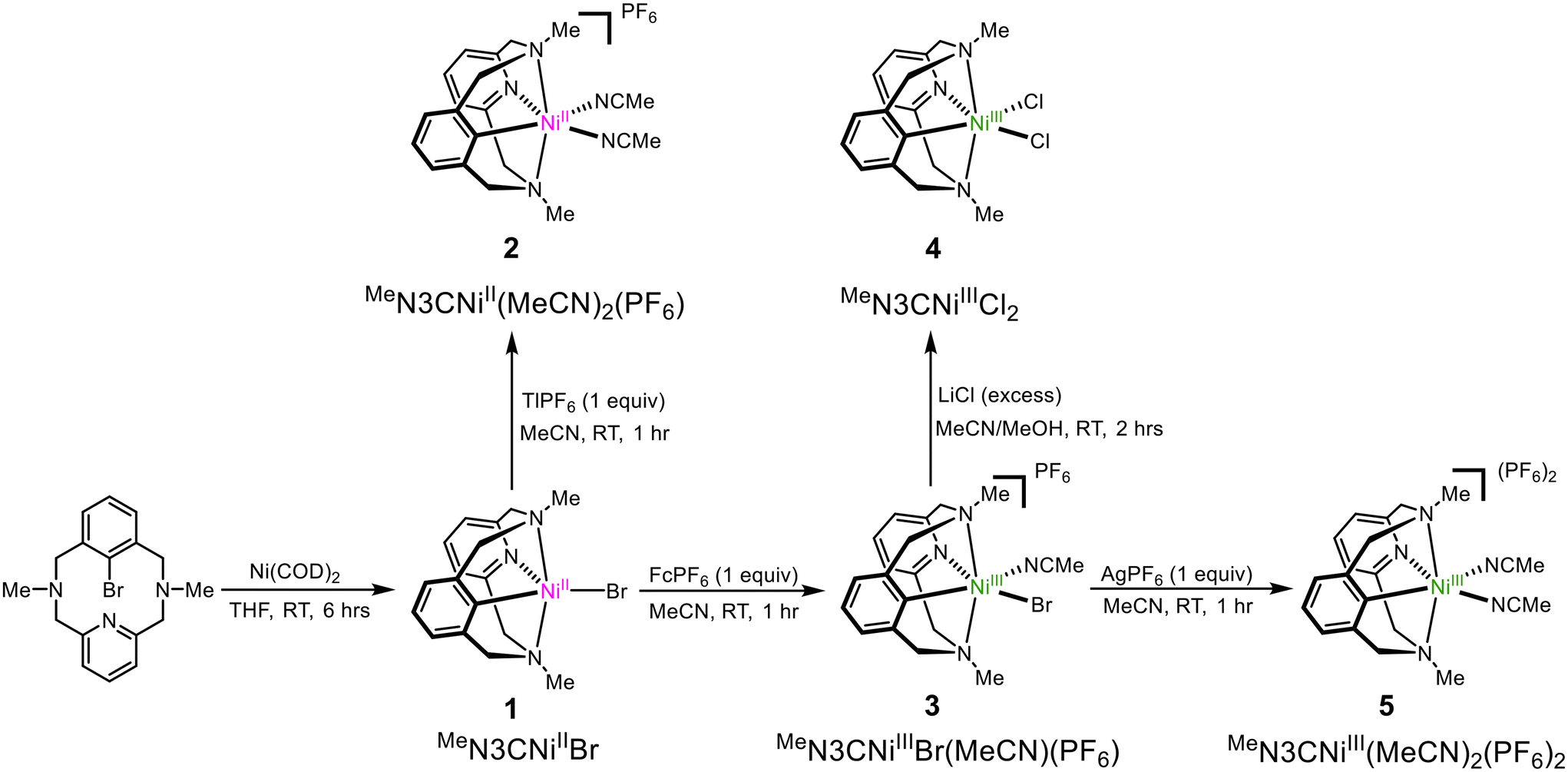
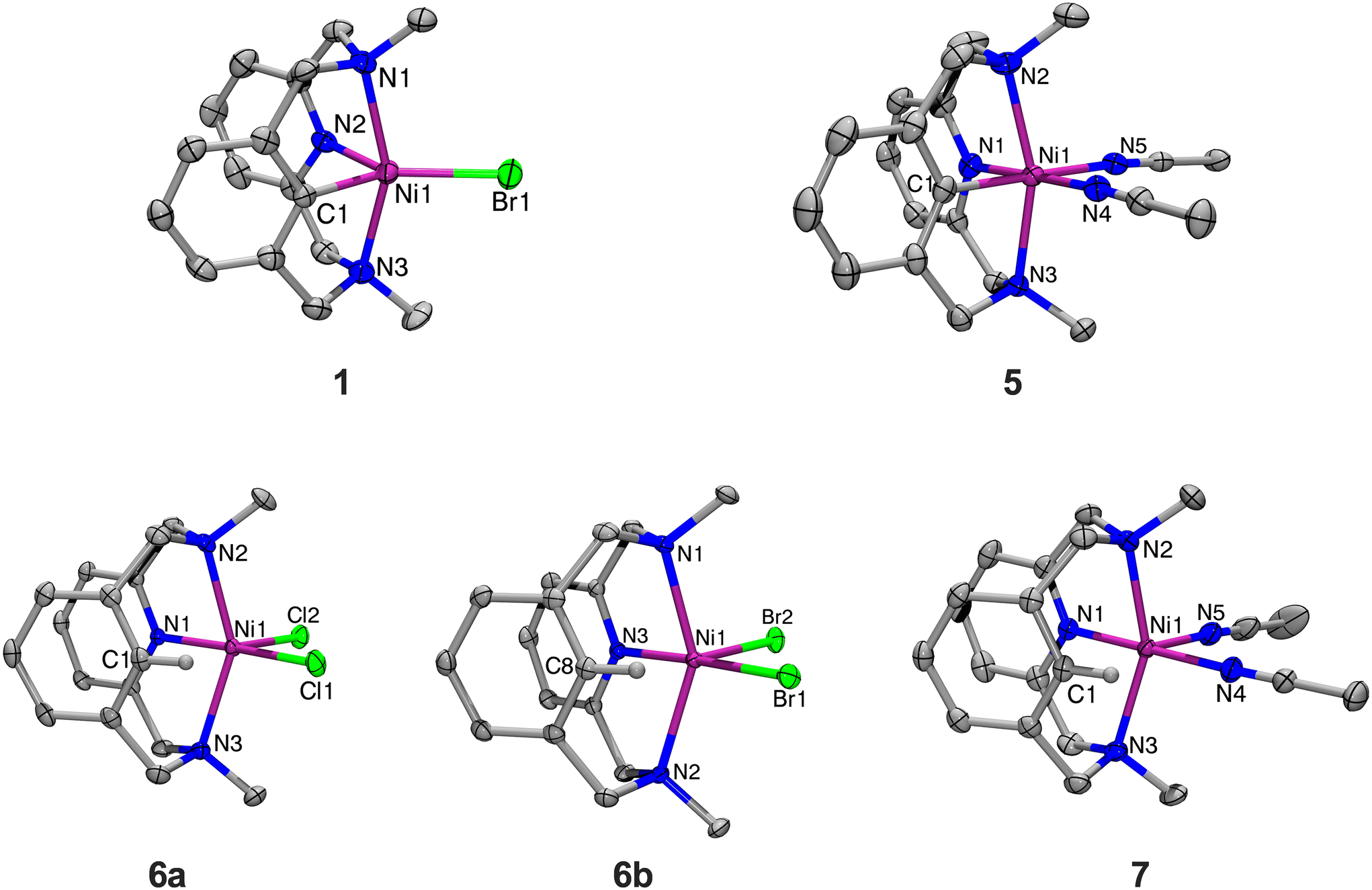
The NiII complex (MeN3C)NiIIBr (1) can be synthesized upon addition of Ni0(COD)2 to MeN3CBr following a modified literature procedure (Scheme 1).31 The crystal structure of 1, which has not been reported previously, reveals a trigonal bipyramidal geometry at the Ni center (Fig. 1), similar to its analog (tBuN3C)NiIIBr reported previously by our group.26 The complex 1 can be oxidized by addition of one equivalent of FcPF6 to yield the NiIII complex [(MeN3C)NiIIIBr(MeCN)]PF6 (3). The solid-state structure of 3 could not be obtained, even upon substitution with different counterions (BArF24−, OTf−, or BF4−), and complex identity was confirmed by ESI-MS and CHN analysis. Halide abstraction of the bromide from 3 by addition of one equivalent of TlPF6, or abstraction and oxidation of 1 by addition of two equivalents of AgPF6 yields the bis-solvento complex [(MeN3C)NiIII(MeCN)2](PF6)2 (5). Both 3 and 5 are stable in air at RT. Evans method analysis of 3 and 5 returns magnetic moments of 1.89 and 1.80μB, indicating that both are paramagnetic with one unpaired electron, as expected for a d7 NiIII center. Structural characterization for 5 reveals an octahedral geometry as expected for d7 ions, with the amine donors in the axial positions (Fig. 1). Their EPR characterizations (77 K, PrCN glass) show the presence of axial signals (Fig. 2) with gave values of 2.125 for 3, along with superhyperfine coupling to the bromide (I = 3/2) is observed in the gy direction, and a value of 2.109 for 5, along with superhyperfine coupling to the two axial N donors (I = 1) in the gz direction. Since the solid-state structure for complex 3 is elusive, excess LiCl was added for halide exchange to give rise to 4.31 The solid-state structure of 4 is also consistent with the previous observations for this system. However, a rhombic EPR signal was observed with gave 2.132, with no superhyperfine coupling to any halide. Taken together, the observed structural and EPR parameters for complexes 3, 4, and 5 strongly suggest the presence of a distorted octahedral NiIII d7 center with a dz ground state.
 |
| | Scheme 1 Synthesis of (MeN3C)Ni complexes. | |
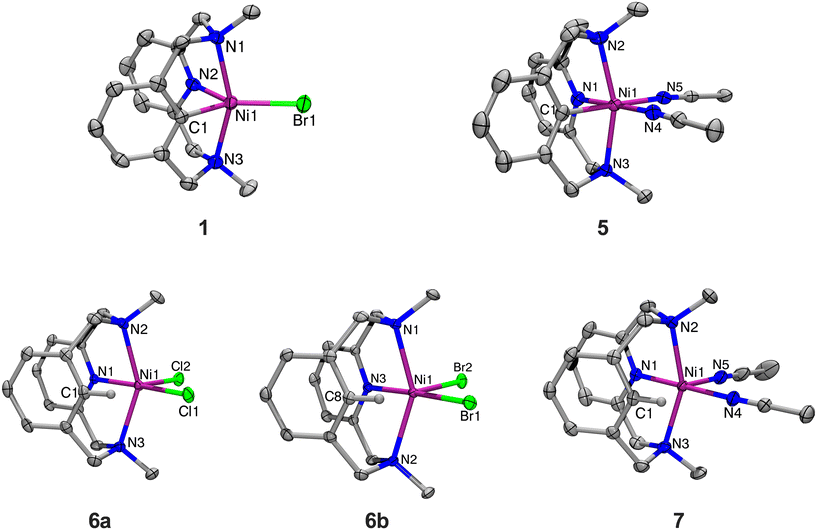 |
| | Fig. 1 ORTEPs (50% probability thermal ellipsoids) of complexes 1, dication of 5, 6a, 6b, and dication of 7. For clarity, hydrogen atoms and counterions have been omitted. Selected bond distances (Å), 1: Ni1–N1, 2.291(8); Ni1–N2, 1.981(7); Ni1–N3, 2.276(8); Ni1–C1, 1.954(9); Ni1–Br1, 2.372(2). 5: Ni1–N1, 1.935(2); Ni1–N2, 2.152(2); Ni1–N3, 2.142(2); Ni1–N4, 1.983(2); Ni1–N5, 1.990(2); Ni1–C1, 1.921(2). 6a: Ni1–N1, 1.993(2); Ni1–N2, 2.233(2); Ni1–N3, 2.242(2); Ni1–C1, 2.539(2); Ni–Cl1, 2.312(5); Ni–Cl2, 2.329(6). 6b: Ni1–N1, 2.223(1); Ni1–N2, 2.255(1); Ni1–N3, 1.985(1); Ni1–C8, 2.482(2); Ni1–Br1, 2.455(4); Ni1–Br2, 2.478(4). 7: Ni1–N1, 1.971(2); Ni1–N2, 2.179(2); Ni1–N3, 2.169(2); Ni1–N4, 2.023(2); Ni1–N5, 2.037(2); Ni1–C1, 2.412(2). | |
 |
| | Fig. 2 The experimental and simulated EPR spectra of 3, 4, and 5 in PrCN at 77 K. Simulations were obtained using the following parameters: 1: gx = 2.233; gy = 2.101 (ABr = 33 G); gz = 2.042; 2: gx = 2.246; gy = 2.106; gz = 2.045; 3: gx = 2.204; gy = 2.077; gz = 2.025 (A2N = 22 G). | |
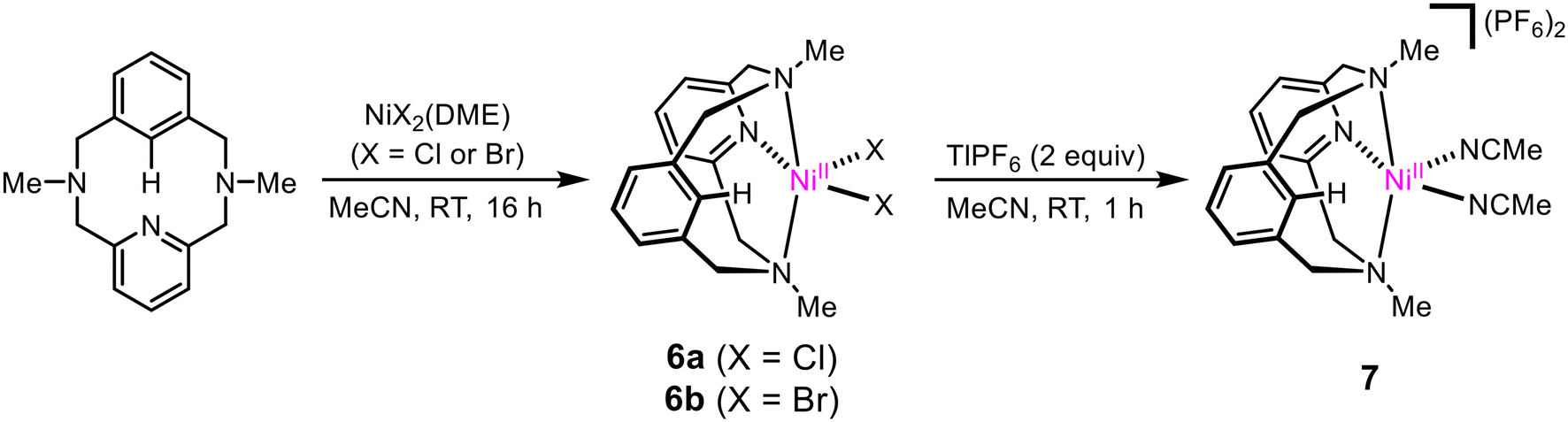
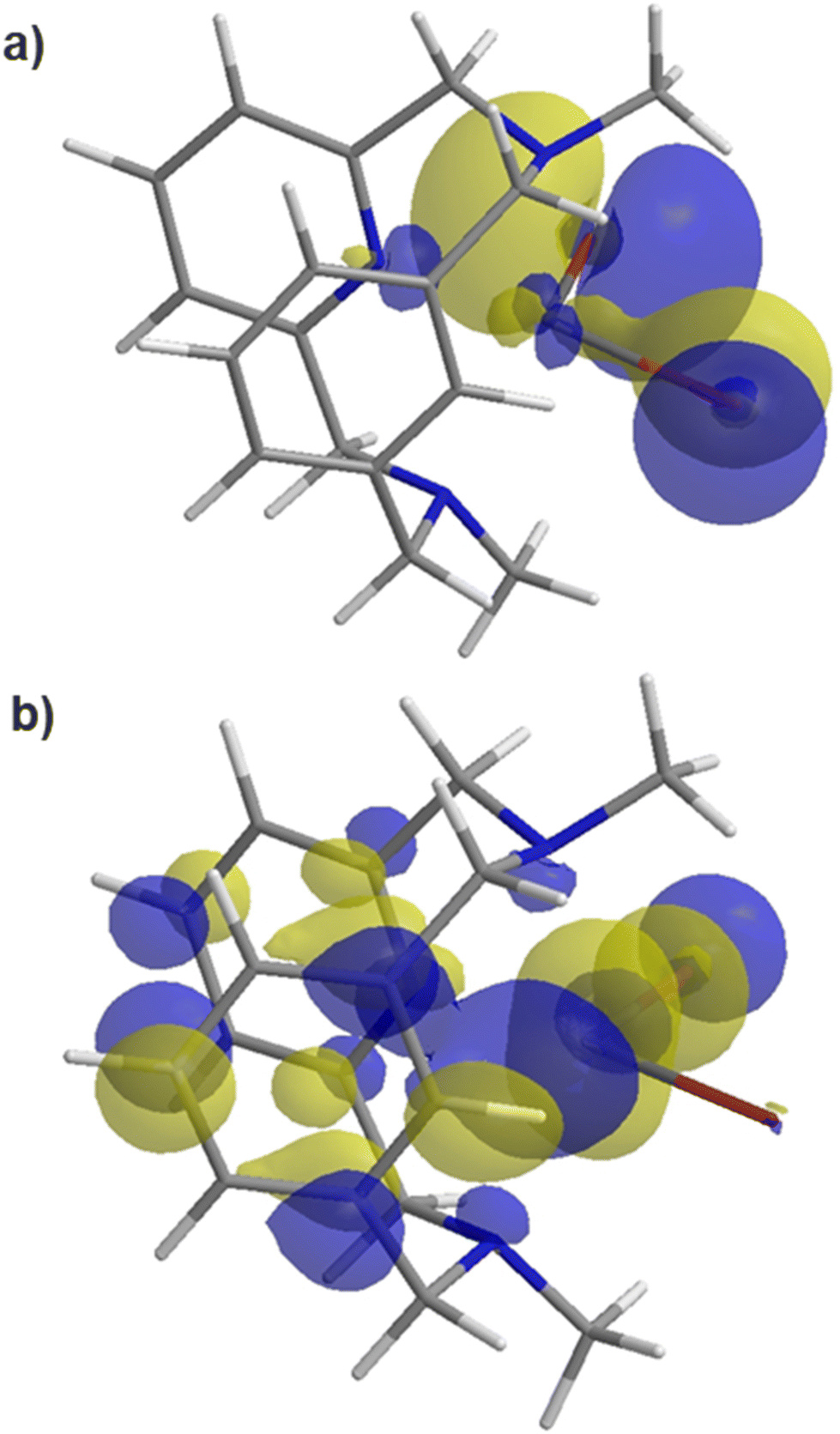
Complexes (MeN3CH)NiIICl2 (6a) and (MeN3CH)NiIIBr2 (6b) were synthesized by stirring the corresponding NiX2(dme) precursor with MeN3CH overnight, in 67% and 67% yields, respectively (Scheme 2). Halide abstraction via addition of 2 equivalents of TlPF6 yielded the bis-solvento complex (MeN3CH)NiII(MeCN)2(PF6)2 (7) in 85% yield, and no Csp2–H activation was observed during the synthesis of 7. Spin state analysis by Evans method of 6a, 6b, and 7 yielded magnetic moments of 3.16μB, 3.12μB, and 2.96μB, respectively, indicating that the three complexes are paramagnetic with two unpaired electrons. The solid-state structures show distorted square pyramidal geometries, with τ5 values of 0.40, 0.42, and 0.41 respectively. Relatively short Ni–C lengths (2.539 Å, 2.482 Å, and 2.416 Å) and narrow Ni–H–C bond angles (94.0°, 96.3°, and 83.5°) are indicative of an agostic interaction in all three (MeN3CH)NiII complexes (Fig. 1). Notably, the Ni–H–C angle and Ni–C bond length increases as the σ-donor ancillary ligands are replaced by π-donor ligands. Even though these complexes are all prone to C–H activation, we postulate that the complex bearing σ-donor ancillary ligands are more prone to activation due to potentially stronger agostic interactions. It is known that C–H agostic interaction is an electronic effect which involves the donation of electron density associated with the C–H bond to a metal center.41 Therefore, computational studies were employed in order to investigate the electronic structures. Density functional theory (DFT) calculations were performed at the B3LYP/def2-TZVPP level of theory, as this combination of hybrid functional and basis set has been previously demonstrated to accurately reproduce experimental parameters for Ni complexes.42–44 The def2-TZVPP basis set was specifically employed to enhance a high level of accuracy in describing electron correlation effects, particularly the non-bonding interactions between the C–H bond and the metal center.45–48 DFT calculations of the gas-phase structure of 6b confirmed the non-bonding interactions of the C–H bond with the metal frontier orbitals (Fig. 3). Natural bond orbital (NBO) analysis of the HOMO further confirmed a weaker overlap between the σ (C–H)-bond and the dz2-orbital of nickel with a weak second order perturbation energy (E2 = 2.3 kcal mol−1). In a reverse fashion, the LUMO analysis shows the interaction of the metal orbital with the antibonding σ(C–H) bond, with an energy of 3.1 kcal mol−1 (Table S4†). We posit that these non-bonding interactions weaken the C–H bond and confirm the agostic interaction observed in this system.
 |
| | Scheme 2 Synthesis of (MeN3CH)Ni complexes. | |
 |
| | Fig. 3 Frontier beta molecular orbitals of 6b (a) HOMO and (b) LUMO, shown as 0.03 isodensity surfaces, obtained by DFT (B3LYP/def2-TZVPP); the LUMO shows the interaction of C–H bonds with the Ni d orbital. | |
Ligand effect on properties of (RN3CX)Ni complexes
Characterization of this series of complexes has allowed us to incorporate the (MeN3CX)Ni series into the metrical and electrochemical trends observed in the (RN3C)Ni systems bearing Np, tBu, and H N-substituents.28,29,49 It was previously noted that moving from the tBuN3C− system to the less sterically hindered NpN3C− system led to an increase in the axial amine donation, as evidenced by a decrease in the average axial Ni–N bond length (2.302 Å vs. 2.239 Å), and an increase in the superhyperfine coupling observed in the gz direction (10 G vs. 14 G). Gratifyingly, the use of the MeN3C− ligand corroborates this finding, bearing a significantly shorter average Ni–N bond distance of 2.147 Å, and a stronger superhyperfine coupling of 22 G with the axial N donors in the gz direction. Finally, the reduction potential for the NiIII/II couple for the MeN3C− system has been lowered by about 100 mV compared to the NpN3C− system. With the tBuN3C− system possessing a less-reeducing NiIII/II redox couple by an additional 150–300 mV, we see a continued trend of decreasing steric bulk on the axial N donors leading to lower reduction potentials. However, unlike the other two systems, MeN3C− complexes exhibit irreversibility with the NiIII/II couple, though 3 demonstrates some quasi-reversibility at higher scan rates (see Fig. S8†). This trend also extends to the (RN3CH)Ni complexes, though it is less pronounced. The (RN3CH)NiBr2 complexes for the N-tBu, N-Np, and N-Me derivatives exhibit average axial Ni–N bond distances of 2.608 Å, 2.309 Å, and 2.239 Å, respectively, demonstrating a clear decrease (Fig. 1). Similarly, when comparing the Ni-bisolvento complexes, the axial N donor atoms move much closer for H (2.158 Å) compared to Me (2.239 Å). However, for comparing Ni–C distances, the trend becomes much less clear. The Np and Me dibromide analogues possess approximately the same Ni–C bond distances at 2.479 Å and 2.482 Å, within the reported error. For the bis-solvento complexes, on the other hand, the Me complex bears a shorter Ni–C distance than the H complex, at 2.416 Å vs. 2.453 Å. This suggests that the MeN3CH complexes may exhibit stronger agostic interactions than the other RN3CH analogues and may also bear a stronger stabilization in the organometallic Ni complex, via donation from the axial N donors. Together, these observations indicate that MeN3CH may be best poised to undergo C–H activation reactivity.
Catalytic trifluoroethoxylation reactivity
To investigate the catalytic properties of these complexes, we initially examined complexes 2, 5, and 6b in the trifluoroalkoxylation reaction with trifluoroethanol (Scheme 3). The Ribas group has recently reported the trifluoro- and difluoroethoxylation of NiII complexes supported by the HN3CH ligand.49 Building on their findings, we sought to assess the steric effects of Ni complexes with the MeN3CH ligand by evaluating the reaction time and yield. The ethoxylation reaction was carried out under air in the presence of trifluoroethanol, FcPF6 as the oxidant, and Cs2CO3 as the base (Table 1). Among the complexes tested, 7 demonstrates catalytic conversion to the ethoxylated product in 49% yield and with a turnover number (∼2) to that reported by the Ribas group. Only stoichiometric amounts of the product were obtained when 2 or 6b was used as the catalyst, suggesting that regeneration of the active catalyst during the catalytic cycle was impeded. The reaction proceeds with similar yields when employing the nickel precursors for 6b and 7, rather than the pre-formed complexes (Table S1†). In contrast, a super-stoichiometric amount of C–O product was obtained for 5, indicating that ethoxylation from a formally NiIII center may be more operative compared to a NiII center as in 2.
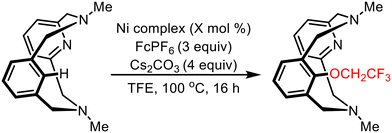 |
| | Scheme 3 General scheme for reactivity of 2,2,2-trifluoroethanol with Ni complexes. | |
Table 1 Catalytic trifluoroethoxylation of MeN3CH by Ni complexes
| Entry |
Complex |
mol % |
Yield (%) |
Conversion (%) |
| 1 |
2
|
30 |
24 |
38 |
| 2 |
5
|
30 |
33 |
45 |
| 3 |
6b
|
30 |
25 |
31 |
| 4 |
7
|
30 |
49 |
>99 |
Stoichiometric C–C bond formation reactivity
We then investigated C–C bond formation using the Ni complexes in various oxidation states. The MeN3CH system supports Ni complexes in different oxidation states, and higher oxidation states of Ni are considered more effective in suppressing β-hydride elimination (βHE) – a common side reaction in C–C cross-coupling employing alkyl substrates. We performed stoichiometric C–C bond formation reactions using the complexes 1 and 3 with an octyl Grignard reagent (Scheme 4). The reaction likely leads to the in situ formation of Ni-alkyl complexes, creating a favorable environment for C–C coupling or βHE at the Ni center. Acidic workup of the reaction mixture results in the release of C–C coupled products, the β-hydride eliminated product octene, the protodemetalated product octane, and thus allowing us to measure the ratio of C–C coupling to βHE. The NiII complex 1 yields 9% of the C–C coupled product, while octene was produced in 37% yield (Table 2). In contrast, the NiIII analog, complex 3, more effectively suppresses βHE and produces a greater yield of the C–C coupled product than 1. Specifically, the yield of MeN3C-octyl was 33%, while octene was produced in 21% yield. Additionally, the homocoupled hexadecane product was observed at a 3% yield with 3, while none was observed for the NiII complex 1. The outcome is likely a result of a monocationic NiII center being unable to bear an additional two anionic ligands, which is supported by a reaction using dicationic 7 (see Table S3†), which yields only 1% of the MeN3C-octyl coupled product but 9% hexadecane. The ratio of C–C coupling to βHE for the NiII complex was 0.24, whereas the NiIII complex showed a much higher ratio of 1.7. Overall, the NiIII complex more efficiently suppressed βHE and yielded the C–C coupled product at a higher efficiency compared to its NiII analogue. Interestingly, complex 3 reports a higher yield of both the MeN3C-octyl and hexadecane coupled products compared to the dichloride derivative 4, even though one might expect more facile transmetallation with two chloride ligands. In this case, the loosely bound acetonitrile ligand may play a role in facilitating transmetallation and reductive elimination, as has been observed recently.5
 |
| | Scheme 4 General scheme for reductive elimination from Ni complexes and products formed. | |
Table 2 C–C bond coupling reaction of NiII (1) and NiIII (3) complexes with octyl Grignard reagent (ND = not detected)
| Complex |
A
|
C
|
E
|
A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E :E |
(A + C):E |
|
1
|
9 ± 2 |
ND |
37 ± 2 |
0.24 ± 0.06 |
0.24 ± 0.06 |
|
3
|
33 ± 6 |
3 ± 1 |
21 ± 7 |
1.6 ± 0.6 |
1.7 ± 0.6 |
Conclusion
In summary, herein we report the synthesis and characterization of a series of NiII and NiIII complexes bearing the MeN3C− or MeN3CH pyridinophane ligand framework. Analysis of the solid-state structures demonstrates the effect of the axial substituent on metrical parameters, particularly the proximity of the Ni center to the Csp2–H bond to support an agostic interaction. Catalytic ethoxylation of the ligand is observed using 2,2,2-trifluoroethanol, demonstrating its competence for C–O bond formation compared to the previously reported HN3CH variant. Reductive elimination studies from activated NiII and NiIII centers shows the relevance for NiIII in suppressing βHE from alkyl coupling partners in favor of productive cross-coupling routes. Overall, this study sheds light on organometallic Ni catalyst design from the ligand and metal perspective for future development of Ni C–H bond catalysts.
Data availability
Data for this article containing synthetic details, spectroscopic characterization, reactivity studies and computational details have been included as part of the ESI.† Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under CCDC 2390120 and 2390122–2390125.†
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We thank the National Science Foundation (CHE 2155160 to L. M. M.) for support. The authors gratefully acknowledge Dr Toby Woods and Dr Danielle Grey of the George L. Clark X-Ray Laboratory for invaluable assistance in collection and interpretation of the crystallographic data. We would also like to thank the NMR laboratory and staff at the University of Illinois Urbana-Champaign for their assistance and expertise.
References
-
N. Heberer, C.-H. Hu and L. M. Mirica, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, pp. 348–374 Search PubMed.
- P. Mondal, P. Pirovano, A. Das, E. R. Farquhar and A. R. McDonald, J. Am. Chem. Soc., 2018, 140, 1834–1841 CrossRef CAS PubMed.
- J. S. Zhang, Y. M. Lee, M. S. Seo, M. Nilajakar, S. Fukuzumi and W. Nam, Inorg. Chem., 2022, 61, 19735–19747 CrossRef CAS PubMed.
- S. K. Kariofillis and A. G. Doyle, Acc. Chem. Res., 2021, 54, 988–1000 CrossRef CAS PubMed.
- L. Griego, J. B. Chae and L. M. Mirica, Chem, 2024, 10, 867–881 CAS.
- M. Carnes, D. Buccella, J. Y. C. Chen, A. P. Ramirez, N. J. Turro, C. Nuckolls and M. Steigerwald, Angew. Chem., Int. Ed., 2009, 48, 290–294 CrossRef CAS PubMed.
- P. J. Alonso, A. B. Arauzo, M. A. García-Monforte, A. Martín, B. Menjón, C. Rillo and M. Tomás, Chem. – Eur. J., 2009, 15, 11020–11030 CrossRef CAS PubMed.
- J. B. Diccianni, C. H. Hu and T. N. Diao, Angew. Chem., Int. Ed., 2017, 56, 3635–3639 CrossRef CAS PubMed.
- N. X. Gu, P. H. Oyala and J. C. Peters, J. Am. Chem. Soc., 2020, 142, 7827–7835 CrossRef CAS PubMed.
- H. Lee, J. Borgel and T. Ritter, Angew. Chem., Int. Ed., 2017, 56, 6966–6969 CrossRef CAS PubMed.
- E. Chong, J. W. Kampf, A. Ariafard, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 6058–6061 CrossRef CAS PubMed.
- C. C. Roberts, E. Chong, J. W. Kampf, A. J. Canty, A. Ariafard and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 19513–19520 CrossRef CAS PubMed.
- J. R. Bour, D. M. Ferguson, E. J. McClain, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 8914–8920 CrossRef CAS PubMed.
- M. W. Milbauer, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2022, 144, 21030–21034 CrossRef CAS PubMed.
- P. Roy, J. R. Bour, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 17382–17387 CrossRef CAS PubMed.
- E. A. Meucci, A. Ariafard, A. J. Canty, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 13261–13267 CrossRef CAS PubMed.
- E. A. Meucci, S. N. Nguyen, N. M. Camasso, E. Chong, A. Ariafard, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 12872–12879 CrossRef CAS PubMed.
- C. C. Roberts, N. M. Camasso, E. G. Bowes and M. S. Sanford, Angew. Chem., Int. Ed., 2019, 58, 9104–9108 CrossRef CAS PubMed.
- N. M. Camasso, A. J. Canty, A. Ariafard and M. S. Sanford, Organometallics, 2017, 36, 4382–4393 CrossRef CAS.
- E. A. Meucci, N. M. Camasso and M. S. Sanford, Organometallics, 2017, 36, 247–250 CrossRef CAS.
- X. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC.
- G. D. Jones, C. McFarland, T. J. Anderson and D. A. Vicic, Chem. Commun., 2005, 4211–4213 RSC.
- O. Vechorkin and X. Hu, Angew. Chem., Int. Ed., 2009, 48, 2937–2940 CrossRef CAS PubMed.
- R. Shi, Z. Zhang and X. Hu, Acc. Chem. Res., 2019, 52, 1471–1483 CrossRef CAS PubMed.
- H. Yin and G. C. Fu, J. Am. Chem. Soc., 2019, 141, 15433–15440 CrossRef CAS PubMed.
- W. Zhou, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2015, 137, 7604–7607 CrossRef CAS PubMed.
- W. Zhou, N. P. Rath and L. M. Mirica, Dalton Trans., 2016, 45, 8693–8695 RSC.
- W. Zhou, S. A. Zheng, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2016, 138, 5777–5780 CrossRef CAS PubMed.
- W. Zhou, M. B. Watson, S. Zheng, N. P. Rath and L. M. Mirica, Dalton Trans., 2016, 137, 15886–15893 RSC.
- A. J. Wessel, J. W. Schultz, F. Tang, H. Duan and L. M. Mirica, Org. Biomol. Chem., 2017, 15, 9923–9931 RSC.
- S. I. Ting, W. L. Williams and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 5575–5582 CrossRef CAS PubMed.
- N. P. Ruhs, J. Khusnutdinova, N. P. Rath and L. M. Mirica, Organometallics, 2019, 38, 3834–3843 CrossRef CAS.
- C. Magallón, O. Planas, S. Roldán-Gómez, J. M. Luis, A. Company and X. Ribas, Organometallics, 2021, 40, 1195–1200 CrossRef PubMed.
- O. Planas, C. J. Whiteoak, V. Martin-Diaconescu, I. Gamba, J. M. Luis, T. Parella, A. Company and X. Ribas, J. Am. Chem. Soc., 2016, 138, 14388–14397 CrossRef CAS PubMed.
- O. Planas, S. Roldán-Gómez, V. Martin-Diaconescu, T. Parella, J. M. Luis, A. Company and X. Ribas, J. Am. Chem. Soc., 2017, 139, 14649–14655 CrossRef CAS PubMed.
- O. Planas, S. Roldan-Gomez, V. Martin-Diaconescu, J. M. Luis, A. Company and X. Ribas, Chem. Sci., 2018, 9, 5736–5746 RSC.
- L. Capdevila, M. Montilla, O. Planas, A. Brotons, P. Salvador, V. Martin-Diaconescu, T. Parella, J. M. Luis and X. Ribas, Inorg. Chem., 2022, 61, 14075–14085 CrossRef CAS PubMed.
- H. M. Dinh, Y. T. He, R. R. Fayzullin, S. Vasylevskyi, E. Khaskin and J. R. Khusnutdinova, Eur. J. Inorg. Chem., 2023, 26, e202300460 CrossRef CAS.
- A. Sarbajna, Y. T. He, M. H. Dinh, O. Gladkovskaya, S. M. W. Rahaman, A. Karimata, E. Khaskin, S. Lapointe, R. R. Fayzullin and J. R. Khusnutdinova, Organometallics, 2019, 38, 4409–4419 CrossRef CAS.
- Y.-T. He, A. Karimata, O. Gladkovskaya, E. Khaskin, R. R. Fayzullin, A. Sarbajna and J. R. Khusnutdinova, Organometallics, 2021, 40, 2320–2331 CrossRef CAS.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- S. Chakrabarti, S. Sinha, G. N. Tran, H. Na and L. M. Mirica, Nat. Commun., 2023, 14, 905 CrossRef CAS PubMed.
- B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035–3039 CrossRef CAS PubMed.
- S. I. Ting, S. Garakyaraghi, C. M. Taliaferro, B. J. Shields, G. D. Scholes, F. N. Castellano and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 5800–5810 CrossRef CAS PubMed.
- E. Sutcliffe, D. A. Cagan and R. G. Hadt, J. Am. Chem. Soc., 2024, 146, 15506–15514 CrossRef CAS PubMed.
- K. J. Nelson, N. P. Kazmierczak, D. A. Cagan, A. H. Follmer, T. R. Scott, S. L. Raj, D. Garratt, N. Powers-Riggs, K. J. Gaffney, R. G. Hadt and A. A. Cordones, J. Phys. Chem. Lett., 2024, 16, 87–94 CrossRef PubMed.
- D. A. Cagan, D. Bím, B. Silva, N. P. Kazmierczak, B. J. McNicholas and R. G. Hadt, J. Am. Chem. Soc., 2022, 144, 6516–6531 CrossRef CAS PubMed.
- Y. R. Poh, D. Morozov, N. P. Kazmierczak, R. G. Hadt, G. Groenhof and J. Yuen-Zhou, J. Am. Chem. Soc., 2024, 146, 15549–15561 CrossRef CAS PubMed.
- L. Capdevila, M. T. G. M. Derks, M. Montilla, J. M. Luis, J. Roithová and X. Ribas, ChemistryEurope, 2024, 2, e202400023 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Yusuff
Moshood‡
and
Liviu M.
Mirica
,
Yusuff
Moshood‡
and
Liviu M.
Mirica