DOI:
10.1039/C8QM00613J
(Research Article)
Mater. Chem. Front., 2019,
3, 284-291
A novel strategy for realizing dual state fluorescence and low-temperature phosphorescence†
Received
28th November 2018
, Accepted 11th December 2018
First published on 11th December 2018
Abstract
We demonstrate a new strategy for the development of dual-state emissive materials by introducing phenyl groups to triphenylpyrrole (TPP) isomers. The phenyl group can increase the rotational barrier and restrict the intramolecular rotation of TPP, thus enhancing the fluorescence efficiency of aggregation induced emission (AIE) materials in solution. Fluorescence of TPP derivatives was not quenched by the drastic intermolecular collision and intramolecular motion at high temperature. On the contrary, the fluorescence of their solutions showed a positive response to temperature. Theoretical calculations demonstrated that the molecules exhibited a higher planar conformation at high temperature. Three donor–acceptor samples with a twisted intramolecular charge transfer (TICT) effect showed a strong phosphorescence with a lifetime up to 10 s at low temperature.
Introduction
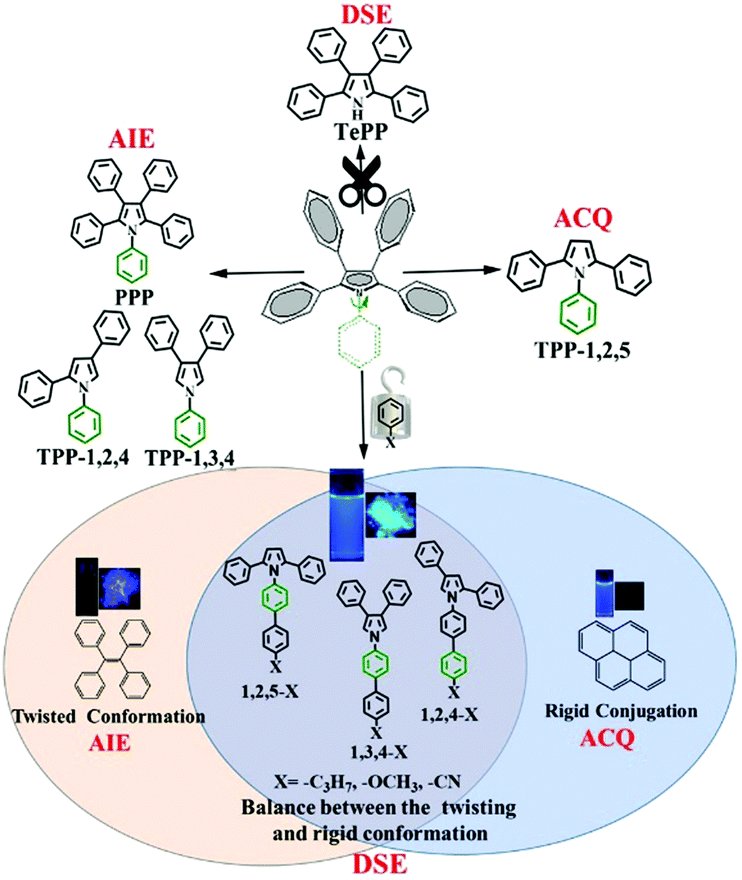
Organic conjugated fluorogens are widely applied in chem/biosensors, bioimaging, and optoelectronics because of their excellent performances, such as facile preparation, easy modification, high fluorescence brightness, and tunable spectral properties.1–6 Most organic fluorogens only exhibit strong luminescence in solution (aggregation-causing quenching, ACQ) or in the solid state form (aggregation-induced emission, AIE), which has prevented further realization of their potential applications.7–13 Recently, researchers developed fluorogens with strong fluorescence in both solid and liquid states, namely, dual-state emission (DSE) materials.14–18 Exploration of DSE materials can overcome the issues associated with ACQ and AIE materials and broaden overall applications of fluorogens in both solution and solid states.14,15 However, how to realize the DSE property and improve their luminescence efficiency is an intriguing question that has no straightforward answers. A rigid conjugation backbone can increase the intrinsic luminescence of organic conjugated molecules. However, this large conjugation resulting in a condensed molecular packing can quench the luminescence in the solid state.7,8 Combinatorial approaches to the discovery of DSE materials have emerged as exciting alternatives for the exploration and optimization of fluorogens. A delicate balance between twisting conformation and rigid conjugation endows organic conjugated molecules with a strong fluorescence intensity in both solution and the solid state.16–18 In addition, high temperature can weaken or even quench the fluorescence due to the increase of intramolecular motion and intermolecular collisions, which is another factor to lower the luminescence efficiency. Thus, it is of great practical significance to study fluorescent materials with positive responses to elevated temperatures.19–24 Several strategies were adopted to avoid the fluorescence quenching at high temperatures. Wang et al. reported a series of PtII-based frameworks with a positive response to temperature because of the intermolecular excimer formation.19 Thermally activated delayed fluorescent (TADF) compounds also showed a higher fluorescence intensity at high temperatures because excitons lose their spin-forbidden states at these temperatures, leading to intersystem crossing (ISC). However, a low ISC efficiency results in low quantum yields of TADF materials.22–24 Despite considerable efforts to develop novel organic fluorogens, DSE materials with a high luminescence efficiency are still underexplored due to their unclear luminescence mechanism. Therefore, in-depth theoretical research and guidance are still needed.
Recently, our group developed a series of polyarylpyrroles such as triphenylpyrrole isomers (TPP-1,2,4, TPP-1,2,5 and TPP-1,3,4), tetraphenylpyrrole (TePP) and pentaphenylpyrrol (PPP) (Scheme 1).25–27 Their AIE and ACQ as well as DSE properties were determined by the position of the corresponding phenyl substitution.27 A phenyl group attached to the 1-position of the pyrrole ring can act as a rotor that consumes the energy of the excited state. Thus, removing a phenyl group attached to the N atom would effectively enhance the molecular emission in solution allowing TePP to have DSE properties.27 On the other hand, as the volume of the group at the 1-position increases, the corresponding rotational barriers also increase, which will effectively restrict the intramolecular motion, allowing materials to have bright fluorescence in solution. Based on this strategy, we designed and synthesized nine compounds demonstrating 33–55% absolute quantum yields in both solution and solid states. The optical properties of these compounds are dependent on the ambient temperature. Fluorescence intensity is significantly enhanced as the temperature increases. In addition, three donor (D)–acceptor (A) compounds show strong phosphorescence in both solution and solid states at low temperatures with a lifetime of up to 10 s.
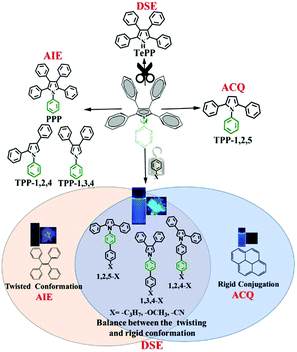 |
| | Scheme 1 The strategy for realizing dual state emission properties. | |
Results and discussion
Synthesis and characteristics of TPP-based compounds
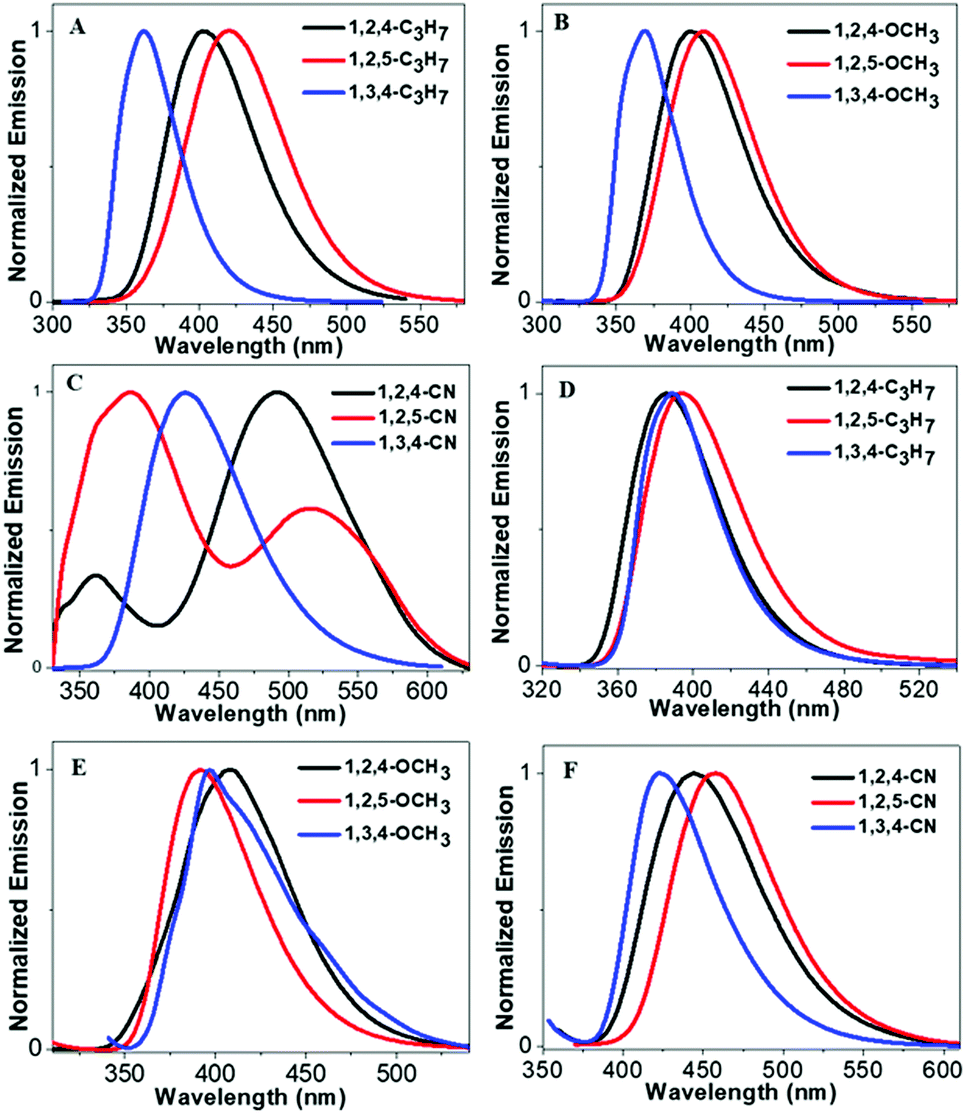
Scheme 2 illustrates molecular structures of the nine triphenylpyrrole (TPP) derivatives. All TPP-based molecules showed absorption bands in the 240–310 nm range and emission bands in the 365–515 nm range (Fig. 1 and Fig. S1, ESI†). The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are shown in Fig. S5 (ESI†) and their energy levels are summarized in Table 1. The HOMO and LUMO of three TPP-based derivatives with CN groups are spatially separated, indicating the existence of intermolecular charge transfer (ICT) (Fig. S2, ESI†).
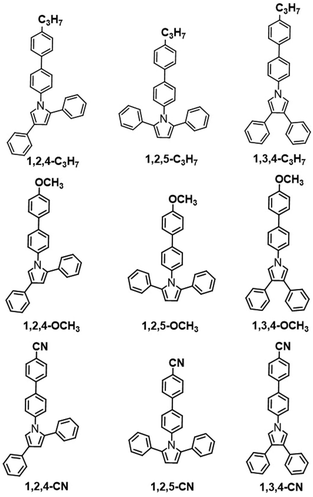 |
| | Scheme 2 Molecular structures of organic conjugated fluorogens. | |
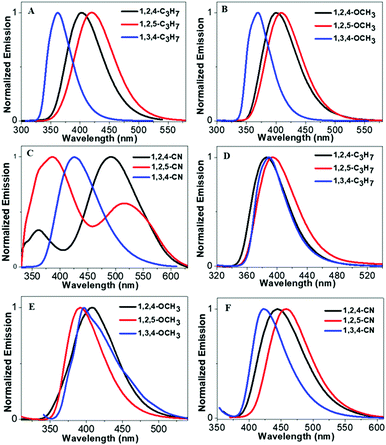 |
| | Fig. 1 (A–C) Emission spectra of TPP-based derivatives in THF solution (concentrations: 10−5 M), and (D–F) emission spectra of TPP-based derivatives in the solid state. | |
Table 1 Photophysical properties of the TPP-based molecules
| Samples |
λ
em (nm) (THF) |
λ
em (nm) (solid) |
Φ
F (%) (THF) |
Φ
F (%) (solid) |
E
g (eV) |
Dipole moment/DB |
τ (ns) (THF) |
τ (ns) (solid) |
k
r [10−8 s−1] (THF) |
k
nr [10−8 s−1] (THF) |
k
r [10−8 s−1] (solid) |
k
nr [10−8 s−1] (solid) |
| 1,2,4-C3H7 |
402 |
389 |
43.2 |
48.2 |
4.25 |
3.09 |
3.06 |
1.71 |
1.41 |
1.86 |
2.82 |
3.03 |
| 1,2,5-C3H7 |
418 |
393 |
32.0 |
42.1 |
4.11 |
2.74 |
3.36 |
2.57 |
0.95 |
2.02 |
1.64 |
2.25 |
| 1,3,4-C3H7 |
365 |
391 |
44.3 |
44.5 |
4.34 |
3.16 |
2.09 |
1.18 |
2.12 |
2.67 |
3.77 |
4.70 |
| 1,2,4-OCH3 |
404 |
399 |
47.1 |
45.7 |
4.21 |
3.48 |
2.32 |
1.17 |
2.03 |
2.28 |
3.91 |
4.64 |
| 1,2,5-OCH3 |
410 |
395 |
36.6 |
40.1 |
4.16 |
3.23 |
2.68 |
1.79 |
1.37 |
2.37 |
2.24 |
3.35 |
| 1,3,4-OCH3 |
367 |
393 |
43.6 |
47.1 |
4.36 |
3.72 |
1.81 |
0.96 |
2.41 |
3.12 |
4.91 |
5.51 |
| 1,2,4-CN |
360/492 |
444 |
47.6 |
53.5 |
3.03 |
3.83 |
5.04 |
6.87 |
0.94 |
1.04 |
0.78 |
0.68 |
| 1,2,5-CN |
386/515 |
458 |
32.8 |
49.6 |
2.95 |
3.62 |
6.72 |
7.23 |
0.49 |
1.00 |
0.69 |
0.70 |
| 1,3,4-CN |
425 |
423 |
55.6 |
44.3 |
3.26 |
3.90 |
4.32 |
5.04 |
1.29 |
1.03 |
0.88 |
1.11 |
DSE properties of TPP-based compounds
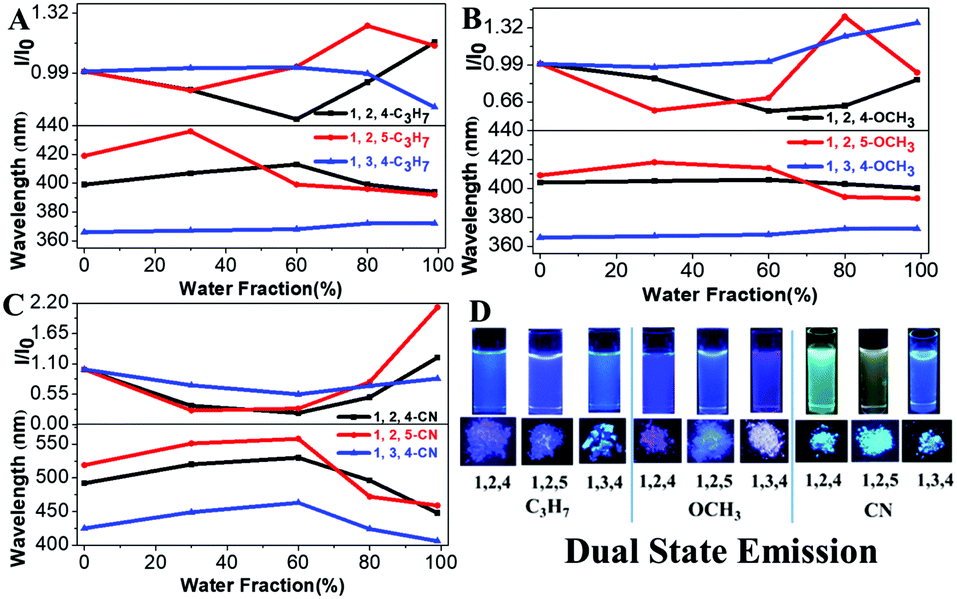
Our previous results demonstrated that the phenyl group at the 1-position of the pyrrole ring acts as a rotor, consumes the excited state energy and lowers the emission in solution.27 Therefore, the increase in the rotational barrier at the 1-position will effectively restrict the intramolecular motion allowing the materials to have bright fluorescence in solution (Scheme 1). This strategy can change the TPP-based molecules with both AIE and ACQ properties to molecules with DSE properties by simply extending the conjugation length at the 1-position of the pyrrole molecule. All molecules showed a high fluorescence QY (33–55%) both in liquid and solid states indicating their DSE properties (Table 1 and Fig. 2). To further study the luminescence properties of compounds in the aggregated state, emission behaviors of their THF/H2O mixtures were investigated.
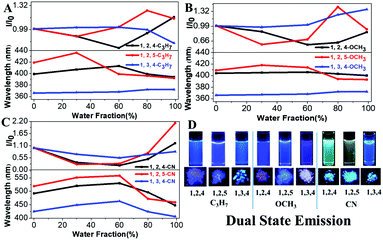 |
| | Fig. 2 (A–C) Fluorescence intensity of TPP derivatives in THF/H2O mixtures with different water fractions. (D) Visible fluorescence of compounds under 302 nm UV light (concentrations: 10−5 M). | |
Fluorescence intensities of the aggregates of all TPP derivatives (measured in THF/99% H2O) were almost identical to those in THF solution, indicating their DSE properties (Fig. S3–S5, ESI†). The maximum emission wavelengths of all molecules redshift as there is an increase in the water fraction, which is likely due to the high polarity of water. However, the maximum emission wavelengths of all molecules except 1,3,4-C3H7 and 1,3,4-OCH3 were then blue-shifted, when the aggregates were formed in higher water fractions. In addition, all compounds showed shorter emission wavelengths in solid states than those in solution except 1,3,4-C3H7 and 1,3,4-OCH3. This could be attributed to the through-space conjugation effect. This effect decreases energy reorganization and suppresses non-radiative decay processes in aggregated states, thereby causing strong and blue-shifted aggregated emissions.28–30 The phenyl rings at the 2,5-positions in 1,2,5-TPP based molecules overcome steric hindrance to form a favorable face-to-face geometry, leading to the longest blue-shifts in the solid state. The blue-shifts of three 1,2,4-TPP based compounds in the solid state are smaller than those of 1,2,5-TPP based molecules because only one face-to-face geometry was formed. 1,3,4-C3H7 and 1,3,4-OCH3 showed red-shifts in the solid state due to the lack of effective through-space conjugation and intramolecular D–A interactions (Fig. S6, ESI†).
Single crystal structures
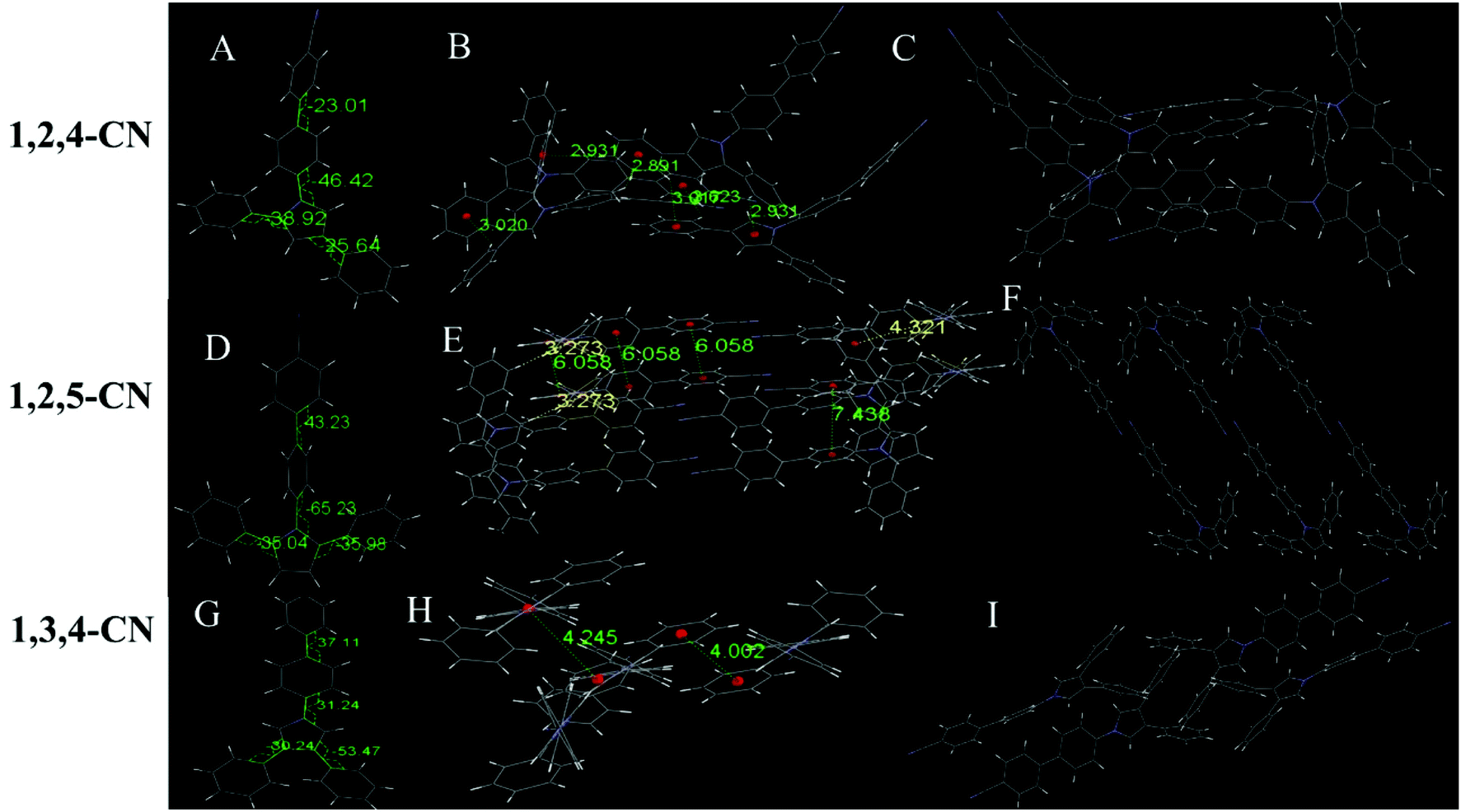
Six of the nine compounds can be crystallized (Tables S3–S8, ESI†), while 1,2,4-C3H7, 1,2,5-C3H7 and 1,2,5-OCH3 did not yield crystals that could be analyzed by XRD. The 1,2,4-OCH3 molecule has a relatively distorted spatial configuration with poor planarity. Crystals of 1,2,4-OCH3 adopted a head-to-head cross-parallel packing pattern along the b-axis. The closest interaction distance of π–π was 5.901 Å and weak C–H⋯π interactions with a 2.977 Å distance were observed (Fig. 3A–C). Crystals of 1,3,4-OCH3 also adopted a head-to-head cross-parallel packing pattern along the b-axis. The closest π–π and C–H⋯π interaction distances were 6.148 and 2.895 Å in the solid state, respectively (Fig. 3E). Compound 1,3,4-C3H7 showed configuration and intermolecular arrangements similar to those for 1,3,4-OCH3 (Fig. 3G–I), suggesting that the two compounds exhibit similar emission wavelengths and fluorescence intensities in the solid state (Table 1). TPP compounds with the −CN group showed different molecular configuration and intermolecular arrangements. The 1,2,4-CN molecule showed a distorted spatial configuration, and its crystal adopted a head-to-tail cross-vertical arrangement pattern along the b-axis. An obvious C–H⋯π interaction was observed with a distance of 2.891 Å and 2.931 Å (Fig. 4A–C). 1,2,5-CN adopted a head-to-head arrangement pattern along the b-axis. The closest interaction distance of the π–π plane was 6.058 Å, and a very weak C–H⋯π interaction in the crystal was observed with a distance of 3.273 Å (Fig. 4D–F). A single crystal structure of 1,3,4-CN adopted a tail-to-tail cross-vertical arrangement pattern along the b-axis. The large twist angles of the 3,4-positioned benzene rings and the pyrrole core make the overall volume of the molecule relatively large. The distance between the closest face-to-face benzene rings reaches 4.002 Å (Fig. 4G–I). Thus, all molecules show no close π–π interactions, resulting in high luminescence efficiency of all the compounds in the solid state. In addition, twisted excited state conformations prevent the corresponding molecules from close packing which results in detrimental formations (such as excimers) and are helpful for aggregate emission. Theoretical calculations showed that the twist angles between the TPP moiety and the biphenyl plane are close to 90° in excited states (Fig. S7, ESI†). Perpendicular conformation of the entire molecule can effectively help to avoid the formation of the excitatory associations, allowing compounds to have strong fluorescence in the aggregated state.31,32
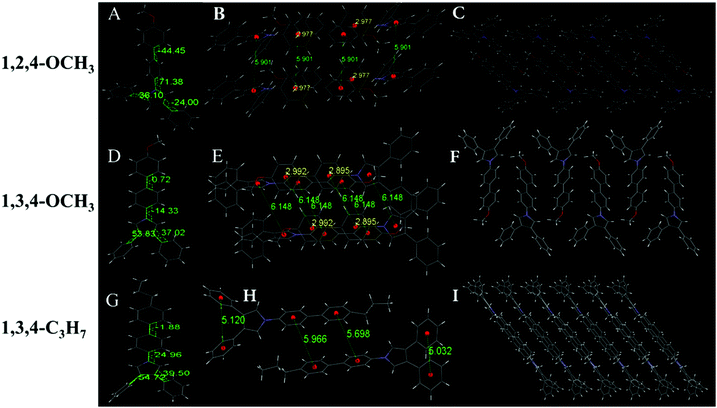 |
| | Fig. 3 (A) Molecule structure, (B) interaction distance of the π⋯π and C–H⋯π interactions and (C) stacking image along the b-axis of 1,2,4-OCH3. (D) Molecular structure, (E) interaction distance of the π⋯π and C–H⋯π interactions and (F) stacking image along the b-axis of 1,3,4-OCH3. (G) Molecular structure, (H) interaction distance of the π⋯π and C–H⋯π interactions and (I) stacking image along the b-axis of 1,3,4-C3H7. Gray color corresponds to carbon, white to hydrogen, red to oxygen and blue to nitrogen. | |
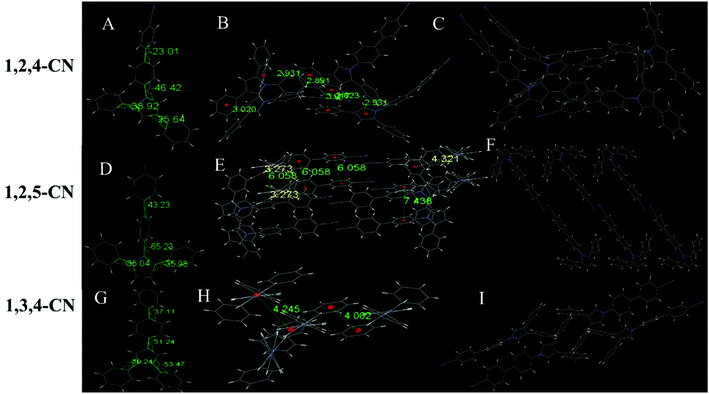 |
| | Fig. 4 (A) Molecular structure, (B) interaction distances of C–H⋯π interactions and (C) stacking image of 1,2,4-CN. (D) Molecular structure, (E) interaction distances of C–H⋯π interactions and (F) stacking image of 1,2,5-CN. (G) Molecular structure, (H) interaction distances of face-to-face phenyl rings and (I) stacking image of 1,3,4-CN. Gray color corresponds to carbon, white to hydrogen and blue to nitrogen. | |
Positive response to temperature
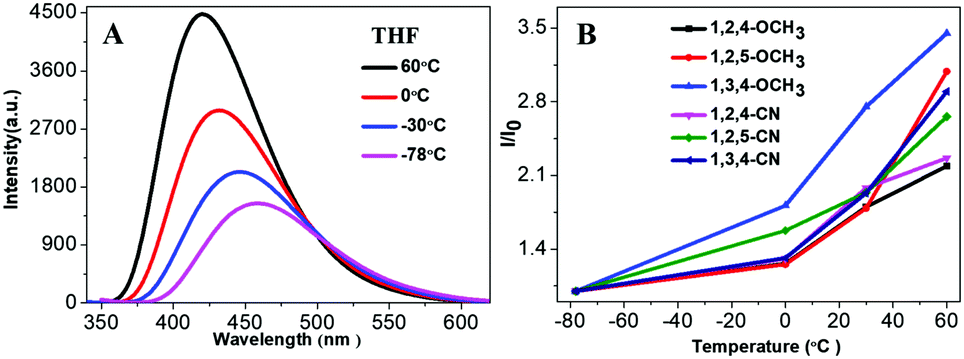
Fluorescence of organic fluorogens is typically quenched at high temperature due to the increase of intramolecular motion and intermolecular collisions. Yet, our TPP-based molecules showed a positive response as temperature increased. Fig. 5A shows the fluorescence intensity of 1,3,4-CN at different temperatures (10−5 M in THF). Fluorescence intensity increased as the temperature increased and decreased when the ambient temperature was lowered (Fig. S8, ESI†). Similar phenomena were also observed for 1,3,4-CN in toluene or DMF solution (Fig. S9, ESI†). However, the fluorescence intensity variations of 1,3,4-CN in toluene or DMF solution were smaller than those in THF solution, indicating that the fluorescence enhancement was influenced by the solvent. All other compounds also demonstrated similar responses to the ambient temperature (Fig. 5B and Fig. S10, ESI†). However, TPP-based derivatives with C3H7/OCH3 groups only showed a variation in fluorescence intensity due to the lack of intramolecular D–A interactions. Emission wavelengths of TPP-based derivatives with CN groups blue-shifted as the temperature increased because polarity of the solvent decreased as temperature increased (Fig. S11, ESI†).33 Since the more polar excited state resulting from a charge transfer is more stable than the less polar ground state in a polar solvent, emission spectra shifted to shorter wavelengths as the temperature increased.33–38
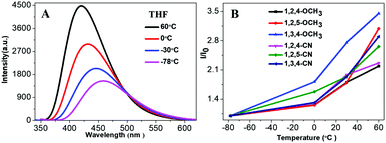 |
| | Fig. 5 (A) PL spectra of 1,3,4-CN solution and (B) relationship between the PL intensities of fluorogens (10−5 M in THF) at different temperatures. | |
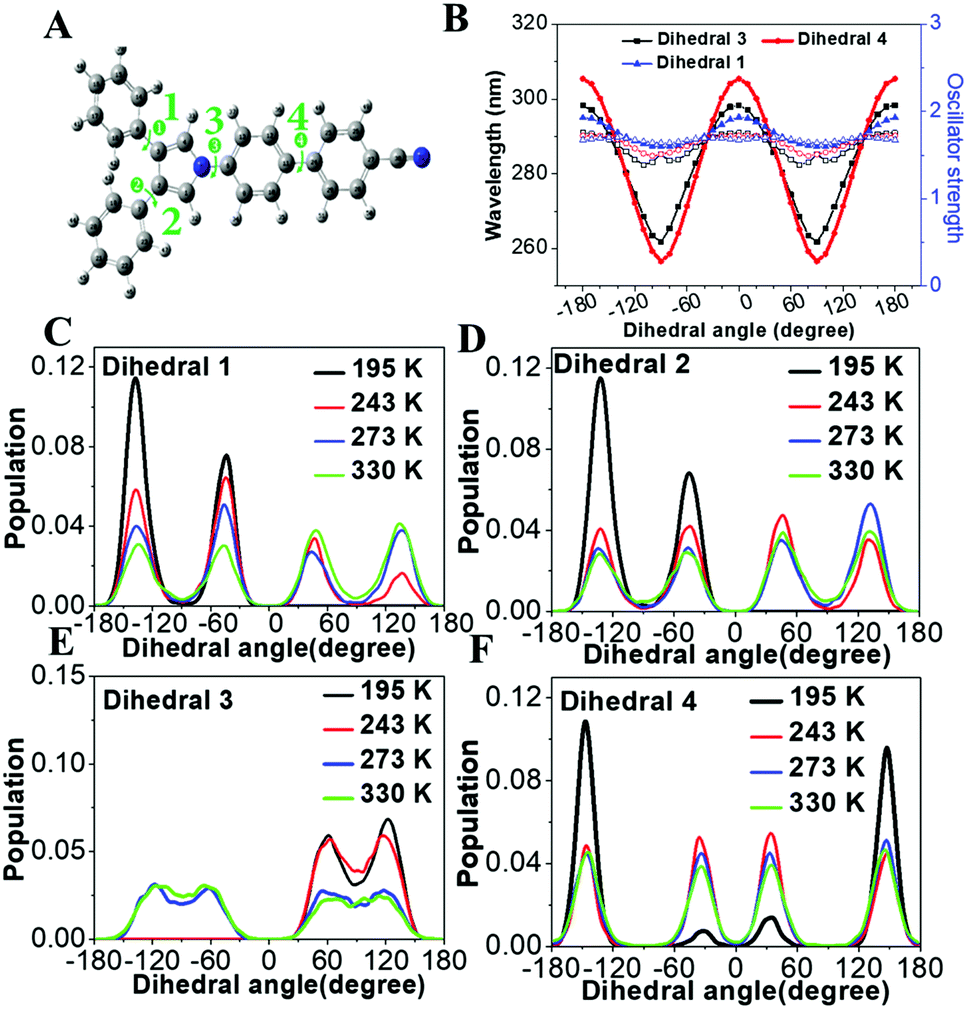
To demonstrate the structure–absorption spectrum relationship for the 1,3,4-CN system, we scanned the profiles of wavelength and oscillator strength as a function of selected key dihedral angles (Fig. 6A and B). All scanned profiles were prepared based on the optimized geometry of 1,3,4-CN in the ground state at the CAM-B3LYP/6-31G(d) level. Except for the selected dihedral angles, all other degrees of freedom were frozen during scanning. Because of the symmetric property of dihedral angles 1 and 2, we only calculated profiles for dihedral angles 1, 3 and 4 (Fig. 6B). We found that: (1) the wavelength profiles of both dihedral angles 3 and 4 fluctuate in much larger amplitude than that of dihedral angle 1, indicating that dihedral angles 3 and 4 have a major impact on the absorption wavelength of 1,3,4-CN. This is also supported by the negligible overlap between the HOMO and LUMO of 1,3,4-CN on the TPP moiety (Fig. S3, ESI†). (2) The oscillator strength, which reflects intensity of the absorption spectrum, fluctuates in a small range between 1.40 and 1.75. (3) For all dihedral angles, the better conjugation rotors show a longer absorption wavelength and the perpendicular rotors give the shortest wavelengths. For example, the wavelengths of dihedral angle 4 were 305 and 256 nm when it equals 0° and 90°, respectively. Therefore, the absorption spectrum of 1,3,4-CN should be determined by the statistical possibility of different conformations.
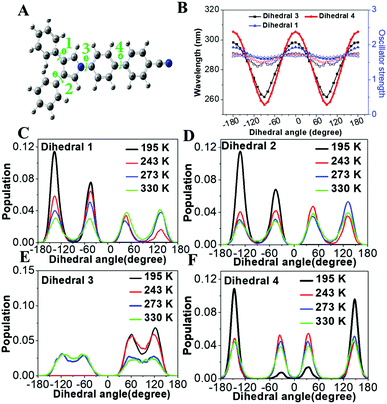 |
| | Fig. 6 (A) Optimized geometry of 1,3,4-CN in the ground state (S0, mim). (B) Wavelength and oscillator strength profiles of 1,3,4-CN as functions of the selected dihedral angles. (C–F) Distributions of selected dihedral angles in the 1,3,4-CN system in THF solution at 195, 243, 263 and 330 K. | |
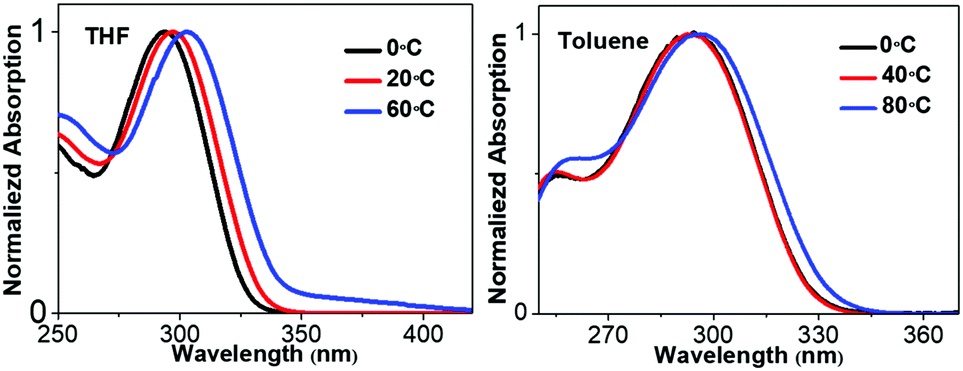
The molecular conformations change in different environments. To get more insights into the relationship between the conformations and absorption spectra of 1,3,4-CN, we performed molecular dynamics (MD) simulations to obtain molecular conformations of 1,3,4-CN at four different temperatures and under two solvent conditions. A single 1,3,4-CN molecule was placed in the center of a cubic box with pre-equilibrated THF or toluene molecules, respectively.39 For each solvent, we setup MD simulations at temperatures: 195, 243, 273 and 330 K, respectively to analyze the temperature effect for the 1,3,4-CN molecule. For each of the eight systems, we first performed energy minimization followed by 500 ps MD simulations under the NVT ensemble with position restraints on 1,3,4-CN. Then we performed two independent 50 ns MD simulations under the NPT ensemble at P = 1 bar and at the target temperature for each system. The temperature and pressure were controlled by the velocity rescaling thermostat and the Parrinello–Rahman barostat. The electrostatic interactions were evaluated using the particle mesh Ewald method. The cutoff distances for both direct space summation and the van de Waals interactions were 1.2 nm. All bond lengths were constrained by the LINCS algorithm.40 Periodic boundary conditions were applied in all three dimensions to minimize the edge effects in a finite system. The time step was 2 fs. The conformations were stored at 40 ps time intervals and in total 2500 MD conformations were collected for each system to get dihedral angle populations. Obtained dihedral angle distributions of 1,3,4-CN in THF and toluene solution are plotted in Fig. 6C–F and Fig. S13 (ESI†), respectively. From Fig. 6C–F, we found that: (1) the dihedral angle distribution is localized in a narrow range at lower temperature (∼195 K). At the same time, at 330 K, dihedral angles are distributed in the whole range: from −180° to 180°. Dihedral angles 1 and 2 play a minor role in the change of absorption spectra (Fig. 6B) and similar distributions at different temperatures are observed. (2) As temperature increased from 195 to 330 K, distributions of dihedral angle 4 between −30° and 30° increased significantly. This is the key factor leading to the obvious red-shift of the absorption spectrum in THF solution (Fig. 7). (3) Distributions of dihedral angles 1, 2 and 3 of 1,3,4-CN in toluene solution were still local in a narrow range even at 343 K reflecting its less sensitive conformational change in toluene than that in THF. Therefore, the absorption spectrum of 1,3,4-CN in toluene was less red-shifted than that in THF solution (Fig. 7). Thus, as temperature increases, the molecular conformation tends to planarize resulting in enhanced fluorescence of the compounds. The twisted conformation of 1,3,4-CN at low temperature will decrease the bandgap and the energy gap between the lowest singlet and the triplet states (ΔEST) and increase the phosphorescence, as discussed below.
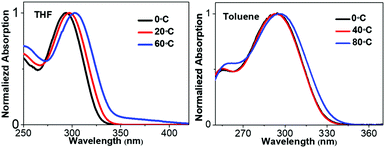 |
| | Fig. 7 Absorption spectra of 1,3,4-CN in the THF and toluene solution at different temperatures (10−5 M). | |
Long lifetime phosphorescence property
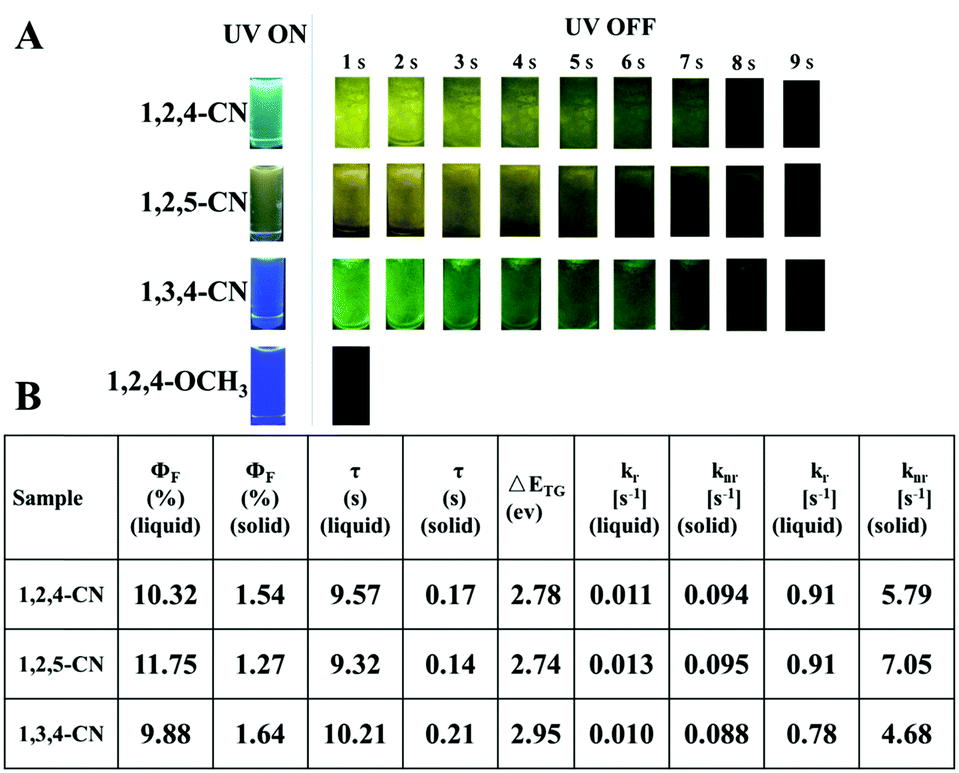
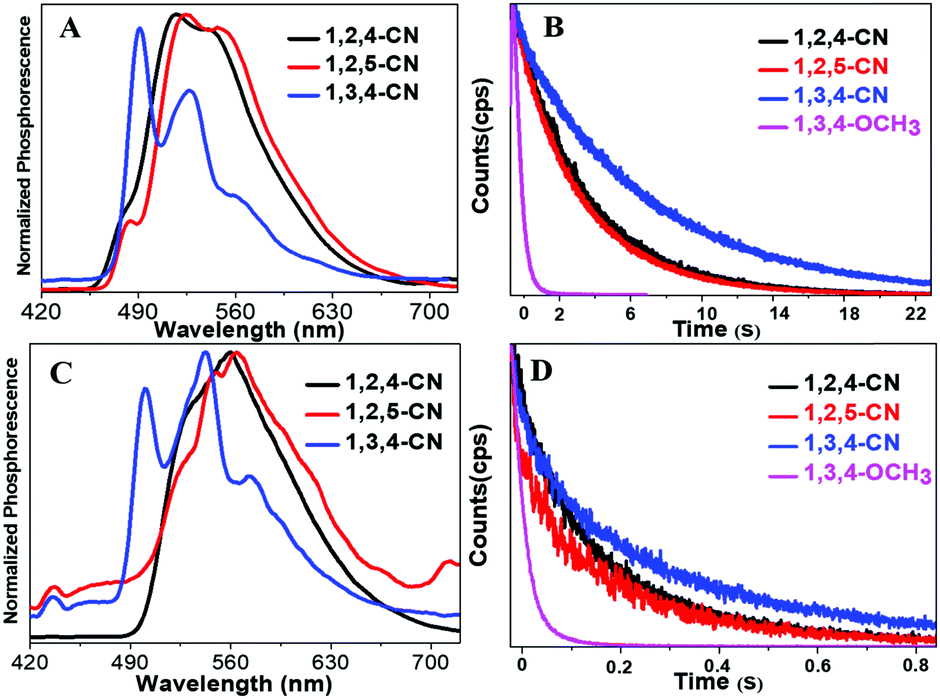
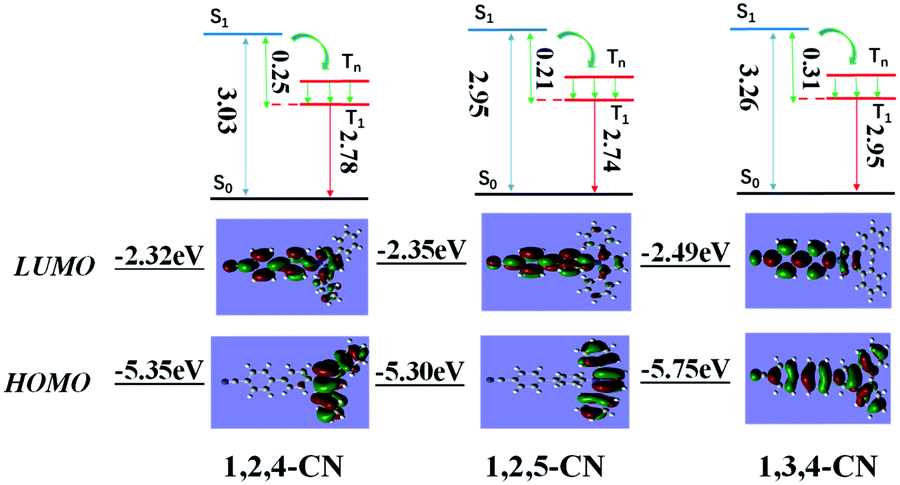
Three TPP derivatives with CN groups showed a more intense fluorescence emission and a shorter emission wavelength at high temperatures. However, they exhibited a pronounced phosphorescence emission at low temperatures both in solution and the solid state (Fig. 8 and 9). The phosphorescence wavelength of 1,2,5-CN in 2-methyl tetrahydrofuran solution was slightly longer than that of 1,2,4-CN. The phosphorescence wavelength for 1,3,4-CN was the shortest (Fig. 9). Three samples in 2-methyl tetrahydrofuran solution showed a phosphorescence lifetime as long as 10 s as well as high excitation a phosphorescence efficiency up to 11.75% (Fig. 8B) because of the small energy gap (ΔEST: the energy gap between the lowest singlet and triplet states). Typically, small values of ΔEST are beneficial for the improvement of the intersystem energy crossing.41–43 Spatial separation of the HOMO and LUMO can lead to small ΔEST in twisted D–A systems.44–46 Therefore, three D–A molecules with ICT effects have excellent low-temperature phosphorescence properties. Phosphorescence wavelengths of three compounds are consistent with their energy gaps between the lowest triplet and the ground states (ΔETG) (Fig. 8B and 10). The other six molecules without ICT effects showed higher ΔEST values (Table S2, ESI†), indicating that it is difficult for them to achieve intersystem crossing, thus showing no phosphorescence emission. In addition, three samples also showed phosphorescence in the solid state at T = 77 K with a short life time and a low QY, probably due to the intermolecular interactions of the compounds in the solid state. This causes the energy of the triplet state to return to the ground state in a non-radiative manner.45–49
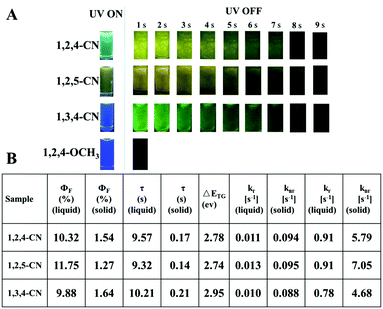 |
| | Fig. 8 (A) Luminescence photos of 1,2,4/1,2,5/1,3,4-CN and 1,3,4-OCH3 in 2-methyltetrahydrofuran solutions (10−5 M) at 77 K before and after UV irradiation at 365 nm. (B) Optical properties of the TPP-based molecules. | |
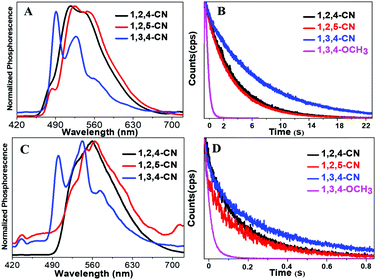 |
| | Fig. 9 Normalized phosphorescence spectra of 1,2,4/1,2,5/1,3,4-CN compounds in solution (A) and in the solid state (C). Time-resolved phosphorescence decay curves of 1,2,4/1,2,5/1,3,4-CN and 1,3,4-OCH3 in 2-methyltetrahydrofuran solutions (10−5 M) at 77 K (B) in the solid state (D). | |
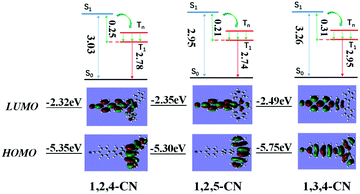 |
| | Fig. 10 Calculated energy diagrams and HOMO/LUMO orbital distributions of TPP based molecules with the CN group. | |
Conclusions
In this work, we synthesized a series of TPP-based molecules, all of which showed DSE properties with quantum yields over 30%. As the temperature increased, the molecules became flatter, which increased their fluorescence intensity. MD simulations showed that the fluorescence increase was caused by the variation of molecular conformation, indicating rigid molecular conformations of DSE molecules. Three TPP molecules with CN groups showed a low-temperature phosphorescence up to 10 s demonstrating a great opportunity for the development of long life-time materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Scientific Foundation of China (Grant No. 51803009, 51673024, 51328302 and 21404010), the Graduate Technological Innovation Project of Beijing Institute of Technology (2018CX10005), the Beijing Institute of Technology Research Fund Program for Young Scholars and the Graduate Technological Innovation Project of Beijing Institute of Technology.
Notes and references
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79–93 RSC.
- Z. X. Cai, N. Zhang, M. A. Awais, A. S. Filatov and L. P. Yu, Angew. Chem., Int. Ed., 2018, 57, 6442–6448 CrossRef CAS PubMed.
- H. Nie, K. Hu, Y. Cai, Q. Peng, Z. Zhao, R. Hu, J. Chen, S. J. Su, A. Qin and B. Z. Tang, Mater. Chem. Front., 2017, 1, 1125–1129 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- Z. X. Cai, W. Y. Lo, T. Y. Zheng, L. W. Li, N. Zhang, Y. B. Hu and L. P. Yu, J. Am. Chem. Soc., 2016, 138, 10630–10635 CrossRef CAS PubMed.
- Y. X. Lei, H. Li, M. C. Liu, J. X. Chen, X. B. Huang and H. Y. Wu, J. Mater. Chem. C, 2014, 2, 7402–7410 RSC.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CrossRef CAS PubMed.
- H. Shi, H. Sun, H. Yang, S. Liu, G. Jenkins, W. Feng, F. Li, Q. Zhao, B. Liu and W. Huang, Adv. Funct. Mater., 2013, 23, 3268–3276 CrossRef CAS.
- Y. X. Lei, Y. Y. Lai, L. C. Dong, J. B. Shi, J. G. Zhi, X. B. Huang and Y. P. Dong, Chem. – Eur. J., 2018, 24, 434–442 CrossRef CAS PubMed.
- G. Chen, W. B. Li, T. R. Zhou, Q. Peng, D. Zhai, W. Z. Yuan, Y. M. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 4496–4501 CrossRef CAS PubMed.
- F. D. Nisi, R. Francischello, A. Battisti, A. Panniello, E. Fanizza, B. Tang and A. Pucci, Mater. Chem. Front., 2017, 1, 1406–1412 RSC.
- B. Chen, Z. J. Zhao and B. Z. Tang, Chin. Sci. Bull., 2016, 61, 3435–3447 Search PubMed.
- Z. Peng, Z. Wang, B. J. Shi, B. Tong and Y. P. Dong, Chin. J. Chem., 2016, 34, 1071–1075 CrossRef CAS.
- T. Beppu, K. Tomiguchi, A. Masuhara, Y. J. Pu and H. Katagiri, Angew. Chem., Int. Ed., 2015, 54, 7332–7335 CrossRef CAS PubMed.
- M. N. Huang, R. N. Yu, S. X. Ye, S. Kang, X. H. Zhu and Y. Q. Wan, Chem. Sci., 2016, 7, 4485–4491 RSC.
- K. Suenaga, K. Tanaka and Y. Chujo, Eur. J. Org. Chem., 2017, 5191–5196 CrossRef CAS.
- M. Li, Y. L. Niu, X. Z. Zhu, Q. Peng, H. Y. Lu, A. D. Xia and C. F. Chen, Chem. Commun., 2014, 50, 2993–2995 RSC.
- Y. Y. Li, Y. X. Lei, L. C. Dong, J. G. Zhi, J. B. Shi, B. Tong and Y. P. Dong, Chem. – Eur. J. DOI:10.1002/chem.201804074.
- L. J. Liu, X. Wang, N. Wang, T. Peng and S. Wang, Angew. Chem., Int. Ed., 2017, 59, 9160–9164 CrossRef PubMed.
- P. O. Sersson, T. Sergeim, R. L. Chen and T. Gillbro, J. Phys. Chem., 1995, 99, 16199–16209 CrossRef.
- Q. Y. Fang, J. W. Li, S. Y. Li, S. Q. Wang, Y. P. Yi, Y. Qian and G. Q. Yang, Chem. Commun., 2017, 53, 5702–5705 RSC.
- Z. Y. Yang, Z. Mao, Z. L. Xie, Y. Zhang, J. Zhao, Z. G. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- T. Hatakeyama, K. Shiren, S. Nomura, S. Nakatsuka, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- A. J. Qin, Y. Zhang, N. Han, J. Mei, S. J. Zhi and B. Z. Tang, Sci. China: Chem., 2012, 55, 772–778 CrossRef CAS.
- L. C. Dong, G. J. Shang, J. B. Shi, J. G. Zhi, B. Tong and Y. P. Dong, J. Phys. Chem. C, 2017, 121, 11658–11664 CrossRef CAS.
- X. Feng, B. Tong, J. B. Shi, L. Chen, J. G. Zhi, P. Lu, Y. G. Ma and Y. P. Dong, J. Phys. Chem. B, 2010, 114, 16731–16736 CrossRef CAS PubMed.
- Y. X. Lei, Q. W. Liu, L. C. Dong, J. B. Shi, J. G. Zhi, Z. X. Cai and Y. P. Dong, Chem. – Eur. J., 2018, 24, 14269–14274 CrossRef CAS PubMed.
- H. Zhang, X. Y. Zheng, N. Xie, Z. K. He, Y. K. Sing, T. K. Kwok, H. H. Y. Sung, I. D. Williams, A. J. Qin and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16264–16272 CrossRef CAS PubMed.
- S. J. Zhen, J. C. Mao, L. Chen, S. Y. Ding, X. S. Zhou, A. J. Qin, Z. J. Zhao and B. Z. Tang, Nano Lett., 2018, 18, 4200–4205 CrossRef CAS PubMed.
- W. W. Luo, H. Nie, B. R. He, Z. J. Zhao, Q. Peng and B. Z. Tang, Chem. – Eur. J., 2017, 23, 18041–18048 CrossRef CAS PubMed.
- Z. Q. Xie, B. Yang, F. Li, G. Cheng, H. Xu, M. Hanif, S. Y. Liu, D. G. Ma and Y. G. Ma, J. Am. Chem. Soc., 2005, 127, 14152–14153 CrossRef CAS PubMed.
- M. Gon, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2018, 57, 6546–6551 CrossRef CAS PubMed.
- K. Li, Y. Y. Liu, Y. Y. Li, Q. Feng, H. W. Hou and B. Z. Tang, Chem. Sci., 2017, 8, 7258–7267 RSC.
- H. Li, Y. Guo, Y. X. Lei, W. X. Gao, M. C. Liu, J. X. Chen, Y. F. Hu, X. B. Huang and H. Y. Wu, Dyes Pigm., 2015, 112, 105–115 CrossRef CAS.
- Y. H. Niu, Q. Wang, H. R. Wu, Y. X. Wang and Y. R. Zhang, New J. Chem., 2017, 41, 9796–9805 RSC.
- X. D. Jiang, X. Liu, T. Fang and C. L. Sun, Dyes Pigm., 2017, 146, 438–444 CrossRef CAS.
- J. Do, J. Huh and E. Kim, Langmuir, 2009, 25, 9405–9412 CrossRef CAS PubMed.
- H. J. Liu, X. J. Xu, Z. J. Shi, K. Q. Liu and Y. Fang, Anal. Chem., 2016, 88, 10167–10175 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- Y. Xiong, Z. Zhao, W. J. Zhao, H. L. Ma, Q. Peng, Z. K. He, X. P. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 7997–8001 CrossRef CAS PubMed.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- Z. F. An, C. Zheng, Y. Tao, R. F. Chen, H. F. Shi, T. Chen, Z. X. Wang, H. H. Li, R. R. Deng and W. Huang, Nat. Mater., 2015, 14, 685–689 CrossRef CAS PubMed.
- L. Gu, H. F. Shi, M. G. Gu, K. Ling, H. L. Ma, S. Z. Cai, L. L. Song, Z. G. Shuai and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 8425–8431 CrossRef CAS PubMed.
- Y. Y. Hu, Z. F. Wang, X. F. Jiang, X. Y. Cai, S. J. Su, F. Huang and Y. Cao, Chem. Commun., 2018, 54, 7850–7853 RSC.
- Y. Gong, L. Zhao, Q. Peng, D. Fan, W. Z. Yuan, Y. Zhang and B. Z. Tang, Chem. Sci., 2015, 6, 4438–4444 RSC.
- Z. Z. Zhang, L. L. Tang, X. J. Fan, Y. H. Wang, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2018, 6, 8984–8989 RSC.
- Y. Takeda, T. Kaihara, M. Okazaki, H. Higginbotham, P. Data, N. Tohnai and S. Minakata, Chem. Commun., 2018, 54, 6847–6850 RSC.
- S. F. Pan, Z. T. Chen, X. L. Zheng, D. H. Wu, H. Feng and Z. S. Qian, J. Phys. Chem. Lett., 2018, 9, 3939–3945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1853745–1853750. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00613j |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Jianbing
Shi
*a,
Jianbing
Shi