
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual experimental–theoretical perspective on ESPT photoacids and their challenges ahead†
Niklas
Sülzner
 *a,
Gregor
Jung
*b and
Patrick
Nuernberger
*c
*a,
Gregor
Jung
*b and
Patrick
Nuernberger
*c
aLehrstuhl für Theoretische Chemie, Ruhr-Universität Bochum, 44780 Bochum, Germany. E-mail: niklas.suelzner@rub.de; Tel: +49 234 32 24523
bBiophysikalische Chemie, Universität des Saarlandes, 66123 Saarbrücken, Germany. E-mail: g.jung@mx.uni-saarland.de; Tel: +49 681 302 71320
cInstitut für Physikalische und Theoretische Chemie, Universität Regensburg, 93040 Regensburg, Germany. E-mail: patrick.nuernberger@ur.de; Tel: +49 941 943 4487
First published on 2nd December 2024
Abstract
Photoacids undergo an increase in acidity upon electronic excitation, enabling excited-state proton transfer (ESPT) reactions. A multitude of compounds that allow ESPT has been identified and integrated in numerous applications, as is outlined by reviewing the rich history of photoacid research reaching back more than 90 years. In particular, achievements together with ambitions and challenges are highlighted from a combined experimental and theoretical perspective. Besides explicating the spectral signatures, transient ion-pair species, and electronic states involved in an ESPT, special emphasis is put on the diversity of methods used for studying photoacids as well as on the effects of the environment on the ESPT, illustrated in detail for 8-hydroxypyrene-1,3,6-trisulfonate (HPTS) and the naphthols as examples of prototypical photoacids. The development of exceptionally acidic super-photoacids and magic photoacids is subsequently discussed, which opens the way to applications even in aprotic solvents and provides additional insight into the mechanisms underlying ESPT. In the overview of highlights from theory, a comprehensive picture of the scope of studies on HPTS is presented, along with the general conceptualization of the electronic structure of photoacids and approaches for the quantification of excited-state acidity. We conclude with a juxtaposition of established applications of photoacids together with potential open questions and prospective research directions.
1 Introduction
Proton transfer (PT) is omnipresent in chemistry and its related fields, including countless examples from biochemistry, chemical synthesis, and catalysis, as well as industrial applications.1–3 It is at the heart of acid–base catalysis4–7 and continues to be relevant in today's research, e.g., in the development of fuel cells.8 In biochemical systems, to name a few examples, the structure and functioning of proteins is determined by the protonation states of the amino acid side chains,9,10 proton gradients along biomolecular membranes act as driving forces for the ATP synthesis,11 and many enzymes either directly catalyze—at least in one step of their catalytic cycle—an acid–base reaction12 or their activity involves proton transfer along a conduction wire, i.e., a preformed hydrogen-bond network of amino acids and water molecules (e.g., in photosystem II13). Proton transfer can also take place in an electronically excited state, e.g., as in fluorescent proteins14–28 or in the chromophore of Firefly's D-luciferin.29–36 Moreover, such an excited-state proton transfer (ESPT) of anthocyanin pigments in plants is biologically relevant for the protection from photodamage since the ESPT provides a fast deactivation channel, efficiently converting the absorbed light energy into heat.37,38Owing to this fundamental importance, there has been a huge interest in the study of proton-transfer reactions since the beginnings of chemistry,39,40 both with respect to the underlying thermodynamics as well as the chemical kinetics. From a thermodynamic perspective, the acid dissociation constant Ka or the pKa, i.e., its negative decadic logarithm, is the most important quantity as it quantifies the Brønsted acid strength.41 While the experimental determination of pKa has nowadays become a standard in aqueous solution owing to a broad variety of well-established techniques (e.g., potentiometric, NMR-spectroscopic, or spectrophotometric titrations),42 the situation is much more complicated in non-aqueous solution since the drastically lower acidity constants in combination with other complications such as low solubilities or homoconjugation challenge the accuracy of experimental detection methods.43,44 Due to these difficulties, computational pKa predictions have become much more important in these environments, providing an alternative path to pKa.45,46
Regarding the kinetics, a molecular probe is typically required to experimentally obtain information on the PT mechanisms including relevant intermediates as well as rate constants for the individual steps. For this task, so-called photoacids have established as particularly powerful tools during the last decades (Fig. 1 and vide infra). These photoacids are molecules that exhibit an increase in acidity (i.e., a decrease in pKa) upon electronic excitation.47,48 In other words, such a photoacid is a stronger acid in its electronically excited state (usually the lowest-lying singlet state S1) compared to the (singlet) ground state S0. Thus, the excited-state pKa (from now on referred to as 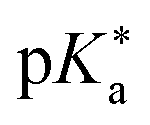 ) is smaller than the ground-state pKa. As a consequence, photo-excitation can trigger an excited-state proton transfer (ESPT)49 from the photoacid to a nearby proton acceptor, e.g., the solvent or any other Brønsted base.50 While most (weak) photoacids
) is smaller than the ground-state pKa. As a consequence, photo-excitation can trigger an excited-state proton transfer (ESPT)49 from the photoacid to a nearby proton acceptor, e.g., the solvent or any other Brønsted base.50 While most (weak) photoacids 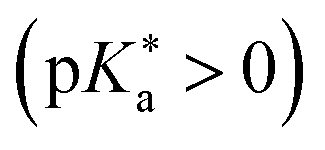 are—due to limited acid strength—restricted to ESPT in neat water51,52 and aqueous solutions containing an additional base53 or co-solvent,54–57 those with a negative
are—due to limited acid strength—restricted to ESPT in neat water51,52 and aqueous solutions containing an additional base53 or co-solvent,54–57 those with a negative  can also perform an ESPT in other protic (e.g., alcohols)58,59 or even aprotic solvents such as dimethyl sulfoxide (DMSO).60,61 In particular, the use of pulsed lasers has made it possible to temporally initiate the ESPT event, which conveniently allows to investigate the associated dynamics in real time using time-resolved spectroscopy.62,63
can also perform an ESPT in other protic (e.g., alcohols)58,59 or even aprotic solvents such as dimethyl sulfoxide (DMSO).60,61 In particular, the use of pulsed lasers has made it possible to temporally initiate the ESPT event, which conveniently allows to investigate the associated dynamics in real time using time-resolved spectroscopy.62,63
 | ||
| Fig. 1 Milestones of experimental research on photoacids exhibiting ESPT. Color coding: studies involving 8-hydroxypyrene-1,3,6-trisulfonate (HPTS) (blue), naphthols (green), or the recent HPTS-based super-photoacids introduced by Jung (orange); contributions from the Huppert group (purple); application of novel experimental techniques (red). Abbreviations: femtosecond (fs), visible (Vis), mid infrared (mIR), femtosecond Raman scattering (FSRS), terahertz (THz), spectroscopy (spec.), solvatochromism (solvato.), super-photoacids (SPAs), new inspection and analysis of the ultrafast time-resolved fluorescence data on HPTS (Huppert Revisited). | ||
Research on light-induced triggering of processes involving proton transfer is rather broad and also covers many phenomena beyond ESPT. The area of metastable-state photoacids64–68 or generally photacid generators (PAGs)69–72 is closely related, and so is the relation between electron and proton transfer73 and proton-coupled electron transfer (PCET)74–78 in the widest sense, however these topics are not within the scope of this review, and neither are triplet photoacids,79–84 Lewis photoacids,85–87 nor intramolecular ESPT (ESIPT).88–90 Rather, within the spectrum of existing photoacid reviews (e.g., ref. 2, 47, 48, 50 and 91–98) the purpose of this article is to give an up-to-date overview on photoacids from an experimental–theoretical perspective (including their upcoming challenges) with a focus on the kinetics and the thermodynamics of the ESPT process.
2 A brief history of photoacids
2.1 The phenomenon of photoacidity
Photoacids have been the subject of ongoing scientific studies (see Fig. 1 for a selection of milestones in experimental photoacid research) since their initial description in the first half of the 20th century by Weber (1932),99 Terenin and Kariakin (1947),100 Petersen (1949),101 and Förster (1949, 1950),102–104 who had reported pH-dependent changes in the fluorescence spectra of several dye molecules; including 1,4-naphthylaminosulfonate, acridine, 8-aminopyrene-1,3,6-trisulfonate,‡ as well as 8-hydroxypyrene-1,3,6-trisulfonate§ and 6,8-dihydroxypyrene-1,3-disulfonate,¶ respectively. Recently afterwards, it has also been Förster who introduced the idea of considering a thermodynamic cycle that takes into account the acid–base equilibria for both the electronic ground state and the excited state to rationalize the phenomenon of photoacidity.102–105 In his memory, this cycle is today referred to as Förster cycle (Fig. 2). This thermodynamic cycle relates ΔpKa, the difference between the excited-state and ground-state (pKa) acidity, to the difference in the Gibbs free electronic excitation energies (ΔGexci) of the protonated (ROH) and deprotonated (RO−) species, as expressed in eqn (1)
and ground-state (pKa) acidity, to the difference in the Gibbs free electronic excitation energies (ΔGexci) of the protonated (ROH) and deprotonated (RO−) species, as expressed in eqn (1) | (1) |
 | ||
| Fig. 2 The Förster cycle. A simple thermodynamic cycle to account for an increased acidity, i.e., a decreased (Gibbs free) acid dissociation energy, ΔGdiss, in the first electronically excited state (S1) compared to the electronic ground state (S0), caused by different relative energies of the acid–base equilibria of the two electronic states. These are coupled through the (Gibbs free) electronic excitation energy, ΔGexci, of the protonated (ROH) and deprotonated species (RO−), respectively. An asterisk (*) is used for referring to the excited state. | ||
If the ground-state pKa is known, eqn (1) can be used to calculate 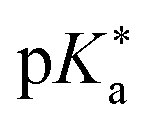 from the electronic transition energies; if the pKa is unknown, still the ΔpKa can be determined. Ideally, 0–0 transition energies should be inserted into eqn (1).106 Owing to its simplicity, the Förster cycle has become the most common route to
from the electronic transition energies; if the pKa is unknown, still the ΔpKa can be determined. Ideally, 0–0 transition energies should be inserted into eqn (1).106 Owing to its simplicity, the Förster cycle has become the most common route to  for both experiments and theory, although also other approaches exist (see Section 7).
for both experiments and theory, although also other approaches exist (see Section 7).
2.2 Different classes of photoacids
ESPT photoacids are typically hydroxyarene or aminoarene compounds and thus the acidic functional group is either a hydroxy (OH) or an amino (NH2) or ammonium (NH3+) group. The most common classes of photoacids include naphthols107–113 (Fig. 3b and c), hydroxyquinolines79,114–117 (Fig. 3e and f), hydroxycoumarins118–127 (Fig. 3d), and hydroxypyrenes61,103,128–131 (Fig. 3g), although also some anilines,132–134 naphthylamines99,101,135–137 and aminopyrenes103,138–144 are known. Phenols (Fig. 3a) also exhibit photoacid character (ΔpKa ∼ −6),145–149 but their UV absorption (∼270 nm) and weak fluorescence make them impractical for applications. Less known photoacids are hydroxyisoquinolines,150 fluorescein derivatives,151–153 hydroxybenzoquinolizinium ions,154,155 and tetracyanohydroquinone.156 The photoacidity of merocyanine-spiropyran systems both for metastable-state and ESPT photoacids has also been explored in more detail recently.68,157–163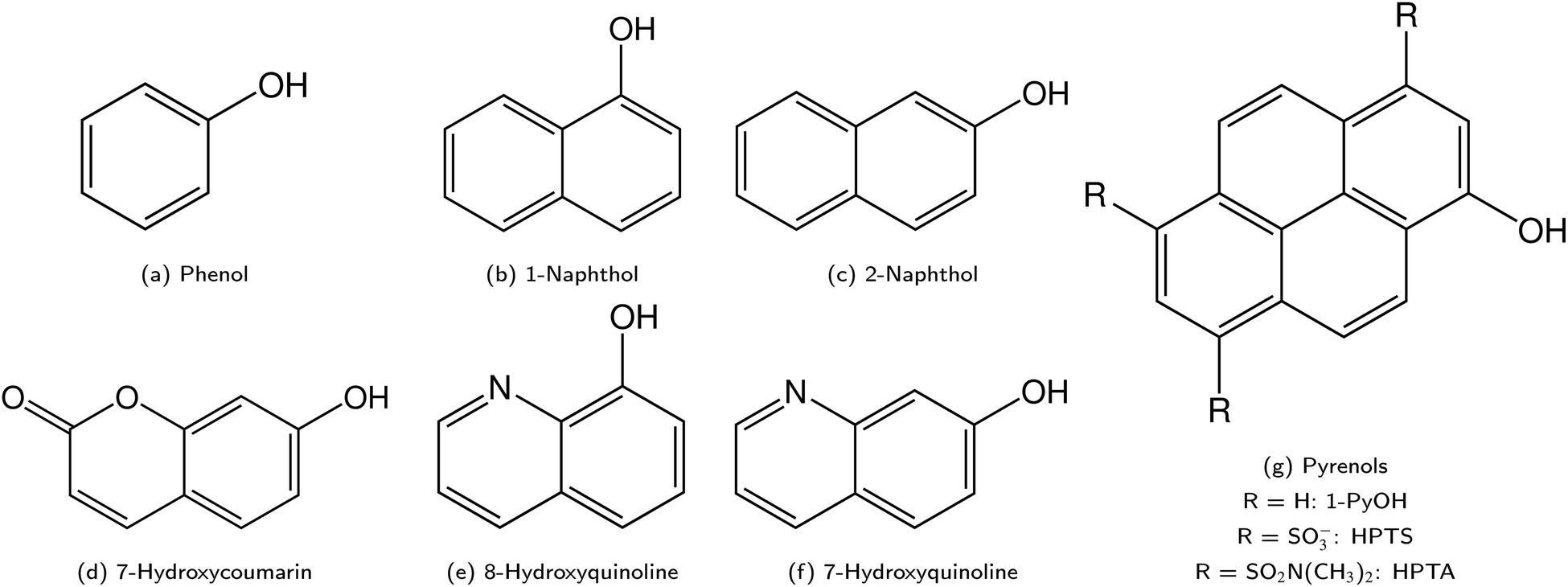 | ||
| Fig. 3 Overview on the different classes of photoacids. The corresponding naphthylamines and aminopyrenes are obtained by substituting the OH group by an NH2/NH3+ in (b and c) or (g), respectively. | ||
The counterparts of photoacids, i.e., photobases, also exist, but have only more recently attracted increased attention in various research fields.164–179 These photobases are compounds that become more basic in the excited state. To rationalize and quantify photobasicity, one simply has to switch the perspective: a photobase is a species whose conjugate acid becomes less acidic in the electronically excited state compared to the ground state. The term photobase is often used incorrectly for the corresponding base of an excited photoacid, which is in fact exactly the opposite of a photobase since these species are particularly weak excited-state bases.
While the aromatic OH group is typical for many photoacids, a common feature of photobases are N-heterocyclic, aromatic frameworks.180,181 A peculiarity is the combination of photoacidic and photobasic functional groups within one molecule.182–187 Here, an ESIPT is possible by contribution of protic solvents, and these systems therefore appear suitable to address the question to which extent water- or proton-wires exist in different solvent compositions.117,188–195
3 Spotlight HPTS—the supposably most studied photoacid
The research on photoacids has been largely stimulated by Huppert and co-workers, reaching back to the 1980s when Huppert—at that time under the supervision of Kolodney—had joined the photoacid community.196,198 After almost 40 years of research, Huppert can be considered one of the leading pioneers in photoacid chemistry. Moreover, he has also catalyzed the huge interest in 8-hydroxypyrene-1,3,6-trisulfonate (HPTS or pyranine), which is one of the first photoacids ever studied.100–102 To date, HPTS has probably become the photoacid investigated in greatest detail in the literature since its first mention in the 1930s,199 owing also to its wide applicability, e.g. as an efficient fluorophore and a ground-state pH sensor,200–203 and to its suitability for use in everyday products like text highlighters and dishwashing detergents. Studying the ESPT of HPTS in water using time-resolved spectroscopy has even been proposed as an educational experiment for students.204 The characteristics of the ESPT of HPTS have been deciphered in a vast number of environments and with many different experimental techniques, as well as with theoretical approaches, as highlighted in the following.3.1 ESPT in dependence on the environment
Aqueous solutions pertain an outstanding role among all studied conditions, and the ESPT to the solvent in bulk water has been analyzed in great detail.51,52,63,140,205–226 Beyond that, also the ESPT to a co-solute base such as acetate53,62,221,227–233 or (per)chloroacetate234–236 could be unveiled, and also the role of co-solvents on the ESPT of HPTS was addressed in solvent mixtures consisting of water and, e.g., methanol197,237–240 or DMSO.54,241 Other parameters include the effect of additional salts198,238,242–245 or the variation of temperature246,247 and pressure248 on the ESPT process. Challenging the limits of ESPT, even the ESPT of HPTS in ice and doped ices,246,249–255 in confined water nanopools of various reverse micelles (with anionic, neutral or cationic surfactants),256–263 or in Nafion fuel cell membranes260,264,265 have been explored. This scope also included many biological environments such as the cavity of cyclodextrin,266–268 other supramolecular assemblies,269 adsorbing bio-materials (hydrophobic insulin amyloid fibrils and hydrophilic cellulose surfaces; chitin, starch, chitosan),270–275 hydrogels,276–278 the presence of proteins,279 or even living cells.280–282 In all the experiments listed above, it was observed that ESPT dynamics of HPTS in aqueous environments can be manipulated through the interaction with adjacent molecules. Apart from these studies where water plays a decisive role, studies on ESPT of HPTS in non-aqueous solution are very scarce though, and for instance involve HPTS and acetate in methanol283 and HPTS in ionic liquids.284,285 Moreover, due to the three- or four-valent anionic nature, the vast majority of studies on HPTS take place only in actual solutions, in contrast to other photoacids (e.g., naphthols that are also studied in gas-phase solvent clusters286–289). One remarkable exception is a photoelectron spectroscopy study that reports a negative electron binding energy for the acid species of HPTS (generated via electrospray), demonstrating the immense reductive potential of such a highly charged anion in the gas phase.2903.2 An arsenal of spectroscopic techniques
Many different spectroscopic techniques have been applied to investigate HPTS and other photoacids, each one with its own advantages and varying accessible information. Changes in the electronic structure upon deprotonation allow spectroscopies in the visible spectral range such as UV/Vis absorption and fluorescence emission to be used for distinguishing the protonated from the deprotonated form. In its simplest form, pH titrations are used for the determination of ground-state pKa values, and fluorescence read-out, i.e. fluorescence excitation spectroscopy, is preferred due to its higher sensitivity.292 Consequently, pH measurements with few or even single molecules are possible, and nanomolar concentration of samples in the focus of a confocal microscope show stochastic fluorescence fluctuations as a result of continuous cycles of deprotonation and protonation events when only one of the ground-state species is excited by an appropriate laser source.291 The kinetics of this interconversion in the thermodynamic equilibrium are then accessible by fluorescence correlation spectroscopy when the photostability is high enough.293 Moreover, since the molecular vibrations are highly specific to intermolecular interactions, mid-infrared (mIR) absorption or Raman scattering spectroscopy can be used to further characterize possible PT intermediates such as ion pairs. In general, time-resolved spectroscopy with high time resolution is unrivaled to obtain information on the dynamics, i.e., the actual kinetics of the ESPT.To briefly visualize the additional information content, steady-state and time-resolved emission are juxtaposed in Fig. 4 for a super-photoacid. Whereas the steady-state fluorescence spectrum foreshadows that more than one emissive species contributes, the time-resolved data discloses which wavelength is emitted at which moment in time after photoexcitation. Thus, the ESPT and its characteristic time scale can be deduced, while more detailed analysis can also provide insight into additional transient species when photoacid and base or the ions of the nascent ion pair interact (vide infra).
 | ||
| Fig. 4 Emission of super-photoacid B (cf.Fig. 13d) in an acetone–water mixture (0.55 M of water). The time-resolved fluorescence (bottom panel) resolves that the steady-state fluorescence (top panel) comprises several contributions with different lifetimes. The displayed data is part of the study in ref. 294. | ||
Steady-state UV/Vis absorption and fluorescence emission spectroscopy have been applied since the early days for a qualitative characterization of the ground-state and excited-state equilibria of HPTS in water,103 but also more recently to its solvatochromism.295,296 The earliest transient absorption experiments of HPTS in aqueous solution were reported by Förster and Völker (1975)297 and Lundy-Douglas and Schelly (1976),298 both using flash photolysis (tens of ns time resolution), followed by laser photolysis experiments by Gutman, Huppert and Pines (1981, 1983).299,300 In parallel, early time-resolved fluorescence spectroscopy on the ps to ns timescale initially based on streak cameras and later time-correlated single photon counting (TCSPC)—both limited by a rather poor ps time resolution based on today's ultrafast standards—was used to study the slower excited-state dynamics,197,198,301 which allowed Agmon, Pines, and Huppert in 1986 to establish their well-known two-step model for the ESPT of HPTS to water (vide infra).205–211
According to the early two-step model for HPTS in water, the excited protonated species (ROH*) of HPTS first transfers its proton to a nearby water molecule under formation of a contact ion pair (CIP) within ∼90 ps. Subsequently, this CIP is separated in a diffusive process, yielding the free deprotonated form (RO−*) within 100 ps.206 Noteworthy, in seminal studies published in 1986, it could be unambiguously shown that the emission dynamics of excited HPTS in water proceed in a non-mono-exponential fashion (see Fig. 5), a consequence of geminate recombination within the ion pair consisting of RO−* and the generated cation. This process may be considered as a quasi-equilibration, while diffusion eventually facilitates the separation of the ions. Modelling of this reprotonation process of RO−* by including a recombination rate and assuming diffusive ion separation allowed a quantitative analysis of the observed dynamics. Furthermore, the discovery of a pronounced recombination contribution could explain why the ion separation is less efficient than expected.297
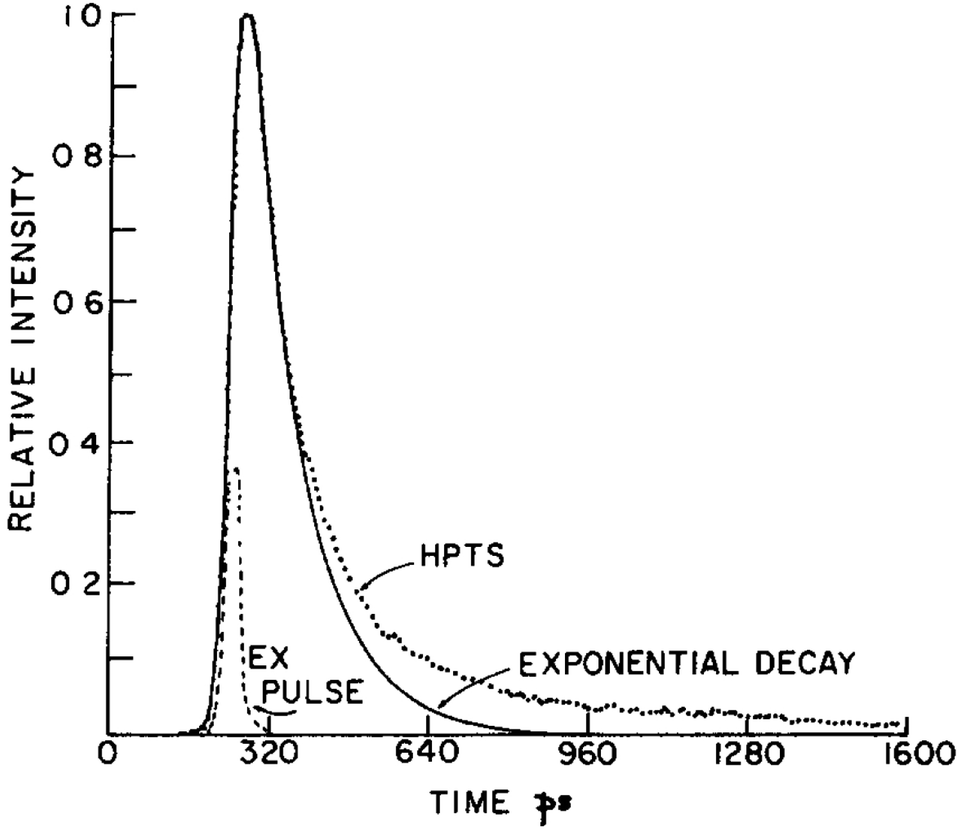 | ||
| Fig. 5 Fluorescence decay of HPTS in water together with an exponential fit, exemplifying that geminate recombination leads to a tail-like emission behavior of the excited protonated solute. The profile of the 25 ps excitation pulse is shown as well. This figure has been reproduced from ref. 206 with the kind consent of E. Pines and with permission from Elsevier, copyright 1986. | ||
In 1996, Tran-Thi and co-workers applied fs fluorescence up-conversion for the first time to study the ultrafast dynamics of HPTS in water213 and later (in 2000) complemented the inferences with results from transient UV/Vis absorption.214 In the same year, ultrafast transient absorption measurements on HPTS were also performed by Huppert et al., but for the bimolecular ESPT of HPTS to acetate.53 Owing to a much better time resolution compared to the previous TCSPC experiments, the results by Tran-Thi et al. revealed the additional involvement of two fast processes in the ESPT kinetics that precede the proton-transfer step, which had been previously unobserved and then started to challenge the two-step model for the ESPT (vide infra).213–216,302
Particularly in the course of this debate but also as a consequence of further issues on HPTS, ultrafast transient mIR absorption62,219–221,229–231,234–236,245,303 and fs stimulated Raman scattering225,226,232,239,304–308 in addition to UV/Vis spectroscopy52,53,140,213,214,217,223,229,233,309 were established as routine methods, and even the use of less conventional techniques such as photon-echo spectroscopy,310 magnetic circular dichroism (MCD),222 Stark spectroscopy,224 or UV-pump-X-ray-probe spectroscopy has been reported. In the latter study, the electronic structure changes of the proton-donating group of the related photoacid 8-aminopyrene-1,3,6-trisulfonic acid (APTS) upon photo-excitation and the subsequent proton transfer dynamics have been visualized.311 Moreover, for photoacids under confinement (vide supra), time-resolved fluorescence anisotropy256,259,263 together with 2D-NMR spectrosopy155,312,313 have become powerful tools for probing the interaction with the environment.
A new perspective became recently available through time-resolved THz spectroscopy in combination with advanced calculations that have been applied by Havenith, Head-Gordon and coworkers to directly study the photo-induced solvent response of water in the vicinity of HPTS, the deprotonated form OPTS and the methoxy derivative MPTS.63 Oscillatory features present in the first ps of the Vis-pump-THz-probe signal could be associated with vibrational energy transfer between solute and adjacent solvent molecules (see Fig. 6). In case of HPTS for which ESPT to the solvent is possible, this transfer of vibrational energy is more efficient and facilitates solvent reorganization, which is a prerequisite for the actual ESPT. Thorough analysis of the data on longer timescales disclosed a dissipation of the proton into the bulk, reflected in a phonon-like propagation behavior.314 In this regard, the ESPT dynamics are monitored from a solvent rather than from a solute response.
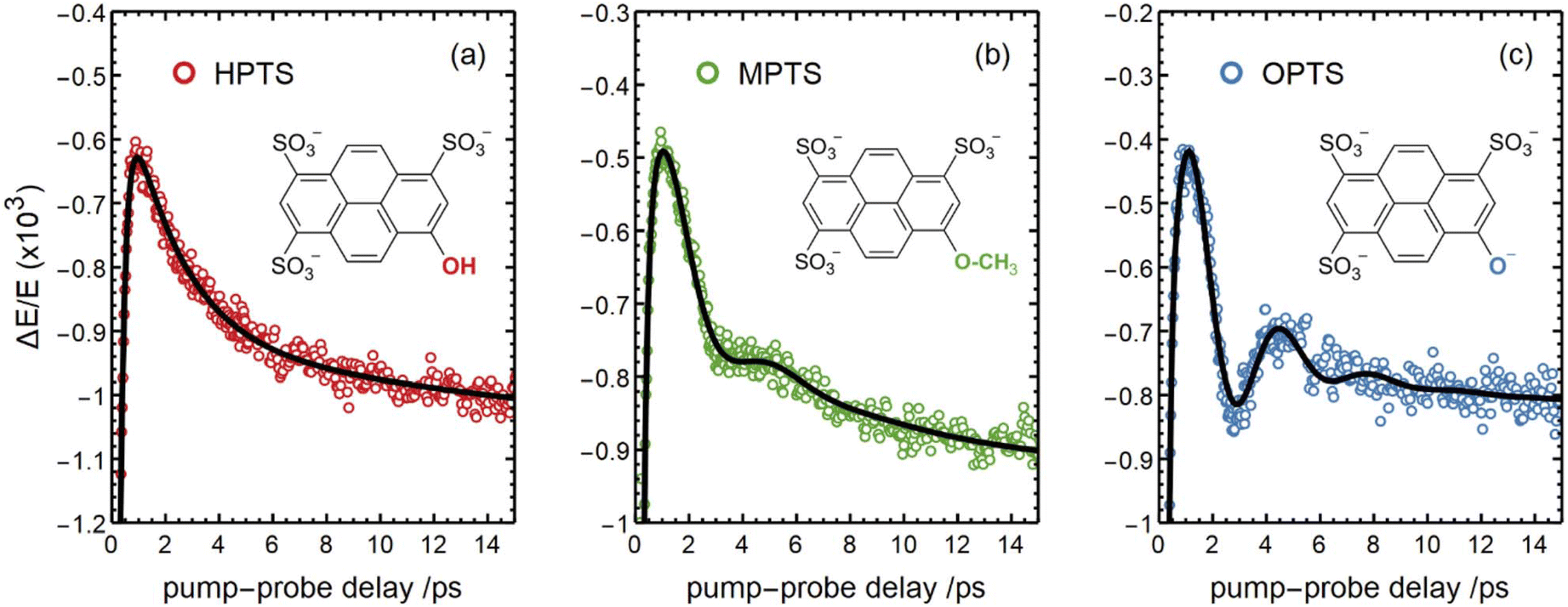 | ||
| Fig. 6 Transient signals observed via Vis-pump-THz-probe spectroscopy applied to a water jet containing solutions of (a) HPTS, (b) MPTS, and (c) OPTS. The oscillatory features originate from vibrational solute–solvent energy transfer, whereas the strong damping in the case of (a) is indicative of a more efficient coupling that paves the way for the subsequent ESPT. This figure has been reproduced from ref. 63 with the kind consent of M. Havenith and with permission from the authors under the terms of CC BY-NC 3.0, copyright 2023. | ||
3.3 Theoretical studies on HPTS
Research groups specialized in theory have continuously put HPTS into the focus of their studies as well, especially in recent years. However, the relatively large size of the HPTS molecule in combination with the fundamental requirement for an accurate description of the electronic excitations hindered the theoretical investigations of photoacids in general and HPTS in particular for a long time. Moreover, for HPTS, the three-valent and four-valent anionic character of the protonated and deprotonated species leads to the additional requirement that an adequate description of the solvent needs to be included in the calculations.This is among the reasons why the early theoretical, and more precisely, quantum-chemical studies rooted in the photoacid community (in the early 2000s) have mostly focused on smaller and simpler photoacids as model systems, such as phenol and cyanophenols (using state-specific CASSCF calculations combined with PCM)302 or cyanonaphthols (using semi-empirical AM1 calculations).315 Apart from other preliminary calculations, e.g., on hydroxyquinoline–water complexes by Fang et al.,316–318 multi-reference methods (including CASSCF and CASPT2) have been applied more broadly to significantly larger photoacids only in more recent times.319,320
The first computational study on HPTS itself has been reported not much later by Hynes and co-workers (in 2002),216 but was still restricted to semi-empirical (CS-INDO) calculations at that time because of relatively high computational costs for a full wavefunction-based treatment using multi-reference methods. In the same year though, Jimenez et al. reported a CIS—i.e., the most simple, non-perturbative correlated single-reference method—study on MPTS, the methoxy analogue of HPTS, addressing its vertical electronic transition energies and the vibrational frequencies.310 About 15 years later, HPTS became the subject of a purely ab initio study in (implicit) solution by Cimino et al. (in 2016),321 using TDDFT as a much cheaper method for electronic excitation energies. Similarly, Graef et al. studied the lowest-energy transitions of pyrene—the main chromophoric core of HPTS—in vacuum and solution.322 During that time, TDDFT—often in combination with some variant of the continuum solvation model PCM—has also been applied to many other photoacids323–341 and particularly also to the prediction of  values342–350 (vide infra).
values342–350 (vide infra).
Over the past five years and besides static, quantum-chemical calculations, HPTS has also been investigated theoretically using molecular dynamics simulations, mostly with quantum-mechanics/molecular-mechanics (QM/MM) approaches for the sake of computational efficiency. One of the first examples is the combined experimental–theoretical study on the ESPT of HPTS to acetate by Heo et al., which revealed a coherent wave packet motion of the reactant (acid) and the product (conjugate base) and identified the vibrational modes that actively participate in this coherent proton transfer.233 The excited-state vibrational dynamics of HPTS have been extensively addressed by Rega and co-workers, e.g., in neat water351,352 and in acetate-containing aqueous solution.353,354 Even the particularly strong so-called super-photoacids (vide infra) have been the subject of theoretical studies, e.g., on the ESPT dynamics of QCy9 in water by Raucci et al.355 or the THz and IR signals of NM6HQ+ in water by Petrone et al.356
Similarly, the ESPT dynamics of HPTS in 1-methylimidazole357 and in water358 have been recently investigated in a joint experimental–theoretical study by Fayer, Martínez and co-workers. In contrast to the previously mentioned earlier quantum chemical study by Cimino et al., in which no ESPT of HPTS had been observed with up to three water molecules,321 the excited-state ab initio MD simulation by Walker et al.358 scrutinize the ESPT of HPTS in bulk water more thoroughly and indicate the involvement of a strong, more extensive hydrogen-bonded network (i.e., a water wire) between the (photo)acidic OH group and one of the three sulfonate groups, e.g., involving eight water molecules (see Fig. 7). In fact, these results allow for a refinement of the existing experimental knowledge on HPTS by providing a clear molecular picture of the intermediates that are involved in the ESPT. They claim that the intermediate time constant of 2–3 ps that had been commonly assigned to a shared proton between HPTS and proximate water molecules213,214,309 rather reflects the deprotonation of HPTS and the corresponding oscillation of the dissociated proton that first remains mobile within the water wire. Only after that, the proton localizes on one of the water molecules and can then diffuse away from the wire, and not just from a single water molecule coordinating the O− group as assumed in previous interpretations, into the bulk solution on a longer timescale (∼80 ps).
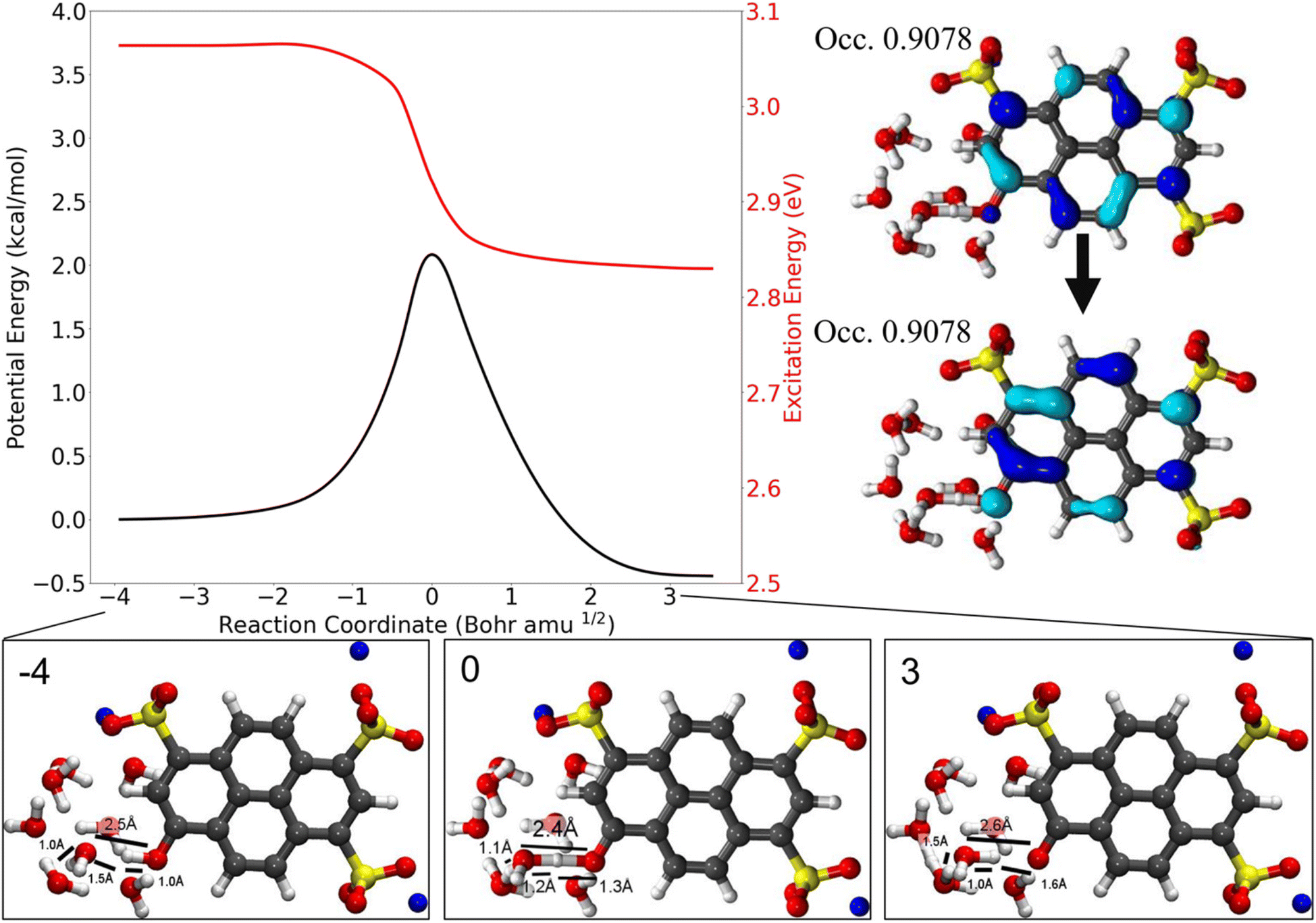 | ||
| Fig. 7 Scan of the potential energy for the first excited state and the vertical excitation energies along the intrinsic reaction coordinate that corresponds to the deprotonation of HPTS. 8H2O molecules and the implicit solvent model PCM have been used in combination with TDDFT. The change in excitation energy corresponds to 4.1 Å in the collective coordinate that quantifies the location of the proton within the water wire. Structures along the path are shown below, with the natural transition orbitals associated with the transition state geometry shown on the right. This figure has been reproduced from ref. 358 with the kind consent of M. D. Fayer and T. Martínez and with permission from the American Chemical Society, copyright 2021. | ||
4 Spotlight—naphthols
Naphthols comprise another important class of photoacids with a long-lasting history and a rich background in many research areas. Particularly important are the two parent compounds: 1-naphthol and 2-naphthol (cf.Fig. 3b and c). Although naphthols do not belong to the very first photoacids studied—which were in fact naphthyl amines (see Section 2)—, their photoacidic character has been first reported by Weller in 1952 and 1954.107,108 Moreover, no research group can be identified who has excelled at studying naphthols in a similar fashion as Huppert and coworkers have for HPTS; rather, the research history of the whole family of naphthols is a collection of many individual contributions. Therefore, this section is less strictly concerned about presenting a complete list, but more about a presentation of exemplary highlights.Typically, the different naphthol derivatives are functionalized with one or multiple sulfonate groups for solubility reasons. For the 1-naphthols, mono-substituted derivatives with a sulfonate group at the ring positions 2,359 3,360 4,361 and 5,56 4-chloro362 and 5-tert-butyl363 and 3,6-disulfonato364 substitution are reported. For the 2-naphthols, monosulfonation at the 6,365 7,366 or 8 (ref. 55) position and disulfonation at the 6,8 (ref. 367) and 3,6 (ref. 301) positions are typical. Even a dihydroxynaphthalene368 and dimers of 2-naphthol369,370 were recently described. In this regard, it is worth noting that 2,2′-dihydroxy-1,1′-naphthaldehyde diazine (pigment yellow 101) can be considered as a bisnaphthol with rich photochemistry beyond ESPT.371 We will not make a particular distinction with respect to the substituents in the following, with the exception of electron-withdrawing groups such as cyano groups, to which a later section is dedicated (cf. Section 5).
4.1 Research highlights
Subsequent to the initial work by Weller,107,108 important earlier contributions (i.e., here prior to 1990) to the research on naphthols and the fundamental understanding of their properties as photoacids were made by Trieff and Sundheim (1965),49 Suzuki and Baba (1967),372 Kuz'min and co-workers (1977–1980),93,373–375 Gutman, Huppert, and co-workers (1979–1981),197,301,376 Schulman et al. (1979),110 Harris and Selinger (1980),111 Tobita, Tsutsumi and Shizuka (1980–1983),112,377–379 Robinson and co-workers,359,380–382 and Webb et al. (1986).113 In more recent times (i.e., after 1990), the ESPT of naphthols in gas-phase clusters has emerged as one particular research area, as extensively, but not exclusively,383–388 studied by Knochenmuss and co-workers.286,287,389–395 Owing to the neutral nature of the naphthol (in contrast to the anionic HPTS), techniques such as mass spectrometry, multi-photon ionization (MPI), time-resolved photo ionization (TRPI), or laser-induced fluorescence (LIF) (cf. ref. 287), could be applied. Even some QM/MM studies have been reported under these conditions, namely for 1-naphthol in liquid-cooled jets.396 Pines and co-workers have also made detailed and influential contributions on naphthols, e.g., on the self-quenching of 1-naphthol,397 the intrinsic proton transfer rate in photoacid–carboxylate pairs,228 the hydrogen-bonding interactions,398 and the role of diffusion-assisted recombination as well as isotope and temperature effects.399–401 Recently, Pines et al. have addressed the role of the sulfonate groups in mono- and di-substituted naphthols as competing protonation sites.402The role of geminate recombination364,403,404 and solvatochromism405,406 was elucidated for naphthols as well by Huppert and coworkers, who intensified their efforts with the advent of naphthol-based super-photoacids which have enhanced photoacidic behavior (see Section 5.2.1). This includes studies on the ESPT of naphthols in neat solvents,407–410 its pressure dependence365,366 and modified ESPT characteristics that arise in binary solvent mixtures, e.g., H2O/D2O,411 methanol–water,56,57,412 or acetonitrile–water.413 Other interesting environments that have been studied using naphthols include ethanol–water,414n-propanol–water,415,416 DMSO–water mixtures,417 ESPT to urea,418 super-critical water,419 micelles,420–425 polymer–surfactant aggregates,426 ionic liquids,427 and high-temperature and high-pressure methanol.428 Theoretical studies have been reported as well.324 Nibbering and co-workers have employed a 2-naphthol derivative for the real-time observation of carbonic acid formation in aqueous solution.429 Interestingly, about 10 years later, Pines et al. have followed the same approach to study carbonic acid, and additionally the physiologically relevant lactic and pyruvic acids, but using a stripped-off version of HPTS (namely, 6-hydroxypyrene-1-sulfonate).430 The Nibbering and Pines groups independently obtained the same pKa value for carbonic acid. Beyond, Cox and Bakker have used naphthol sulfonates to study the deuteron transfer dynamics through short-living water wires.431
4.2 Differences in the electronic structure
 | ||
| Fig. 8 Classification of the two lowest-lying excited singlet states of benzene (D6h point group) and naphthalene (D2h point group), as 1La (a and c) and 1Lb (b and d), respectively, based on the distribution of effective charges according to Platt. Visualization of the transition density with contours in red and blue referring to opposite signs. | ||
Although not part of Platt's original discussion, a fundamental consequence of this definition is that a and b each also determine an intramolecular axis that directly reflects the orientation of the corresponding transition dipole moment (TDM), i.e., the polarization, for the transitions from S0 into the 1La and 1Lb states, respectively. Due to the different orientation and magnitude of their TDMs, these two states depend differently on substituents or the solvent.433,436 The brighter 1La state is said to have a higher charge-transfer (CT) character (i.e., to be more polar), while the darker 1Lb state is more covalent in nature. Therefore, the 1La state is assumed to be stabilized more strongly compared to the 1Lb state by polar substituents. In an extreme, a level inversion between these states can also occur.
For linear polycyclic aromatic hydrocarbons (PAHs) including naphthalene and the acenes (e.g., anthracene), the a axis, which is aligned with the TDM of the strong S0 → 1La transition, can be identified as the shorter axis, while the b axis, which is aligned with the TDM of the weak (quasi-forbidden) S0 → 1Lb transition, is the longer axis, with both axes being perpendicular to each other (cf.Fig. 9b). However, this assignment of the a and b axes as the short and long intramolecular axes does not hold generally and depends on the specific compound under study. For example and in contrast to the acenes, the a axis is identified as the longer axis according to the orientation of the TDM in the simple benzene (cf.Fig. 9a) and some non-linear PAHs like pyrene (cf.Fig. 9c), allowing for two important observations: first, in all of these examples, the a axis turns out to be the one passing through ring carbon atoms, while the b axis is the one cutting carbon–carbon bonds. Second, the character and the symmetry of the 1La and 1Lb states is preserved among these compounds.
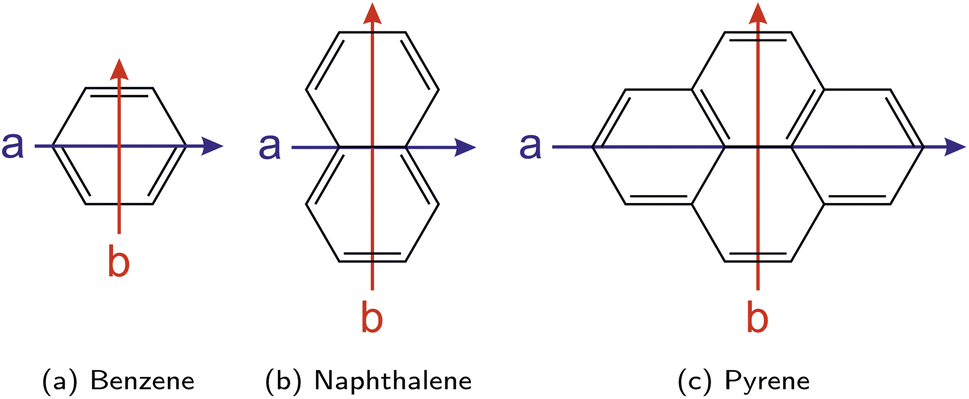 | ||
| Fig. 9 Orientation of the molecular axes a (in blue) and b (in red) according to Platt's notation for the examples benzene (a), naphthalene (b), and pyrene (c). | ||
Let us briefly compare Platt's a and b axes with the geometrical orientation of these molecules as typically used in physical and theoretical chemistry. Importantly, due to their relation to electronic states, Platt's a and b axes must not be mistaken for the principal axes of inertia carrying the same labels, which is an occasional misconception in the literature and potential source of confusion. According to Mulliken's recommendation for the notation of spectra of polyatomic molecules, for planar molecules with D2h symmetry, the x axis is chosen perpendicular to the molecular plane, while the z axis should be chosen passing through the largest number of atoms or, if ambiguous, dissecting the largest number of bonds.437 Hence, for both the selected examples of linear (i.e., naphthalene and the acenes) and non-linear (i.e., pyrene) PAHs with D2h symmetry discussed here, it is the z axis that corresponds to the b axis and the y axis that corresponds to the a axis. With this choice of the axes, 1La and 1Lb have B2u and B1u symmetry, respectively438,439 (note that sometimes the z axis is chosen perpendicular to the molecular plane, then the symmetry species would be B2u and B3u).440,441
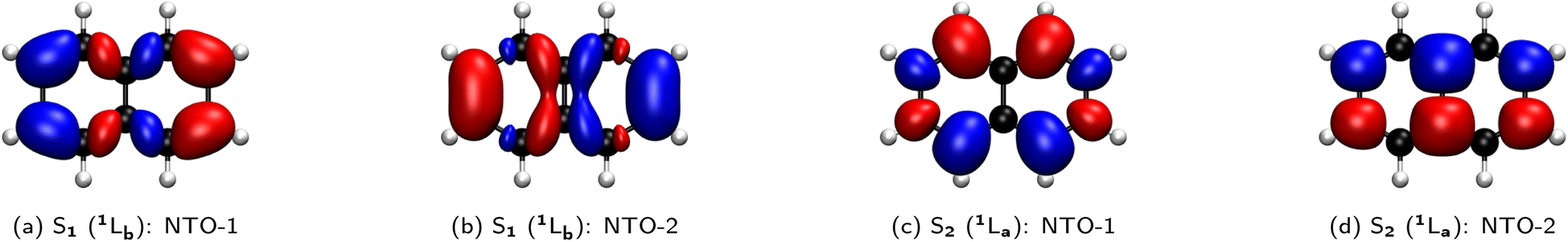
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of the HOMO → LUMO+1 and HOMO−1 → LUMO transitions. A perfect example is the simple naphthalene, as illustrated by the natural transition orbitals (NTOs) for the electronic transitions from the ground state (S0) to the two lowest-lying excited states (S1, S2), which can be either 1La, 1Lb, or a mixture (Fig. 10).
:50 mixture of the HOMO → LUMO+1 and HOMO−1 → LUMO transitions. A perfect example is the simple naphthalene, as illustrated by the natural transition orbitals (NTOs) for the electronic transitions from the ground state (S0) to the two lowest-lying excited states (S1, S2), which can be either 1La, 1Lb, or a mixture (Fig. 10).
 | ||
| Fig. 10 Analysis of the two lowest-lying electronically excited states and assignment of the 1La and 1Lb character for prototypical compounds. For each state, the two dominant natural transition orbital (NTO) pairs together with their weights (in parentheses) are given. The electronic transitions are calculated in the gas phase with ADC(2)/aug-cc-pVDZ at MP2/cc-pVDZ geometries, except for the charged 4sPy and HPTS that refer to implicit solution (water) using COSMO-ADC(2) energies at COSMO-MP2 geometries. | ||
Originating from a singular value decomposition of the transition density matrix, NTOs provide a more compact representation of single excitations compared to the canonical MO basis, becoming particularly powerful when many MOs contribute to a given transition. Thereby, the NTOs allow to visualize and rationalize the changes in the electronic distribution taking place upon excitation. NTOs as the (left and right) singular vectors always come in pairs with a so-called occupied (or hole; i.e., left after excitation) and virtual (or particle; i.e. the excited electron) part, representing the probability distribution of the electron prior and after the transition, respectively. Moreover, the corresponding singular values determine weights describing how strongly each NTO pair contributes to a given transition.
In the case of naphthalene, the NTO analysis verifies that the S0 → 1La transition is dominated by a single NTO pair whose—based on an additional comparison that is not shown here—occupied and virtual parts resemble the HOMO and LUMO, respectively. Similarly, the two NTO pairs that contribute equally (ca. 50%) to the S0 → 1Lb transition can be identified with the HOMO and LUMO+1 as well as the HOMO−1 and LUMO based on the occupied and virtual parts of the first and second NTO pairs, respectively. However, it needs to be emphasized here that this correlation of 1La and 1Lb with the frontier orbitals is specific to a given molecular class only and can hence be different for other PAHs (e.g., for different ring sizes or condensation types), but particularly if substituents are introduced to the aromatic rings.
The NTOs also allow for a direct assignment of 1La and 1Lb based on pure considerations of charge polarization according to Platt's fundamental definition (vide supra). For this, the product of the occupied and virtual parts of the dominant NTOs needs to be considered and analyzed with respect to the location of effective charges and nodes (e.g., see Fig. 11 for naphthalene). Since all NTO pairs for a given transition have the same symmetry, it is furthermore sufficient to consider only a specific pair, e.g., one that appears easiest to interpret. For example, it is quite simple to see that the product of the occupied and virtual parts of the second NTO pair for the S0 → S1 transition of naphthalene (Fig. 11b) mainly consists of ππ* contributions from the two outmost left and right neighboring C atoms (i.e., C2–C3 and C6–C7). The coefficients at the other C atoms basically vanish in the product due to the presence of vertical nodal planes in either NTO part. The presence of effective charges located at the bonds is indicate for an 1Lb and the opposite phase between the coefficients at the terminal C atoms indicates that this transition is polarized along the longer, horizontal axis that becomes assigned as the b axis. In analogy, the product of the occupied and virtual parts of the second NTO pair for the S0 → S2 transition (Fig. 11d) dissects into pure contributions with coefficients at the atoms, indicative for 1La, with a polarization along the shorter, vertical axis, thus labelled a. Despite their dominant weight, the NTO pairs that are shown in the first column of Fig. 10 might be somewhat more difficult to interpret with respect to the charge polarization since their products are harder to imagine (cf.Fig. 11a and c). However, we have observed the following rule of thumb that allows for an even faster assignment of the polarization axes: For naphthalene, but also for the other PAH examples in Fig. 10, if there is a common nodal plane (through the center) present in both occupied and virtual parts of a given NTO pair, the nodal plane contains the a or b axis, respectively. For example, for the 1Lb state one can see that the b axis is the common nodal plane in NTO pair 1, while it is the a axis for 1La as seen in NTO pair 1.
 | ||
| Fig. 11 Visualization of the product of the occupied and virtual parts of the first (NTO-1) and second (NTO-2) dominant NTO pairs for the transition into the first (S1, 1Lb; a and b) and second (S2, 1La; c and d) excited state of naphthalene, respectively. | ||
4.2.3.1 Phenol. Although extensively used in the past to describe the electronic structure of various hydroxyarene photoacids, Platt's assignment of the 1La and 1Lb states is strictly valid only for a limited number of unsubstituted arenes, mainly because (i) (polar) substituents can enhance mixing between the two states and (ii) the perimeter model itself is no longer suitable for larger ring systems (e.g., pyrene). However, care must be taken when trying to quantify this state mixing, e.g., by comparing the weights of the NTOs. For example, already phenol as the simplest hydroxyarene (see Fig. 10) demonstrates that the orbital transitions involving the HOMO contribute much stronger for both 1La and 1Lb, as judged based on higher NTO weights compared to the parent compound benzene. In particular, the character of the contributions to these transitions and the orientation of the TDMs remain mostly unaltered for phenol, as can be seen by the almost identical NTOs, excluding any state mixing between 1La and 1Lb. Thus, the higher weights for the NTOs involving the HOMO just indicate that these transition become HOMO-dominant in the case of phenol, in line with a lower HOMO energy due to the participation of the O atom from the OH group. It should be also noted here that these orbital considerations differ even more strongly from the PAHs in case of the deprotonated species of the photoacids (e.g., phenolate) since the HOMO is dominated even more by the O atom due to the negative charge. However, a detailed discussion of the electronic structure of these corresponding bases is not part of this review.
4.2.3.2 Naphthols. The classical examples to look at in the context of photoacids are 1-naphthol (1N) and 2-naphthol (2N). 1N, despite having an energetically lower 1Lb state, is considered an 1La photoacid, while 2N, also with a lower 1Lb state, is regarded an 1Lb photoacid (cf.Fig. 10). Thus, this classification as 1La or 1Lb photoacids is not simply based on the energetic order of these states, but also considers which of them is relevant for the photoacidity, i.e., the accessibility in terms of the brightness of these states. For both 1N and 2N, the introduction of the OH group as a substituent breaks the initial D2h symmetry of the parent naphthalene, reducing it to Cs. As an important consequence, both 1La and 1Lb have the same symmetry (A′) in the Cs point group, enabling a mixing of these two states. For 1N that is closer to the unsubstituted naphthalene, the lower-lying 1Lb state is less bright, rendering the brighter 1La the relevant state, while for 2N, which apparently deviates more strongly from naphthalene, the 1Lb state is much brighter due significant 1La–1Lb state mixing.
The two compounds 1N and 2N differ remarkably in their photoacidic behavior. Both photoacids show almost the same pKa values in the ground state (9.3 for 1N, 9.6 for 2N) in water, but 1N (−1.0) is much more acidic in its 1La state, compared to 2N (3.3) in its 1Lb state.432 Unveiling the reasons behind this puzzling observation has lead to a debate in the literature. Initially, gas-phase experiments in solvent clusters by Knochenmuss and co-workers287,390,396 as well as theoretical studies by Tran-Thi, Hynes, and co-workers214–216 have largely shaped the view that the polar 1La state, although higher in energy in less polar environments, should be the relevant state for photoacidity in more polar solvents, whereas the less-polar, energetically lower 1Lb state dominates in less polar solvents. Hence, a so-called level inversion in polar solvents has been suggested as an additional mechanism.442 Later, Pines has emphasized that the electronic structure of 2N is in fact much more congested than sometimes assumed, meaning that both the 1La and the 1Lb state need to be considered.39,432 Indications for this are also observed here (see Fig. 10) as pure 1La character is neither observed for 1N nor for 2N, with particularly strong mixing for 2N. Moreover, to explain the overall increased photoacidity of 1N over 2N, Pines suggested to take the effects on both species into consideration, the naphthol (acid) and naphtholate anion (base), thereby inferring a stronger stabilization on the base side.39 More recently, Nibbering, Chergui and co-workers have performed time-resolved fluorescence and mIR spectroscopy experiments that compare 1N and 2N in chloroform (as an unpolar solvent) and DMSO (as a polar solvent), demonstrating that the solvent changes the character of the heavily mixed electronic states in the case of 1N (see Fig. 12).443 For 2N, the fractional 1La character remains low (∼10%) and constant over time after excitation (in accordance with a higher 1Lb character for 2N, cf.Fig. 10), while, for 1N, this fraction also remains constant (∼20%) in the less polar chloroform, but increases within 0.5 ps from ∼20% to ∼40% in the more polar DMSO.
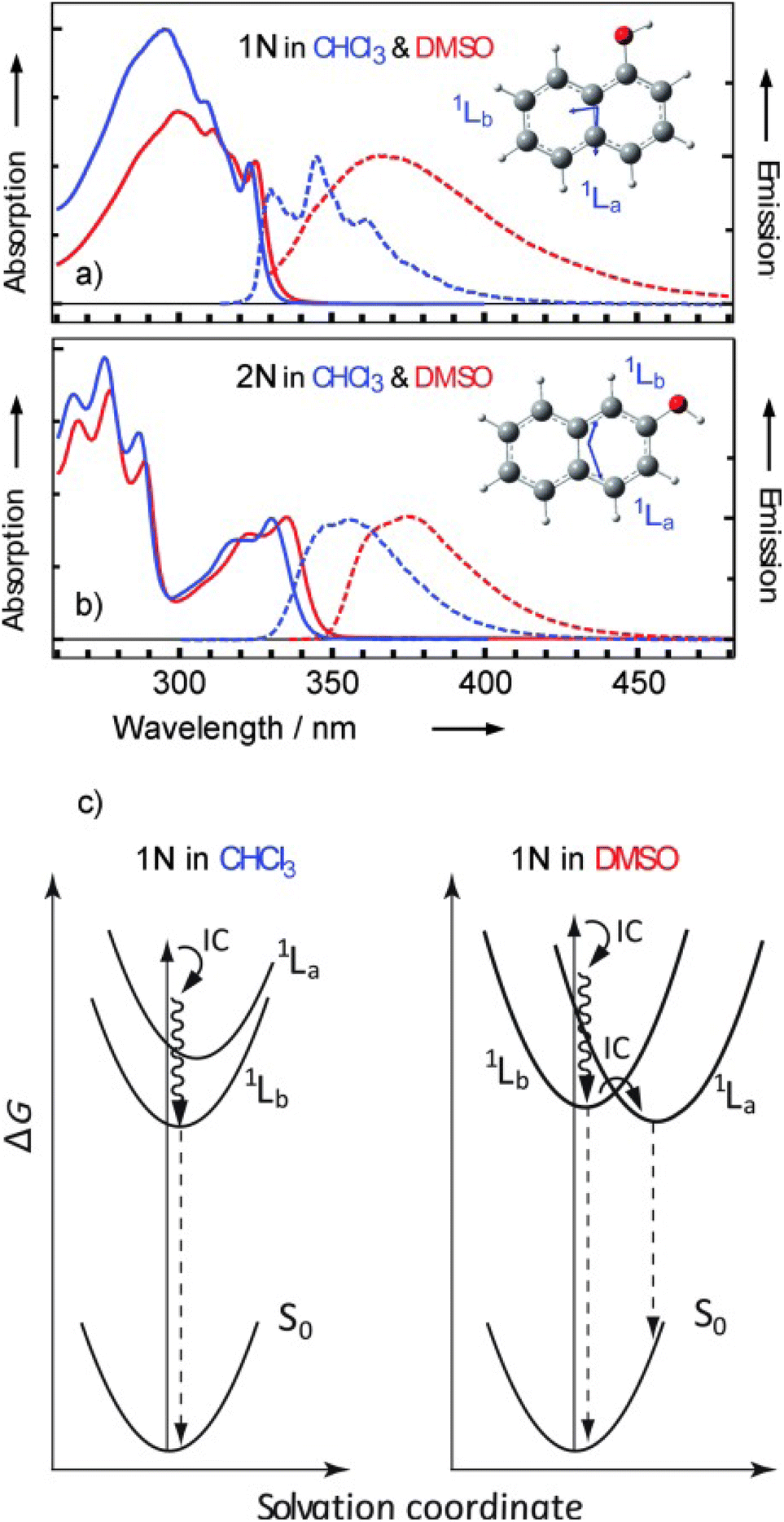 | ||
| Fig. 12 Normalized static optical absorption (solid lines) and emission (dashed lines) of: (a) 1N and (b) 2N. Insets: molecular structures and transition dipole moments (blue arrows) of the 1La and 1Lb transitions of 1N and 2N, predicted by TDDFT for the molecules in gas phase. (c) Scheme of the dynamics of 1N after photoexcitation. This figure has been reproduced from ref. 443 with the kind consent from E. T. J. Nibbering and M. Chergui and with permission from Wiley-VCH, copyright 2013. | ||
4.2.3.3 Pyrenols. Similar attempts have also been made to rationalize the photoacidity of HPTS in terms of its electronic structure,140,214–216,220,222,223 but the experimental and theoretical evidence is not fully conclusive and hence a strict assignment of these two states might not even be possible (vide supra), which sometimes is empirically described by heavy 1La–1Lb mixing. Although Platt's notation does not strictly apply to pyrene that as a peri-condensed hydrocarbon lacks cata-condensation, its electronically excited states can still be nicely characterized as 1La and 1Lb. For example, the calculations on pyrene presented here (Fig. 10) show that S1 has roughly equal contributions from two NTO pairs with a transition dipole moment aligned along the b axis, indicative for 1Lb character, while S2 is dominated by a single NTO pair with a TDM aligned along the a axis, characteristic for 1La. However, as for the naphthols, symmetry breaking via substitution introduces state mixing. Interestingly, the effect from OH substitution is rather small as the character and relative order of S1 and S2 remain mostly the same (i.e., moving from pyrene to 1-pyrenol). Quadruple substitution with sulfonate groups does not affect the character since the symmetry is maintained, but alters the energetic order shifting 1La below 1Lb (i.e., moving from pyrene to 4sPy), as judged based on NTO analysis. Similar as the changes from pyrene to 1-pyrenol, substitution of one of the four sulfonates by an OH group (i.e., moving from 4sPy to HPTS) does again break the symmetry, causing distortion of the NTOs, but also maintains the energetic order. However, while the first excited state (S1) in HPTS has still dominantly 1La character, the second excited state (S2) reveals a significant 1La–1Lb mixing. Using the difference between the NTO weights (or their deviation from 50%) as a rough estimate, the degree of mixing appears similar for HPTS (76%, 22%) as for 1-pyrenol (77%, 21%), and thus somewhat larger than for 1-naphthol (67%, 32%), but considerably less than for 2-naphthol (83%, 15%).
5 Spotlight—super-photoacids
5.1 Initial naphthol derivatives
Despite this wealth in photoacid research, HPTS and most other early photoacids, especially naphthols,55–57,363,364,431 are mainly restricted to perform an ESPT in aqueous solutions due to limited acid strength, i.e., too large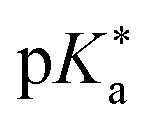 values. To overcome this, i.e., to enhance the photoacidity by lowering the
values. To overcome this, i.e., to enhance the photoacidity by lowering the 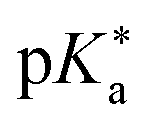 value, Tolbert and Haubrich (in 1990) had the idea to substitute 2-naphthol with electron-withdrawing cyano (CN) groups at different ring positions.444 The CN group was chosen as a compromise between strength and stability since the commonly used sulfonate (SO3−) group is only a weak electron acceptor, whereas the nitro (NO2) group is one of the strongest electron acceptors, but nitro-substituted chromophores are typically photo-labile and tend to weakly fluoresce.445 Owing to the drastically enhanced photoacidity for some of these cyanonaphthols, particularly with the CN at the 5 or 7 position (shifting the
value, Tolbert and Haubrich (in 1990) had the idea to substitute 2-naphthol with electron-withdrawing cyano (CN) groups at different ring positions.444 The CN group was chosen as a compromise between strength and stability since the commonly used sulfonate (SO3−) group is only a weak electron acceptor, whereas the nitro (NO2) group is one of the strongest electron acceptors, but nitro-substituted chromophores are typically photo-labile and tend to weakly fluoresce.445 Owing to the drastically enhanced photoacidity for some of these cyanonaphthols, particularly with the CN at the 5 or 7 position (shifting the  to −1.2 and −1.3, respectively), Tolbert and Haubrich also introduced the term super-photoacids, from then on a general term for photoacids with a negative
to −1.2 and −1.3, respectively), Tolbert and Haubrich also introduced the term super-photoacids, from then on a general term for photoacids with a negative 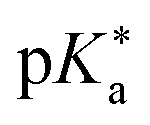 value.
value.
Driven by the enhancement in photoacidity, the ESPT of such super-photoacids is strongly accelerated, leading to an ultrafast deprotonation in water and also rendering the ESPT possible in non-aqueous solution (vide infra). For example, as one of the first super-photoacids, the above-mentioned compound 5-cyano-2-naphthol (5CN2, Fig. 13a) has been thoroughly studied with respect to its ESPT in many different solvents including water,408 alcohols,409 DMSO,60 solvent mixtures400,404,412 and gas-phase solvent clusters286 as well as to its solvatochromism in a variety of organic solvents.406,409
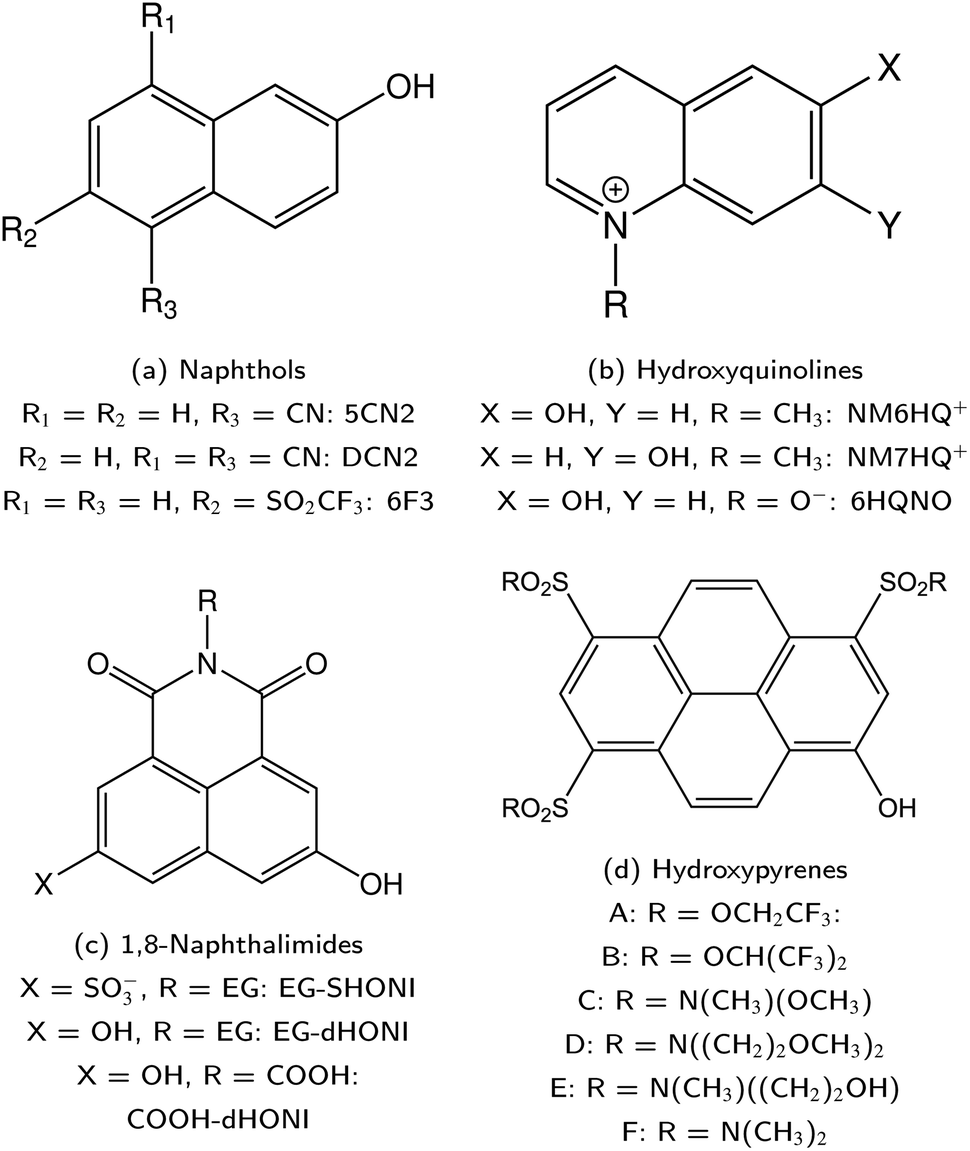 | ||
| Fig. 13 A selection of super-photoacids I. Naphthol (a), hydroxyquinoline (b), 1,8-naphthalimide (c), and hydroxypyrene (d) derivatives. [Abbreviation: ethylene glycol residue (EG = CH2CH2OCH2CH2OH)]. | ||
5.2 The race toward strongest photoacids
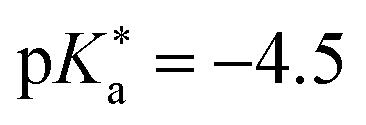 446) has become most thoroughly studied, e.g., regarding the ESPT in water446 and various alcohols,407,447,448 under high pressures,366,419,428,449–452 or even in ionic liquids.427 Although 2-naphthols are weaker photoacids compared to 1-naphthols (vide supra), almost exclusively 2-naphthol derivatives (with multiple cyano groups) are known as super-photoacids, which is related to weak fluorescence and facile deactivation of cyano-1-naphthols via proton quenching.113
446) has become most thoroughly studied, e.g., regarding the ESPT in water446 and various alcohols,407,447,448 under high pressures,366,419,428,449–452 or even in ionic liquids.427 Although 2-naphthols are weaker photoacids compared to 1-naphthols (vide supra), almost exclusively 2-naphthol derivatives (with multiple cyano groups) are known as super-photoacids, which is related to weak fluorescence and facile deactivation of cyano-1-naphthols via proton quenching.113
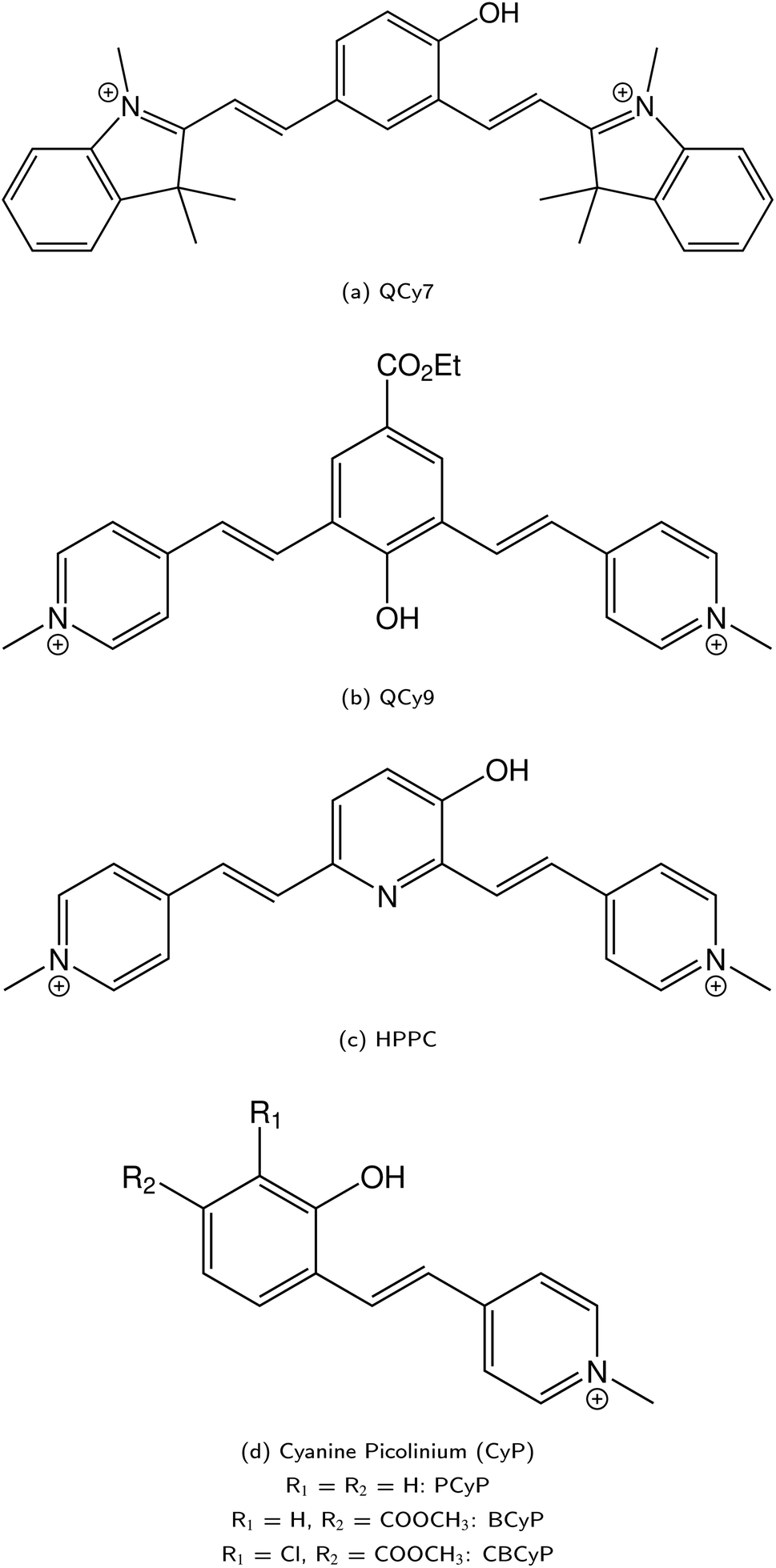 | ||
| Fig. 14 A selection of super-photoacids II. Quinone cyanine dyes: QCy7 (a), QCy9 (b), HPPC (c), and CyP (d). | ||
Upon the hydroyxyquinolines, NM6HQ+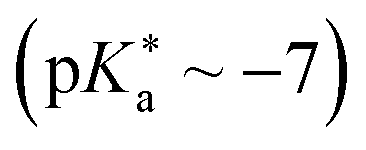 455 has been studied experimentally in water and in various alcohols by Popov and colleagues,455,456 and the related (non-N-methylated) 6HQ as well as NM6HQ+ in the same solvents and additionally in water–acetonitrile mixtures by Pérez Lustres et al.457–459 Moreover, the Kwon group has studied ESPT of NM7HQ+ (
455 has been studied experimentally in water and in various alcohols by Popov and colleagues,455,456 and the related (non-N-methylated) 6HQ as well as NM6HQ+ in the same solvents and additionally in water–acetonitrile mixtures by Pérez Lustres et al.457–459 Moreover, the Kwon group has studied ESPT of NM7HQ+ ( 460) in hydrogen-bonded complexes with methanol, ethanol, DMSO, and N-methylbenzamide in the solvent acetonitrile.460–465 Similarly, the group has employed the photoacids 7HQ and NM7HQ+ in solutions of diols with varying chain length to study the role of hydrogen-bond bridging on the ESPT.194,466 Very recently, Choi et al. have presented a new method for determining the equilibrium constant for the ground-state hydrogen-bond formation via time-resolved instead of steady-state spectroscopy, which is superior to the traditional determination in cases when the addition of the HB acceptor (base) impacts the bulk polarity of the solution, as for NM7HQ+ and DMSO in acetonitrile.464
460) in hydrogen-bonded complexes with methanol, ethanol, DMSO, and N-methylbenzamide in the solvent acetonitrile.460–465 Similarly, the group has employed the photoacids 7HQ and NM7HQ+ in solutions of diols with varying chain length to study the role of hydrogen-bond bridging on the ESPT.194,466 Very recently, Choi et al. have presented a new method for determining the equilibrium constant for the ground-state hydrogen-bond formation via time-resolved instead of steady-state spectroscopy, which is superior to the traditional determination in cases when the addition of the HB acceptor (base) impacts the bulk polarity of the solution, as for NM7HQ+ and DMSO in acetonitrile.464
 )469 and QCy9 (Fig. 14b,
)469 and QCy9 (Fig. 14b, 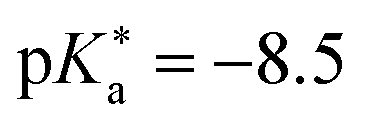 ).470 Both are able to readily perform an ESPT in liquid469–471 or even solid472,473 H2O (or D2O) as well as in typical alcohols such as methanol and ethanol,58 and also in ethanol–trifluoroethanol solvent mixtures in the case of QCy9.474 Due to its immense (photo-)acidity, the ESPT rate of QCy9, which continues to be the strongest photoacid among the hydroxyaromatics to date, reaches a value of kPT ∼ 1013 s−1 (i.e., a lifetime of 100 fs) that is identical in water, methanol, and ethanol, indicating ESPT kinetics beyond the solvent-controlled regime.58 Even more recently, Huppert and co-workers have continued to fine-tune the properties of these dyes and reported newly synthesized
).470 Both are able to readily perform an ESPT in liquid469–471 or even solid472,473 H2O (or D2O) as well as in typical alcohols such as methanol and ethanol,58 and also in ethanol–trifluoroethanol solvent mixtures in the case of QCy9.474 Due to its immense (photo-)acidity, the ESPT rate of QCy9, which continues to be the strongest photoacid among the hydroxyaromatics to date, reaches a value of kPT ∼ 1013 s−1 (i.e., a lifetime of 100 fs) that is identical in water, methanol, and ethanol, indicating ESPT kinetics beyond the solvent-controlled regime.58 Even more recently, Huppert and co-workers have continued to fine-tune the properties of these dyes and reported newly synthesized
• 3-hydroxypyridine-dipicolinium cyanine (HPPC, Fig. 14c; 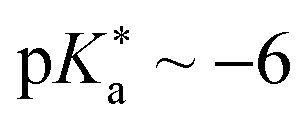 ),475
),475
• phenol cyanine picolinium (PCyP, Fig. 14d;  ),476
),476
• chloro benzoate cyanine picolinium (CBCyP, Fig. 14d; 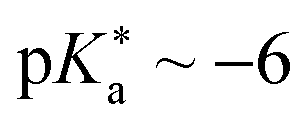 ),59,477 and
),59,477 and
• phenol benzoate cyanine picolinium (BCyP, Fig. 14d; 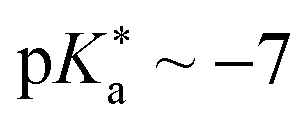 )478
)478
salt super-photoacids. Apart from the common solvents for ESPT (i.e., water, methanol, and ethanol), CBCyP is one of the very few photoacids that has also been studied in acetonitrile–water mixtures.413
5.3 Naphthalimides of Brouwer & Kumpulainen
As highlighted above, the design of new super-photoacids mainly pursued the route of functionalizing already known weaker photoacids or promising candidates (e.g., fluorescent dyes) with electron-withdrawing groups in order to shift the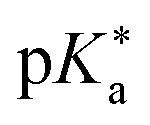 into the negative regime. This strategy also applies to the super-photoacidic HPTS derivatives developed by Jung and co-workers,61 to which the subsequent section has been dedicated. However, as the final example here, the super-photoacids based on 1,8-naphthalimide introduced by Kumpulainen et al. are presented.
into the negative regime. This strategy also applies to the super-photoacidic HPTS derivatives developed by Jung and co-workers,61 to which the subsequent section has been dedicated. However, as the final example here, the super-photoacids based on 1,8-naphthalimide introduced by Kumpulainen et al. are presented.
Substituted 1,8-naphthalimides (NI) had been known as fluorescent probes and sensors for quite some time479–481 and even the excited-state ion-pair formation of 3- and 4-hydroxy-N-methyl-1,8-naphthalimides in organic solvents had been studied via fluorescence.482–484 However, due to the missing data for aqueous solutions in combination with the promising properties of these compounds, Kumpulainen, Brouwer, et al. have developed three new NI-based super-photoacids (see Fig. 13c) with  values ranging from −1.2 to −1.9.485 These compounds differ only in the number of OH groups and in the substituent at the N atom of the NI framework. EG-SHONI
values ranging from −1.2 to −1.9.485 These compounds differ only in the number of OH groups and in the substituent at the N atom of the NI framework. EG-SHONI 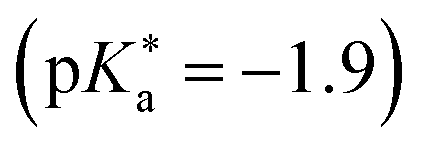 contains an ethylene glycol (EG) moiety at the N, one OH (HO) group in the first aromatic ring and a sulfonate (S) group in the second ring. In contrast, this sulfonate group is replaced by another OH group in EG-dHONI
contains an ethylene glycol (EG) moiety at the N, one OH (HO) group in the first aromatic ring and a sulfonate (S) group in the second ring. In contrast, this sulfonate group is replaced by another OH group in EG-dHONI 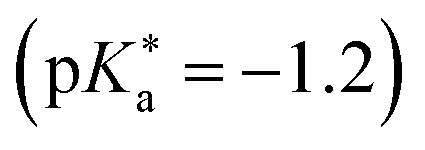 , having two (dual) OH groups in total. Lastly, COOH-dHONI
, having two (dual) OH groups in total. Lastly, COOH-dHONI  is similar to EG-dHONI, but the ethylene glycol is replaced by a carboxy (COOH) group. All three compounds exhibit deprotonation upon excitation in alcohols and in DMSO.485 EG-dHONI has been subject to more detailed kinetic studies in water, which revealed that ESPT follows a two-step model involving a short-range proton transfer under formation of a contact ion pair and its diffusive separation.485 Moreover, the complex formation and the ESPT of other dHONI photoacids and different organic bases in acetonitrile and benzonitrile solution has been studied, strongly indicating a three-step model for the ESPT in these environments involving hydrogen-bonded complexes and a solvent-separated ion pair as additional intermediates.486 Later, EG-SHONI has been studied more detailed using a combination of fluorescence up-conversion, transient UV/Vis absorption, and transient mIR absorption to decipher the ultrafast ESPT in DMSO, revealing not only a reversibility of the initial PT step that yields the contact ion pair, but also a breakdown of the (two-step) Eigen–Weller model—despite excellent agreement with the experimental data—due to a solvent control via solvent relaxation.487,488
is similar to EG-dHONI, but the ethylene glycol is replaced by a carboxy (COOH) group. All three compounds exhibit deprotonation upon excitation in alcohols and in DMSO.485 EG-dHONI has been subject to more detailed kinetic studies in water, which revealed that ESPT follows a two-step model involving a short-range proton transfer under formation of a contact ion pair and its diffusive separation.485 Moreover, the complex formation and the ESPT of other dHONI photoacids and different organic bases in acetonitrile and benzonitrile solution has been studied, strongly indicating a three-step model for the ESPT in these environments involving hydrogen-bonded complexes and a solvent-separated ion pair as additional intermediates.486 Later, EG-SHONI has been studied more detailed using a combination of fluorescence up-conversion, transient UV/Vis absorption, and transient mIR absorption to decipher the ultrafast ESPT in DMSO, revealing not only a reversibility of the initial PT step that yields the contact ion pair, but also a breakdown of the (two-step) Eigen–Weller model—despite excellent agreement with the experimental data—due to a solvent control via solvent relaxation.487,488
5.4 Pyrene derivatives in the new millennium
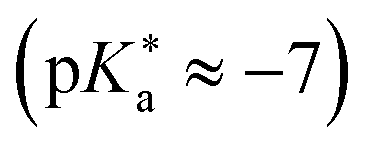 224,489 is frequently referred to in the literature as a cationic photoacid and HPTS as a neutral photoacid, despite their net negative charges in solution.140 In contrast, HPTA is a truly non-charged photoacid and exhibits a slightly higher excited-state acidity
224,489 is frequently referred to in the literature as a cationic photoacid and HPTS as a neutral photoacid, despite their net negative charges in solution.140 In contrast, HPTA is a truly non-charged photoacid and exhibits a slightly higher excited-state acidity 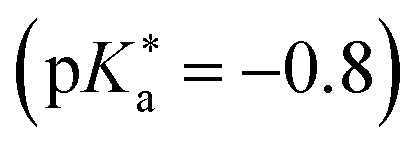 140 compared to the parent compound HPTS
140 compared to the parent compound HPTS  .207
.207
 by up to 5 units by changing the ground-state pKa, yielding a set of photoacids with
by up to 5 units by changing the ground-state pKa, yielding a set of photoacids with 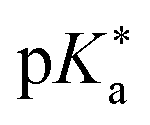 between −0.8 and −3.9. Owing to the larger effect of the fluorinated esters, compounds A and B are stronger photoacids compared to C–F, while B is the strongest
between −0.8 and −3.9. Owing to the larger effect of the fluorinated esters, compounds A and B are stronger photoacids compared to C–F, while B is the strongest 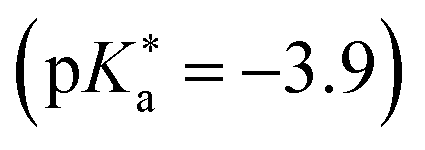 of this class. The N atom of the amide group(s) in compound C is directly attached to a methoxy (OCH3) group, which makes C slightly more acidic compared to photoacids D–F. However, compounds D and E, which turned out to have similar strengths as the previoulsy reported F (HPTA), were actually (successfully) designed to improve the water solubility compared to F, whereas C shows a high permeability through biological membranes. Aiming at even stronger photoacids that are capable of performing an ESPT in even more exotic environments, Maus et al. recently synthesized an APTS-based analogue of compound A, which is strong enough
of this class. The N atom of the amide group(s) in compound C is directly attached to a methoxy (OCH3) group, which makes C slightly more acidic compared to photoacids D–F. However, compounds D and E, which turned out to have similar strengths as the previoulsy reported F (HPTA), were actually (successfully) designed to improve the water solubility compared to F, whereas C shows a high permeability through biological membranes. Aiming at even stronger photoacids that are capable of performing an ESPT in even more exotic environments, Maus et al. recently synthesized an APTS-based analogue of compound A, which is strong enough 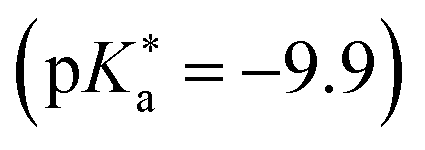 to perform ESPT in concentrated sulfuric acid (H2SO4), hence pointing to the development of magic photoacids.491 Here, the actual proton acceptor is bisulfate (HSO4−). Further attempts to generate even stronger photoacids by electrophilic substitution of diazapyrene492 failed so far.493 Unrelated to the Jung group, Lennox et al. reported an even newer pyrene-based photoacid, i.e., N,N,N′,N′,N′′,N′′-hexaethyl-8-hydroxypyrene-1,3,6-trisulfonamide (EtHPTA), derived from HPTS by conversion of the sulfonate into diethyl sulfonamide groups, with a pKa drop of ca. −11 (in acetonitrile) and exhibiting a long-lived (80 μs) triplet state.131 Besides, Hu et al. developed 2-(benzothiazol-2-yl)pyren-1-ol (P3-NS), a new hydroxypyrene derivative bearing benzothiazole, as a fluorescent sensor for nitroaromatic compounds (e.g., picric acid) that undergoes ESIPT in nonpolar or weakly polar solvents.494
to perform ESPT in concentrated sulfuric acid (H2SO4), hence pointing to the development of magic photoacids.491 Here, the actual proton acceptor is bisulfate (HSO4−). Further attempts to generate even stronger photoacids by electrophilic substitution of diazapyrene492 failed so far.493 Unrelated to the Jung group, Lennox et al. reported an even newer pyrene-based photoacid, i.e., N,N,N′,N′,N′′,N′′-hexaethyl-8-hydroxypyrene-1,3,6-trisulfonamide (EtHPTA), derived from HPTS by conversion of the sulfonate into diethyl sulfonamide groups, with a pKa drop of ca. −11 (in acetonitrile) and exhibiting a long-lived (80 μs) triplet state.131 Besides, Hu et al. developed 2-(benzothiazol-2-yl)pyren-1-ol (P3-NS), a new hydroxypyrene derivative bearing benzothiazole, as a fluorescent sensor for nitroaromatic compounds (e.g., picric acid) that undergoes ESIPT in nonpolar or weakly polar solvents.494
Whereas most standard photoacids cannot transfer their proton to DMSO, the super-photoacid B shows indications to do so even in the aprotic, weakly basic solvent acetone.61 Acetone appears to be a solvent that has not been the subject of any ESPT study until recently. In particular, impurities of water are known to interfere with the ESPT in such aprotic environments, e.g., as known for 6-hydroxyquinolinium in acetonitrile–water mixtures.458 In order to address the ESPT of photoacid B in acetone, we recently first performed a systematic study on the ESPT in acetone–water mixtures,294 prior to the experiments in neat acetone.499 These studies revealed that the ESPT in acetone–water mixtures follows a three-step Eigen–Weller model (see Section 6.2) with a reaction complex (CPX) between the photoacid and water, formed either already in the ground state or at least in the first step after excitation, and a hydrogen-bonded ion pair (HBIP) as central intermediates. Only the CPX can effectively perform an ESPT, passing through the HBIP, and finally yielding the free RO−* species if sufficient water molecules are present. Overall, the effective ESPT rate constant in acetone takes a value of 0.25 × 109 s−1, approaching the fluorescence lifetime and being slower compared to all other studied solvents (vide supra). Experiments with the strongest available ammonium-based photoacids in acetone in order to observe, e.g., keto–enol tautomerism by time-resolved IR spectroscopy failed as the mandatory ground-state protonation of the aminopyrene derivative initiated polymerization of acetone.493
While association constants could be determined from the changes in the UV/Vis absorption spectra, more detailed spectroscopic information on these intermediates was not directly accessible. Similarly, the study in neat acetone did not fully answer the question regarding the actual proton acceptor, keeping the possibility of ESPT to acetone still not fully settled. To fill this gap, complementary theoretical studies on the thermodynamics, i.e., the 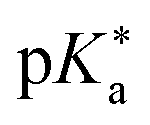 value in DMSO500 and acetone501 have been performed as well. These further support that the experimentally observed peak progression in the UV/Vis absorption spectra of photoacid B upon addition of water can only be explained based on specific intermolecular interactions, as well as that B remains the strongest photoacid among A–F even in acetone (with
value in DMSO500 and acetone501 have been performed as well. These further support that the experimentally observed peak progression in the UV/Vis absorption spectra of photoacid B upon addition of water can only be explained based on specific intermolecular interactions, as well as that B remains the strongest photoacid among A–F even in acetone (with  ).501
).501
6 Variations of the ESPT kinetics with photoacid strength and solvent
6.1 Separating different timescales
ESPT reactions are the outcome of numerous consecutive elementary processes that in total cover a wide range of timescales, from the sub-fs to the ns regime, comprising:48,502• electronic redistribution upon excitation (sub-fs),
• hydrogen-bond rearrangement near the (photo)acidic functional group (fs),
• proton dissociation and proton solvation (ps), and
• geminate recombination and excited-state decay (ns).
Huppert and colleagues proposed a classification of the photoacids into four regimes:503
(I)  (Weak photoacids): relatively slow ESPT (kPT ∼ 1010 s−1), efficient only in water; limited by covalent interactions within the proton-transferring complex; e.g., HPTS.
(Weak photoacids): relatively slow ESPT (kPT ∼ 1010 s−1), efficient only in water; limited by covalent interactions within the proton-transferring complex; e.g., HPTS.
(II)  (Typical super-photoacids): faster ESPT in water, also more efficient in many protic solvents; e.g., HPTS derivatives (HPTA, A–E).
(Typical super-photoacids): faster ESPT in water, also more efficient in many protic solvents; e.g., HPTS derivatives (HPTA, A–E).
(III) 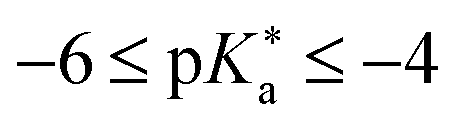 (Strong super-photoacids): rapid ESPT under solvent control in water, efficient in aprotic solvents; limited by solvent re-orientation; e.g., NM6HQ+.
(Strong super-photoacids): rapid ESPT under solvent control in water, efficient in aprotic solvents; limited by solvent re-orientation; e.g., NM6HQ+.
(IV) 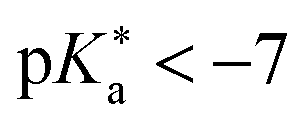 (Strongest super-photoacids): ultrafast ESPT beyond solvent control in water, methanol, and ethanol (kPT ≈ 1013 s−1); limited by donor–acceptor distance within complex; e.g., QCy7.
(Strongest super-photoacids): ultrafast ESPT beyond solvent control in water, methanol, and ethanol (kPT ≈ 1013 s−1); limited by donor–acceptor distance within complex; e.g., QCy7.
When moving from regime I to IV, the photoacid strength and ESPT rate increase, i.e. the capability to transfer protons to the solvent grows. In a nutshell, this classification reflects the competition between the ESPT rate constant, which is determined by the 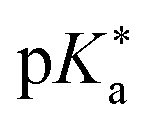 of a given photoacid and the solvent, and the radiative decay rate. Only if the photoacid is sufficiently strong, the ESPT is fast enough to happen within the typical fluorescence lifetime of a few ns. This distinction also highlights that the exact mechanism of the ESPT depends on the photoacid strength.
of a given photoacid and the solvent, and the radiative decay rate. Only if the photoacid is sufficiently strong, the ESPT is fast enough to happen within the typical fluorescence lifetime of a few ns. This distinction also highlights that the exact mechanism of the ESPT depends on the photoacid strength.
6.2 Multi-step models for ESPT in water
The Förster cycle (cf.Fig. 2) marks the simplest starting point for any description of the ESPT process, involving only the protonated species (ROH*) and the fully separated ion pair (FIP) consisting of the deprotonated species (RO−*) in the excited state and the dissociated proton (attached to some proton acceptor). However, since this one-step model was proposed based on a purely thermodynamic view that does not necessarily correspond to any mechanism, it often constitutes a drastic oversimplification. Instead, owing to the pioneering work of Eigen4,504,505 and Weller,107–109 a multi-step model for acid dissociation (in water) has been established that involves multiple proton-transfer intermediates in a step-wise dissociation. With respect to photoacids, the Eigen–Weller model has become particularly popular due to the work by Huppert and co-workers who have extensively applied it to the ESPT kinetics of HPTS205–212,237 and other weak (anionic) photoacids (e.g., naphthol sulfonates)364,400,404,409,412 in aqueous solution. Consisting of only two steps in the early description, this model has been extended over the years to a total of four steps, i.e., involving five species, in its most general formulation (see Fig. 15).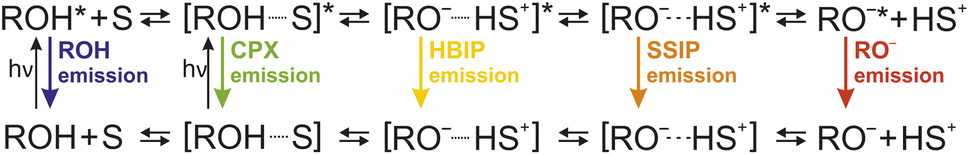 | ||
| Fig. 15 Four-step Eigen–Weller model. Five reactive species are considered: the free photoacid (ROH, blue), the reaction complex (CPX, green), the hydrogen-bonded ion pair (HBIP, yellow), the solvent-separated ion pair (SSIP, orange), and the free anion (RO−, red) [or fully separated ion pair (FIP), red]. | ||
According to the early two-step model for HPTS in water, the ROH* species first transfers its proton to a nearby water molecule under formation of a (hydrogen-bonded) contact ion pair (CIP or HBIP) within 90 ps.205,206 Only in a second step, this CIP is separated in a diffusive process, yielding the free RO−*. It is important to note here that diffusion plays a central role in the special case of HPTS. Due to its quadruple negative charge, geminate recombination of RO−* and the dissociated proton is largely enhanced by the coulomb attraction, giving rise to non-exponential (de-)population kinetics (Fig. 5) for ROH* and RO−*, manifesting in a characteristic long-time tail with a t−3/2 power-law asymptote.211,212 Thus, a mathematical treatment of the ESPT kinetics requires a (numerical) solution to the Debye–Smoluchowski equation (DSE).207–210 However, for most strong neutral or even cationic photoacids, solving the DSE is not necessary as diffusion has only a minor effect.58,456,470 Particularly, in the case of cationic photoacids, the lack of the negative charge in the corresponding base leads to the formation of a so-called Eigen complex instead of a HBIP, which is held together by dipole–dipole rather than coulomb interactions.465,506
Later, the rise of ultrafast time-resolved measurements revealed additional contributions to the ESPT of HPTS in water on the timescale of ∼300 fs and ∼3 ps, preceding the actual proton-transfer step, which challenged the existing two-step model.213–216 While the first (fastest) contribution was originally assigned by Tran-Thi et al. to solvation dynamics of an initial locally excited (LE) state and has been widely accepted,214 the interpretation of the second (intermediate) contribution became heavily debated. Interpretations are multifold and range from a conversion of an initial LE state into another electronic state (Tran-Thi),216 a reorganization of the surrounding water molecules to form a water wire (Bakker),231 a slow charge redistribution prior to the PT step (Spry/Fayer),140,223,224 the formation of a solvent-separated ion pair (Huppert),217,218 the hydration of the proton during deprotonation (Fang),226 and vibrational energy transfer into the first hydration shell (Havenith).63 In particular, the assignment to a solvent-separated ion pair (SSIP) as an additional intermediate between the CIP and the FIP by Huppert and co-workers lead to an expansion of their two-step into a three-step Eigen–Weller model217,218 (compare Fig. 15). Moreover, in light of the new knowledge on HPTS during 30 years of research and specifically a discrepancy regarding recent results by Lawler and Fayer259 for HPTS in water, Simkovitch et al. have recently presented a retrospective on their analysis.51,52 By repeating the experiments using newer equipment with better time resolution, they reaffirmed their previous reports regarding the t−3/2 power-law behavior, but they adopt the interpretation by Spry and Fayer for the 3 ps contribution as a slow charge rearrangement.
6.3 ESPT in non-aqueous solution
Several characteristics of the ESPT change when moving from aqueous to non-aqueous solutions. For example, in contrast to the most protic solvent, water, excited ion pairs such as the HBIP and SSIP are no longer short-lived in alcohols458,488,496 and even longer-lived in aprotic solvents,458,485–488 owing to lower proton conductivities (as argued by Kumpulainen et al.486). Moreover, diffusion-assisted geminate recombination usually has a negligible effect in aprotic solvents and thus the DSE does not need be considered, as demonstrated by Agmon and co-workers who discussed a (general) kinetic transition in reversible excited state reactions as a function of the excited-state lifetimes of the involved species.60,403,507–509 Besides, it has been argued that the solvent has a decisive effect on the order of the two lowest-lying electronic states, which can change when moving from aqueous to non-aqueous solutions.220,443,510 However, the reduced proton-acceptor ability in the case of protic or polar-aprotic solvents (e.g., DMSO) raises in parallel the need for complex formation prior to the ESPT. Therefore, if the photoacid and the proton acceptor are not in direct proximity prior to excitation, the (diffusive) formation of an encounter complex will typically constitute the first step, which requires another expansion of the kinetic model into a four-step Eigen–Weller model458,486 (Fig. 15). In the extreme, further restraining proton dissociation by changing to even less polar solvents (e.g., acetonitrile) finally completely prevents the ESPT unless a strong base is present for this complexation.460,486 Very recently, Lee et al. have demonstrated that the ESPT of the cationic photoacid NM7HQ+ (cf. Section 5.2.2) to DMSO in acetonitrile solution involves an Eigen complex and a Zundel-type ion, [(DMSO)2·H]+, for the solvated proton species.465 The importance of the transient Eigen complex as a short-lived, rarely observed ESPT intermediate has been recently highlighted in ref. 495. Similarly, also in aqueous solution this demand for pre-coordination with a base is reflected in the formation of water wires during (or prior to) the ESPT in water-co-solvent mixtures.56,57,413If a hydrogen-bonded pre-assembly between the photoacid and the proton-accepting base is present in aprotic solvents which themselves are bad proton acceptors, it is possible to isolate the characteristic emission spectra of the different species of the Eigen–Weller model in an elegant way. This was exemplarily shown for a complex of compound A (see Fig. 13d) and tri-n-octylphosphine oxide (TOPO), for which a deconvolution of the fluorescence spectrum into four contributing species could be realized in a systematic study using aprotic solvents with strongly varying polarity.511 It could be shown for a sample in acetonitrile that 7% of the integrated emission originate from ROH and RO− each, 39% from CPX, and 47% from HBIP (Fig. 16); whereas, no distinction between an SSIP as another potential intermediate and the FIP was possible. The formation of HBIP was proven by another change of the static dipole moment in the order of 3 debye. Time-resolved studies further revealed that the emission signals do not all occur simultaneously, but also not purely sequentially, as an equilibrium between CPX and HBIP is established faster than the excited-state lifetime.512 The subsequent irreversible dissociation of the HBIP takes place on the nanosecond timescale.
 | ||
| Fig. 16 Emission spectrum (dots) from a sample in which a complex between compound A and TOPO is present (solvent acetonitrile, 400 nm excitation). The spectrum is comprised of contributions from non-complexed ROH, a hydrogen-bonded complex CPX, a hydrogen-bonded ion pair HBIP, and the anionic species after the ion pair has separated. The displayed data is part of the study in ref. 511. | ||
6.4 Theories for the kinetics of elementary proton-transfer reactions
In the preceding sections of this review, ESPT to solvent was described as a sequential process with several steps. However, once SSIP is formed, no influence of the proton in aprotic solvents or only little influence in protic solvents on the electronic properties of the remaining base molecule (the former photoacid in its excited state) is expected.235 Therefore, differences in the ESPT kinetics of photoacids are assumed to be dominated by the initial proton-transfer step, that is the formation of HBIP from CPX, and, to a lower extent, by the subsequent transition from HBIP to SSIP. Kinetics models for the proton-transfer step were developed already in the past century and have been assessed elsewhere,88,503 and isotope effects turned out as worthwhile tools to verify the different models.513–516Shortly, these theories rely on intersecting harmonic electronic potentials in the diabatic regime (Marcus theory) which split up in the adiabatic limit, resulting in the well-accepted one dimensional double-well potential of proton transfer reactions.517,518 Both general approaches predict a correlation between the thermodynamic free energy of the reaction and the rate constant for the proton hopping along the reaction coordinate (free-energy correlation).513,519 Maximum rate constants are found in the range of 1012 to 1013 s−1,235,496,503 and the interpretation of this maximum value depends on the investigated system: solvent relaxation dynamics359,520 or the frequency of the intermolecular vibration of the donor and acceptor heavy atoms framing the hydrogen-transfer coordinate were held responsible for this limit.503 Irrespective of the accuracy of these interpretations, these findings revealed that analyses based on the reaction proceeding solely along a hydrogen-bond coordinate are an oversimplification and that taking into account additional contributions might be necessary.521 Modifications still in terms of an one-dimensional model are accomplished by considering an activation term, the work function, which does not contribute to the overall reaction energy but comprises geometric changes facilitating the proton transfer.50 This correction term for the simplified Marcus theory can become even larger than the intrinsic barrier of the proton transfer.522 Alternatively, further coordinates spanning potential energy surfaces for the proton transfer reaction are developed, which allow for considering tunneling as well.514,515
A peculiarity of Marcus theory is that it predicts a decreasing proton transfer rate constant for very large exergonicities, whereas, models in the adiabatic regime like the semi-empirical bond-energy bond-order (BEBO) model level off. So far, it is still debated whether multidimensional configurations, required to promote proton transfer, are compatible with this so-called inverted Marcus regime.522,523 First studies in this direction have been reported,518 and magic photoacids might be appropriate to provide additional insight into this highly topical facet of ESPT.
7 Quantification of excited-state acidity
7.1 The molecular origin of photoacidity
The most fundamental origin of photoacidity can be found in the electronic charge redistribution upon excitation (cf.Fig. 17). As commonly explained, photoacidity is caused by an intramolecular charge transfer (ICT) in the excited state of the ROH species from the O atom in the OH group to the aromatic ring, more precisely the distal ring in the case of condensed aromatics.94,113 In terms of molecular orbitals, this ICT corresponds to a transition of an electron from the non-bonding MO (NBMO) localized at the O atom into the lowest unoccupied MO (LUMO) that does not or less extend over the O atom (cf. Saway et al.7). Accordingly, the decreased electronic charge density at the OH group enhances its acidity. However, despite its plausibility, this explanation is incomplete since it does not take into account the corresponding base RO−, as pointed out by Agmon.48 As for the ground-state, (photo)acidity is the mutual outcome of both a destabilization of ROH and a stabilization of RO−. If no charge transfer was found for RO−, the proposed ICT of ROH would actually lead to an increased ROH–RO− energy gap that would make the excited-state deprotonation less favored. Thus, an even larger ICT with the same character must occur for the base, which is indeed confirmed by experiments, e.g. in solvatochromism295,296,405,406,524 or Stark spectroscopy.223 Therefore, photoacidity can be exemplified as the net effect of an excess RO− stabilization. | ||
| Fig. 17 Differences in the electron density between the first excited state S1 and the ground state S0 for the acid (a) and corresponding base (b) species of photoacid F (cf.Fig. 14d). Densities calculated with ADC(2)/aug-cc-pVDZ at optimized MP2/cc-pVDZ geometries and plotted at isosurface values of ±5 × 10−4 (−: blue; +: red). The displayed data is part of the study in ref. 500. | ||
Another possibility to rationalize this phenomenon is in terms of Hückel's (anti)aromaticity,525 as originally proposed by Agmon48,315 and recently reviewed by Wen526 and Yan et al.527 Aromatic molecules experience an energetic stabilization due to a beneficial electronic configuration, whereas molecules that would be classified as antiaromatic are energetically destabilized leading to structural perturbations, e.g., non-planarity or bond length alternation, in order to avoid the antiaromatic structure.528 For both excited triplet and singlet states, it has been shown that the situation changes and Hückel aromatics become less aromatic, i.e., more antiaromatic, and vice versa antiaromatics become more aromatic.529–531 Prototypic hydroxyarene photoacids such as phenols or naphthols are aromatic in their protonated form. However, after deprotonation the negative charge is stabilized by delocalizing it in the conjugated π system at the expense of including an additional electron pair in the conjugation and lowering its aromatic character. Due to the reversal of aromaticity and antiaromaticity in the S1 state, the protonated species is less aromatic in the excited state (S1) and hence becomes destabilized compared the ground state (S0). Even more important, in the excited state, the deprotonated species increases in aromaticity corresponding to a stabilization in accordance with the strong ICT. These differences in the aromatic character of the acid and the base and between S1 and S0 typically lead to larger Stokes shifts for the acid compared to the base and correspondingly stronger structural changes for the acid.302,315
7.2 Different ways to determine 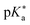

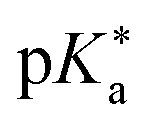 in both experimental and theoretical investigations.532 It has been critically reviewed many times, e.g., by Pines et al.39,432 and Grabowski et al.105,533 From an experimental perspective, UV/Vis absorption and fluorescence emission spectroscopy are typically used to determine the band maximum positions for the first electronic transition of the two species and then the average absorption–emission energy is used in eqn (1), as an approximation to the true 0–0 transition energy.47,105 This implicitly assumes that the shape of the potential energy surfaces in both electronic states are similar (i.e., the harmonic force constants are alike), allowing the vibrational contributions to the vibronic 0–0 transition and the differences between absorption and fluorescence emission to effectively cancel out in the average.534 However, the main problem arises from the use of the fluorescence energies in the first place since full equilibration is not generally given in the excited state, depending on the lifetime, which can cause
in both experimental and theoretical investigations.532 It has been critically reviewed many times, e.g., by Pines et al.39,432 and Grabowski et al.105,533 From an experimental perspective, UV/Vis absorption and fluorescence emission spectroscopy are typically used to determine the band maximum positions for the first electronic transition of the two species and then the average absorption–emission energy is used in eqn (1), as an approximation to the true 0–0 transition energy.47,105 This implicitly assumes that the shape of the potential energy surfaces in both electronic states are similar (i.e., the harmonic force constants are alike), allowing the vibrational contributions to the vibronic 0–0 transition and the differences between absorption and fluorescence emission to effectively cancel out in the average.534 However, the main problem arises from the use of the fluorescence energies in the first place since full equilibration is not generally given in the excited state, depending on the lifetime, which can cause 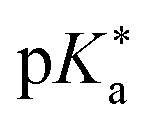 to be effectively treated as a non-equilibrium quantity. Moreover, the Förster cycle requires knowledge on pKa, which is usually straightforward in aqueous solution, but becomes more difficult in non-aqueous solution (vide supra). Alternatively,
to be effectively treated as a non-equilibrium quantity. Moreover, the Förster cycle requires knowledge on pKa, which is usually straightforward in aqueous solution, but becomes more difficult in non-aqueous solution (vide supra). Alternatively, 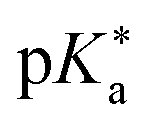 can also be determined experimentally by use of fluorescence titration,535,536 which identifies
can also be determined experimentally by use of fluorescence titration,535,536 which identifies 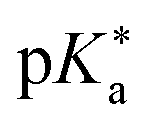 as the inflection point in the plot of the fluorescence intensity ratio I(ROH)/(I(ROH) + I(RO−)) against pH, or by direct kinetic measurements of the excited-state proton dissociation and recombination rate constants48 (see Fig. 18); both with their own limitations. Fluorescence titration is virtually limited to aqueous solutions due to the requirement of having a defined pH, which becomes non-trivial in non-aqueous solutions where no unified pH has yet been established.538–541 The direct kinetic method is in principle most accurate, but such measurements are technically more demanding and also the (missing) reversibility of the ESPT process can become an issue again.542 Therefore, the Förster cycle continues to be the standard route for experimental
as the inflection point in the plot of the fluorescence intensity ratio I(ROH)/(I(ROH) + I(RO−)) against pH, or by direct kinetic measurements of the excited-state proton dissociation and recombination rate constants48 (see Fig. 18); both with their own limitations. Fluorescence titration is virtually limited to aqueous solutions due to the requirement of having a defined pH, which becomes non-trivial in non-aqueous solutions where no unified pH has yet been established.538–541 The direct kinetic method is in principle most accurate, but such measurements are technically more demanding and also the (missing) reversibility of the ESPT process can become an issue again.542 Therefore, the Förster cycle continues to be the standard route for experimental 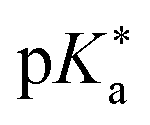 determination. Recently, Bhide et al. have reported a method for the experimental quantification of acidity for weak photoacids—i.e., when the ESPT is slow—using steady-state photoluminescence spectroscopy and a driving-force-dependent kinetic theory.543
determination. Recently, Bhide et al. have reported a method for the experimental quantification of acidity for weak photoacids—i.e., when the ESPT is slow—using steady-state photoluminescence spectroscopy and a driving-force-dependent kinetic theory.543
 | ||
Fig. 18 Three main experimental methods for determining the excited-state acidity,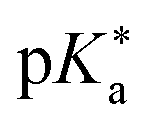 , using coumarin 183 as an example: (a) fluorescence titration that measures the fluorescence spectra of the excited ROH* and RO−* species at varying pH; (b) the Förster cycle that shifts the ground-state pKa by the change in acidity , using coumarin 183 as an example: (a) fluorescence titration that measures the fluorescence spectra of the excited ROH* and RO−* species at varying pH; (b) the Förster cycle that shifts the ground-state pKa by the change in acidity  determined from electronic transition (i.e., absorption and/or emission) energies; (c) time-resolved experiments that measure the transient signals of the excited species after electronic excitation, allowing for determination of the rate constants involved in the ESPT based on kinetic models. This figure has been reproduced from ref. 537 with the kind consent from S. Park and with permission from the Royal Society of Chemistry, copyright 2022. determined from electronic transition (i.e., absorption and/or emission) energies; (c) time-resolved experiments that measure the transient signals of the excited species after electronic excitation, allowing for determination of the rate constants involved in the ESPT based on kinetic models. This figure has been reproduced from ref. 537 with the kind consent from S. Park and with permission from the Royal Society of Chemistry, copyright 2022. | ||
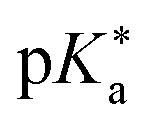 when comparing experimental and theoretical
when comparing experimental and theoretical 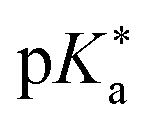 , and second,
, and second,  will often not be a pure equilibrium quantity.
will often not be a pure equilibrium quantity.
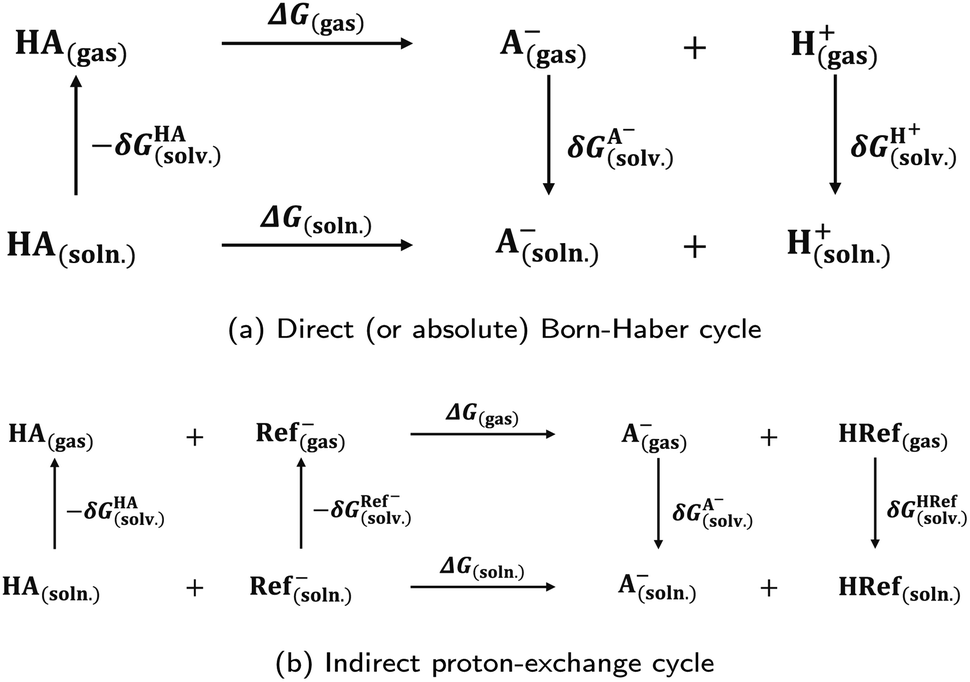 | ||
Fig. 19 Different thermodynamic cycles for the calculation of (ground-state) pKa values. The direct cycle (a) and the proton-exchange cycle (b) as an example of an indirect cycle. These cycles can be equally applied to electronically excited states for the prediction of  . This figure is based on ref. 544. . This figure is based on ref. 544. | ||
7.3 Existing theoretical studies on 
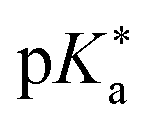 in particular is still very sparse. Early calculations by Gao et al. used a QM/MM formalism—treating the solute at the Complete Active Space Self Consistent Field (CASSCF) and multi-reference second-order perturbation theory (CASPT2) levels and the solvent classically—to determine the ΔpKa of phenol in water.561 Later, Granucci et al. also applied CASSCF and CASPT2, but in combination with the Polarizable Continuum Model (PCM)562 for implicit solvation to study the excited-state acidity of phenol and cyanophenols in the gas phase and in water.302 The use of such high-level multi-reference (MR) methods is generally restricted to smaller molecules due to the size of the active space. This is the reason why larger systems such as pyrenols216 or anthocyanins,563 but even naphthols315 could only be studied within semi-empirical frameworks for some time. In line with this, Szczepanik et al. also discussed the effect of the cyano (CN) group on pKa and
in particular is still very sparse. Early calculations by Gao et al. used a QM/MM formalism—treating the solute at the Complete Active Space Self Consistent Field (CASSCF) and multi-reference second-order perturbation theory (CASPT2) levels and the solvent classically—to determine the ΔpKa of phenol in water.561 Later, Granucci et al. also applied CASSCF and CASPT2, but in combination with the Polarizable Continuum Model (PCM)562 for implicit solvation to study the excited-state acidity of phenol and cyanophenols in the gas phase and in water.302 The use of such high-level multi-reference (MR) methods is generally restricted to smaller molecules due to the size of the active space. This is the reason why larger systems such as pyrenols216 or anthocyanins,563 but even naphthols315 could only be studied within semi-empirical frameworks for some time. In line with this, Szczepanik et al. also discussed the effect of the cyano (CN) group on pKa and  in phenol, naphthol, biphenyl, and aniline derivatives using AM1 and single excitation CI (SECI).532,564 In addition, also some molecular simulation studies on
in phenol, naphthol, biphenyl, and aniline derivatives using AM1 and single excitation CI (SECI).532,564 In addition, also some molecular simulation studies on 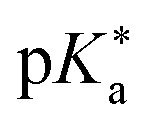 , e.g., on pyrimidine in aqueous solution by Gao et al. (using semi-empirical AM1 for the electronic structure)565 or the biologically relevant phytochromobilin chromophore by Borg et al. (using TDDFT in combination with IEF-PCM for the electronic excitations; tested against CIS, CIS(D), SAC-CI, and CASSCF)566,567 have been reported.
, e.g., on pyrimidine in aqueous solution by Gao et al. (using semi-empirical AM1 for the electronic structure)565 or the biologically relevant phytochromobilin chromophore by Borg et al. (using TDDFT in combination with IEF-PCM for the electronic excitations; tested against CIS, CIS(D), SAC-CI, and CASSCF)566,567 have been reported.
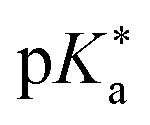 for coumarins that is purely based on TDDFT in combination with PCM.342 This approach has been further extended by Houari et al. to the classes of naphthols and cyanonaphthols by critically comparing the results for the Förster cycle and the direct (Born–Haber) cycle as well as for the linear response (LR) and state-specific (SS) variants of PCM.346 Later, Houari et al. have applied their improved protocol for
for coumarins that is purely based on TDDFT in combination with PCM.342 This approach has been further extended by Houari et al. to the classes of naphthols and cyanonaphthols by critically comparing the results for the Förster cycle and the direct (Born–Haber) cycle as well as for the linear response (LR) and state-specific (SS) variants of PCM.346 Later, Houari et al. have applied their improved protocol for 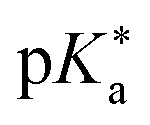 prediction based on TDDFT and (SS)-PCM again to the coumarins, while also addressing the effect of explicit solvent (water) in the first solvation shell of these photoacids.328 Recently, Aquino and co-workers have used TDDFT in combination with PCM or the Conductor-Like Screening Model (COSMO) to study the photoacidity of two different hydroxyflavylium cations.348,350 Moreover, Joung et al. have used (TD)DFT as a reference for evaluating different experimental methods for the determination of
prediction based on TDDFT and (SS)-PCM again to the coumarins, while also addressing the effect of explicit solvent (water) in the first solvation shell of these photoacids.328 Recently, Aquino and co-workers have used TDDFT in combination with PCM or the Conductor-Like Screening Model (COSMO) to study the photoacidity of two different hydroxyflavylium cations.348,350 Moreover, Joung et al. have used (TD)DFT as a reference for evaluating different experimental methods for the determination of 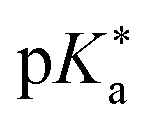 , revealing the usage of optical bandgaps that are defined by the absorption and emission edges as transition energies in the Förster cycle to be most reliable.
, revealing the usage of optical bandgaps that are defined by the absorption and emission edges as transition energies in the Förster cycle to be most reliable.
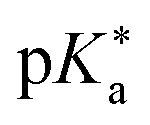 , e.g., regarding the reaction mechanism of the ESPT (kinetics) mechanism,68,191,321,325–330,332,335,574–576 spectral characterization and insights into the origin of photoacidity through the electronic structure,322,331,334,336,471,577 or as the method of choice for treating the electronic excitations in numerous QM/MM molecular dynamics (MD) studies (see Section 3).193,351–355,357,358,578–580 TDDFT has been also applied to study the proton transfer within the biologically relevant green fluorescent proteins.27,28 Only a few AIMD studies using MR methods instead of TDDFT have been reported.581–583 While the use of TDDFT for electronic excitation energies is for sure not an issue on its own—TDDFT has established as a rich field that continuously develops and validates new methods—,584–586 standard DFT methods can still not be said to be fully generally reliable.587,588 Prominent examples of such failures for TDDFT include charge-transfer or Rydberg excitations, and these have been addressed in the literature, e.g., by using range-separated functionals such as CAM-B3-LYP.589 However, TDDFT sometimes gives wrong shapes for the potential energy surfaces of the excited states590 and suffers from relatively low accuracies for vertical excitations (±0.3 eV).591 In an extreme case, TDDFT can even predict a wrong order of the electronic states under certain circumstances. Recently, Acharya et al. have demonstrated that TDDFT fails to predict the correct order of the 1La and 1Lb states of the photoacids 1- and 2-naphthol by comparison with Equation-Of-Motion Coupled Cluster Singles and Doubles (EOM-CCSD) (all calculations using C-PCM) and with experimental results.592 Similar observations had been previously made by Grimme and Parac,593 who assigned this imbalanced description of the two states with TDDFT to an underestimation of the interaction of the ionic components in the corresponding 1La-state wave functions with current standard functionals, and by Prlj et al.,594 who extended the scope of this issue to a set of fused heteroaromatics and demonstrated that the 1Lb state is highly sensitive to correlation effects, while the 1La state suffers from the same drawbacks as charge transfer excitations.
, e.g., regarding the reaction mechanism of the ESPT (kinetics) mechanism,68,191,321,325–330,332,335,574–576 spectral characterization and insights into the origin of photoacidity through the electronic structure,322,331,334,336,471,577 or as the method of choice for treating the electronic excitations in numerous QM/MM molecular dynamics (MD) studies (see Section 3).193,351–355,357,358,578–580 TDDFT has been also applied to study the proton transfer within the biologically relevant green fluorescent proteins.27,28 Only a few AIMD studies using MR methods instead of TDDFT have been reported.581–583 While the use of TDDFT for electronic excitation energies is for sure not an issue on its own—TDDFT has established as a rich field that continuously develops and validates new methods—,584–586 standard DFT methods can still not be said to be fully generally reliable.587,588 Prominent examples of such failures for TDDFT include charge-transfer or Rydberg excitations, and these have been addressed in the literature, e.g., by using range-separated functionals such as CAM-B3-LYP.589 However, TDDFT sometimes gives wrong shapes for the potential energy surfaces of the excited states590 and suffers from relatively low accuracies for vertical excitations (±0.3 eV).591 In an extreme case, TDDFT can even predict a wrong order of the electronic states under certain circumstances. Recently, Acharya et al. have demonstrated that TDDFT fails to predict the correct order of the 1La and 1Lb states of the photoacids 1- and 2-naphthol by comparison with Equation-Of-Motion Coupled Cluster Singles and Doubles (EOM-CCSD) (all calculations using C-PCM) and with experimental results.592 Similar observations had been previously made by Grimme and Parac,593 who assigned this imbalanced description of the two states with TDDFT to an underestimation of the interaction of the ionic components in the corresponding 1La-state wave functions with current standard functionals, and by Prlj et al.,594 who extended the scope of this issue to a set of fused heteroaromatics and demonstrated that the 1Lb state is highly sensitive to correlation effects, while the 1La state suffers from the same drawbacks as charge transfer excitations.
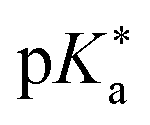 (vide supra), ADC(2) and CC2 are much more applicable in a black-box fashion with respect to the computational costs compared to aforementioned multi-reference methods.
(vide supra), ADC(2) and CC2 are much more applicable in a black-box fashion with respect to the computational costs compared to aforementioned multi-reference methods.
Until recently, the only study that had used ADC(2) in the calculations of the excitation energies for (photoacid)  has been the work by Wang et al. on the photoacidity of cationic hydroxypyranoanthocyanines.349 However, in the actual computation of
has been the work by Wang et al. on the photoacidity of cationic hydroxypyranoanthocyanines.349 However, in the actual computation of  values, they restrict themselves to B3-LYP TDDFT instead of ADC(2) despite—or due to—a close similarity between the results for these two methods. Related studies by the same groups (i.e., Aquino, Quina, and co-workers) on similar pyranoanthocyanines337,338 or other hydroxyflavylium cations339,340 also use ADC(2) in combination with COSMO for describing the electronically excited states, but do not explore
values, they restrict themselves to B3-LYP TDDFT instead of ADC(2) despite—or due to—a close similarity between the results for these two methods. Related studies by the same groups (i.e., Aquino, Quina, and co-workers) on similar pyranoanthocyanines337,338 or other hydroxyflavylium cations339,340 also use ADC(2) in combination with COSMO for describing the electronically excited states, but do not explore 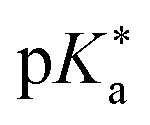 values. Recently, Khodia et al. have also used ADC(2) in a combined experimental–computational study on the ESIPT of a pyridylbenzimidazole photoacid within a water complex.595 Still, to the best of our knowledge, there had still been basically no approach to
values. Recently, Khodia et al. have also used ADC(2) in a combined experimental–computational study on the ESIPT of a pyridylbenzimidazole photoacid within a water complex.595 Still, to the best of our knowledge, there had still been basically no approach to 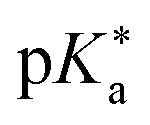 beyond TDDFT that actually uses accurate, but easy-to-apply WFT methods like ADC(2) or CC2. This gap regarding WFT studies on photoacid
beyond TDDFT that actually uses accurate, but easy-to-apply WFT methods like ADC(2) or CC2. This gap regarding WFT studies on photoacid 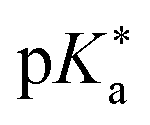 values has recently been addressed by Hättig and co-workers.
values has recently been addressed by Hättig and co-workers.
Ghiami-Shomami and Hättig performed extensive benchmark calculations of Förster-cycle-based 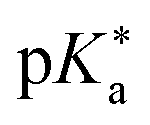 values of 12 photoacids (phenols, naphthols, and hydroxycoumarins) and 8 photobases (quinolines and acridine) in water,596 covering a broad variety of methods for describing the electronic transitions (including 8 simple up to state-of-the-art DFT functionals, ADC(2), and CC2) and obtaining the corresponding ground-state pKa values with COSMO-RS597–599 at the DFT BP86 level. Their results indicate that such a simple protocol based on an implicit solvent model and vertical excitation energies is in general not sufficient to predict
values of 12 photoacids (phenols, naphthols, and hydroxycoumarins) and 8 photobases (quinolines and acridine) in water,596 covering a broad variety of methods for describing the electronic transitions (including 8 simple up to state-of-the-art DFT functionals, ADC(2), and CC2) and obtaining the corresponding ground-state pKa values with COSMO-RS597–599 at the DFT BP86 level. Their results indicate that such a simple protocol based on an implicit solvent model and vertical excitation energies is in general not sufficient to predict 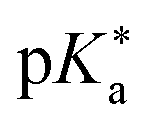 values with an accuracy comparable with that achieved by the above mentioned protocols for the ground state pKa values. Following the same approach, Sülzner and Hättig have studied the
values with an accuracy comparable with that achieved by the above mentioned protocols for the ground state pKa values. Following the same approach, Sülzner and Hättig have studied the 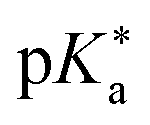 values of the hydroxypyrene derivatives discussed in Section 5.4 in the aprotic solvents DMSO500 and acetone501 using ADC(2) and CC2 in combination with COSMO. Prior to
values of the hydroxypyrene derivatives discussed in Section 5.4 in the aprotic solvents DMSO500 and acetone501 using ADC(2) and CC2 in combination with COSMO. Prior to 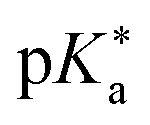 prediction in acetone, the study in DMSO first assessed the performance of the selected computational methods for the full set of photoacids A–F (cf.Fig. 13d) by validation against experimental data.500 Additionally, with COSMO-RS as the underlying method for the ground-state pKa values in this computational protocol, the (previously missing) corresponding linear free-energy relationship parameters for pKa prediction in acetone also needed to be determined.600 Overall, the results on these photoacids in DMSO and acetone (cf.Fig. 20) show that whenever the photoacidic OH group can form hydrogen bonds either with solvent molecules or by self-association, the errors of a purely implicit solvation model limit the accuracy of the calculations. However, and in line with the previous results by Ghiami-Shomami and Hättig, the second-order methods ADC(2) and CC2 significantly outperform TDDFT for the prediction of ΔpKa, i.e., the only contribution to
prediction in acetone, the study in DMSO first assessed the performance of the selected computational methods for the full set of photoacids A–F (cf.Fig. 13d) by validation against experimental data.500 Additionally, with COSMO-RS as the underlying method for the ground-state pKa values in this computational protocol, the (previously missing) corresponding linear free-energy relationship parameters for pKa prediction in acetone also needed to be determined.600 Overall, the results on these photoacids in DMSO and acetone (cf.Fig. 20) show that whenever the photoacidic OH group can form hydrogen bonds either with solvent molecules or by self-association, the errors of a purely implicit solvation model limit the accuracy of the calculations. However, and in line with the previous results by Ghiami-Shomami and Hättig, the second-order methods ADC(2) and CC2 significantly outperform TDDFT for the prediction of ΔpKa, i.e., the only contribution to  including the excitation energies according to this protocol. For reliable calculations of the absolute
including the excitation energies according to this protocol. For reliable calculations of the absolute 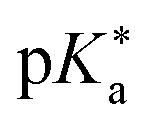 , but also in general for the description of excited-state proton-transfer reactions in solution, a more accurate approach seems required, with MD simulations comprising explicit solvent being among the desiderata for future studies.
, but also in general for the description of excited-state proton-transfer reactions in solution, a more accurate approach seems required, with MD simulations comprising explicit solvent being among the desiderata for future studies.
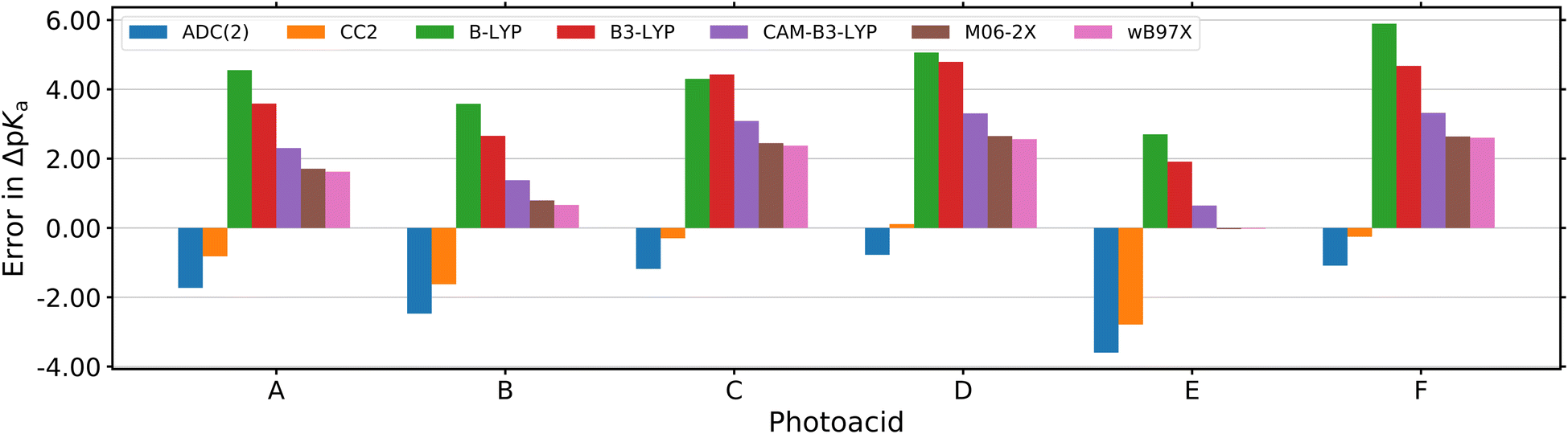 | ||
| Fig. 20 Comparison of the prediction error in Förster-cycle ΔpKa values (cf.eqn (1)) of Jung's super-photoacids A–F (cf.Fig. 13d) in the aprotic solvent acetone for different quantum-chemical methods with respect to the experiment. In total, the wavefunction-based methods ADC(2) and CC2 perform much better compared to TDDFT. The slightly worse performance for photoacids A and B is attributed to the higher need for explicit solvent interactions that are missing with an implicit solvent model. The distinct deviations for E are due to intermolecular hydrogen bonding between two photoacid molecules. The displayed data is part of the study in ref. 501. | ||
8 Conclusion and outlook
8.1 The theoretical future of photoacids
Among the theoretical studies presented and discussed here, we would like to conclude with some highlights that are particularly interesting to us. First, the ability to obtain direct insights into the electronic structure of photoacids via quantum-chemical calculations remains a fundamental advantage from the theoretical perspective, as the character of the excited states is known to play a role for the photoacidity.39,432 This is not only important for the comparison of calculated with experimental spectra, but can, vice versa, even help with the interpretation of experimental data. Platt's notation for the two lowest-lying excited states, 1La and 1Lb, although being conceptually very useful, quickly reaches its limit, e.g., for highly-substituted systems whose broken symmetry can introduce mixing between these two states. Such a mixing already occurs for the naphthols443 and attempts to assign the 1La and 1Lb states of HPTS have led to an apparently unresolved debate.214,216–218,220,223,224,226,230 Although new high-level calculations on HPTS could contribute to resolving this debate, forcing such an assignment of 1La and 1Lb might not be meaningful due the potential limitations of that model when exceeding its intended range of molecules. Second, even outside the picture of Platt's 1La and 1Lb states, an accurate description of the electronically excited states is important. In this regard, a general gap of WFT studies can be made out in the literature since most studies use (TD)DFT, which can be potentially problematic since the order of the these two low-lying states is sensitive to the amount of exact exchange in the DFT functional.593 Particularly, the purpose of a theoretical study might be to actually predict the order of these states, requiring extra caution from the user in these cases.An improved protocol for the prediction of ΔpKa, i.e., the acidity change upon excitation, still remains a future goal. Easy-to-use protocols developed so far typically rely on cheap methods like TDDFT combined with an implicit solvent model.342,346,347 Methodologically, not only a reliable and accurate electronic structure method (e.g., coupled cluster theory instead of TDDFT), but also a proper solvation treatment are required for this. While preceding studies have shown that the implicit solvent model COSMO in combination with ADC(2) or CC2 can yield reasonably good accuracy in the prediction of Förster-cycle-based ΔpKa values in the aprotic solvents DMSO and acetone,500,501 implicit solvation is not sufficient in the protic solvent water.501,596 Improving the description of solvation must not directly lead to incorporating lots of explicit solvent molecules and running an MD simulation, but could also first try to use a more accurate model within these two extremes of implicit and fully explicit solvation [e.g., the embedded cluster reference interaction site model (EC-RISM)].601,602 However, when aiming at best compatibility between theory and experiments, such MD simulations in solution will eventually become inevitable since, even apart from solvent effects, the vertical approximation has already been identified as the (other) main limitation (cf. acetone and DMSO).500,501 In contrast to static calculations (often on isolated molecules in vacuum), these MD simulations are capable of explicitly accounting for the vibrational contributions to the vibronic transitions (e.g., via an ensemble average), as further discussed in the following.
On the one hand, the sampling for generating representative solvent configurations around the photoacid as the solute is best performed using ab initio MD (AIMD) instead of classical MD techniques to avoid a systematic bias on the electronic excitation energies through the geometries as affected by the underlying method for calculating the energy gradients. While such AIMD simulations are practically restricted to DFT as the electronic structure method for the time propagation, a different (higher-level) method could be used for calculating the vertical excitation energies at selected snapshots taken from the trajectory (e.g., see references in ref. 603). On the other hand, the comparison of theoretical and experimental ΔpKa values will also involve the fluorescence because an average of the absorption and emission band maxima is typically used in the experiments as an approximation to the 0–0 transition energies. This makes things even worse since already a ground-state AIMD simulation can be a demanding task for photoacid molecules of the size of HPTS with explicit solvent, but an equivalent description of fluorescence requires capturing the excited-state dynamics. Due to the higher computational demands compared to the ground state, such MD simulations for the excited state are basically restricted to TDDFT as a cheap method, with the potential disadvantages discussed earlier (see Section 7.3.3).
Mentioning excited-state dynamics brings us to our final highlight from a theoretical perspective: The allegate dream of simulating the full ESPT process of a photoacid in solution. Technically, this still requires an AIMD simulation for sampling excited-state dynamics, again with the associated requirements and limitations discussed above. If level crossing is involved, e.g., an inversion of the 1La and 1Lb states that is frequently discussed for the naphthols (cf. ref. 39 and 432 and references therein), surface-hopping techniques,** (e.g., Tully's fewest switches surface hopping, FSSH)605 are needed to account for transitions between the excited states. Important examples of such surface-hopping AIMD studies on photoacids, which are still quite rare in general, include the work on the ESIPT of 2-phenylphenol582 and 4-(2′-hydroxyphenyl)pyridine583 by Cui, Thiel, and co-workers; and that on salicylidene methylamine Schiff-base photoswitches606 by Barbatti and co-workers. Importantly, in these non-adiabatic excited-state dynamics studies, the choice of initial points has been generally found a rather important topic in theoretical-computational studies, from spectrum simulation to solute–solvent configuration.607–610 Moreover, a fundamental problem arises from the different timescales of the ESPT process itself (cf. Section 6.1). Since such AIMD simulations in the excited state are typically restricted to a few tens or hundreds of ps, only the initial processes contributing to the ESPT might be observable apart from ultrafast solvation dynamics; including (1) the possible conversion from a locally excited (LE) state into a charge-transfer (CT) state (i.e., 1La/1Lb conversion) of the ROH* species prior to the proton transfer (PT) and (2) the PT event itself yielding a contact ion pair (CIP) as the first intermediate. The subsequent separation of the CIP into free ions, with other hydrogen-bonded species as additional intermediates in terms of a multi-step Eigen–Weller model are out of reach for excited-state AIMD. However, it can be argued that the separation of the CIP is less unique—and hence less interesting—for photoacids as this resembles the normal dissociation of ionic species in solution that is characteristic for a given solvent. Among the few AIMD studies on photoacids in the literature, the two recent ones on the excited-state and ESPT of HPTS in 1-methylimidazole (by Thomaz et al.)357 and water (by Walker et al.)358 appear impressive examples to us, demonstrating how the technical frontiers are evolving.
8.2 Established and future-oriented applications of photoacids
Among all photoacids, HPTS is certainly the most widely used and commercially available fluorescence marker due to its strong visible fluorescence. Some applications rely on its multiple negative charges,611 a property which is, however, not specific and other fluorophores might be equally useful, e.g., for the delivery of electrons upon photoexcitation.612,613 Its usefulness as paradigmatic†† pH indicator was noticed already many decades ago (see references in ref. 203), but an application to cellular systems is limited by the restricted permeation through biological interfaces due to the high number of charges. Therefore, removal of these by substitution drastically accelerates the uptake,61 and similar synthetic modifications enable the incorporation into nanoparticles making them pH-sensitive.615 Also the related ESIPT photoacids,616 metastable-state photoacids,617,618 and photo-acid generators619 have proven successful for controlling (intracellular) pH. The advantage of photoacids over other pH-sensitive fluorophores is the possible ratiometric readout turning these pH-indicators more robust against concentration variations.620 One might argue here that the intensity ratio is obtained by exciting both the neutral and deprotonated state. Such a readout consequently calls for two excitation light sources which is less beneficial than those systems where the intensity ratio results from two detection channels.291 Fluorescence titrations of photoacids can make use of this experimental approach if the kinetics of de- and reprotonation are faster than the excited-state lifetime.621Truly dual-emissive conditions are met in the ESPT reaction itself, and several groups have pointed out that a minimum number of water molecules is necessary for ESPT in otherwise spectroscopically silent solvents or environments (see also the discussion in ref. 97).286,294,622 Such a strategy has been exploited to assess the water content in acetonitrile,623 or even in solid phases like metal–organic frameworks,624 silica materials and phosphate matrices.615,625 Likewise, hydrogen isotopes can be distinguished in protic solvents due to the kinetic isotope effect when the rate constant for the overall ESPT is in same range as the time constant for fluorescence emission.491,626
Another well-known way to make use of the dual-emissive properties is to inhibit the ESPT reaction by chemical substitution of the proton. A variety of substrates for enzyme reactions with low substrate specificity has been realized in the past.200,201,627 Removal of the negative charges allows for visualizing enzymatic reactions in the cellular environment as exemplified in the classification of the most abundant brain tumor meningioma by its phosphatase activity.628 Even the activity of two enzymes, independently from each other, can be probed by four emission colors in a confocal microscope: color tuning is achieved by playing with the electron-withdrawing capability of one substituent.184 As previous work has demonstrated the high photostability and large fluorescence quantum yields of these pyrene-based new millennium dyes,61 transfer of this dual-emissive readout to single-molecule microscopy enabled to track individual, Pd-induced deprotection reactions. This example of single-molecule chemistry can be operated in a fluorogenic approach or by simultaneously recording trajectories of substrate and product fluorescence.629,630
ESPT is often exploited for mechanistic studies of proton transfer in general and may complement findings by other spectroscopies. Therefore, ESPT is studied in solid materials625 as well as in unconventional liquids like concentrated sulfuric acid,491 liquid crystals,631 or ionic liquids.285,427 Among these, the luminescent ionic liquid with HPTS as anion is worth being highlighted;632 we are curious about the properties of cationic photoacids with weakly coordinating counter anions once they are available. Studies of ESPT on material surfaces or membranes are focused on characterizing proton diffusion with geometrical restrictions.633,634 The generation of protons on the surface of photocatalytically active nanoparticles even allows for manipulating their motion due to a change of the charge.635 More generally, owing to their property as light-controlled proton sources, photoacids can be used in photo-catalysis (see ref. 7 for a recent review), e.g., as initiators for polymerization,636,637 esterification,638 acetalization,639,640 glycosylation,641 olefin hydroarylation,313 indane synthesis,642 and self-propulsion of oil droplets;636 as photosensitizers (e.g., for singlet oxygen generation),643 or, for photoswitching enzyme activity.644 Other, more technically oriented applications of the photoinduced generation of protons aim at the release of CO2 from captured CO2 (as HCO3−)645–647 or the build-up of a pH gradient for a photo-electrochemical cell.648–650 The performance of these devices, however, is facilitated when the deprotonated state lasts longer, which explains the preference yet for metastable-state photoacids. Another electrochemical application of ESPT is light-controlled proton conduction.651,652 As free protons require additional water molecules for stabilization, ionization of photoacids buried in polymeric structures is supposed to lead to larger structures, and an application as carrier for therapeutics which release the cargo by a light stimulus on demand is foreseen.653–658 On the other hand, proton release in free solution apparently leads to a volume shrinkage.659 In conclusion, the swelling behaviour of photoacids in polymers is more complicated.163
Further research areas will be addressed once more regioselective substitution of pyrene-based photoacids or even stronger photoacids than those reported before are available: specific modifications on the pyrene core430,502 appear challenging according to our experience.
8.3 Open questions—new aspects to be explored by excited-state proton transfer
While some of the latter described applications certainly deserve further development for generating technologically relevant devices, we focus here on fundamental aspects of proton transfer reactions. Although the proton transfer is one of the best investigated reactions, not all aspects of this elementary reaction are known. For example, we wonder how the proton jumps away from the HBIP to the next intermediate of the reaction chain depicted in Fig. 15 and which molecular rearrangement or solvent properties assist this process. Is tunneling playing a significant role in ESPT?626,660 Actually, few low-temperature studies of ESPT in protic solvents are known and point into that direction.253,254,456,472,661ESPT is a paradigmatic charge transfer reaction including the transfer of a mass particle (compared to electron transfer), and findings about it may have impact for other chemical reactions.662,663 Even the important questions of chirality can be addressed.369,664 However, some peculiarities may be met due to the low mass (compared to the transfer of other atoms), and one may look for further evidence of the inverted Marcus-regime in proton transfer reactions.518
Finally, we emphasize that ESPT is one of the few adiabatic photochemical reactions—if not the only one useful. The adiabatic character becomes superior to the other well-studied charge transfer reaction, i.e., the electron transfer, when light emission is mandatory for detection like in single-molecule fluorescence spectroscopy. ESPT is quasi-reversible and can be monitored multiple times due to the ground-state reprotonation, in contrast to irreversible reactions. Hence, we expect to detect even intermediates and to characterize the influence of the environment on the different reaction steps which makes this kind of reaction unique. So far, single-molecule ESPT experiments in solution have been performed,497,498 but the transfer to individual molecules, e.g., the continuous observation of ESPT in solid DMSO, broke down yet. Our current interpretation is that infrequent formation of FIP, but still too frequent for single-molecule studies, leads to an intermittent disappearance of the photoacid state until the proton accidently is recaptured by the left negative charge. Other combinations of quantum-optical phenomena with photochemistry include different strengths of light-matter coupling,654,665,666 and we attempt to realize different logical gates for the generation of photons by means of photoacids.184
Data availability
No primary research results, software or code have been included as part of this review.Author contributions
Niklas Sülzner: conceptualization, methodology, validation, investigation, visualization, writing – original draft. Gregor Jung: conceptualization, supervision, resources, funding acquisition, writing – review & editing. Patrick Nuernberger: conceptualization, visualization, supervision, project administration, resources, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Christof Hättig and Dr Roger Jan Kutta for many inspiring discussions and their continuous support. NS thanks Ömer Tiska for enduring long discussions and going through many confusion during the assignment of the electronic states of phenol (and other) photoacids. We are indebted to many colleagues for fruitful debates and input, and especially those who granted us permission to use some of their research results for visualizing the different aspects of photoacidity in this manuscript. We have tried to give an appealing and potentially comprehensive overview of the topic, but we also want to apologize to those researchers whose studies might not have made it into our review of the field. We are grateful for the support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation): RTG 2620 “Ion Pair Effects in Molecular Reactivity” project 426795949 (PN); project JU650/12-1 (GJ); under Germany's Excellence Strategy EXC 2033–390677874–RESOLV (NS).References
- P.-T. Chou and K. M. Solntsev, J. Phys. Chem. B, 2015, 119, 2089 CrossRef PubMed.
- H. Kagel, M. Frohme and J. Glökler, J. Cell. Biotechnol., 2018, 4, 23–30 Search PubMed.
- S. Hammes-Schiffer, J. Phys. Chem. B, 2021, 125, 3725–3726 Search PubMed.
- M. Eigen, Angew Chem. Int. Ed. Engl., 1964, 3, 1–19 Search PubMed.
- H. Hattori and Y. Ono, Metal Oxides in Heterogeneous Catalysis, Elsevier, 2018, pp. 133–209 Search PubMed.
- P. Wan and D. Shukla, Chem. Rev., 1993, 93, 571–584 CrossRef.
- J. Saway, Z. M. Salem and J. J. Badillo, Synthesis, 2021, 53, 489–497 CrossRef.
- M. Shabani, H. Younesi, M. Pontié, A. Rahimpour, M. Rahimnejad and A. A. Zinatizadeh, J. Cleaner Prod., 2020, 264, 121446 CrossRef.
- M. Petukh, S. Stefl and E. Alexov, Curr. Pharm. Des., 2013, 19, 4182–4190 CrossRef PubMed.
- G. S. Longo, N. A. Pérez-Chávez and I. Szleifer, Curr. Opin. Colloid Interface Sci., 2019, 41, 27–39 Search PubMed.
- P. Neupane, S. Bhuju, N. Thapa and H. K. Bhattarai, Biomol. Concepts, 2019, 10, 1–10 Search PubMed.
- A. J. Kirby, eLS, John Wiley & Sons, Ltd, 2010 Search PubMed.
- I. Ghosh, S. Khan, G. Banerjee, A. Dziarski, D. J. Vinyard, R. J. Debus and G. W. Brudvig, J. Phys. Chem. B, 2019, 123, 8195–8202 CrossRef PubMed.
- M. Chattoraj, B. A. King, G. U. Bublitz and S. G. Boxer, Proc. Natl. Acad. Sci., 1996, 93, 8362–8367 CrossRef.
- H. Lossau, A. Kummer, R. Heinecke, F. Pöllinger-Dammer, C. Kompa, G. Bieser, T. Jonsson, C. M. Silva, M. M. Yang, D. C. Youvan and M. E. Michel-Beyerle, Chem. Phys., 1996, 213, 1–16 CrossRef.
- P. Leiderman, R. Gepshtein, I. Tsimberov and D. Huppert, J. Phys. Chem. B, 2008, 112, 1232–1239 CrossRef.
- P. Leiderman, D. Huppert, S. J. Remington, L. M. Tolbert and K. M. Solntsev, Chem. Phys. Lett., 2008, 455, 303–306 CrossRef.
- K. M. Solntsev, O. Poizat, J. Dong, J. Rehault, Y. Lou, C. Burda and L. M. Tolbert, J. Phys. Chem. B, 2008, 112, 2700–2711 CrossRef.
- J. N. Henderson, M. F. Osborn, N. Koon, R. Gepshtein, D. Huppert and S. J. Remington, J. Am. Chem. Soc., 2009, 131, 13212–13213 CrossRef.
- S. P. Laptenok, J. Conyard, P. C. Bulman Page, Y. Chan, M. You, S. R. Jaffrey and S. R. Meech, Chem. Sci., 2016, 7, 5747–5752 RSC.
- C. Chen, L. Zhu, M. S. Baranov, L. Tang, N. S. Baleeva, A. Y. Smirnov, I. V. Yampolsky, K. M. Solntsev and C. Fang, J. Phys. Chem. B, 2019, 123, 3804–3821 CrossRef.
- Z. Wang, Y. Zhang, C. Chen, R. Zhu, J. Jiang, T.-C. Weng, Q. Ji, Y. Huang, C. Fang and W. Liu, Angew. Chem., Int. Ed., 2023, 62, e202212209 CrossRef PubMed.
- J. J. v. Thor and P. M. Champion, Annu. Rev. Phys. Chem., 2023, 74, 123–144 CrossRef.
- A. Acharya, A. M. Bogdanov, B. L. Grigorenko, K. B. Bravaya, A. V. Nemukhin, K. A. Lukyanov and A. I. Krylov, Chem. Rev., 2017, 117, 758–795 CrossRef.
- V. Helms and W. Gu, Fluorescent Proteins I: From Understanding to Design, Springer, Berlin, Heidelberg, 2012, pp. 171–181 Search PubMed.
- G. Jung, Fluorescent Analogues of Biomolecular Building Blocks: Design and Applications, John Wiley & Sons, Ltd, 2016, pp. 55–90 Search PubMed.
- G. Donati, A. Petrone, P. Caruso and N. Rega, Chem. Sci., 2018, 9, 1126–1135 RSC.
- A. Petrone, P. Cimino, G. Donati, H. P. Hratchian, M. J. Frisch and N. Rega, J. Chem. Theory Comput., 2016, 12, 4925–4933 CrossRef PubMed.
- Y. Erez and D. Huppert, J. Phys. Chem. A, 2010, 114, 8075–8082 CrossRef PubMed.
- I. Presiado, Y. Erez and D. Huppert, J. Phys. Chem. A, 2010, 114, 13337–13346 CrossRef PubMed.
- I. Presiado, Y. Erez and D. Huppert, J. Phys. Chem. A, 2010, 114, 9471–9479 CrossRef PubMed.
- Y. Erez, I. Presiado, R. Gepshtein and D. Huppert, J. Phys. Chem. A, 2011, 115, 1617–1626 CrossRef PubMed.
- I. Presiado, R. Gepshtein, Y. Erez and D. Huppert, J. Phys. Chem. A, 2011, 115, 7591–7601 CrossRef.
- J. Kuchlyan, D. Banik, A. Roy, N. Kundu and N. Sarkar, J. Phys. Chem. B, 2014, 118, 13946–13953 CrossRef.
- J. Kuchlyan, D. Banik, N. Kundu, S. Ghosh, C. Banerjee and N. Sarkar, J. Phys. Chem. B, 2014, 118, 3401–3408 CrossRef PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. J. DeSa, P. Naumov and K. M. Solntsev, J. Am. Chem. Soc., 2016, 138, 16252–16258 CrossRef PubMed.
- V. O. Silva, A. A. Freitas, A. L. Maçanita and F. H. Quina, J. Phys. Org. Chem., 2016, 29, 594–599 CrossRef.
- F. H. Quina, P. F. Moreira, C. Vautier-Giongo, D. Rettori, R. F. Rodrigues, A. A. Freitas, P. F. Silva and A. L. Maçanita, Pure Appl. Chem., 2009, 81, 1687–1694 CrossRef.
- D. Pines and E. Pines, Hydrogen-Transfer Reactions, John Wiley & Sons, Ltd, 2006, pp. 377–415 Search PubMed.
- Proton-Transfer Reactions, ed. E. Caldin and V. Gold, Springer US, Boston, MA, 1975 Search PubMed.
- J. Reijenga, A. van Hoof, A. van Loon and B. Teunissen, Anal. Chem. Insights, 2013, 8, ACIS12304 CrossRef PubMed.
- B. Pathare, V. Tambe and V. P. Patil, Int. J. Pharm. Pharm. Sci., 2014, 6, 26–34 Search PubMed.
- L. Barcza and Á. Buvári-Barcza, J. Chem. Educ., 2003, 80, 822 CrossRef.
- A. Buvári-Barcza and L. Barcza, Die Pharmazie, 2005, 60, 243–246 Search PubMed.
- K. S. Alongi and G. C. Shields, Annual Reports in Computational Chemistry, Elsevier, 2010, vol. 6, pp. 113–138 Search PubMed.
- P. G. Seybold and G. C. Shields, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 290–297 Search PubMed.
- J. F. Ireland and P. A. H. Wyatt, Advances in Physical Organic Chemistry, Academic Press, 1976, vol. 12, pp. 131–221 Search PubMed.
- N. Agmon, J. Phys. Chem. A, 2005, 109, 13–35 CrossRef PubMed.
- N. M. Trieff and B. R. Sundheim, J. Phys. Chem., 1965, 69, 2044–2059 CrossRef.
- L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol., A, 1993, 75, 1–20 CrossRef.
- R. Simkovitch, D. Pines, N. Agmon, E. Pines and D. Huppert, J. Phys. Chem. B, 2016, 120, 12615–12632 CrossRef PubMed.
- R. Simkovitch, G. G. Rozenman and D. Huppert, J. Photochem. Photobiol., A, 2017, 344, 15–27 CrossRef.
- L. Genosar, B. Cohen and D. Huppert, J. Phys. Chem. A, 2000, 104, 6689–6698 CrossRef.
- A. A. Awasthi and P. K. Singh, ChemPhysChem, 2018, 19, 198–207 CrossRef PubMed.
- O. Gajst, O. Green, L. Pinto da Silva, J. C. G. Esteves da Silva, D. Shabat and D. Huppert, J. Phys. Chem. A, 2018, 122, 8126–8135 CrossRef PubMed.
- O. Gajst, L. Pinto da Silva, J. C. G. Esteves da Silva and D. Huppert, J. Phys. Chem. A, 2018, 122, 4704–4716 CrossRef PubMed.
- O. Gajst, L. Pinto da Silva, J. C. G. Esteves da Silva and D. Huppert, J. Phys. Chem. A, 2019, 123, 48–58 CrossRef.
- R. Simkovitch, S. Shomer, R. Gepshtein, M. E. Roth, D. Shabat and D. Huppert, J. Photochem. Photobiol., A, 2014, 277, 90–101 CrossRef.
- O. Gajst, O. Green, R. Simkovitch, D. Shabat and D. Huppert, J. Photochem. Photobiol., A, 2018, 353, 546–556 CrossRef.
- I. V. Gopich, K. M. Solntsev and N. Agmon, J. Chem. Phys., 1999, 110, 2164–2174 CrossRef.
- B. Finkler, C. Spies, M. Vester, F. Walte, K. Omlor, I. Riemann, M. Zimmer, F. Stracke, M. Gerhards and G. Jung, Photochem. Photobiol. Sci., 2014, 13, 548 CrossRef PubMed.
- M. Rini, B.-Z. Magnes, E. Pines and E. T. J. Nibbering, Science, 2003, 301, 349–352 CrossRef PubMed.
- C. Hoberg, J. J. Talbot, J. Shee, T. Ockelmann, D. D. Mahanta, F. Novelli, M. Head-Gordon and M. Havenith, Chem. Sci., 2023, 14, 4048–4058 RSC.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef PubMed.
- L. A. Tatum, J. T. Foy and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 17438–17441 CrossRef PubMed.
- Y. Liao, Acc. Chem. Res., 2017, 50, 1956–1964 CrossRef PubMed.
- Y. Liao, Phys. Chem. Chem. Phys., 2022, 24, 4116–4124 RSC.
- Y.-Z. Ma, U. I. Premadasa, V. S. Bryantsev, A. R. Miles, I. N. Ivanov, A. Elgattar, Y. Liao and B. Doughty, Phys. Chem. Chem. Phys., 2024, 26, 4062–4070 RSC.
- M. Shirai and M. Tsunooka, Bull. Chem. Soc. Jpn., 1998, 71, 2483–2507 CrossRef CAS.
- C. J. Martin, G. Rapenne, T. Nakashima and T. Kawai, J. Photochem. Photobiol., C, 2018, 34, 41–51 CrossRef CAS.
- N. Zivic, P. K. Kuroishi, F. Dumur, D. Gigmes, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2019, 58, 10410–10422 CrossRef CAS PubMed.
- N. A. Kuznetsova, G. V. Malkov and B. G. Gribov, Russ. Chem. Rev., 2020, 89, 173 CrossRef CAS.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- R. I. Cukier and D. G. Nocera, Annu. Rev. Phys. Chem., 1998, 49, 337–369 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. V. Soudackov, J. Phys. Chem. B, 2008, 112, 14108–14123 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- N. Berg, S. Bergwinkl, P. Nuernberger, D. Horinek and R. M. Gschwind, J. Am. Chem. Soc., 2021, 143, 724–735 CrossRef CAS PubMed.
- D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1069–1081 CrossRef CAS PubMed.
- S.-I. Lee and D.-J. Jang, J. Phys. Chem., 1995, 99, 7537–7541 CrossRef CAS.
- G. Jackson and G. Porter, Proc. R. Soc. London, Ser. A, 1997, 260, 13–30 Search PubMed.
- K. Park, K. J. Shin and H. Kim, J. Chem. Phys., 2009, 131, 154105 CrossRef.
- S. Goia, M. A. P. Turner, J. M. Woolley, M. D. Horbury, A. J. Borrill, J. J. Tully, S. J. Cobb, M. Staniforth, N. D. M. Hine, A. Burriss, J. V. Macpherson, B. R. Robinson and V. G. Stavros, Chem. Sci., 2022, 13, 486–496 RSC.
- X. Pan, T. Han, J. Long, B. Xie, Y. Du, Y. Zhao, X. Zheng and J. Xue, Phys. Chem. Chem. Phys., 2022, 24, 18427–18434 RSC.
- X. Pan, J. Long, Y. Du, X. Zheng and J. Xue, Chin. J. Chem. Phys., 2023, 36, 50–56 CrossRef CAS.
- J. D. Henrich, S. Suchyta and B. Kohler, J. Phys. Chem. B, 2015, 119, 2737–2748 CrossRef CAS.
- E. G. Hohenstein, J. Am. Chem. Soc., 2016, 138, 1868–1876 CrossRef.
- N. Iibuchi, T. Eto, M. Aoyagi, R. Kurinami, H. Sakai, T. Hasobe, D. Takahashi and K. Toshima, Org. Biomol. Chem., 2020, 18, 851–855 RSC.
- S. J. Formosinho and L. G. Arnaut, J. Photochem. Photobiol., A, 1993, 75, 21–48 CrossRef.
- V. I. Tomin, A. P. Demchenko and P.-T. Chou, J. Photochem. Photobiol., C, 2015, 22, 1–18 CrossRef.
- R. K. Venkatraman and A. J. Orr-Ewing, Acc. Chem. Res., 2021, 54, 4383–4394 CrossRef PubMed.
- E. Vander Donckt, Progress in Reaction Kinetics, Pergamon Press, Oxford, 1st edn, 1970, ch. 5, vol. 5, pp. 273–299 Search PubMed.
- S. G. Schulman, Modern Fluorescence Spectroscopy, Plenum Pres, New York, 1st edn, 1976, ch. 6, vol. 2, pp. 239–275 Search PubMed.
- I. Y. Martynov, A. B. Demyashkevich, B. M. Uzhinov and M. G. Kuz’min, Russ. Chem. Rev., 1977, 46, 1 CrossRef.
- H. Shizuka, Acc. Chem. Res., 1985, 18, 141–147 CrossRef.
- E. M. Kosower and D. Huppert, Annu. Rev. Phys. Chem., 1986, 37, 127–156 CrossRef.
- E. Pines and D. Pines, Ultrafast Hydrogen Bonding Dynamics and Proton Transfer Prosesses in the Condensed Phase, Springer Netherlands, Dordrecht, 2002, pp. 155–184 Search PubMed.
- L. M. Tolbert and K. M. Solntsev, Acc. Chem. Res., 2002, 35, 19–27 CrossRef PubMed.
- E. Pines, Patai's Chemistry of Functional Groups, John Wiley & Sons, Ltd, 2009 Search PubMed.
- K. Weber, Z. Phys. Chem., 1932, 15B, 18–44 CrossRef.
- A. Terenin and A. Kariakin, Nature, 1947, 159, 881–882 CrossRef PubMed.
- S. Petersen, Angew. Chem., 1949, 61, 17–19 CrossRef CAS.
- T. Förster, Naturwissenschaften, 1949, 36, 186–187 CrossRef.
- T. Förster, Z. Elektrochem. Angew. Phys. Chem., 1950, 54, 42–46 CrossRef.
- T. Förster, Z. Elektrochem. Angew. Phys. Chem., 1950, 54, 531–535 CrossRef.
- Z. R. Grabowski and A. Grabowska, Z. Phys. Chem., 1976, 101, 197–208 CrossRef CAS.
- W. H. Mulder, J. Photochem. Photobiol., A, 2003, 161, 21–25 CrossRef.
- A. Weller and Z. Elektrochem, Ber. Bunsenges. Phys. Chem., 1952, 56, 662–668 CrossRef.
- A. Weller and Z. Elektrochem, Ber. Bunsenges. Phys. Chem., 1954, 58, 849–853 CrossRef.
- A. Weller, Z. Phys. Chem., 1958, 17, 224–245 CrossRef.
- S. G. Schulman, L. S. Rosenberg and W. R. Vincent, J. Am. Chem. Soc., 1979, 101, 139–142 CrossRef.
- C. M. Harris and B. K. Selinger, J. Phys. Chem., 1980, 84, 1366–1371 CrossRef.
- K. Tsutsumi and H. Shizuka, Z. Phys. Chem., 1980, 122, 129–142 CrossRef.
- S. P. Webb, L. A. Philips, S. W. Yeh, L. M. Tolbert and J. H. Clark, J. Phys. Chem., 1986, 90, 5154–5164 CrossRef.
- T. Nakagawa, S. Kohtani and M. Itoh, J. Am. Chem. Soc., 1995, 117, 7952–7957 CrossRef.
- E. Bardez, I. Devol, B. Larrey and B. Valeur, J. Phys. Chem. B, 1997, 101, 7786–7793 CrossRef.
- T. G. Kim and M. R. Topp, J. Phys. Chem. A, 2004, 108, 10060–10065 CrossRef.
- M. Ekimova, F. Hoffmann, G. Bekçioğlu-Neff, A. Rafferty, O. Kornilov, E. T. J. Nibbering and D. Sebastiani, J. Am. Chem. Soc., 2019, 141, 14581–14592 CrossRef.
- D. W. Fink and W. R. Koehler, Anal. Chem., 1970, 42, 990–993 CrossRef.
- G. J. Yakatan, R. J. Juneau and S. G. Schulman, Anal. Chem., 1972, 44, 1044–1046 CrossRef.
- S. G. Schulman and L. S. Rosenberg, J. Phys. Chem., 1979, 83, 447–451 CrossRef.
- R. Simkovitch and D. Huppert, J. Phys. Chem. B, 2015, 119, 14683–14696 CrossRef.
- J. Sérgio Seixas de Melo and A. L. Maçanita, J. Phys. Chem. B, 2015, 119, 2604–2610 CrossRef PubMed.
- R. Simkovitch, L. Pinto da Silva, J. C. G. Esteves da Silva and D. Huppert, J. Phys. Chem. B, 2016, 120, 10297–10310 CrossRef PubMed.
- R. Simkovitch and D. Huppert, J. Phys. Chem. B, 2017, 121, 129–142 CrossRef.
- J. F. Joung, S. Kim and S. Park, J. Phys. Chem. B, 2015, 119, 15509–15515 CrossRef.
- J. F. Joung, S. Kim and S. Park, Phys. Chem. Chem. Phys., 2017, 19, 25509–25517 RSC.
- J. F. Joung, S. Kim and S. Park, J. Phys. Chem. B, 2018, 122, 5087–5093 CrossRef.
- B. H. Milosavljevic and J. K. Thomas, Photochem. Photobiol. Sci., 2002, 1, 100–104 CrossRef.
- E. Pines, D. Pines, Y.-Z. Ma and G. R. Fleming, ChemPhysChem, 2004, 5, 1315–1327 CrossRef.
- M. Lukeman, M.-D. Burns and P. Wan, Can. J. Chem., 2011, 89, 433–440 CrossRef.
- J. Christian Lennox, E. O. Danilov and J. L. Dempsey, Phys. Chem. Chem. Phys., 2019, 21, 16353–16358 RSC.
- S. Shiobara, S. Tajima and S. Tobita, Chem. Phys. Lett., 2003, 380, 673–680 CrossRef.
- J. Oshima, S. Shiobara, H. Naoumi, S. Kaneko, T. Yoshihara, A. K. Mishra and S. Tobita, J. Phys. Chem. A, 2006, 110, 4629–4637 CrossRef PubMed.
- J. Oshima, T. Yoshihara and S. Tobita, Chem. Phys. Lett., 2006, 423, 306–311 CrossRef.
- K. Tsutsumi and H. Shizuka, Chem. Phys. Lett., 1977, 52, 485–488 CrossRef.
- A. A. El-Rayyes, H. P. Perzanowski, S. A. I. Barri and U. K. A. Klein, J. Phys. Chem. A, 2001, 105, 10169–10175 CrossRef.
- A.-R. Al-Betar, A. El-Rayyes and U. K. A. Klein, J. Fluoresc., 2005, 15, 689–696 CrossRef PubMed.
- H. Shizuka, K. Tsutsumi, H. Takeuchi and I. Tanaka, Chem. Phys., 1981, 59, 183–190 CrossRef.
- E. Pines and G. R. Fleming, J. Phys. Chem., 1991, 95, 10448–10457 CrossRef.
- D. B. Spry and M. D. Fayer, J. Chem. Phys., 2008, 128, 084508 CrossRef.
- P. Daublain, A. K. Thazhathveetil, Q. Wang, A. Trifonov, T. Fiebig and F. D. Lewis, J. Am. Chem. Soc., 2009, 131, 16790–16797 CrossRef.
- C.-H. Ho, B. R. Clark, M. R. Guerin, B. D. Barkenbus, T. K. Rao and J. L. Epler, Mutat. Res., Environ. Mutagen. Relat. Subj., 1981, 85, 335–345 Search PubMed.
- N. Acar and Ö. Koçak, Turkish J. Chem., 2002, 26, 201–211 Search PubMed.
- H. Örücü and N. Acar, J. Mol. Struct., 2018, 1174, 43–51 CrossRef.
- E. L. Wehry and L. B. Rogers, J. Am. Chem. Soc., 1965, 87, 4234–4238 CrossRef CAS.
- I. Avigal, J. Feitelson and M. Ottolenghi, J. Chem. Phys., 1969, 50, 2614–2617 CrossRef CAS.
- S. G. Schulman, W. R. Vincent and W. J. M. Underberg, J. Phys. Chem., 1981, 85, 4068–4071 CrossRef CAS.
- J. Steadman and J. A. Syage, J. Chem. Phys., 1990, 92, 4630–4632 CrossRef CAS.
- J. A. Syage and J. Steadman, J. Chem. Phys., 1991, 95, 2497–2510 CrossRef CAS.
- R. F. Salikov, A. Y. Belyy, K. P. Trainov, J. A. Velmiskina, M. G. Medvedev, V. M. Korshunov, I. V. Taydakov, D. N. Platonov and Y. V. Tomilov, J. Photochem. Photobiol., A, 2022, 427, 113808 CrossRef CAS.
- J. Yguerabide, E. Talavera, J. M. Alvarez and B. Quintero, Photochem. Photobiol., 1994, 60, 435–441 CrossRef CAS.
- A. Orte, E. M. Talavera, A. L. Maçanita, J. C. Orte and J. M. Alvarez-Pez, J. Phys. Chem. A, 2005, 109, 8705–8718 CrossRef CAS.
- D. Surzhikova, M. Gerasimova, V. Tretyakova, A. Plotnikov and E. Slyusareva, J. Photochem. Photobiol., A, 2021, 413, 113233 CrossRef CAS.
- H. Ihmels and K. Schäfer, Photochem. Photobiol. Sci., 2009, 8, 309–311 CrossRef CAS.
- K. Schäfer, H. Ihmels, C. Bohne, K. P. Valente and A. Granzhan, J. Org. Chem., 2016, 81, 10942–10954 CrossRef.
- M. Zahid, G. Grampp, A. Mansha, I. A. Bhatti and S. Asim, J. Fluoresc., 2013, 23, 829–837 CrossRef CAS PubMed.
- Y. Luo, C. Wang, P. Peng, M. Hossain, T. Jiang, W. Fu, Y. Liao and M. Su, J. Mater. Chem. B, 2013, 1, 997–1001 RSC.
- T. Halbritter, C. Kaiser, J. Wachtveitl and A. Heckel, J. Org. Chem., 2017, 82, 8040–8047 CrossRef CAS PubMed.
- C. Kaiser, T. Halbritter, A. Heckel and J. Wachtveitl, Chem.–A Eur. J., 2021, 27, 9160–9173 CrossRef CAS.
- C. R. Aldaz, T. E. Wiley, N. A. Miller, N. Abeyrathna, Y. Liao, P. M. Zimmerman and R. J. Sension, J. Phys. Chem. B, 2021, 125, 4120–4131 CrossRef.
- V. J. Périllat, C. Berton and C. Pezzato, Mater. Today Chem., 2022, 25, 100918 CrossRef.
- C. Berton and C. Pezzato, Eur. J. Org Chem., 2023, 26, e202300070 CrossRef.
- A. Zika, M. Agarwal, W. Zika, D. M. Guldi, R. Schweins and F. Gröhn, Nanoscale, 2024, 16, 923–940 RSC.
- V. L. Shapovalov, A. B. Demyashkevich and M. G. Kuz’min, J. Appl. Spectrosc., 1980, 33, 1362–1366 CrossRef.
- B. Zelent, J. M. Vanderkooi, R. G. Coleman, I. Gryczynski and Z. Gryczynski, Biophys. J., 2006, 91, 3864–3871 CrossRef PubMed.
- N. V. Nucci, B. Zelent and J. M. Vanderkooi, J. Fluoresc., 2008, 18, 41–49 CrossRef PubMed.
- B. Zelent, J. M. Vanderkooi, N. V. Nucci, I. Gryczynski and Z. Gryczynski, J. Fluoresc., 2009, 19, 21–31 CrossRef PubMed.
- E. W. Driscoll, J. R. Hunt and J. M. Dawlaty, J. Phys. Chem. Lett., 2016, 7, 2093–2099 CrossRef PubMed.
- E. W. Driscoll, J. R. Hunt and J. M. Dawlaty, J. Phys. Chem. A, 2017, 121, 7099–7107 CrossRef PubMed.
- J. R. Hunt and J. M. Dawlaty, J. Phys. Chem. A, 2018, 122, 7931–7940 CrossRef PubMed.
- W. Sheng, M. Nairat, P. D. Pawlaczyk, E. Mroczka, B. Farris, E. Pines, J. H. Geiger, B. Borhan and M. Dantus, Angew. Chem., Int. Ed., 2018, 57, 14742–14746 CrossRef CAS PubMed.
- J. Lahiri, M. Moemeni, J. Kline, B. Borhan, I. Magoulas, S. H. Yuwono, P. Piecuch, J. E. Jackson, M. Dantus and G. J. Blanchard, J. Phys. Chem. B, 2019, 123, 8448–8456 CrossRef CAS.
- S. Roy, S. Ardo and F. Furche, J. Phys. Chem. A, 2019, 123, 6645–6651 CrossRef CAS.
- J. F. Joung, J. Lee, J. Hwang, K. Choi and S. Park, New J. Chem., 2020, 44, 668–673 RSC.
- M. Sittig, J. C. Tom, J. K. Elter, F. H. Schacher and B. Dietzek, Chem.–A Eur. J., 2021, 27, 1072–1079 CrossRef CAS.
- M. J. Voegtle and J. M. Dawlaty, J. Am. Chem. Soc., 2022, 144, 8178–8184 CrossRef CAS PubMed.
- R. Mathew, P. Verma, A. Barak and Y. Adithya Lakshmanna, J. Phys. Chem. A, 2023, 127, 7419–7428 CrossRef CAS.
- B. Antalicz, J. Versluis and H. J. Bakker, J. Am. Chem. Soc., 2023, 145, 6682–6690 CrossRef CAS PubMed.
- J. R. Hunt, J. Hecht, C. Goolsby, J. Hagihara, M. Loza and S. del Pozo, J. Phys. Chem. A, 2024, 128(30), 6199–6207 CrossRef CAS.
- S. F. Alamudun, K. Tanovitz, A. Fajardo, K. Johnson, A. Pham, T. Jamshidi Araghi and A. S. Petit, J. Phys. Chem. A, 2020, 124, 2537–2546 CrossRef CAS PubMed.
- S. F. Alamudun, K. Tanovitz, L. Espinosa, A. Fajardo, J. Galvan and A. S. Petit, J. Phys. Chem. A, 2021, 125, 13–24 CrossRef CAS.
- J. Ditkovich, T. Mukra, D. Pines, D. Huppert and E. Pines, J. Phys. Chem. B, 2015, 119, 2690–2701 CrossRef CAS PubMed.
- J. Ditkovich, D. Pines and E. Pines, Phys. Chem. Chem. Phys., 2016, 18, 16106–16115 RSC.
- B. Finkler, I. Riemann, M. Vester, A. Grüter, F. Stracke and G. Jung, Photochem. Photobiol. Sci., 2016, 15, 1544–1557 CrossRef CAS.
- R. Hilal and S. G. Aziz, Mol. Simul., 2019, 45, 165–177 CrossRef CAS.
- T. Stoerkler, T. Pariat, A. D. Laurent, D. Jacquemin, G. Ulrich and J. Massue, Molecules, 2022, 27, 2443 CrossRef CAS PubMed.
- K. U. Jagushte, N. Sadhukhan, H. P. Upadhyaya and S. Dutta Choudhury, J. Phys. Chem. B, 2023, 127, 9788–9801 CrossRef CAS.
- Q.-S. Li, W.-H. Fang and J.-G. Yu, J. Phys. Chem. A, 2005, 109, 3983–3990 CrossRef CAS PubMed.
- W. Qin, A. Vozza and A. M. Brouwer, J. Phys. Chem. C, 2009, 113, 11790–11795 CrossRef CAS.
- Y. Wang, H. Yin, Y. Shi, M. Jin and D. Ding, New J. Chem., 2014, 38, 4458–4464 RSC.
- Y. Cui, H. Zhao, J. Zhao, P. Li, P. Song and L. Xia, New J. Chem., 2015, 39, 9910–9917 RSC.
- G. Bekçioğlu, C. Allolio, M. Ekimova, E. T. J. Nibbering and D. Sebastiani, Phys. Chem. Chem. Phys., 2014, 16, 13047–13051 RSC.
- G. Bekçioğlu, C. Allolio and D. Sebastiani, J. Phys. Chem. B, 2015, 119, 4053–4060 Search PubMed.
- Y.-J. Kim and O.-H. Kwon, Phys. Chem. Chem. Phys., 2016, 18, 32826–32839 Search PubMed.
- Y.-H. Liu, S.-M. Wang, C. Zhu and S. H. Lin, New J. Chem., 2017, 41, 8437–8442 Search PubMed.
- O.-H. Kwon, Y.-S. Lee, B. K. Yoo and D.-J. Jang, Angew. Chem., Int. Ed., 2006, 45, 415–419 CrossRef PubMed.
- D. Huppert and E. Kolodney, Chem. Phys., 1981, 63, 401–410 CrossRef.
- D. Huppert, E. Kolodney, M. Gutman and E. Nachliel, J. Am. Chem. Soc., 1982, 104, 6949–6953 CrossRef.
- E. Tietze and O. Bayer, Adv. Cycloaddit., 1939, 540, 189–210 Search PubMed.
- O. S. Wolfbeis, E. Fürlinger, H. Kroneis and H. Marsoner, Fresenius Z. Anal. Chem., 1983, 314, 119–124 CrossRef.
- O. S. Wolfbeis and E. Koller, Anal. Biochem., 1983, 129, 365–370 CrossRef.
- Y. Avnir and Y. Barenholz, Anal. Biochem., 2005, 347, 34–41 CrossRef PubMed.
- R. Nandi and N. Amdursky, Acc. Chem. Res., 2022, 55, 2728–2739 CrossRef.
- P. Changenet, T. Gustavsson and I. Lampre, J. Chem. Educ., 2020, 97, 4482–4489 CrossRef.
- E. Pines and D. Huppert, J. Chem. Phys., 1986, 84, 3576–3577 CrossRef.
- E. Pines and D. Huppert, Chem. Phys. Lett., 1986, 126, 88–91 CrossRef.
- N. Agmon, E. Pines and D. Huppert, J. Chem. Phys., 1988, 88, 5631–5638 CrossRef.
- N. Agmon, J. Chem. Phys., 1988, 88, 5639–5642 CrossRef.
- N. Agmon, J. Chem. Phys., 1988, 89, 1524–1528 CrossRef.
- E. Pines, D. Huppert and N. Agmon, J. Chem. Phys., 1988, 88, 5620–5630 CrossRef.
- D. Huppert, E. Pines and N. Agmon, J. Opt. Soc. Am. B, 1990, 7, 1545–1550 CrossRef.
- D. Huppert, S. Y. Goldberg, A. Masad and N. Agmon, Phys. Rev. Lett., 1992, 68, 3932–3935 CrossRef.
- C. Prayer, T. Gustavsson and T.-H. Tran-Thi, AIP Conf. Proc., 1996, 364, 333–339 CrossRef.
- T. H. Tran-Thi, T. Gustavsson, C. Prayer, S. Pommeret and J. T. Hynes, Chem. Phys. Lett., 2000, 329, 421–430 CrossRef.
- J. T. Hynes, T.-H. Tran-Thi and G. Granucci, J. Photochem. Photobiol., A, 2002, 154, 3–11 CrossRef.
- T.-H. Tran-Thi, C. Prayer, P. Millié, P. Uznanski and J. T. Hynes, J. Phys. Chem. A, 2002, 106, 2244–2255 CrossRef.
- P. Leiderman, L. Genosar and D. Huppert, J. Phys. Chem. A, 2005, 109, 5965–5977 CrossRef PubMed.
- R. Gepshtein, P. Leiderman, L. Genosar and D. Huppert, J. Phys. Chem. A, 2005, 109, 9674–9684 CrossRef.
- O. F. Mohammed, D. Pines, J. Dreyer, E. Pines and E. T. J. Nibbering, Science, 2005, 310, 83–86 CrossRef PubMed.
- O. F. Mohammed, J. Dreyer, B.-Z. Magnes, E. Pines and E. T. J. Nibbering, ChemPhysChem, 2005, 6, 625–636 CrossRef.
- B. J. Siwick and H. J. Bakker, J. Am. Chem. Soc., 2007, 129, 13412–13420 CrossRef PubMed.
- D. B. Spry, A. Goun, C. B. Bell and M. D. Fayer, J. Chem. Phys., 2006, 125, 144514 CrossRef.
- D. B. Spry and M. D. Fayer, J. Chem. Phys., 2007, 127, 204501 CrossRef.
- L. N. Silverman, D. B. Spry, S. G. Boxer and M. D. Fayer, J. Phys. Chem. A, 2008, 112, 10244–10249 CrossRef PubMed.
- F. Han, W. Liu and C. Fang, Chem. Phys., 2013, 422, 204–219 CrossRef.
- W. Liu, Y. Wang, L. Tang, B. G. Oscar, L. Zhu and C. Fang, Chem. Sci., 2016, 7, 5484–5494 RSC.
- S. Y. Goldberg, E. Pines and D. Huppert, Chem. Phys. Lett., 1992, 192, 77–81 CrossRef CAS.
- E. Pines, B.-Z. Magnes, M. J. Lang and G. R. Fleming, Chem. Phys. Lett., 1997, 281, 413–420 CrossRef.
- M. Rini, D. Pines, B.-Z. Magnes, E. Pines and E. T. J. Nibbering, J. Chem. Phys., 2004, 121, 9593–9610 CrossRef PubMed.
- B. J. Siwick, M. J. Cox and H. J. Bakker, J. Phys. Chem. B, 2008, 112, 378–389 CrossRef.
- M. J. Cox, R. L. A. Timmer, H. J. Bakker, S. Park and N. Agmon, J. Phys. Chem. A, 2009, 113, 6599–6606 CrossRef PubMed.
- W. Liu, F. Han, C. Smith and C. Fang, J. Phys. Chem. B, 2012, 116, 10535–10550 CrossRef PubMed.
- W. Heo, N. Uddin, J. W. Park, Y. M. Rhee, C. H. Choi and T. Joo, Phys. Chem. Chem. Phys., 2017, 19, 18243–18251 RSC.
- O. F. Mohammed, D. Pines, E. T. J. Nibbering and E. Pines, Angew. Chem., 2007, 119, 1480–1483 CrossRef.
- O. F. Mohammed, D. Pines, E. Pines and E. T. J. Nibbering, Chem. Phys., 2007, 341, 240–257 CrossRef.
- M. J. Cox and H. J. Bakker, J. Chem. Phys., 2008, 128, 174501 CrossRef PubMed.
- N. Agmon, D. Huppert, A. Masad and E. Pines, J. Phys. Chem., 1991, 95, 10407–10413 CrossRef.
- P. Leiderman, R. Gepshtein, A. Uritski, L. Genosar and D. Huppert, J. Phys. Chem. A, 2006, 110, 5573–5584 CrossRef.
- B. G. Oscar, W. Liu, N. D. Rozanov and C. Fang, Phys. Chem. Chem. Phys., 2016, 18, 26151–26160 RSC.
- N. Nandi and K. Sahu, J. Photochem. Photobiol., A, 2019, 374, 138–144 CrossRef CAS.
- S. G. Schulman, R. N. Kelly and N. J. Gonzalez, Pure Appl. Chem., 1987, 59, 655–662 CAS.
- E. Pines and D. Huppert, J. Am. Chem. Soc., 1989, 111, 4096–4097 CrossRef CAS.
- E. Pines, D. Huppert and N. Agmon, J. Phys. Chem., 1991, 95, 666–674 CrossRef CAS.
- N. Agmon, S. Y. Goldberg and D. Huppert, J. Mol. Liq., 1995, 64, 161–195 CrossRef CAS.
- M. J. Cox, B. J. Siwick and H. J. Bakker, ChemPhysChem, 2009, 10, 236–244 CrossRef CAS PubMed.
- E. Pines, B. Cohen and D. Huppert, Isr. J. Chem., 1999, 39, 347–360 CrossRef.
- P. Leiderman, R. Gepshtein, A. Uritski, L. Genosar and D. Huppert, J. Phys. Chem. A, 2006, 110, 9039–9050 CrossRef CAS.
- D. H. Huppert, A. Jayaraman, R. G. Maines, D. W. Steyert and P. M. Rentzepis, J. Chem. Phys., 1984, 81, 5596–5600 CrossRef CAS.
- E. Pines and D. Huppert, Chem. Phys. Lett., 1985, 116, 295–301 CrossRef CAS.
- A. Uritski, P. Leiderman and D. Huppert, J. Phys. Chem. A, 2006, 110, 13686–13695 CrossRef CAS PubMed.
- A. Uritski, P. Leiderman and D. Huppert, J. Phys. Chem. C, 2007, 111, 8856–8865 CrossRef CAS.
- P. Leiderman, A. Uritski and D. Huppert, J. Phys. Chem. A, 2007, 111, 4998–5007 CrossRef CAS.
- A. Uritski and D. Huppert, J. Phys. Chem. A, 2008, 112, 4415–4425 CrossRef CAS PubMed.
- A. Uritski, I. Presiado, Y. Erez, R. Gepshtein and D. Huppert, J. Phys. Chem. C, 2009, 113, 17915–17926 CrossRef CAS.
- R. L. A. Timmer, M. J. Cox and H. J. Bakker, J. Phys. Chem. A, 2010, 114, 2091–2101 CrossRef CAS PubMed.
- K. J. Tielrooij, M. J. Cox and H. J. Bakker, ChemPhysChem, 2009, 10, 245–251 CrossRef CAS PubMed.
- T. Mondal, A. K. Das, D. K. Sasmal and K. Bhattacharyya, J. Phys. Chem. B, 2010, 114, 13136–13142 CrossRef CAS.
- A. K. Mandal, S. Ghosh, A. K. Das, T. Mondal and K. Bhattacharyya, ChemPhysChem, 2013, 14, 788–796 CrossRef CAS.
- C. Lawler and M. D. Fayer, J. Phys. Chem. B, 2015, 119, 6024–6034 CrossRef CAS PubMed.
- D. B. Spry, A. Goun, K. Glusac, D. E. Moilanen and M. D. Fayer, J. Am. Chem. Soc., 2007, 129, 8122–8130 CrossRef CAS PubMed.
- G. Ghosh, R. Ghosh, D. Mukherjee, M. N. Hasan, N. Pan, L. Roy, S. Biswas, R. Das and S. Kumar Pal, J. Mol. Liq., 2024, 407, 125181 CrossRef CAS.
- G. Ghosh, D. S. Roy, R. Ghosh, D. Mukherjee, S. Biswas, L. Roy, A. Chattopadhyay, R. Das and S. K. Pal, ChemPhysChem, 2024, 25, e202300635 CrossRef PubMed.
- T. Jang, S. Lee and Y. Pang, Phys. Chem. Chem. Phys., 2024, 26, 11283–11294 RSC.
- D. B. Spry and M. D. Fayer, J. Phys. Chem. B, 2009, 113, 10210–10221 CrossRef.
- J. E. Thomaz, C. M. Lawler and M. D. Fayer, J. Phys. Chem. B, 2017, 121, 4544–4553 CrossRef.
- S. K. Mondal, K. Sahu, P. Sen, D. Roy, S. Ghosh and K. Bhattacharyya, Chem. Phys. Lett., 2005, 412, 228–234 CrossRef.
- S. K. Mondal, K. Sahu, S. Ghosh, P. Sen and K. Bhattacharyya, J. Phys. Chem. A, 2006, 110, 13646–13652 CrossRef PubMed.
- R. Gepshtein, P. Leiderman, D. Huppert, E. Project, E. Nachliel and M. Gutman, J. Phys. Chem. B, 2006, 110, 26354–26364 CrossRef PubMed.
- S. Ghosh, S. Dey, U. Mandal, A. Adhikari, S. K. Mondal and K. Bhattacharyya, J. Phys. Chem. B, 2007, 111, 13504–13510 CrossRef PubMed.
- N. Amdursky, R. Simkovitch and D. Huppert, J. Phys. Chem. B, 2014, 118, 13859–13869 CrossRef PubMed.
- R. Chowdhury, A. Saha, A. K. Mandal, B. Jana, S. Ghosh and K. Bhattacharyya, J. Phys. Chem. B, 2015, 119, 2149–2156 Search PubMed.
- R. Simkovitch and D. Huppert, J. Phys. Chem. A, 2015, 119, 641–651 Search PubMed.
- R. Simkovitch and D. Huppert, J. Phys. Chem. B, 2015, 119, 11684–11694 Search PubMed.
- R. Simkovitch and D. Huppert, J. Phys. Chem. B, 2015, 119, 9795–9804 Search PubMed.
- H. Peretz-Soroka, A. Pevzner, G. Davidi, V. Naddaka, M. Kwiat, D. Huppert and F. Patolsky, Nano Lett., 2015, 15, 4758–4768 CrossRef.
- N. Amdursky, R. Orbach, E. Gazit and D. Huppert, J. Phys. Chem. C, 2009, 113, 19500–19505 CrossRef.
- R. Simkovitch, O. Gajst, E. Zelinger, O. Yarden and D. Huppert, J. Photochem. Photobiol., B, 2017, 174, 1–9 CrossRef CAS PubMed.
- S. Chakraborty, S. Nandi, K. Bhattacharyya and S. Mukherjee, ChemPhysChem, 2019, 20, 3221–3227 CrossRef CAS.
- I. Das, S. Panja and M. Halder, J. Phys. Chem. B, 2016, 120, 7076–7087 CrossRef PubMed.
- S. S. Mojumdar, R. Chowdhury, A. K. Mandal and K. Bhattacharyya, J. Chem. Phys., 2013, 138, 215102 CrossRef PubMed.
- M. A. Amin, S. Nandi, P. Mondal, T. Mahata, S. Ghosh and K. Bhattacharyya, Phys. Chem. Chem. Phys., 2017, 19, 12620–12627 RSC.
- S. Nandi, S. Ghosh and K. Bhattacharyya, J. Phys. Chem. B, 2018, 122, 3023–3036 CrossRef.
- S. K. Mondal, S. Ghosh, K. Sahu, P. Sen and K. Bhattacharyya, J. Chem. Sci., 2007, 119, 71–76 CrossRef.
- S. Sen Mojumdar, T. Mondal, A. K. Das, S. Dey and K. Bhattacharyya, J. Chem. Phys., 2010, 132, 194505 CrossRef.
- S. Maiti, S. Mitra, C. A. Johnson, K. C. Gronborg, S. Garrett-Roe and P. M. Donaldson, J. Phys. Chem. Lett., 2022, 13, 8104–8110 CrossRef PubMed.
- R. Knochenmuss, K. M. Solntsev and L. M. Tolbert, J. Phys. Chem. A, 2001, 105, 6393–6401 CrossRef.
- R. Knochenmuss and I. Fischer, Int. J. Mass Spectrom., 2002, 220, 343–357 CrossRef.
- A. J. Fleisher, P. J. Morgan and D. W. Pratt, J. Chem. Phys., 2009, 131, 211101 CrossRef PubMed.
- A. Melnichuk and R. J. Bartlett, J. Chem. Phys., 2011, 134, 244303 CrossRef.
- J. Yang, X.-P. Xing, X.-B. Wang, L.-S. Wang, A. P. Sergeeva and A. I. Boldyrev, J. Chem. Phys., 2008, 128, 091102 CrossRef PubMed.
- J. E. Whitaker, R. P. Haugland and F. G. Prendergast, Anal. Biochem., 1991, 194, 330–344 CrossRef CAS.
- J. Widengren, B. Terry and R. Rigler, Chem. Phys., 1999, 249, 259–271 CrossRef CAS.
- F. H. C. Wong and C. Fradin, J. Fluoresc., 2011, 21, 299–312 CrossRef CAS PubMed.
- N. Sülzner, B. Geissler, A. Grandjean, G. Jung and P. Nuernberger, ChemPhotoChem, 2022, 6, e202200041 CrossRef.
- N. Barrash-Shiftan, B. B. Brauer and E. Pines, J. Phys. Org. Chem., 1998, 11, 743–750 CrossRef CAS.
- G. Jung, S. Gerharz and A. Schmitt, Phys. Chem. Chem. Phys., 2009, 11, 1416 RSC.
- T. Förster and S. Völker, Chem. Phys. Lett., 1975, 34, 1–6 CrossRef.
- K. Lundy-Douglas and Z. A. Schelly, Adv. Mol. Relax. Processes, 1976, 8, 79–86 CrossRef CAS.
- M. Gutman, D. Huppert and E. Pines, J. Am. Chem. Soc., 1981, 103, 3709–3713 CrossRef CAS.
- E. Pines and D. Huppert, J. Phys. Chem., 1983, 87, 4471–4478 CrossRef.
- K. K. Smith, K. J. Kaufmann, D. Huppert and M. Gutman, Chem. Phys. Lett., 1979, 64, 522–527 CrossRef.
- G. Granucci, J. T. Hynes, P. Millié and T.-H. Tran-Thi, J. Am. Chem. Soc., 2000, 122, 12243–12253 CrossRef.
- J. Shin, C. H. Lim and M. Lim, Photochem. Photobiol. Sci., 2022, 21, 1419–1431 CrossRef PubMed.
- Y. Wang, W. Liu, L. Tang, B. Oscar, F. Han and C. Fang, J. Phys. Chem. A, 2013, 117, 6024–6042 CrossRef.
- W. Liu, L. Tang, B. G. Oscar, Y. Wang, C. Chen and C. Fang, J. Phys. Chem. Lett., 2017, 8, 997–1003 CrossRef PubMed.
- B. G. Oscar, C. Chen, W. Liu, L. Zhu and C. Fang, J. Phys. Chem. A, 2017, 121, 5428–5441 CrossRef PubMed.
- L. Tang, Y. Wang, L. Zhu, C. Lee and C. Fang, J. Phys. Chem. Lett., 2018, 9, 2311–2319 CrossRef PubMed.
- L. Tang, L. Zhu, Y. Wang and C. Fang, J. Phys. Chem. Lett., 2018, 9, 4969–4975 CrossRef PubMed.
- D. B. Spry, A. Goun and M. D. Fayer, J. Phys. Chem. A, 2007, 111, 230–237 CrossRef PubMed.
- R. Jimenez, D. A. Case and F. E. Romesberg, J. Phys. Chem. B, 2002, 106, 1090–1103 CrossRef.
- S. Eckert, M.-O. Winghart, C. Kleine, A. Banerjee, M. Ekimova, J. Ludwig, J. Harich, M. Fondell, R. Mitzner, E. Pines, N. Huse, P. Wernet, M. Odelius and E. T. J. Nibbering, Angew. Chem., Int. Ed., 2022, 61, e202200709 CrossRef PubMed.
- M. Sedgwick, R. L. Cole, C. D. Rithner, D. C. Crans and N. E. Levinger, J. Am. Chem. Soc., 2012, 134, 11904–11907 CrossRef PubMed.
- G. Ferrino, M. De Rosa, P. Della Sala, C. Gaeta, C. Talotta, A. Soriente, Z. Cao, B. Maity, L. Cavallo and P. Neri, Chem.–A Eur. J., 2024, 30, e202303678 CrossRef PubMed.
- T. Ockelmann, C. Hoberg, A. Buchmann, F. Novelli and M. Havenith, J. Phys. Chem. B, 2023, 127, 9560–9565 CrossRef PubMed.
- N. Agmon, W. Rettig and C. Groth, J. Am. Chem. Soc., 2002, 124, 1089–1096 CrossRef PubMed.
- W.-H. Fang, J. Am. Chem. Soc., 1998, 120, 7568–7576 CrossRef.
- W.-H. Fang, J. Phys. Chem. A, 1999, 103, 5567–5573 CrossRef.
- Q.-S. Li and W.-H. Fang, Chem. Phys. Lett., 2003, 367, 637–644 CrossRef.
- B.-b. Xie, C.-x. Li, G.-l. Cui and Q. Fang, Chin. J. Chem. Phys., 2016, 29, 38–46 CrossRef.
- X.-F. Tang, P.-K. Jia, Y. Zhao, J. Xue, G. Cui and B.-B. Xie, Phys. Chem. Chem. Phys., 2022, 24, 20517–20529 RSC.
- P. Cimino, U. Raucci, G. Donati, M. G. Chiariello, M. Schiazza, F. Coppola and N. Rega, Theor. Chem. Acc., 2016, 135, 117 Search PubMed.
- E. L. Graef and J. B. L. Martins, J. Mol. Model., 2019, 25, 183 CrossRef PubMed.
- A. Kyrychenko and J. Waluk, J. Phys. Chem. A, 2006, 110, 11958–11967 CrossRef PubMed.
- H. Wang, X. Wang, X. Li and C. Zhang, J. Mol. Struct.:THEOCHEM, 2006, 770, 107–110 CrossRef.
- M. Savarese, P. A. Netti, C. Adamo, N. Rega and I. Ciofini, J. Phys. Chem. B, 2013, 117, 16165–16173 CrossRef PubMed.
- M. Savarese, P. A. Netti, N. Rega, C. Adamo and I. Ciofini, Phys. Chem. Chem. Phys., 2014, 16, 8661–8666 RSC.
- A. D. Laurent, Y. Houari, P. H. P. R. Carvalho, B. A. D. Neto and D. Jacquemin, RSC Adv., 2014, 4, 14189–14192 RSC.
- Y. Houari, S. Chibani, D. Jacquemin and A. D. Laurent, J. Phys. Chem. B, 2015, 119, 2180–2192 CrossRef PubMed.
- U. Raucci, M. Savarese, C. Adamo, I. Ciofini and N. Rega, J. Phys. Chem. B, 2015, 119, 2650–2657 CrossRef PubMed.
- J. Zhao, H. Yao, J. Liu and M. R. Hoffmann, J. Phys. Chem. A, 2015, 119, 681–688 CrossRef PubMed.
- Z. Qu, P. Li, X. Zhang, E. Wang, Y. Wang and P. Zhou, J. Lumin., 2016, 177, 197–203 CrossRef.
- M. Savarese, U. Raucci, R. Fukuda, C. Adamo, M. Ehara, N. Rega and I. Ciofini, J. Comput. Chem., 2017, 38, 1084–1092 CrossRef.
- D. Yang, J. Zhao, M. Jia and X. Song, RSC Adv., 2017, 7, 34034–34040 RSC.
- P. Krishnakumar, R. Kar and D. K. Maity, J. Phys. Chem. A, 2018, 122, 929–936 CrossRef PubMed.
- P. Zhou and K. Han, Acc. Chem. Res., 2018, 51, 1681–1690 CrossRef PubMed.
- A. A. Freitas, K. Shimizu, L. G. Dias and F. H. Quina, J. Braz. Chem. Soc., 2007, 18, 1537–1546 CrossRef.
- F. Siddique, C. P. Silva, G. T. M. Silva, H. Lischka, F. H. Quina and A. J. A. Aquino, Photochem. Photobiol. Sci., 2019, 18, 45–53 CrossRef PubMed.
- C. P. Silva, G. T. M. Silva, T. d. S. Costa, V. M. T. Carneiro, F. Siddique, A. J. A. Aquino, A. A. Freitas, J. A. Clark, E. M. Espinoza, V. I. Vullev and F. H. Quina, Pure Appl. Chem., 2020, 92, 255–263 CrossRef.
- J. He, X. Li, G. T. M. Silva, F. H. Quina and A. J. A. Aquino, J. Braz. Chem. Soc., 2019, 30, 492–498 Search PubMed.
- G. T. M. Silva, C. P. Silva, K. M. Silva, R. M. Pioli, T. S. Costa, V. V. Marto, A. A. Freitas, J. Rozendo, L. M. O. S. Martins, V. F. Cavalcante, L. Sun, A. J. A. Aquino, V. M. T. Carneiro and F. H. Quina, Photochem, 2022, 2, 423–434 CrossRef.
- T. Udagawa, I. Hattori, Y. Kanematsu, T. Ishimoto and M. Tachikawa, Int. J. Quantum Chem., 2022, 122, e26962 CrossRef.
- D. Jacquemin, E. A. Perpète, I. Ciofini and C. Adamo, J. Phys. Chem. A, 2008, 112, 794–796 CrossRef PubMed.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Phys. Chem. A, 2010, 114, 5932–5939 CrossRef PubMed.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef.
- É. Brémond, M. E. Alberto, N. Russo, G. Ricci, I. Ciofini and C. Adamo, Phys. Chem. Chem. Phys., 2013, 15, 10019 RSC.
- Y. Houari, D. Jacquemin and A. D. Laurent, Phys. Chem. Chem. Phys., 2013, 15, 11875 RSC.
- Y. Houari, D. Jacquemin and A. D. Laurent, Chem. Phys. Lett., 2013, 583, 218–221 CrossRef CAS.
- A. Aqdas, F. Siddique, R. Nieman, F. H. Quina and A. J. A. Aquino, Photochem. Photobiol., 2019, 95, 1339–1344 CrossRef CAS PubMed.
- J. Wang, F. Siddique, A. A. Freitas, C. P. Silva, G. T. M. Silva, F. H. Quina, H. Lischka and A. J. A. Aquino, Theor. Chem. Acc., 2020, 139, 117 Search PubMed.
- J. Cui, F. Siddique, R. Nieman, G. T. M. Silva, F. H. Quina and A. J. A. Aquino, Theor. Chem. Acc., 2021, 140, 90 Search PubMed.
- M. G. Chiariello and N. Rega, J. Phys. Chem. A, 2018, 122, 2884–2893 CrossRef PubMed.
- M. G. Chiariello, G. Donati and N. Rega, J. Chem. Theory Comput., 2020, 16, 6007–6013 CrossRef.
- M. G. Chiariello, U. Raucci, G. Donati and N. Rega, J. Phys. Chem. A, 2021, 125, 3569–3578 CrossRef PubMed.
- M. G. Chiariello, G. Donati, U. Raucci, F. Perrella and N. Rega, J. Phys. Chem. B, 2021, 125, 10273–10281 CrossRef.
- U. Raucci, M. G. Chiariello and N. Rega, J. Chem. Theory Comput., 2020, 16, 7033–7043 CrossRef.
- A. Petrone, G. Donati, P. Caruso and N. Rega, J. Am. Chem. Soc., 2014, 136, 14866–14874 CrossRef PubMed.
- J. E. Thomaz, A. R. Walker, S. J. Van Wyck, J. Meisner, T. J. Martinez and M. D. Fayer, J. Phys. Chem. B, 2020, 124, 7897–7908 CrossRef PubMed.
- A. R. Walker, B. Wu, J. Meisner, M. D. Fayer and T. J. Martínez, J. Phys. Chem. B, 2021, 125, 12539–12551 CrossRef PubMed.
- G. W. Robinson, J. Phys. Chem., 1991, 95, 10386–10391 CrossRef.
- A. Uritski, I. Presiado and D. Huppert, J. Phys. Chem. C, 2008, 112, 11991–12002 CrossRef.
- A. Uritski, I. Presiado and D. Huppert, Isr. J. Chem., 2009, 49, 235–249 CrossRef.
- I. Sarkar, New J. Chem., 2016, 40, 6666–6674 RSC.
- M. Prémont-Schwarz, T. Barak, D. Pines, E. T. J. Nibbering and E. Pines, J. Phys. Chem. B, 2013, 117, 4594–4603 CrossRef PubMed.
- A. Masad and D. Huppert, Chem. Phys. Lett., 1991, 180, 409–415 CrossRef CAS.
- P. Leiderman, L. Genosar, N. Koifman and D. Huppert, J. Phys. Chem. A, 2004, 108, 2559–2566 CrossRef CAS.
- L. Genosar, P. Leiderman, N. Koifman and D. Huppert, J. Phys. Chem. A, 2004, 108, 1779–1789 CrossRef CAS.
- E. Pines, D. Pines, O. Gajst and D. Huppert, J. Chem. Phys., 2020, 152, 074205 CrossRef CAS PubMed.
- G. R. Han, E. Lim, J. Kang, D. Hwang, J. Heo, S. K. Kim and J. W. Lee, J. Phys. Chem. A, 2023, 127, 7884–7891 CrossRef CAS PubMed.
- K. M. Solntsev, E.-A. Bartolo, G. Pan, G. Muller, S. Bommireddy, D. Huppert and L. M. Tolbert, Isr. J. Chem., 2009, 49, 227–233 CrossRef CAS PubMed.
- M. Sambol, K. Ester, S. Landgraf, B. Mihaljević, M. Cindrić, M. Kralj and N. Basarić, Photochem. Photobiol. Sci., 2019, 18, 1197–1211 CrossRef CAS PubMed.
- L. Lorenz, J. Plötner, V. V. Matylitsky, A. Dreuw and J. Wachtveitl, J. Phys. Chem. A, 2007, 111, 10891–10898 CrossRef PubMed.
- S. Suzuki and H. Baba, Bull. Chem. Soc. Jpn., 1967, 40, 2199–2200 CrossRef.
- I. Y. Martynov, N. K. Zaitsev, I. V. Soboleva, B. M. Uzhinov and M. G. Kuz’min, J. Appl. Spectrosc., 1978, 28, 732–736 CrossRef.
- A. B. Demjaschkewitch, N. K. Zaitsev and M. G. Kuzmin, Chem. Phys. Lett., 1978, 55, 80–83 CrossRef.
- H. Lemmetyinen, A. B. Demyashkevich and M. G. Kuzmin, Chem. Phys. Lett., 1980, 73, 98–101 CrossRef.
- M. Gutman and D. Huppert, J. Biochem. Biophys. Methods, 1979, 1, 9–19 CrossRef PubMed.
- S. Tobita and H. Shizuka, Chem. Phys. Lett., 1980, 75, 140–144 CrossRef.
- H. Shizuka and S. Tobita, J. Am. Chem. Soc., 1982, 104, 6919–6927 CrossRef.
- H. Shizuka and K. Tsutsumi, Bull. Chem. Soc. Jpn., 1983, 56, 629–630 CrossRef.
- J. Lee, G. W. Robinson, S. P. Webb, L. A. Philips and J. H. Clark, J. Am. Chem. Soc., 1986, 108, 6538–6542 CrossRef.
- G. W. Robinson, P. J. Thistlethwaite and J. Lee, J. Phys. Chem., 1986, 90, 4224–4233 CrossRef.
- G. W. Robinson, J. Lee and M.-P. Bassez, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2351–2359 RSC.
- O. Cheshnovsky and S. Leutwyler, J. Chem. Phys., 1988, 88, 4127–4138 CrossRef.
- J. J. Breen, L. W. Peng, D. M. Willberg, A. Heikal, P. Cong and A. H. Zewail, J. Chem. Phys., 1990, 92, 805–807 CrossRef.
- T. C. Swinney and D. F. Kelley, J. Phys. Chem., 1991, 95, 2430–2434 CrossRef.
- S. K. Kim, J.-K. Wang and A. H. Zewail, Chem. Phys. Lett., 1994, 228, 369–378 CrossRef.
- S. K. Kim, J. J. Breen, D. M. Willberg, L. W. Peng, A. Heikal, J. A. Syage and A. H. Zewail, J. Phys. Chem., 1995, 99, 7421–7435 CrossRef.
- Y. Matsumoto, T. Ebata and N. Mikami, J. Chem. Phys., 1998, 109, 6303–6311 CrossRef.
- R. Knochenmuss, O. Cheshnovsky and S. Leutwyler, Chem. Phys. Lett., 1988, 144, 317–323 CrossRef.
- R. Knochenmuss and S. Leutwyler, J. Chem. Phys., 1989, 91, 1268–1278 CrossRef.
- R. Knochenmuss, G. R. Holtom and D. Ray, Chem. Phys. Lett., 1993, 215, 188–192 CrossRef.
- R. D. Knochenmuss and D. E. Smith, J. Chem. Phys., 1994, 101, 7327–7336 CrossRef.
- R. Knochenmuss, Chem. Phys. Lett., 1998, 293, 191–196 CrossRef CAS.
- R. Knochenmuss, Chem. Phys. Lett., 1999, 311, 439–445 CrossRef CAS.
- R. Knochenmuss, I. Fischer, D. Lührs and Q. Lin, Isr. J. Chem., 1999, 39, 221–230 CrossRef CAS.
- R. Knochenmuss, P. L. Muiño and C. Wickleder, J. Phys. Chem., 1996, 100, 11218–11227 CrossRef CAS.
- E. Pines and G. R. Fleming, Chem. Phys., 1994, 183, 393–402 CrossRef CAS.
- B.-Z. Magnes, D. Pines, N. Strashnikova and E. Pines, Solid State Ionics, 2004, 168, 225–233 CrossRef CAS.
- E. Pines, D. Tepper, B.-Z. Magnes, D. Pines and T. Barak, Ber. Bunsenges. Phys. Chem., 1998, 102, 504–510 CrossRef CAS.
- E. Pines, D. Pines, T. Barak, B.-Z. Magnes, L. M. Tolbert and J. E. Haubrich, Ber. Bunsenges. Phys. Chem., 1998, 102, 511–517 CrossRef CAS.
- E. Pines, B.-Z. Magnes and T. Barak, J. Phys. Chem. A, 2001, 105, 9674–9680 CrossRef CAS.
- D. Pines, D. Eliovich, D. Aminov, M. Sigalov, D. Huppert and E. Pines, Spectroscopy and Computation of Hydrogen-Bonded Systems, John Wiley & Sons, Ltd, 2023, pp. 261–292 Search PubMed.
- I. V. Gopich and N. Agmon, J. Chem. Phys., 1999, 110, 10433–10444 CrossRef CAS.
- K. M. Solntsev and N. Agmon, Chem. Phys. Lett., 2000, 320, 262–268 CrossRef CAS.
- K. M. Solntsev, D. Huppert and N. Agmon, J. Phys. Chem. A, 1998, 102, 9599–9606 CrossRef CAS.
- K. M. Solntsev, D. Huppert, L. M. Tolbert and N. Agmon, J. Am. Chem. Soc., 1998, 120, 7981–7982 CrossRef CAS.
- I. Carmeli, D. Huppert, L. Tolbert and J. Haubrich, Chem. Phys. Lett., 1996, 260, 109–114 CrossRef CAS.
- D. Huppert, L. M. Tolbert and S. Linares-Samaniego, J. Phys. Chem. A, 1997, 101, 4602–4605 CrossRef CAS.
- K. M. Solntsev, D. Huppert and N. Agmon, J. Phys. Chem. A, 1999, 103, 6984–6997 CrossRef CAS.
- C. Clower, K. M. Solntsev, J. Kowalik, L. M. Tolbert and D. Huppert, J. Phys. Chem. A, 2002, 106, 3114–3122 CrossRef CAS.
- O. Gajst, G. G. Rozenman and D. Huppert, J. Phys. Chem. A, 2018, 122, 209–216 CrossRef CAS PubMed.
- K. M. Solntsev, D. Huppert, N. Agmon and L. M. Tolbert, J. Phys. Chem. A, 2000, 104, 4658–4669 CrossRef CAS.
- O. Gajst, L. Pinto da Silva, J. C. G. Esteves da Silva and D. Huppert, J. Phys. Chem. A, 2018, 122, 6166–6175 CrossRef CAS PubMed.
- M. Than Htun, A. Suwaiyan and U. K. A. Klein, Chem. Phys. Lett., 1995, 243, 506–511 CrossRef CAS.
- M. Than Htun, A. Suwaiyan and U. K. A. Klein, Chem. Phys. Lett., 1995, 243, 512–518 CrossRef CAS.
- M. T. Htun, Chem. Phys. Lett., 2011, 510, 73–77 CrossRef CAS.
- R. Yang and S. G. Schulman, Talanta, 2003, 60, 535–542 CrossRef PubMed.
- M. Than Htun, Chem. Phys. Lett., 2000, 328, 437–445 CrossRef.
- I. Kobayashi, M. Terazima and Y. Kimura, J. Phys. Chem. B, 2012, 116, 1043–1052 CrossRef PubMed.
- S. Abou-Al Einin, A. K. Zaitsev, N. K. Zaitsev and M. G. Kuzmin, J. Photochem. Photobiol., A, 1988, 41, 365–373 CrossRef.
- Y. V. Il’ichev, A. B. Demyashkevich and M. G. Kuzmin, J. Phys. Chem., 1991, 95, 3438–3444 CrossRef.
- Y. V. Il’ichev, A. B. Demyashkevich, M. G. Kuzmin and H. Lemmetyinen, J. Photochem. Photobiol., A, 1993, 74, 51–63 CrossRef.
- K. M. Solntsev, Y. V. Il’ichev, A. B. Demyashkevich and M. G. Kuzmin, J. Photochem. Photobiol., A, 1994, 78, 39–48 CrossRef.
- K. M. Solntsev, S. A. Al.Ainain, Y. V. Il’ichev and M. G. Kuzmin, J. Phys. Chem. A, 2004, 108, 8212–8222 CrossRef.
- D. Mandal, S. K. Pal and K. Bhattacharyya, J. Phys. Chem. A, 1998, 102, 9710–9714 CrossRef.
- P. Dutta, A. Halder, S. Mukherjee, P. Sen, S. Sen and K. Bhattacharyya, Langmuir, 2002, 18, 7867–7871 CrossRef.
- K. Fujii, Y. Yasaka, M. Ueno, Y. Koyanagi, S. Kasuga, Y. Matano and Y. Kimura, J. Phys. Chem. B, 2017, 121, 6042–6049 CrossRef PubMed.
- K. Fujii, M. Aramaki and Y. Kimura, J. Phys. Chem. B, 2018, 122, 12363–12374 CrossRef PubMed.
- K. Adamczyk, M. Prémont-Schwarz, D. Pines, E. Pines and E. T. J. Nibbering, Science, 2009, 326, 1690–1694 CrossRef PubMed.
- D. Pines, J. Ditkovich, T. Mukra, Y. Miller, P. M. Kiefer, S. Daschakraborty, J. T. Hynes and E. Pines, J. Phys. Chem. B, 2016, 120, 2440–2451 CrossRef PubMed.
- M. J. Cox and H. J. Bakker, J. Phys. Chem. A, 2010, 114, 10523–10530 CrossRef PubMed.
- E. Pines, in The Chemistry of Phenols, ed. Z. Rappoport, 2003, John Wiley & Sons: Hoboken, NJ, pp. 491–527 Search PubMed.
- J. R. Platt, J. Chem. Phys., 1949, 17, 484–495 CrossRef.
- A. T. Balaban and F. Harary, Tetrahedron, 1968, 24, 2505–2516 CrossRef.
- Systematics of the Electronic Spectra of Conjugated Molecules: A Source Book, Papers of the Chicago group, 1949-1964, ed. J. R. Platt and U. of Chicago, Laboratory of Molecular Structure and Spectra, Dept. of Physics, University of Chicago, Chicago, 1964 Search PubMed.
- E. B. Guidez and C. M. Aikens, J. Phys. Chem. C, 2013, 117, 21466–21475 CrossRef.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1997–2011 CrossRef.
- M. Rubio, M. Merchán, E. Ortí and B. O. Roos, Chem. Phys., 1994, 179, 395–409 CrossRef.
- M. K. Roos, S. Reiter and R. de Vivie-Riedle, Chem. Phys., 2018, 515, 586–595 CrossRef.
- S. Knippenberg, J. H. Starcke, M. Wormit and A. Dreuw, Mol. Phys., 2010, 108, 2801–2813 CrossRef.
- F. Bettanin, L. F. A. Ferrão, M. J. Pinheiro, A. J. A. Aquino, H. Lischka, F. B. C. Machado and D. Nachtigallova, J. Chem. Theory Comput., 2017, 13, 4297–4306 CrossRef PubMed.
- B.-Z. Magnes, N. V. Strashnikova and E. Pines, Isr. J. Chem., 1999, 39, 361–373 CrossRef.
- F. Messina, M. Prémont-Schwarz, O. Braem, D. Xiao, V. S. Batista, E. T. J. Nibbering and M. Chergui, Angew. Chem., 2013, 125, 7009–7013 CrossRef.
- L. M. Tolbert and J. E. Haubrich, J. Am. Chem. Soc., 1990, 112, 8163–8165 CrossRef.
- Y. Shioya, M. Yagi and J. Higuchi, Chem. Phys. Lett., 1989, 154, 25–28 CrossRef.
- L. M. Tolbert and J. E. Haubrich, J. Am. Chem. Soc., 1994, 116, 10593–10600 CrossRef.
- B. Cohen and D. Huppert, J. Phys. Chem. A, 2000, 104, 2663–2667 CrossRef.
- B. Cohen, J. Segal and D. Huppert, J. Phys. Chem. A, 2002, 106, 7462–7467 CrossRef.
- N. Koifman, B. Cohen and D. Huppert, J. Phys. Chem. A, 2002, 106, 4336–4344 CrossRef.
- B. Cohen, P. Leiderman and D. Huppert, J. Lumin., 2003, 102–103, 676–681 CrossRef.
- L. Genosar, T. Lasitza, R. Gepshtein, P. Leiderman, N. Koifman and D. Huppert, J. Phys. Chem. A, 2005, 109, 4852–4861 CrossRef PubMed.
- R. M. D. Nunes, L. G. Arnaut, K. M. Solntsev, L. M. Tolbert and S. J. Formosinho, J. Am. Chem. Soc., 2005, 127, 11890–11891 CrossRef.
- H. Yu, O.-H. Kwon and D.-J. Jang, J. Phys. Chem. A, 2004, 108, 5932–5937 CrossRef.
- K. M. Solntsev, C. E. Clower, L. M. Tolbert and D. Huppert, J. Am. Chem. Soc., 2005, 127, 8534–8544 CrossRef.
- A. V. Popov, E.-A. Gould, M. A. Salvitti, R. Hernandez and K. M. Solntsev, Phys. Chem. Chem. Phys., 2011, 13, 14914 RSC.
- E.-A. Gould, A. V. Popov, L. M. Tolbert, I. Presiado, Y. Erez, D. Huppert and K. M. Solntsev, Phys. Chem. Chem. Phys., 2012, 14, 8964 RSC.
- J. L. Pérez Lustres, S. A. Kovalenko, M. Mosquera, T. Senyushkina, W. Flasche and N. P. Ernsting, Angew. Chem., Int. Ed., 2005, 44, 5635–5639 CrossRef.
- J. L. Pérez-Lustres, F. Rodriguez-Prieto, M. Mosquera, T. A. Senyushkina, N. P. Ernsting and S. A. Kovalenko, J. Am. Chem. Soc., 2007, 129, 5408–5418 CrossRef.
- M. Veiga-Gutiérrez, A. Brenlla, C. Carreira Blanco, B. Fernández, S. A. Kovalenko, F. Rodríguez-Prieto, M. Mosquera and J. L. P. Lustres, J. Phys. Chem. B, 2013, 117, 14065–14078 CrossRef PubMed.
- Y. M. Lee, S.-Y. Park, H. Kim, T. G. Kim and O.-H. Kwon, Methods Appl. Fluoresc., 2016, 4, 024004 CrossRef PubMed.
- S.-Y. Park, T. G. Kim, M. J. Ajitha, K. Kwac, Y. M. Lee, H. Kim, Y. Jung and O.-H. Kwon, Phys. Chem. Chem. Phys., 2016, 18, 24880–24889 RSC.
- S.-Y. Park, Y. M. Lee, K. Kwac, Y. Jung and O.-H. Kwon, Chem.–A Eur. J., 2016, 22, 4340–4344 CrossRef CAS PubMed.
- Y. Choi, H. Kim and O. Kwon, Bull. Korean Chem. Soc., 2022, 43, 501–507 CrossRef CAS.
- Y.-J. Choi, H.-W. Nho, Y.-J. Kim and O.-H. Kwon, ChemPhotoChem, 2024, 8, e202300164 CrossRef CAS.
- S.-H. Lee, Y.-J. Choi, Y.-J. Kim, J.-M. Kee and O.-H. Kwon, Cell Rep. Phys. Sci., 2024, 5, 102155 CrossRef CAS.
- Y.-J. Kim, S. Rakshit, G. Y. Jin, P. Ghosh, Y. M. Lee, W.-W. Park, Y. S. Kim and O.-H. Kwon, Chem.–A Eur. J., 2017, 23, 17179–17185 CrossRef CAS PubMed.
- N. Karton-Lifshin, E. Segal, L. Omer, M. Portnoy, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2011, 133, 10960–10965 CrossRef CAS PubMed.
- N. Karton-Lifshin, L. Albertazzi, M. Bendikov, P. S. Baran and D. Shabat, J. Am. Chem. Soc., 2012, 134, 20412–20420 CrossRef CAS PubMed.
- N. Karton-Lifshin, I. Presiado, Y. Erez, R. Gepshtein, D. Shabat and D. Huppert, J. Phys. Chem. A, 2012, 116, 85–92 CrossRef CAS PubMed.
- R. Simkovitch, E. Kisin-Finfer, S. Shomer, R. Gepshtein, D. Shabat and D. Huppert, J. Photochem. Photobiol., A, 2013, 254, 45–53 CrossRef CAS.
- C. Lee, S. Chung, H. Song, Y. M. Rhee, E. Lee and T. Joo, ChemPhotoChem, 2021, 5, 245–252 CrossRef CAS.
- R. Simkovitch, S. Shomer, R. Gepshtein, D. Shabat and D. Huppert, J. Phys. Chem. A, 2013, 117, 3925–3934 CrossRef CAS PubMed.
- R. Simkovitch, K. Akulov, S. Shomer, M. E. Roth, D. Shabat, T. Schwartz and D. Huppert, J. Phys. Chem. A, 2014, 118, 4425–4443 CrossRef CAS PubMed.
- R. Simkovitch, S. Shomer, R. Gepshtein, D. Shabat and D. Huppert, J. Phys. Chem. A, 2014, 118, 1832–1840 CrossRef CAS PubMed.
- O. Green, R. Simkovitch, L. Pinto da Silva, J. C. G. Esteves da Silva, D. Shabat and D. Huppert, J. Phys. Chem. A, 2016, 120, 6184–6199 CrossRef CAS PubMed.
- L. Pinto da Silva, O. Green, O. Gajst, R. Simkovitch, D. Shabat, J. C. G. Esteves da Silva and D. Huppert, ACS Omega, 2018, 3, 2058–2073 CrossRef CAS PubMed.
- O. Green, O. Gajst, R. Simkovitch, D. Shabat and D. Huppert, J. Photochem. Photobiol., A, 2017, 349, 230–237 CrossRef CAS.
- O. Green, O. Gajst, R. Simkovitch, D. Shabat and D. Huppert, J. Phys. Chem. A, 2017, 121, 3079–3087 CrossRef CAS PubMed.
- Z. Li, Q. Yang, R. Chang, G. Ma, M. Chen and W. Zhang, Dyes Pigm., 2011, 88, 307–314 CrossRef CAS.
- B. Zhu, C. Gao, Y. Zhao, C. Liu, Y. Li, Q. Wei, Z. Ma, B. Du and X. Zhang, Chem. Commun., 2011, 47, 8656–8658 RSC.
- Z. Luo, B. Yang, C. Zhong, F. Tang, M. Yuan, Y. Xue, G. Yao, J. Zhang and Y. Zhang, Dyes Pigm., 2013, 97, 52–57 CrossRef CAS.
- L. Biczók, P. Valat and V. Wintgens, Phys. Chem. Chem. Phys., 1999, 1, 4759–4766 RSC.
- L. Biczók, P. Valat and V. Wintgens, Phys. Chem. Chem. Phys., 2001, 3, 1459–1464 RSC.
- L. Biczók, P. Valat and V. Wintgens, Photochem. Photobiol. Sci., 2003, 2, 230–235 CrossRef PubMed.
- T. Kumpulainen, B. H. Bakker, M. Hilbers and A. M. Brouwer, J. Phys. Chem. B, 2015, 119, 2515–2524 CrossRef CAS PubMed.
- T. Kumpulainen, B. H. Bakker and A. M. Brouwer, Phys. Chem. Chem. Phys., 2015, 17, 20715–20724 RSC.
- T. Kumpulainen, A. Rosspeintner, B. Dereka and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 4516–4521 CrossRef CAS PubMed.
- P. Verma, A. Rosspeintner, B. Dereka, E. Vauthey and T. Kumpulainen, Chem. Sci., 2020, 11, 7963–7971 RSC.
- A. Yamaguchi, M. Namekawa, T. Kamijo, T. Itoh and N. Teramae, Anal. Chem., 2011, 83, 2939–2946 CrossRef CAS PubMed.
- B. Hinkeldey, A. Schmitt and G. Jung, ChemPhysChem, 2008, 9, 2019–2027 CrossRef CAS PubMed.
- D. Maus, A. Grandjean and G. Jung, J. Phys. Chem. A, 2018, 122, 9025–9030 CrossRef CAS PubMed.
- R. F. Robbins, J. Chem. Soc., 1960, 2553–2556 RSC.
- D. Maus, PhD Thesis, Universität des Saarlandes, Saarbrücken, Germany, 2021 Search PubMed.
- Y. Hu, J. F. Joung, J.-E. Jeong, Y. Jeong, H. Y. Woo, Y. She, S. Park and J. Yoon, Sens. Actuators, B, 2019, 280, 298–305 CrossRef CAS.
- C. Spies, B. Finkler, N. Acar and G. Jung, Phys. Chem. Chem. Phys., 2013, 15, 19893 RSC.
- C. Spies, S. Shomer, B. Finkler, D. Pines, E. Pines, G. Jung and D. Huppert, Phys. Chem. Chem. Phys., 2014, 16, 9104 RSC.
- M. Vester, T. Staut, J. Enderlein and G. Jung, J. Phys. Chem. Lett., 2015, 6, 1149–1154 CrossRef CAS PubMed.
- M. Vester, A. Grueter, B. Finkler, R. Becker and G. Jung, Phys. Chem. Chem. Phys., 2016, 18, 10281–10288 RSC.
- J. Knorr, N. Sülzner, B. Geissler, C. Spies, A. Grandjean, R. J. Kutta, G. Jung and P. Nuernberger, Photochem. Photobiol. Sci., 2022, 21, 2179–2192 CrossRef CAS PubMed.
- N. Sülzner and C. Hättig, J. Phys. Chem. A, 2022, 126, 5911–5923 CrossRef PubMed.
- N. Sülzner and C. Hättig, Phys. Chem. Chem. Phys., 2023, 25, 11130–11144 RSC.
- N. Agmon, J. Mol. Liq., 2000, 85, 87–96 CrossRef CAS.
- R. Simkovitch, S. Shomer, R. Gepshtein and D. Huppert, J. Phys. Chem. B, 2015, 119, 2253–2262 CrossRef CAS PubMed.
- M. Eigen, L. De Maeyer and Z. Elektrochem, Ber. Bunsenges. Phys. Chem., 1955, 59, 986–993 CrossRef.
- M. Eigen, W. Kruse, G. Maass and L. Demaeyer, Prog. React. Kinet. Mech., 1964, 2, 285 Search PubMed.
- N. Sülzner, Chem, 2024, 10, 3276–3278 Search PubMed.
- N. Agmon, J. Chem. Phys., 1999, 110, 2175–2180 CrossRef.
- N. Agmon and I. V. Gopich, Chem. Phys. Lett., 1999, 302, 399–404 CrossRef.
- K. M. Solntsev, D. Huppert and N. Agmon, Phys. Rev. Lett., 2001, 86, 3427–3430 CrossRef PubMed.
- E. M. Espinoza, J. A. Clark, C. P. d. Silva, J. B. Derr, G. T. d. M. Silva, M. K. Billones, M. Morales, F. H. Quina and V. I. Vullev, J. Photochem. Photobiol., 2022, 10, 100110 CrossRef.
- A. Grandjean, J. L. Pérez Lustres, S. Muth, D. Maus and G. Jung, J. Phys. Chem. Lett., 2021, 12, 1683–1689 CrossRef PubMed.
- A. Grandjean, J. L. Pérez Lustres and G. Jung, ChemPhotoChem, 2021, 5, 1094–1105 Search PubMed.
- M. Barroso, L. G. Arnaut and S. J. Formosinho, J. Phys. Chem. A, 2007, 111, 591–602 CrossRef PubMed.
- H.-H. Limbach, K. B. Schowen and R. L. Schowen, J. Phys. Org. Chem., 2010, 23, 586–605 CrossRef.
- P. M. Kiefer and J. T. Hynes, J. Phys. Org. Chem., 2010, 23, 632–646 Search PubMed.
- A. J. Kresge, D. S. Sagatys and H. L. Chen, J. Am. Chem. Soc., 1977, 99, 7228–7233 CrossRef.
- R. A. Marcus, J. Phys. Chem., 1968, 72, 891–899 CrossRef.
- A. Das, P. Ghosh, A. Dutta and P. Sen, Chem. Phys. Impact, 2021, 3, 100044 CrossRef.
- A. J. Kresge, Acc. Chem. Res., 1975, 8, 354–360 CrossRef.
- P. M. Kiefer and J. T. Hynes, Isr. J. Chem., 2004, 44, 171–184 CrossRef.
- K. Yates and J. Phys, Org. Chem., 1989, 2, 300–322 Search PubMed.
- D. N. Silverman, Biochimica et Biophys. Acta (BBA) – Bioenerg., 2000, 1458, 88–103 CrossRef PubMed.
- K. S. Peters, Acc. Chem. Res., 2009, 42, 89–96 CrossRef PubMed.
- A. Shabashini, S. Kumar Panja, A. Biswas, S. Bera and G. Chandra Nandi, J. Photochem. Photobiol., A, 2022, 432, 114087 CrossRef.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- Z. Wen, L. José Karas, C.-H. Wu and J. I-Chia Wu, Chem. Commun., 2020, 56, 8380–8383 RSC.
- J. Yan, T. Slanina, J. Bergman and H. Ottosson, Chem.–Eur. J., 2023, 29, e202203748 CrossRef PubMed.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef PubMed.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef.
- B. J. Lampkin, Y. H. Nguyen, P. B. Karadakov and B. VanVeller, Phys. Chem. Chem. Phys., 2019, 21, 11608–11614 RSC.
- C.-H. Wu, L. J. Karas, H. Ottosson and J. I.-C. Wu, Proc. Natl. Acad. Sci., 2019, 116, 20303–20308 CrossRef.
- B. Szczepanik, J. Mol. Struct., 2015, 1099, 209–214 CrossRef.
- Z. R. Grabowski, W. Rubaszewska and J. Chem, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 11–28 RSC.
- E. R. Davidson and A. A. Jarzęcki, Chem. Phys. Lett., 1998, 285, 155–159 CrossRef.
- A. Weller, P. React. Kinetics Mech., 1961, 1, 187 Search PubMed.
- S. Schulman and Q. Fernando, Tetrahedron, 1968, 24, 1777–1783 CrossRef.
- J. F. Joung, M. Jeong and S. Park, Phys. Chem. Chem. Phys., 2022, 24, 21714–21721 RSC.
- D. Himmel, S. K. Goll, I. Leito and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 6885–6888 CrossRef CAS.
- F. Bastkowski, S. Seitz, A. Heering, R. Born, S. Lainela, I. Leito, J. Nerut, J. Saame, V. Radtke, D. Nagy, Z. S. Nagyne, L. Szucs, M. Rozikova, M. Vičarova, L. C. Deleebeeck, A. Snedden, B. Anes, R. Bettencourt, M. F. Camoes, R. Quendera, L. F. Ribeiro, D. Stoica, T. Naykki, L. Liv, E. Uysal and G. Yerleşkesi, Messunsicherheit praxisgerecht bestimmen – Prüfprozesse in der industriellen Praxis 2021, VDI Verlag, 2021, pp. 13–22 Search PubMed.
- L. Deleebeeck, A. Snedden, D. Nagy, Z. Szilágyi Nagyné, M. Roziková, M. Vičarová, A. Heering, F. Bastkowski, I. Leito, R. Quendera, V. Cabral and D. Stoica, Sensors, 2021, 21, 3935 CrossRef CAS PubMed.
- V. Radtke, D. Stoica, I. Leito, F. Camões, I. Krossing, B. Anes, M. Roziková, L. Deleebeeck, S. Veltzé, T. Näykki, F. Bastkowski, A. Heering, N. Dániel, R. Quendera, L. Liv, E. Uysal and N. Lawrence, Pure Appl. Chem., 2021, 93, 1049–1060 CrossRef CAS.
- K. M. Solntsev, D. Huppert and N. Agmon, J. Phys. Chem. A, 2001, 105, 5868–5876 CrossRef CAS.
- R. Bhide, C. N. Feltenberger, G. S. Phun, G. Barton, D. Fishman and S. Ardo, J. Am. Chem. Soc., 2022, 144, 14477–14488 CrossRef PubMed.
- J. Ho and M. L. Coote, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 649–660 Search PubMed.
- M. D. Liptak and G. C. Shields, Int. J. Quantum Chem., 2001, 85, 727–741 Search PubMed.
- J. R. Pliego, Chem. Phys. Lett., 2003, 367, 145–149 CrossRef.
- J. Ho and M. L. Coote, Theor. Chem. Acc., 2009, 125, 3 Search PubMed.
- R. Casasnovas, J. Ortega-Castro, J. Frau, J. Donoso and F. Muñoz, Int. J. Quantum Chem., 2014, 114, 1350–1363 CrossRef.
- I. A. Topol, G. J. Tawa, S. K. Burt and A. A. Rashin, J. Chem. Phys., 1999, 111, 10998–11014 CrossRef.
- M. D. Liptak and G. C. Shields, J. Am. Chem. Soc., 2001, 123, 7314–7319 CrossRef.
- A. M. Toth, M. D. Liptak, D. L. Phillips and G. C. Shields, J. Chem. Phys., 2001, 114, 4595–4606 CrossRef.
- M. D. Liptak, K. C. Gross, P. G. Seybold, S. Feldgus and G. C. Shields, J. Am. Chem. Soc., 2002, 124, 6421–6427 CrossRef PubMed.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, J. Phys. Chem. A, 1998, 102, 7787–7794 CrossRef.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2007, 111, 408–422 CrossRef PubMed.
- E. Rossini and E.-W. Knapp, J. Comput. Chem., 2016, 37, 1082–1091 CrossRef PubMed.
- E. Rossini, A. D. Bochevarov and E. W. Knapp, ACS Omega, 2018, 3, 1653–1662 CrossRef CAS PubMed.
- L. C. Kröger, S. Müller, I. Smirnova and K. Leonhard, J. Phys. Chem. A, 2020, 124, 4171–4181 CrossRef.
- M. Busch, E. Ahlberg, E. Ahlberg and K. Laasonen, ACS Omega, 2022, 7, 17369–17383 CrossRef CAS.
- J. W. Zheng and W. H. Green, J. Phys. Chem. A, 2023, 127, 10268–10281 CrossRef CAS.
- J. W. Zheng, E. A. Ibrahim and W. H. Green, pKa Prediction in Non-Aqueous Solvents, chemRxiv, preprint, 2024, https://chemrxiv.org/engage/chemrxiv/article-details/663e53e521291e5d1df186b5 Search PubMed.
- J. Gao, N. Li and M. Freindorf, J. Am. Chem. Soc., 1996, 118, 4912–4913 CrossRef CAS.
- B. Mennucci, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 386–404 Search PubMed.
- P. F. Moreira, L. Giestas, C. Yihwa, C. Vautier-Giongo, F. H. Quina, A. L. Maçanita and J. C. Lima, J. Phys. Chem. A, 2003, 107, 4203–4210 CrossRef.
- B. Szczepanik, S. Styrcz and M. Góra, Spectrochim. Acta, Part A, 2008, 71, 403–409 CrossRef PubMed.
- J. Gao and K. Byun, Theor. Chem. Acc., 1997, 96, 151–156 Search PubMed.
- O. A. Borg and B. Durbeej, J. Phys. Chem. B, 2007, 111, 11554–11565 CrossRef PubMed.
- O. Anders Borg and B. Durbeej, Phys. Chem. Chem. Phys., 2008, 10, 2528 RSC.
- D. Jacquemin and C. Adamo, Density-Functional Methods for Excited States, Springer International Publishing, Cham, 2016, pp. 347–375 Search PubMed.
- D. Jacquemin, B. Mennucci and C. Adamo, Phys. Chem. Chem. Phys., 2011, 13, 16987–16998 RSC.
- E. Brémond, M. Savarese, C. Adamo and D. Jacquemin, J. Chem. Theory Comput., 2018, 14, 3715–3727 CrossRef PubMed.
- P.-F. Loos, A. Scemama and D. Jacquemin, J. Phys. Chem. Lett., 2020, 11, 2374–2383 CrossRef PubMed.
- D. Jacquemin, E. A. Perpète, I. Ciofini and C. Adamo, Acc. Chem. Res., 2009, 42, 326–334 CrossRef PubMed.
- D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef PubMed.
- Y. Wang, H. Li and Y. Shi, New J. Chem., 2015, 39, 7026–7032 RSC.
- J. Yi and H. Fang, J. Mol. Model., 2017, 23, 312 CrossRef PubMed.
- J. Yi and H. Fang, Struct. Chem., 2018, 29, 1341–1350 CrossRef.
- D. Jacquemin, E. A. Perpète, G. Scalmani, I. Ciofini, C. Peltier and C. Adamo, Chem. Phys., 2010, 372, 61–66 CrossRef.
- M. Guglielmi, I. Tavernelli and U. Rothlisberger, Phys. Chem. Chem. Phys., 2009, 11, 4549–4555 RSC.
- G. Bekçioğlu, F. Hoffmann and D. Sebastiani, J. Phys. Chem. A, 2015, 119, 9244–9251 CrossRef.
- Y. Li, F. Siddique, A. J. A. Aquino and H. Lischka, J. Phys. Chem. A, 2021, 125, 5765–5778 CrossRef PubMed.
- J. D. Coe, B. G. Levine and T. J. Martínez, J. Phys. Chem. A, 2007, 111, 11302–11310 CrossRef.
- S.-H. Xia, B.-B. Xie, Q. Fang, G. Cui and W. Thiel, Phys. Chem. Chem. Phys., 2015, 17, 9687–9697 RSC.
- W.-W. Guo, X.-Y. Liu, W.-K. Chen and G. Cui, RSC Adv., 2016, 6, 85574–85581 RSC.
- D. Jacquemin, I. Duchemin and X. Blase, J. Phys. Chem. Lett., 2017, 8, 1524–1529 CrossRef.
- C. A. Guido, B. Mennucci, G. Scalmani and D. Jacquemin, J. Chem. Theory Comput., 2018, 14, 1544–1553 CrossRef.
- R. Sarkar, M. Boggio-Pasqua, P.-F. Loos and D. Jacquemin, J. Chem. Theory Comput., 2021, 17, 1117–1132 CrossRef PubMed.
- A. D. Laurent and D. Jacquemin, Int. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123–135 CrossRef PubMed.
- A. L. Sobolewski, W. Domcke and C. Hättig, J. Phys. Chem. A, 2006, 110, 6301–6306 CrossRef PubMed.
- C. Suellen, R. G. Freitas, P.-F. Loos and D. Jacquemin, J. Chem. Theory Comput., 2019, 15, 4581–4590 CrossRef PubMed.
- A. Acharya, S. Chaudhuri and V. S. Batista, J. Chem. Theory Comput., 2018, 14, 867–876 CrossRef.
- S. Grimme and M. Parac, ChemPhysChem, 2003, 4, 292–295 CrossRef PubMed.
- A. Prlj, M. E. Sandoval-Salinas, D. Casanova, D. Jacquemin and C. Corminboeuf, J. Chem. Theory Comput., 2016, 12, 2652–2660 CrossRef.
- S. Khodia and S. Maity, Phys. Chem. Chem. Phys., 2022, 24, 12043–12051 RSC.
- A. Ghiami-Shomami and C. Hättig, J. Comput. Chem., 2023, 44, 1941–1955 CrossRef PubMed.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef.
- A. Klamt, V. Jonas, T. Bürger and J. C. W. Lohrenz, J. Phys. Chem. A, 1998, 102, 5074–5085 Search PubMed.
- A. Klamt and F. Eckert, Fluid Phase Equilib., 2000, 172, 43–72 CrossRef.
- N. Sülzner, J. Haberhauer, C. Hättig and A. Hellweg, J. Comput. Chem., 2022, 43, 1011–1022 CrossRef.
- T. Kloss, J. Heil and S. M. Kast, J. Phys. Chem. B, 2008, 112, 4337–4343 Search PubMed.
- F. Hoffgaard, J. Heil and S. M. Kast, J. Chem. Theory Comput., 2013, 9, 4718–4726 Search PubMed.
- S. Tsuru, B. Sharma, D. Marx and C. Hättig, J. Chem. Theory Comput., 2023, 19, 2291–2303 Search PubMed.
- J. M. Toldo, M. T. d. Casal, E. Ventura, S. A. d. Monte and M. Barbatti, Phys. Chem. Chem. Phys., 2023, 25, 8293–8316 RSC.
- J. C. Tully, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef.
- J. Jankowska, M. Barbatti, J. Sadlej and A. L. Sobolewski, Phys. Chem. Chem. Phys., 2017, 19, 5318–5325 RSC.
- M. Barbatti and K. Sen, Int. J. Quantum Chem., 2016, 116, 762–771 CrossRef.
- A. Petrone, F. Perrella, F. Coppola, L. Crisci, G. Donati, P. Cimino and N. Rega, Chem. Phys. Rev., 2022, 3, 021307 CrossRef.
- M. Ruckenbauer, M. Barbatti, T. Müller and H. Lischka, J. Phys. Chem. A, 2010, 114, 6757–6765 CrossRef.
- M. Ruckenbauer, M. Barbatti, T. Müller and H. Lischka, J. Phys. Chem. A, 2013, 117, 2790–2799 CrossRef PubMed.
- D. B. Cordes, S. Gamsey, Z. Sharrett, A. Miller, P. Thoniyot, R. A. Wessling and B. Singaram, Langmuir, 2005, 21, 6540–6547 CrossRef PubMed.
- A. B. Kotlyar, N. Borovok, S. Raviv, L. Zimanyi and M. Gutman, Photochem. Photobiol., 1996, 63, 448–454 CrossRef.
- M. Schmitz, M.-S. Bertrams, A. C. Sell, F. Glaser and C. Kerzig, J. Am. Chem. Soc., 2024, 146, 25799–25812 CrossRef PubMed.
- F. Johannsen, L. Williams, M. H. Chak and M. Drescher, J. Phys. Chem. Lett., 2024, 15, 7069–7074 CrossRef PubMed.
- A. Clasen, S. Wenderoth, I. Tavernaro, J. Fleddermann, A. Kraegeloh and G. Jung, RSC Adv., 2019, 9, 35695–35705 RSC.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- L. Wimberger, J. Andréasson and J. E. Beves, Chem. Commun., 2022, 58, 5610–5613 RSC.
- Y. Cheng, X. Ma, J. Zhai and X. Xie, Chem. Commun., 2023, 59, 1805–1808 RSC.
- P. Utroša, J. A. Carroll, E. Žagar, D. Pahovnik and C. Barner-Kowollik, Chem.–A Eur. J., 2024, 30, e202400820 CrossRef PubMed.
- A. P. Demchenko, J. Mol. Struct., 2014, 1077, 51–67 CrossRef.
- M. Lawrence, C. J. Marzzacco, C. Morton, C. Schwab and A. M. Halpern, J. Phys. Chem., 1991, 95, 10294–10299 CrossRef CAS.
- L. M. Tolbert, L. C. Harvey and R. C. Lum, J. Phys. Chem., 1993, 97, 13335–13340 CrossRef CAS.
- K. Schäfer and H. Ihmels, J. Fluoresc., 2017, 27, 1221–1224 CrossRef PubMed.
- J. Othong, J. Boonmak, F. Kielar and S. Youngme, ACS Appl. Mater. Interfaces, 2020, 12, 41776–41784 CrossRef CAS.
- F. A. d. C. Silva, E. T. Rezende, D. B. Filho, D. d. Brito Rezende, I. M. Cuccovia, L. F. Gome, M. F. P. d. Silva and M. J. Politi, J. Lumin., 2014, 146, 57–63 CrossRef CAS.
- P. M. Kiefer and J. T. Hynes, J. Phys. Chem. A, 2003, 107, 9022–9039 CrossRef CAS.
- E. Sato, K. Chiba, M. Hoshi and Y. Kanaoka, Chem. Pharm. Bull., 1992, 40, 786–788 CrossRef CAS.
- S. Hemmer, H. Hui, J. Draeger, J. Menges, E. Schwarz, A. Wrede, J. Oertel, L. Kaestner, G. Jung and S. Urbschat, A novel fluorescent diagnostic probe as a potential Point-of-Care diagnostic tool to estimate recurrence risk of meningiomas, 2024 Search PubMed , submitted.
- J. A. Menges, A. Grandjean, A. Clasen and G. Jung, ChemCatChem, 2020, 12, 2630–2637 CrossRef CAS.
- J. A. Menges, A. Clasen, M. Jourdain, J. Beckmann, C. Hoffmann, J. König and G. Jung, Langmuir, 2019, 35, 2506–2516 CrossRef CAS PubMed.
- D. Han, C. Li, X. Jin, J. Zhou, Y. Xu, T. Jiao and P. Duan, Adv. Photonics Res., 2022, 3, 2100287 CrossRef CAS.
- K. Y. Yung, A. J. Schadock-Hewitt, N. P. Hunter, F. V. Bright and G. A. Baker, Chem. Commun., 2011, 47, 4775–4777 RSC.
- N. Amdursky, Y. Lin, N. Aho and G. Groenhof, Proc. Natl. Acad. Sci., 2019, 116, 2443–2451 CrossRef CAS PubMed.
- Y. Sha, Z. Guo, Y. Han, Z. Xue, M. Li, Y. Wan, W. Yang and X. Ma, J. Phys. Chem. C, 2023, 127, 14458–14467 CrossRef CAS.
- S. Heckel, J. Hübner, A. Leutzgen, G. Jung and J. Simmchen, Catalysts, 2021, 11, 599 CrossRef CAS.
- A. Yucknovsky, B. B. Rich, S. Gutkin, A. Ramanthrikkovil Variyam, D. Shabat, B. Pokroy and N. Amdursky, J. Phys. Chem. B, 2022, 126, 6331–6337 CrossRef PubMed.
- P. Sun, Z. Li, X. Zhang, Y. Liao and S. Liao, Macromol. Rapid Commun., 2024, 45, 2400054 CrossRef CAS PubMed.
- J. Tripathi, H. Gupta and A. Sharma, Esterification of Carboxylic Acids utilizing Eosin Y as a Photoacid Catalyst, chemRxiv, preprint, 2024, https://chemrxiv.org/engage/chemrxiv/article-details/663218ca91aefa6ce1e0070c Search PubMed.
- H. Liu, Y. Chen, D. An, X. Zhang and S. Liao, Synlett, 2022, 33, 800–804 CrossRef CAS.
- J. Saway, A. F. Pierre and J. J. Badillo, Org. Biomol. Chem., 2022, 20, 6188–6192 RSC.
- N. Otani, K. Higashiyama, H. Sakai, T. Hasobe, D. Takahashi and K. Toshima, Eur. J. Org Chem., 2023, 26, e202300287 CrossRef CAS.
- B. Yang, K. Dong, X.-S. Li, L.-Z. Wu and Q. Liu, Org. Lett., 2022, 24, 2040–2044 CrossRef CAS.
- A. M. M. Alazaly, A. S. I. Amer, A. M. Fathi and A. A. Abdel-Shafi, J. Photochem. Photobiol., A, 2018, 364, 819–825 CrossRef CAS.
- S. Kohse, A. Neubauer, A. Pazidis, S. Lochbrunner and U. Kragl, J. Am. Chem. Soc., 2013, 135, 9407–9411 CrossRef CAS.
- A. M. Alfaraidi, B. Kudisch, N. Ni, J. Thomas, T. Y. George, K. Rajabimoghadam, H. J. Jiang, D. G. Nocera, M. J. Aziz and R. Y. Liu, J. Am. Chem. Soc., 2023, 145, 26720–26727 CrossRef CAS PubMed.
- O. Alghazwat, A. Elgattar and Y. Liao, Photochem. Photobiol. Sci., 2023, 22, 2573–2578 CrossRef CAS.
- A. de Vries, K. Goloviznina, M. Reiter, M. Salanne and M. R. Lukatskaya, Chem. Mater., 2024, 36, 1308–1317 CrossRef CAS PubMed.
- J. Bae, H. Lim, J. Ahn, Y. H. Kim, M. S. Kim and I.-D. Kim, Adv. Mater., 2022, 34, 2201734 CrossRef CAS.
- A. Yucknovsky, Y. Shlosberg, N. Adir and N. Amdursky, Angew. Chem., Int. Ed., 2023, 62, e202301541 CrossRef CAS.
- G. S. Phun, R. Bhide and S. Ardo, Energy Environ. Sci., 2023, 16, 4593–4611 RSC.
- S. Haghighat, S. Ostresh and J. M. Dawlaty, J. Phys. Chem. B, 2016, 120, 1002–1007 CrossRef CAS PubMed.
- S.-F. Zhou, G.-M. Wu, C.-X. Zhang and Q.-L. Wang, Adv. Mater. Interfaces, 2022, 9, 2101247 CrossRef CAS.
- F. Wendler, M. Sittig, J. C. Tom, B. Dietzek and F. H. Schacher, Chem.–A Eur. J., 2020, 26, 2365–2379 CrossRef CAS.
- M. Bitsch, A. K. Boehm, A. Grandjean, G. Jung and M. Gallei, Molecules, 2021, 26, 7350 CrossRef CAS PubMed.
- H. Chen, K. Wen, Y. Lu, X. Zhang, Y. Shi, Q. Shi, H. Ma, Q. Peng and H. Huang, Sci. China:Chem., 2022, 65, 2528–2537 CrossRef CAS.
- L. Kang, H. Zhao, S. Liu, Y. Liu, Y. Liu, D. Chen, H. Qiu, J. Yang, Y. Gu and Y. Zhao, Eur. J. Med. Chem., 2022, 242, 114669 CrossRef CAS PubMed.
- L. I. Kaberov, M. Sittig, A. Chettri, A. Ibrahim, B. Dietzek-Ivanšić and F. H. Schacher, Polym. Chem., 2023, 14, 3453–3464 RSC.
- A. Chettri, L. I. Kaberov, N. Klosterhalfen, S. Perera, M. Jamshied, F. H. Schacher and B. Dietzek-Ivanšić, Chem.–A Eur. J., 2024, 30, e202401047 CrossRef CAS.
- A. Losi and C. Viappiani, Chem. Phys. Lett., 1998, 289, 500–506 CrossRef CAS.
- P. M. Kiefer and J. T. Hynes, J. Phys. Chem. A, 2004, 108, 11793–11808 CrossRef CAS.
- T. B. McAnaney, X. Shi, P. Abbyad, H. Jung, S. J. Remington and S. G. Boxer, Biochemistry, 2005, 44, 8701–8711 CrossRef.
- P. M. Kiefer and J. T. Hynes, J. Phys. Chem. A, 2002, 106, 1834–1849 CrossRef CAS.
- P. M. Kiefer and J. T. Hynes, J. Phys. Chem. A, 2002, 106, 1850–1861 CrossRef CAS.
- K. M. Solntsev, L. M. Tolbert, B. Cohen, D. Huppert, Y. Hayashi and Y. Feldman, J. Am. Chem. Soc., 2002, 124, 9046–9047 CrossRef CAS PubMed.
- F. Pavošević, S. Hammes-Schiffer, A. Rubio and J. Flick, J. Am. Chem. Soc., 2022, 144, 4995–5002 CrossRef.
- I. Sokolovskii and G. Groenhof, Nanophotonics, 2024, 13(14), 2687–2694 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the late Professor Dan Huppert on the occasion of his 80th birthday. |
| ‡ Initially referred to as 3-aminopyrene-5,8,10-trisulfonate. |
| § Initially referred to as 3-hydroxypyrene-5,8,10-trisulfonate. |
| ¶ Initially referred to as 3,5-dihydroxypyrene-8,10-disulfonate. |
| || E.g., see ref. 434 for a definition of the different condensation types for polycyclic aromatic compounds. |
| ** E.g., see ref. 604 for a recent perspective on the use of surface hopping modeling for describing charge (including proton) and energy transfer by Barbatti and co-workers. |
| †† See ref. 614 for an example of real-time monitoring of the photo-induced pH jump using a diamagnetic pH indicator consisting of a mestable-state photoacid and an EPR spin probe. |
| This journal is © The Royal Society of Chemistry 2025 |