
Evaporated manganese films as a starting point for the preparation of thin-layer MnOx water-oxidation anodes†
Carolin E.
Frey‡
a,
Frances
Kwok‡
b,
Diego
Gonzáles-Flores
c,
Jonas
Ohms
a,
Kayla A.
Cooley
b,
Holger
Dau
 c,
Ivelina
Zaharieva
*c,
Timothy N.
Walter
b,
Hamed
Simchi
b,
Suzanne E.
Mohney
*b and
Philipp
Kurz
*a
c,
Ivelina
Zaharieva
*c,
Timothy N.
Walter
b,
Hamed
Simchi
b,
Suzanne E.
Mohney
*b and
Philipp
Kurz
*a
aInstitut für Anorganische und Analytische Chemie and Freiburger Materialforschungszentrum (FMF), Albert-Ludwigs-Universität Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: philipp.kurz@ac.uni-freiburg.de
bMaterials Research Institute, The Pennsylvania State University, N-209 Millennium Science Complex, University Park, PA 16802, USA. E-mail: mohney@ems.psu.edu
cFachbereich Physik, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany. E-mail: ivelina.zaharieva@physik.fu-berlin.de
First published on 3rd May 2017
Abstract
A novel method to prepare anodes for water electrolysis cells has been developed, which starts from layers of elemental manganese deposited by physical vapour deposition (PVD) on indium-doped tin oxide (ITO). Oxidation in dry air at 300 °C transforms this metallic Mn layer into a manganese(II)-rich MnOx coating (x = 1–1.3), which also contains a buried layer of an In–Sn alloy originating from reactions with the ITO support. The MnOx films are well connected to the underlying substrate and act as efficient catalysts for water-oxidation catalysis (WOC) at neutral pH. Detailed post-operando analyses using XRD, SEM, TEM and XAS revealed that the dense MnO/Mn3O4 film is virtually not affected by 2 h of electrochemical WOC at E ≈ +1.8 V vs. RHE, corresponding well to the observed good stability of catalytic currents, which is unusual for such thin layers of a MnOx catalyst. The current densities during electrolyses are so far low (i ≈ 50–100 μA cm−2 at pH 7), but optimization of the preparation process may allow for significant improvements. This new, rather easy, and adaptable preparation method for stable, thin-layer MnOx water-oxidation anodes could thus prove to be very useful for a variety of applications.
Introduction
The oxidation of water to molecular oxygen (eqn (1)) is a key reaction of most schemes for the storage of renewable energy into molecular fuels.1 Water-oxidation is a mechanistically complicated process taking place at demanding oxidation potentials.2 As a result, efficient water-oxidation catalysts (WOC) are needed to achieve useful reaction rates. Furthermore, the design of devices to produce renewable fuels (e.g. electrolysers driven by electricity from renewable sources or photo-electrochemical cells, PECs) usually requires that such catalysts are integrated into anodes to run the water-oxidation reaction electrochemically. In the case of PECs, the anodes usually have complicated multi-layer architectures.3,4| 2H2O → O2 + 4H+ + 4e− | (1) |
Ru- or Ir-based materials generally show the highest WOC activities at low overpotentials,5,6 but these metals are rare and thus probably not suitable for large-scale applications. Among the more abundant metals, cobalt and nickel oxides have attracted much attention in recent years, and very high activities have in particular been found for mixed Ni/Fe-oxides operated in strongly alkaline electrolytes (e.g. η < 300 mV for i = 10 mA cm−2 in 1 M KOH).7,8 It has also been known for decades that manganese oxides can be successfully used as WOC materials, offering the possibility to develop robust and especially affordable catalysts.9–18 Though generally not as active as the materials mentioned before,5,6,8 MnOx-anodes often suffer less from corrosion and can also be used in neutral or even acidic electrolytes,9,15 where especially Ni-based anodes show little activity. The stability of MnOx over a larger pH range is already visible when comparing the Pourbaix diagram for the system Mn/H2O with those of, e.g., Ni or Co.19 For mixed manganese oxides, which for example contain s-block metal ions like K+ or Ca2+ as part of their structures, stabilities must be even better as no dissolved manganese can be found in the reaction media even if these (K, Ca)MnOx catalysts are operated for 1 h or more at high electrochemical potential E and/or low pH.14,15,20 Finally, biological water-oxidation catalysis by the enzyme Photosystem II, where a CaMn4Ox-cluster forms the active site, is by now mechanistically very well understood, offering the unique possibility to understand and optimize Mn-based WOC in a bio-inspired fashion.21
Due to their generally poor mechanical stabilities and low electric conductivities, the manufacturing of the entire electrode from MnOx (even though attempted22) or a simple anodization of metallic Mn rods or plates are not viable strategies. Instead, 0.1–10 μm thin layers of manganese oxides are usually deposited onto conductive supports like carbon, silicon, TiO2 or doped tin(IV) oxides. Electric currents can still pass through these rather thin coatings against an acceptable resistance, and such “MnOx-coated” anodes can therefore be used for electrocatalytic H2O-oxidation.
Due to the fact that (depending on definitions) ∼10–30 different manganese oxides are known, which can contain Mn2+, Mn3+ and/or Mn4+ (also together with a number of other cations) in various ratios,23 the possibilities for the preparation of “MnOx” catalyst layers are virtually unlimited. Currently, the most common methods are (a) electrodeposition from Mn2+-containing solutions,9,16,24 (b) calcination of surfaces impregnated by manganese salts17 and (c) “printing” of inks containing pre-synthesised MnOx-particles.15,18 Anodes for electrochemical WOC have been successfully prepared via all three routes. However, for (a) and (b) it is often difficult to control the morphology of the deposited oxide, while layers obtained by route (c) can suffer from poor mechanical and/or electrical contacts to the conductive support. Additionally, all three methods often result in the formation of porous materials, which is beneficial for catalysis as the catalyst layer has a large, accessible surface area. On the other hand, such MnOx-coatings offer only limited protection against corrosion for the underlying substrate (e.g., Si-based light absorbers in the case of certain PECs). Therefore, the development of new deposition methods is still much needed to enlarge the available “tool box” for the preparation of manganese-based water-oxidation anodes.
In addition to the strategies mentioned above, other techniques yielding thin MnOx films have been explored. Atomic layer deposition (ALD) is an established method to prepare catalytically active coatings of controlled composition and nanostructure. For example, in 2012 Bent and co-workers used the organometallic Mn(II) precursor [Mn(EtCp)2] together with H2O to cover glassy carbon supports with layers of Mn(II) oxide (MnO) up to 40 nm thick.25 After heating the oxide for 10 h at 480 °C in air, the material was converted to Mn2O3. These annealed electrodes showed promising properties for electrochemical WOC in 0.1 M KOH (detected by cyclic voltammetry, CV) with current densities of up to 5 mA cm−2 at +1.7 V (overpotential η ≈ 470 mV, note: all potentials in this report are given vs. RHE). In recent years, this general method has been modified by a number of groups by using different Mn precursors, nanostructured substrates, additional metals like titanium, etc.26–29 However, ALD is a very slow deposition technique, and the MnOx-coatings accessible by ALD in a reasonable time were thus only few to tens of nanometres thick. From WOC experiments using electrodeposited films it is known that corrosion and mechanical detachment of the catalysts become significant for such very thin layers.16 It is therefore probably no surprise that only one of the reports concerning MnOx-catalysts prepared in this way shows experimental data which proves good long-term stability.29
In contrast to ALD, physical vapour deposition (PVD) methods have so far not been used in the context of WOC by MnOx. To us, it seemed possible that they might be suitable for this task as they generally offer faster deposition rates (>1 Å s−1) and thus the option to prepare thicker, more durable catalyst layers. PVD can also be tuned to produce films of different morphologies and compositions and might thus be suitable to synthesise different manganese oxide phases in well-defined nano-architectures. However, for a start we first had a look at a rather simple, straightforward PVD approach: manganese metal evaporated onto a conductive oxide support and its conversion into active WOC anodes by oxidation in air (Fig. 1). As will be presented in the following, this method proved to be an advantageous way to obtain 200–500 nm films of MnOx of remarkable stability at WOC-conditions. We were able to characterize the oxide layer before and after catalysis in detail using various analytical tools. On the other hand, the current densities for water-oxidation obtained are so far low, so our results should be viewed as a proof-of-concept and only as first steps towards more optimized WOC anodes.
 | ||
| Fig. 1 Schematic representation of the two-step preparation method for manganese oxide coated electrodes. | ||
Results and discussion
Preparation and characterization of MnOx layers on ITO
Using electron beam evaporation, elemental Mn was deposited directly onto indium tin oxide (ITO) glass substrates, which are commonly used supports for WOC studies15,16,30 (see ESI† for experimental details). This method yielded a smooth, shiny Mn layer covering the ITO. Depending on the deposition time, the layer thicknesses varied (as determined by profilometry), and samples with 50, 150 or 235 nm thick Mn coatings were prepared. Scanning electron microscopy (SEM) of the 150 nm thick Mn film revealed that the surface had a cauliflower-like appearance (Fig. 2A), with two predominant length scales: hundreds of nanometres for clusters or domains comprised of smaller grains only tens of nanometres across. The surface morphology of the ITO/glass substrate shows similar features (ESI, Fig. S1†) and might thus induce this morphology in the deposited material. A cross-sectional SEM image of the film suggests a columnar microstructure (with column diameters again on a length scale of ∼100 nm, Fig. 2B), and also confirms that the films are dense. | ||
| Fig. 2 (A) Surface and (B) cross-sectional SEM images of 150 nm thick layers of metallic manganese films prepared by electron-beam evaporation on an ITO-coated glass substrate. Images (C) and (D) show the related micrographs for the MnOx coating formed from such 150 nm Mn films by annealing in air for 300 °C. Layer compositions were identified by TEM analysis (see below, Fig. 4). | ||
Next, the Mn was oxidized in dry air for 16 h at 300 °C, which yielded iridescent, golden-brown layers. As expected for a conversion of Mn metal into MnOx, a significant increase of the total layer thicknesses to 110, 265 or 405 nm was found by profilometry after the annealing step. A linear relationship between the thicknesses of related Mn- and MnOx- films was found (see ESI, Fig. S2†), indicating that similar conversion reactions occur in all three cases. SEM also shows that the material resulting from the air annealing is still a compact film, but one can also detect the formation of flakes reaching out from the layer's surface (Fig. 2C and D). The observations described here for a film of 150 nm starting thickness are very similar to those made for both thinner (50 nm) and thicker (235 nm) elemental Mn coatings (see ESI, Fig. S3†).
We used grazing incidence X-ray diffraction (GIXRD) to identify the dominant manganese phases of the layers. In all cases, reflexions caused by the ITO substrate are clearly visible (marked by circles in Fig. 3) and demonstrate that GIXRD probes the entire layer volume here. Unsurprisingly, the XRD results show that the material initially deposited by PVD is α-Mn (Fig. 3, bottom), the most common polymorph of elemental manganese. Using the density of α-Mn and the profilometry data given above, one can calculate that with increasing thickness the coatings correspond to about 0.7, 2.0 and 3.1 μmolMn cm−2, respectively.
 | ||
| Fig. 3 X-ray diffractograms detected at an incidence angle of Ω = 1.0° for electrodes coated by PVD-deposited Mn-films of different thicknesses (bottom) and the same electrodes after being annealed in air for 16 h at 300 °C (top). | ||
After annealing the layers in air, the reflexions of α-Mn are not visible any longer and instead a mixture of at least two manganese oxides can be detected by GIXRD: manganosite (MnIIO) and hausmannite (MnII,III3O4). From a thermodynamic point of view, this result is unexpected, as one would anticipate that the MnIV-oxide pyrolusite (β-MnO2), the most stable manganese oxide in air at 300 °C,31 would be the main product. Indeed, the formation of β-MnO2 has been observed in a related study when Mn films were oxidized in air at 300–500 °C.32 However, in the prior study, atmospheric conditions were used rather than dry synthetic air which might result in a different rate of oxidation due to the influence of humidity. In addition to the reaction with oxygen, the metallic Mn film also reduces the ITO layer. As described in more detail for the following TEM results, a solid-solution of In–Sn exists within the film. Its presence can also be detected here by weak XRD reflexions that suggest the same tetragonal crystal structure as indium (see also ESI†).
Overall, the GIXRD data indicates a complete conversion of the metallic Mn precursor layers into Mn oxides during the reaction with air at 300 °C. Interestingly, the presented preparation method does not yield the most stable oxide MnO2 (which is known to be inactive in WOC13,20) but instead a mixed MnOx film with MnII as the main manganese oxidation state.
Cross-sectional TEM confirmed the polycrystalline nature of the films but also revealed a surprising and more complicated structure in the electrodes than was evident from XRD or SEM. Rather than forming a simple pair of layers on the glass substrate (layer sequence MnOx/ITO/glass), oxidation of the Mn/ITO/glass samples resulted in additional layers, as seen in Fig. 4A. Energy dispersive spectroscopy (EDS) indicated these layers to be MnOx/In–Sn/MnOx/ITO/glass (Fig. 4B–E). XRD signals for In with dissolved Sn corroborate this finding (Fig. 3).
 | ||
| Fig. 4 (A) Bright field TEM micrograph of manganese film cross-sections for a 50 nm electrode after annealing in air. The dark layer on the surface is a protective Au layer added only for TEM measurements. (B) HAADF STEM image. An EDS map of the film allowed elemental identification of the five distinct layers, with (C) Mn, (D) O, (E) In, and (F) Sn. | ||
This unusual layered structure is the result of two competing oxidation processes: while Mn atoms near the surface are oxidized by air during annealing, Mn atoms near the ITO layer are oxidized by In3+/Sn4+. Indium and tin remain together in roughly the same proportion as in the original ITO and are fully reduced to form an In–Sn alloy, as evidenced by the lack of O (green) in the In-rich (red) areas in the EDS map (Fig. 4D–E). This conclusion could be confirmed by calculations, which showed that the reductions of In2O3 and SnO2 by Mn metal (resulting in In–Sn and MnO) is thermodynamically favoured when oxygen is scarce (see ESI, Fig. S4†).
Concerning the position of the layers, it appears that Mn diffuses into the ITO and reduces it to form In and Sn, resulting in a MnOx layer inside the ITO and leaving the In and Sn in their original positions. The co-existence of In and Sn in the same proportion after reduction points to the low mobility of In and Sn, so that the alternating layer sequence MnOx/In–Sn/MnOx/ITO seems to be the result of the greater mobility of Mn compared to In and Sn during solid-state diffusion.
Despite the fact that ITO is a common substrate for WOC electrocatalysts, this phenomenon has not been reported for MnOx films prepared by other methods and is most likely the consequence of the unusual fact that our route involves a metallic intermediate with manganese in the oxidation state Mn0. However, unusual alternating layers in diffusion couples have been observed in unrelated studies and were in these cases also attributed to large differences in the mobility of species in a multi-component system.33
Regardless of the described multi-layer structure, the EDS map provides evidence that only the elements Mn and O are present at the top ∼50 nm of the electrodes (where catalysis takes place). This region can therefore still be best described as a manganese oxide MnOx, and the internal reduction of In and Sn is not expected to affect catalytic performance. Nevertheless, this unexpected finding suggests that other materials (which do not react with manganese oxides) might be better choices as conductive supports for MnOx catalyst films.
Electrochemical properties and water-oxidation catalysis
The electrochemical properties of the air-annealed electrodes were investigated using a sequence of cyclic voltammetry (CV) and chronoamperometry measurements in aqueous phosphate buffer at pH 7 (see ESI† for details). We note that a neutral electrolyte is not ideal to reach maximum WOC current densities. Nevertheless, water electrolysis at pH 7 has been widely studied recently, for example with the aim to develop catalysts for PEC devices (“artificial leaves”). So far, such water-splitting PECs cannot be immersed into corrosive electrolytes because of their complex multi-layer architectures. They also operate at moderate current densities of 1–10 mA cm−2 due to the limitations of their light-absorbing components.3,4 Additionally, WOC at non-alkaline conditions has to be mastered if one aims to couple water oxidation to CO2 reduction, which has to take place at the near-neutral pH of a HCO3−-buffered electrolyte.34 In view of these points, WOC studies at pH 7 are an important addition to “normal” WOC studies at pH > 12. As already mentioned in the introduction, this might generally be the pH-regime especially suitable for Mn-based catalysts, and thus we chose a neutral phosphate electrolyte as reaction medium for this study.Fig. 5 shows CV scans recorded for a “50 nm-electrode” before and after a conditioning cycle involving oxidizing potentials well above +1.9 V. The sharp rise of the current at ∼+1.7 V and the small differences between outward and reverse scans are very similar to e.g. a platinum metal foil at this potential window. Catalytic waves for H2O oxidation can be detected for E > ∼+1.6 V, while the “pre-waves”, characteristic of bulk redox-state changes of the Mn ions, which are often found for electrodeposited or printed MnOx-electrodes,15,16,24 are comparably small. Furthermore, the fact that the CV scans recorded before and after ∼30 min of conditioning time at E > 1.6 V are very similar indicates that the material is changed only slightly by applying oxidizing potentials. The fraction of ‘redox-active’ manganese ions undergoing oxidation state changes within the relevant potential range can be roughly quantified by analysing the reductive currents of the cathodic CV scan for electrode potentials ranging from 900 to 1600 mV: the current density of ∼−50 μA cm−2 recorded for 35 s correspond to the transfer of ∼18 nmol of electrons per cm2. Even if all these reducing equivalents were used for the re-reduction of previously oxidized manganese centres, this only corresponds to a fraction of less than 3% of the Mn atoms changing their oxidation states by one unit (compare values for μmolMn cm−2 calculated above). Therefore, it seems safe to conclude that the Mn oxidation state of the bulk of the oxide layer does not change in response to the applied electrochemical potential. This is in clear contrast to electrodeposited birnessite type MnOx, where typically about half of the manganese ions undergo MnIII to MnIV oxidation state changes within the potential range of 900–1600 mV.12 Accordingly, there may be a redox-active phase of amorphous birnessite in the “50 nm-electrode”, but it could comprise only about 6% of the manganese ions.
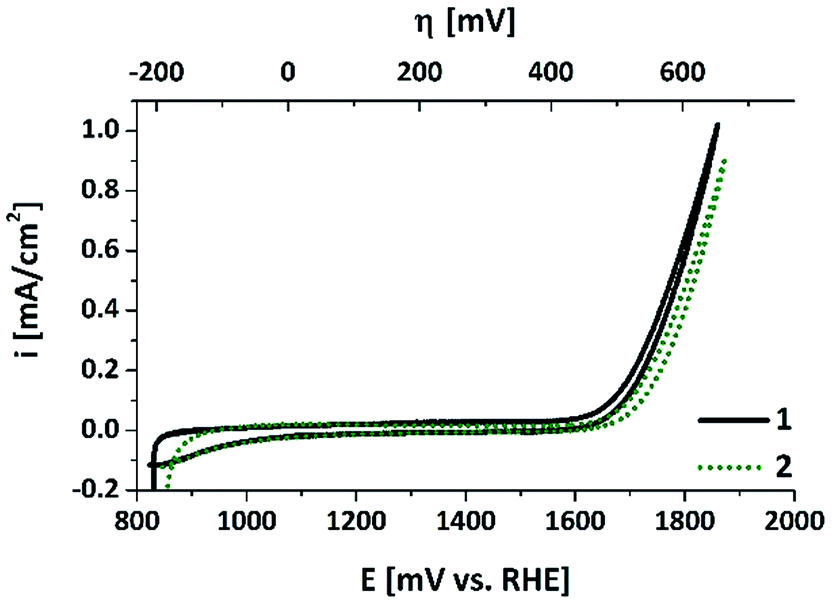 | ||
| Fig. 5 CV scans for an annealed “50 nm-electrode” in aqueous phosphate buffer (pH 7, overpotential η for water-oxidation indicated at the top). The two curves show data for CV scans recorded before (1) and after (2) a “Tafel plot sequence” involving about 30 min operation at potentials >1.6 V (see ESI† for details). Sweep rate: 20 mV s−1. | ||
Finally, we would like to note that in the CV scans, current densities of up to 1 mA cm−2 are reached with these electrodes, so our fabrication method seems to result in at least a reasonable electrical contact between the MnOx-film and the ITO support. The electrochemical properties described so far are also found for the anodes bearing thicker oxide layers with two important differences (see ESI, Fig. S5†): (a) for 150 nm- or 250 nm-electrodes, the catalytic currents for E > 1.6 V are much smaller (about one fifth of the values for the 50 nm film); (b) the current densities during the reverse sweeps are also lower (∼−25 μA cm−2 for 1.6 > E > 0.9 V), meaning that the fraction of potentially redox-active Mn ions is even smaller in these cases.
Chronoamperometry experiments were carried out to probe the ability of the electrodes to sustain electrochemical WOC over extended time periods. As shown in Fig. 6, significant water-oxidation currents could be detected for at least 2 h when the MnOx-electrodes prepared by Mn PVD and air-annealing were used as anodes in a water-electrolysis cell at pH 7 and η = 540 mV.
 | ||
| Fig. 6 Chronoamperometry traces for MnOx-electrodes of different film thicknesses operated for 2 h as water-oxidation anodes at E = +1.77 V (η = 540 mV) in aqueous 1/15 M phosphate buffer (pH 7). | ||
For all three investigated film thicknesses we find a marked current decrease during the first ∼20 min of electrolysis. This phenomenon is well documented also for other types of MnOx-electrodes and might be explained by capacitive charging and surface rearrangement reactions, which contribute to the current density (i) during the first period of operation, followed by the “pure” water-oxidation current detected afterwards. For the thicker oxide films, the latter phase is characterized by a very stable WOC current density. However, i is rather low – less than 50 μA cm−2 – and thus a factor of ∼20 smaller than for e.g. anodes coated by μm-thick layers of the amorphous MnIII,IV oxide birnessite.15 On the other hand, it should be noted that film stability is excellent, especially when compared with thin films prepared by electrodeposition methods.16 The situation is different for the thinnest coating of 50 nm MnOx: here, much higher current densities of >100 μA cm−2 are found, but this comes at a price of reduced stability, indicated by continuously decreasing water-oxidation currents (Fig. 6).
The current density trends described here for 2 h of electrolysis at constant potential are mirrored by the Tafel analysis of potential-step experiments. Depending on the applied potential, i varies in the range of 5–500 μA cm−2, and again the values observed for the 50 nm-film are much higher (but also much more unstable) than for thicker layers (see SI, Fig. S6†). We are aware of the fact that WOC current densities well below 10 mA cm−2 are most probably too low for practical applications and that higher values have already been reached for MnOx at neutral conditions. On the other hand, we would like to stress that the anodes described here show encouraging stability during long-term operation. Though a behaviour as shown in Fig. 6 is definitely not optimal yet, it is to the best of our knowledge among the best for thin film MnOx-electrodes, where data for extended operation is either missing or often shows a rapid loss of activity on the time-scale of minutes.16,26,28,29,35
Post operando investigations by XRD, TEM and XAS
GIXRD at Ω = 1.0° was used again to examine the oxidized electrode originally coated with 150 nm of Mn after electrolysis to observe any significant changes in the film. We identified the same phases in the film after electrolysis, as shown in Fig. 7. Scanning electron microscopy of the surface and a cleaved cross-section of the same films revealed a similar thickness and surface morphology after extended electrochemical testing (see ESI, Fig. S7†) as before testing (Fig. 2, C/D), albeit with a few localized regions of possible electrochemical corrosion.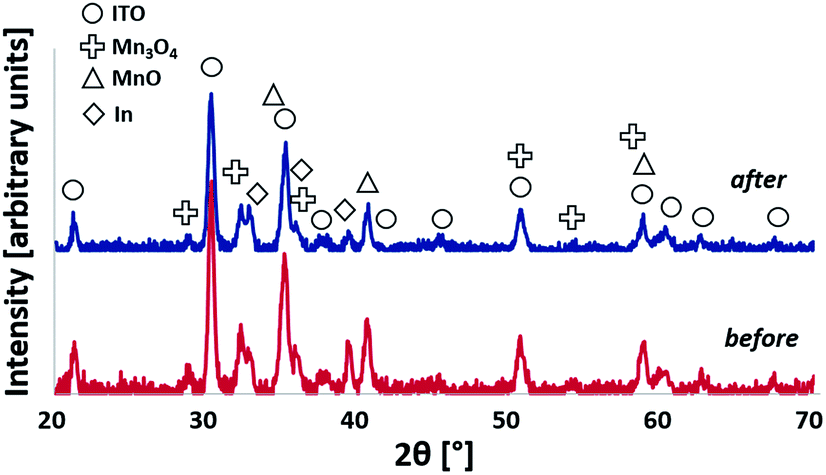 | ||
| Fig. 7 X-ray diffractograms detected at an incidence angle of Ω = 1.0° of 150 nm Mn-coated electrode before (bottom) and after (top) electrolysis (2 h at E = +1.77 V). | ||
The TEM examination of an electrode after WOC operation demonstrated the same layered structure as the previously characterized sample, where MnOx layers alternate with In–Sn layers (Fig. 8). This sample, however, also exhibited areas where ITO was completely reduced down to the substrate. This difference compared to the thinner layer shown in Fig. 4 is likely due to a thicker starting Mn layer allowing the Mn to fully reduce the ITO. Regardless of the internal structure, the near-surface region of the electrodes where catalysis takes place can be described as an In- and Sn-free MnOx.
 | ||
| Fig. 8 (A) Bright field TEM micrograph of manganese film cross-sections for a 150 nm electrode after 2 h of operation as WOC anode (E = +1.77 V). The dark layer on the surface is a protective Au layer added during sample preparation. (B) HAADF STEM image. An EDS map of the film allowed elemental identification of the five distinct layers, with (C) Mn, (D) O, (E) In, and (F) Sn. | ||
Furthermore, we used X-ray absorption spectroscopy (XAS) to study the effect of annealing and water-oxidation electrocatalysis on the atomic structure of the deposited films. In Fig. 9A, X-ray absorption near edge spectra (XANES) for the Mn K-edge are shown. The XANES spectrum of the as deposited material (green line) corresponds to Mn metal as it is very similar to the reference spectrum of α-Mn. Annealing at 300 °C for 16 h in air results in a shift of the position of the absorption edge to higher energies, indicating an oxidation of the metallic phase. The XANES spectrum of this oxidized material can be assigned to manganosite (MnIIO, rock salt structure) as the main phase by comparison to a reference spectrum. The formation of MnO is confirmed by the EXAFS spectra (X-ray absorption fine structure, Fig. 9B), where peaks up to a reduced distance of 6 Å indicate a well-ordered crystalline material.
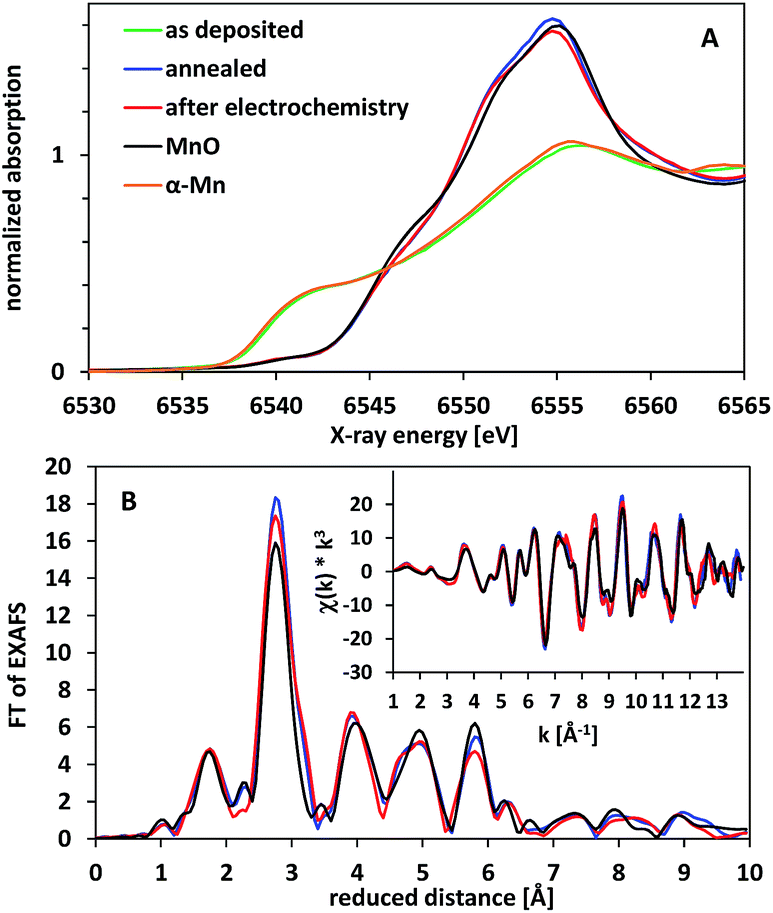 | ||
| Fig. 9 X-ray absorption spectra of electrodes coated by thin Mn/MnOx layers. (A) XANES spectra. (B) Fourier transformed EXAFS spectra (original EXAFS spectra shown as inset). Spectra for α-Mn (orange) and manganosite (MnO, black) reference materials are shown for comparison. | ||
The oxide material formed after annealing shows a high stability at WOC conditions. When exposed to an oxidation potential of E = +1.77 V (η = 540 mV) for 2 h, the XAS spectra show that the bulk material is hardly changed (red lines in Fig. 9). This is to some extent surprising, as MnO should not be stable at these E-/pH-values according to the Pourbaix diagram.14,19 Manganosite is additionally known to be catalytically inactive in WOC when Ce4+ is used as an oxidation agent.20 However, some of us reported previously that MnO nanoparticles can be oxidized by the strong chemical oxidant Ce4+, resulting in the formation of an active, birnessite-type catalyst with a layered MnOx structure and a Mn oxidation state above +3.5.10
In the light of this observation and also the XRD and TEM data presented above, we thus suspect that in our case a partial oxidation of the electrode surface might occur upon exposure to oxidizing potentials, resulting in a very thin layer of Mn2O3 and/or birnessite on top of (and protecting) the MnO phase below. At a thickness of 260 nm for the oxide layer, XAS can typically not detect additional phases (especially amorphous ones) if they are present in an amount below 10% of the volume of the investigated material. Here, the amount of material converted to Mn2O3 and/or birnessite must be even much smaller, as we would otherwise also detect pronounced changes by XRD and/or electron microscopy for the anodes after electrolysis.
Conclusions
In conclusion, we find that physical vapour deposition of Mn metal followed by annealing in air at 300 °C is a promising new route to prepare MnOx-coated anodes for water electrolysis. The oxide layers obtained in this way are in very good electrical contact with the underlying ITO substrate and can be characterised as compact mixtures of the nanocrystalline MnII/III-oxides manganosite (MnIIO) and hausmannite (MnII,III3O4). Unlike common other methods to coat surfaces with crystalline MnII/MnIII-oxide catalysts, this new route does not involve temperatures of 500–1000 °C, which are prohibitively high for many support materials. It also results in dense layers of homogeneous thicknesses.For the most part, the films' compositions are not changed even after 2 h of continuous WOC operation at ∼+1.8 V vs. RHE in water (pH 7). However, as it is known that MnO and Mn3O4 are hardly active in WOC, we suspect that a very thin layer of amorphous MnIII/IV-oxide forms at the surface of the coating and acts as the real water-oxidation catalyst. This has been reported before in related studies, where birnessite-type MnOx (with average Mn oxidation states of +3.5–3.9 and known for their good WOC activities) were formed starting from MnII/III-oxides at oxidising conditions.10,18,36 Such a surface-amorphisation might also occur here and could explain the small morphology changes detected by SEM for anode surfaces after electrochemical operation (ESI, Fig. S7†).
Water oxidation at near-neutral pH is of high interest regarding electrocatalytic CO2 reduction. For insufficiently understood reasons (one might actually be the beneficial effect of carbonate-containing electrolytes37), the current densities for water oxidation at alkaline pH are generally much higher when compared to neutral conditions at the same overpotential. The WOC current densities reached by the herein presented anodes in long-term experiments at pH 7 (2 h) are so far small (i ≈ 50–100 μA cm−2 for η = 540 mV), but it should be noted that in our cases they originate from thin, smooth catalyst layers. A normalization of the catalytic activities per Mn and/or per electrochemically active surface area might thus put the performance anodes in a more favourable light, but as only very little “normalized data” for MnOx catalyst layers exists, a comparison of the numbers did not appear feasible to us at the moment.
Furthermore, we would like to mention that both the PVD process and the annealing steps allow for multiple ways to optimize the properties of the catalytic layer (nanostructuring, increase of the MnIII-percentage, etc.) and we are therefore optimistic that higher current densities can be reached without sacrificing stability. The PVD approach also allows to deposit thin MnOx layers on a wide variety of materials in a very controlled way. As we find that the ITO support used here is most likely not the best choice (side reactions with Mn and low surface area of this material), we suspect that a screening of alternative back-contacts might also allow significant improvements in performance. Finally, we would like to note that the presented PVD method is also very suitable for the coating of the surfaces of PEC devices (“artificial leaves”) and the preparation of “study-objects” for mechanistic investigations on Mn-based water-oxidation catalysis.
Experimental section
Electrode preparation by PVD/air-annealing
Manganese films were deposited by electron beam evaporation on indium tin oxide films on glass obtained from Sigma Aldrich. Manganese pieces with 99.95% purity (metals basis) were obtained from Alfa Aesar as the source material. Deposition was performed at a rate of ∼1 Å s−1 in a Kurt J. Lesker Axxis multi-purpose physical vapour deposition system with a base pressure < 2 × 10−7 Torr (<2.7 × 10−7 mbar). Samples were annealed for 16 h at 300 °C in a tube furnace in dry synthetic air from a gas cylinder flowing at rate of 100 sccm.Profilometry
Film thicknesses were measured using a KLA-Tencor P-10 surface profiler and are the average of three separate measurements made across a step created on a calibration sample using Kapton tape to mask part of the sample.SEM measurements
Micrographs of the surfaces of the films and of cleaved cross-sections were obtained in a Leo 1530 (operated at 5 kV) or Zeiss Merlin (operated at 10 kV) field emission scanning electron microscope.X-ray powder diffractometry
Grazing incidence X-ray diffraction was performed on a PANalytical X'Pert Pro multipurpose diffractometer. X-radiation from a Cu source was supplied at angle of incidence (Ω) of 1° with a 0.027 mm collimator, and data were collected over 2θ angles from 20–70°.Electrochemical measurements
A typical three-electrode set-up was used to carry out cyclic voltammetry and chronoamperometry experiments. It consisted of a Ag/AgCl reference electrode (ALS, saturated KCl system), a Pt rod as counter electrode (Metrohm, diameter ∼2 mm) and the Mn-coated ITO electrode (active geometric area ∼1.3 cm2) as working electrode. 50 mL of an air-saturated phosphate buffer (pH 7, prepared from KH2PO4 and Na2HPO4·2H2O, total phosphorous concentration 66 mM) served as the electrolyte solution. All electrochemical measurements were carried out in air at room temperature using a computer-controlled VersaSTAT 4 potentiostat/galvanostat from Princeton Applied Research. Concerning the conversion of reference electrode potentials, please see ESI.†X-ray absorption spectroscopy
X-ray absorption spectra were measured at the BESSY II synchrotron of the Helmholtz-Zentrum für Materialien und Energie (HZB, Berlin, Germany) at 20 K in a liquid-helium cryostat as described previously.38 The spectra of the coated electrodes and the α-Mn reference foil (10 μm thick, Goodfellow) were collected at the XPP/KMC-3 beamline, with Mn foil recorded in absorption mode with a homemade ionisation chamber and the spectra of the electrodes measured in fluorescence mode using a single channel energy-resolving Ge detector (Canberra). The spectrum of the MnO reference powder (Sigma Aldrich) had been previously recorded at the KMC-1 beamline of BESSY II in absorption mode after dilution of MnO with boron nitride in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 volume ratio. For all samples, KMnO4 powder was measured simultaneously in transmission mode and used as energy calibration standard (the pre-edge maximum was set to 6543.3 eV). Other details about data analysis and simulations are provided elsewhere.38
:20 volume ratio. For all samples, KMnO4 powder was measured simultaneously in transmission mode and used as energy calibration standard (the pre-edge maximum was set to 6543.3 eV). Other details about data analysis and simulations are provided elsewhere.38
Transmission electron microscopy and energy dispersive spectroscopy
Cross-sectional TEM samples were prepared with the Helios Nanolab 660 or the Quanta 200 3D Dual Beam, both scanning electron microscopes coupled with a focused ion beam (FIB). The samples were imaged and analysed using the Talos F200X with SuperX EDS detector (operated at 200 kV).Acknowledgements
Special funds from the International Office of the ALU Freiburg financed a two-month research stay of C. E. F. at PSU that kick-started this project. S. E. M. thanks the Penn State Institutes for Energy and the Environment and F. K. thanks Penn State's College of Engineering Research Institute for funding. Ivo Zizak from the Helmholtz-Zentrum Berlin provided excellent technical support at the XPP/KMC-3 beamline of the BESSY II synchrotron. For the German groups, financial support by the Federal Ministry of Education and Research (BMBF cluster project MANGAN and project IN-SITU-XAS/05K16KE2) and the German Research Foundation (DFG priority program SPP1613, grants KU2885/2-2 and DA402/7-2; project A4 in CRC 1078 hosted by Freie Univ. Berlin) are gratefully acknowledged.Notes and references
- (a) Solar energy for fuels, ed. H. Tüysüz and C. K. Chan, Springer, Heidelberg, New York, 2016, vol. 371 Search PubMed; (b) Chemical energy storage, ed. R. Schlögl, de Gruyter, Berlin, 2013 Search PubMed; (c) Energy production and storage, ed. R. H. Crabtree, Wiley, Chichester, 2010 Search PubMed; (d) Molecular water oxidation catalysis, ed. A. Llobet, Wiley, Chichester, 2014 Search PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767 CrossRef CAS PubMed.
- K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., Int. Ed., 2013, 52, 10426 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120 CrossRef CAS PubMed.
- B. M. Hunter, J. D. Blakemore, M. Deimund, H. B. Gray, J. R. Winkler and A. M. Müller, J. Am. Chem. Soc., 2014, 136, 13118 CrossRef CAS PubMed.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737 CrossRef CAS PubMed.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002 CrossRef CAS PubMed.
- A. Indra, P. W. Menezes, I. Zaharieva, E. Baktash, J. Pfrommer, M. Schwarze, H. Dau and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 13206 CrossRef CAS PubMed.
- (a) A. Iyer, J. Del-Pilar, C. K. King'ondu, E. Kissel, H. F. Garces, H. Huang, A. M. El-Sawy, P. K. Dutta and S. L. Suib, J. Phys. Chem. C, 2012, 116, 6474 CrossRef CAS; (b) M. M. Najafpour, D. J. Sedigh, B. Pashaei and S. Nayeri, New J. Chem., 2013, 37, 2448 RSC; (c) M. Wiechen, M. M. Najafpour, S. I. Allakhverdiev and L. Spiccia, Energy Environ. Sci., 2014, 7, 2203 RSC.
- I. Zaharieva, D. González-Flores, B. Asfari, C. Pasquini, M. R. Mohammadi, K. Klingan, I. Zizak, S. Loos, P. Chernev and H. Dau, Energy Environ. Sci., 2016, 9, 2433 CAS.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494 CrossRef CAS PubMed.
- P. Kurz, Top. Curr. Chem., 2016, 371, 49 CrossRef CAS PubMed.
- S. Y. Lee, D. González-Flores, J. Ohms, T. Trost, H. Dau, I. Zaharieva and P. Kurz, ChemSusChem, 2014, 7, 3442 CrossRef CAS PubMed.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081 CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 1519 CrossRef CAS PubMed.
- M. Fekete, R. K. Hocking, S. L. Y. Chang, C. Italiano, A. F. Patti, F. Arena and L. Spiccia, Energy Environ. Sci., 2013, 6, 2222 CAS.
- N. Takeno, Atlas of Eh-pH Diagrams, Geological Survey of Japan, published online, 2005 Search PubMed.
- C. E. Frey and P. Kurz, Chem.–Eur. J., 2015, 21, 14958 CrossRef CAS PubMed.
- (a) N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588 CrossRef CAS PubMed; (b) M. M. Najafpour, G. Renger, M. Holynska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886 CrossRef CAS PubMed.
- M. Morita, C. Iwakura and H. Tamura, Electrochim. Acta, 1979, 24, 357 CrossRef CAS.
- J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447 CrossRef CAS.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525 CrossRef CAS PubMed.
- K. L. Pickrahn, S. W. Park, Y. Gorlin, H.-B.-R. Lee, T. F. Jaramillo and S. F. Bent, Adv. Energy Mater., 2012, 2, 1269 CrossRef CAS.
- K. L. Pickrahn, Y. Gorlin, L. C. Seitz, A. Garg, D. Nordlund, T. F. Jaramillo and S. F. Bent, Phys. Chem. Chem. Phys., 2015, 17, 14003 RSC.
- (a) K. L. Pickrahn, A. Garg and S. F. Bent, ACS Catal., 2015, 5, 1609 CrossRef CAS; (b) H. Jin, D. Hagen and M. Karppinen, Dalton Trans., 2016, 45, 18737 RSC; (c) Y. W. Li, Q. Qiao, J. Z. Zhang, Z. G. Hu and J. H. Chu, Thin Solid Films, 2015, 574, 115 CrossRef CAS.
- F. Mattelaer, T. Bosserez, J. Rongé, J. A. Martens, J. Dendooven and C. Detavernier, RSC Adv., 2016, 6, 98337 RSC.
- N. C. Strandwitz, D. J. Comstock, R. L. Grimm, A. C. Nichols-Nielander, J. Elam and N. S. Lewis, J. Phys. Chem. C, 2013, 117, 4931 CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- A. F. Holleman and E. Wiberg, Lehrbuch der Anorganischen Chemie, de Gruyter, Berlin, 2007 Search PubMed.
- R. Baca, Mater. Sci. Semicond. Process., 2013, 16, 1280 CrossRef CAS.
- C. R. Kao and Y. A. Chang, Acta Metall. Mater., 1993, 41, 3463 CrossRef CAS.
- (a) B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541 CrossRef CAS PubMed; (b) M. Schreier, L. Curvat, F. Giordano, L. Steier, A. Abate, S. M. Zakeeruddin, J. Luo, M. T. Mayer and M. Gratzel, Nat. Commun., 2015, 6, 7326 CrossRef CAS PubMed.
- X.-F. Luo, J. Wang, Z.-S. Liang, S.-Z. Chen, Z.-L. Liu and C.-W. Xu, Int. J. Hydrogen Energy, 2017, 42, 7151 CrossRef CAS.
- A. Ramírez, P. Hillebrand, D. Stellmach, M. M. May, P. Bogdanoff and S. Fiechter, J. Phys. Chem. C, 2014, 118, 14073 Search PubMed.
- B. M. Hunter, W. Hieringer, J. R. Winkler, H. B. Gray and A. M. Müller, Energy Environ. Sci., 2016, 9, 1734 CAS.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details; additional figures on results from profilometry, electron microscopy and electrochemical measurements. See DOI: 10.1039/c7se00172j |
| ‡ The first two authors contributed equally to this work as main experimental investigators. |
| This journal is © The Royal Society of Chemistry 2017 |