
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diverse metastable diarylacetonitrile radicals generated by polymer mechanochemistry†
Takumi
Yamamoto
 a,
Daisuke
Aoki
a,
Koichiro
Mikami
b and
Hideyuki
Otsuka
*ac
a,
Daisuke
Aoki
a,
Koichiro
Mikami
b and
Hideyuki
Otsuka
*ac
aDepartment of Chemical Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayma, Meguro-ku, Tokyo 152-8550, Japan. E-mail: otsuka@mac.titech.ac.jp
bEngineering Division, Process Device Innovation Center, Panasonic Industry Co., Ltd., 1006 Kadoma, Kadoma City, Osaka 571-8506, Japan
cLiving Systems Materialogy (LiSM) Research Group, International Research Frontiers Initiative (IRFI), Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan
First published on 14th February 2024
Abstract
Fluorescent radicals have attracted great attention as luminescent materials, mostly on account of their potential to achieve higher luminescence efficiency than closed-shell molecules. However, analyzing fluorescent radicals at ambient conditions remains a challenging task, because radicals are usually unstable in air. In addition, to the best of our knowledge, research aimed at controlling fluorescence wavelengths through substituent changes has not yet been accomplished. Here, we report diverse metastable diarylacetonitrile (DAAN) radicals, which contain different substituents, generated by polymeric mechano-chemical reactions. The DAAN radicals, generated by ball-milling powdered polystyrene together with DAAN derivatives, were dispersed within the polystyrene matrix, where they retained their radical state, which allowed measuring solid-state fluorescence spectra. These measurements revealed that a wide range of fluorescence wavelengths from green to red (λem,max = 517–635 nm) can be achieved only by changing the substituents on the aromatic rings in these DAAN radicals. This phenomenon has not been observed for the well-studied triarylmethyl radicals. The fluorescence wavelength of these DAAN radicals can be precisely estimated by time-dependent density-functional theory (TD-DFT) calculations. The amount of DAAN radicals generated upon ball-milling is discussed in conjunction with DFT calculations and experimental results. Our results suggest that the orbital interactions with polymeric mechanoradicals, the bond-dissociation enthalpy, and the steric protection of the radical center are of paramount importance for the generation of DAAN radicals. The results of this study can be expected to provide useful guidelines for the development of advanced fluorescent radicals.
Introduction
Recently, studies on the luminescence properties of stable carbon-centred radicals have attracted great attention.1,2 Generally, they exhibit unusual photophysical properties such as long-wavelength emission,3–7 and high electro-luminescence quantum efficiency.8–11 However, most free radicals are short-lived because they are incredibly reactive due to the presence of unpaired electrons.12,13 In addition, most free radicals usually exhibit weak or no luminescence under ambient conditions and have long been used as quenchers of luminophores.14 Thus, the number of organic luminescent radical species that persist under ambient conditions remains limited. Only a few free radicals that can stably luminesce at room temperature, e.g., triarylmethyl radical derivatives15–19 and N-heterocyclic carbene-based radicals have been reported so far.20 However, the design, synthesis, and purification of these radicals is relatively complicated given their high reactivity.21 This complexity has so far hampered progress in the design and development of fluorescent radicals. Hence, developing new strategies to generate and analyse fluorescent radicals remains highly desirable.In this context, we focused on the mechanochemical generation of diarylacetonitrile (DAAN) radicals in a polymer matrix. Although DAAN radicals are known to be fluorescent,22–24 their luminescence properties have not yet been adequately studied. DAAN derivatives with a variety of substituents can be synthesized in one or two steps from commercially available precursors, which may allow the systematic evaluation of the effects of substituents, a task that has proven difficult for conventional fluorescent radicals. In the present study, we conducted a comprehensive study on DAAN radicals generated by polymeric mechanochemical reactions (Fig. 1). In general, it is easier, both experimentally and computationally, to evaluate the fluorescence properties of radicals in the solid state than in solution because the effect of solvent does not have to be considered. Moreover, the effect of coupling reactions between the generated radicals, which cannot be neglected in solution, can be suppressed by the rigid polymer matrix, allowing unexplored fluorescent radicals to be discovered and investigated systematically. The purpose of this study is to (1) establish the concept of using mechanochemical reactions of polymer chains as a useful tool to generate and characterize metastable fluorescent radicals in the bulk, and to (2) clarify the luminescence properties of DAAN radicals by combining experimental and computational chemistry. For this purpose, twenty different DAAN derivatives were prepared as fluorescent radical precursors, and the radical species generated in the polystyrene matrix by ball-milling tests were systematically evaluated using solid-state fluorescence spectroscopy, electron-paramagnetic-resonance (EPR) measurements, and density-functional-theory (DFT) calculations. The present study can be expected to inspire the development of a design strategy for fluorescent radicals as well as an approach for the visual and quantitative evaluation of short-lived radicals, which would be highly desirable in order to improve our understanding of fluorescent radical species.
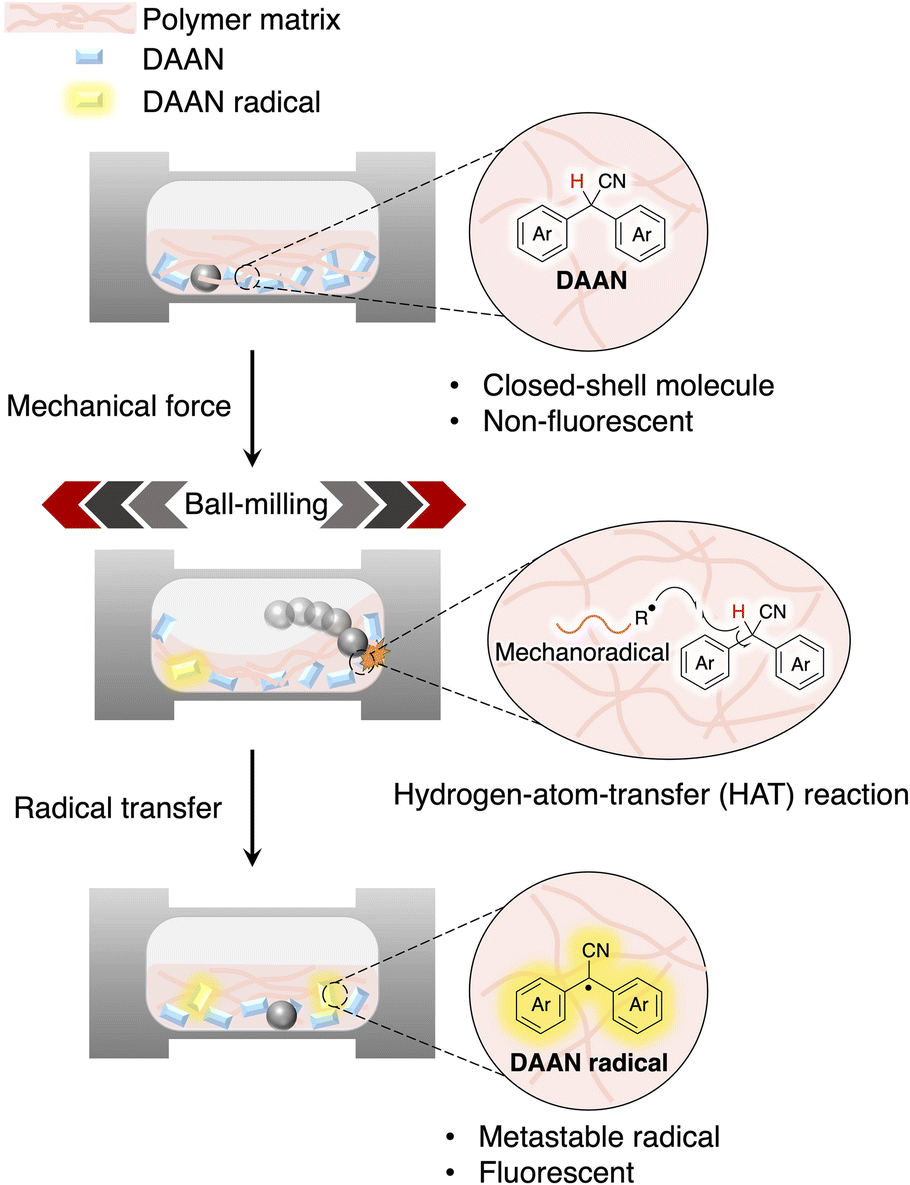 | ||
| Fig. 1 Schematic illustration of the analytical methods used for the characterization of DAAN radicals within this study. | ||
Results and discussion
Synthesis of the DAAN derivatives
To evaluate the substituent effect in DAAN radicals, a series of nineteen DAAN derivatives with different substituents, which are precursors for fluorescent radicals via polymer-matrix-assisted radicalization, was synthesized.25–27 Each compound was analysed using 1H NMR, 13C NMR, and FT-IR spectroscopy as well as mass spectrometry (MS). The detailed experimental procedures for the synthesis of the compounds and their spectral data are described in the ESI† (Fig. S2–S39†). The DAAN derivatives are denoted as R1/R2, where R1 and R2 refer to the substituents. In addition to the nineteen synthesized DAAN compounds, a commercially available DAAN derivative without any substituents on the aromatic ring (H/H) was used for evaluating the fluorescence properties. These DAAN derivatives were color-coded into four groups according to the type of substituent: derivatives for which a sufficient captodative effect, which refers to the stabilization of radicals by a synergistic effect of an electron-withdrawing substituent and an electron-donating substituent,28,29 cannot be expected (grey), derivatives with one or two electron-donating groups on the aromatic ring (red), derivatives with one electron-donating group and one electron-withdrawing group on the aromatic ring (blue), and derivatives with substituents at positions other than the para-position (green) (Fig. 2). | ||
| Fig. 2 Chemical structures of the DAAN derivatives used in this study. | ||
Fluorescence properties of the DAAN radicals
To investigate the fluorescence properties of the DAAN radicals, ball-milling experiments were carried out using a Retch MM400 mixer mill (10 mL stainless-steel milling jar using one stainless-steel ball with a diameter of 5 mm). Polystyrene (Mn = 66.0 kg mol−1; Mw/Mn = 1.46; 100 mg) was ground in the presence of each DAAN derivative (50 μmol) via ball-milling at 30 Hz for 10 min. Then, the solid-state fluorescence spectra of the samples were measured. The maximum fluorescence wavelength (λem,max) and the appearance of each sample after grinding are summarized in Table 1 (for the fluorescence spectra, see Fig. S40–S59†). Under irradiation from UV light, fluorescence was observed for all samples except for NO2/OMe. Surprisingly, this result clearly demonstrates that the wavelength of the fluorescence originating from the radicals can be tuned from 519 nm to 635 nm only by changing the substituents. It is important to note here that the fluorescence wavelength of triarylmethyl radical derivatives has been controlled in many cases by extending the conjugation length.30–32 This should probably be interpreted in terms of the cyano group of DAAN radicals, which allows the two aromatic rings to adopt a near-planar conformation and thus amplifies the resonance effect of the substituents. These results represent an important aspect of the design and control of the fluorescence wavelength of fluorescent radicals.| λ em,max/nm | Under UV light (λex = 365 nm) | |
|---|---|---|
| a Fluorescence was not observed. | ||
| F/F | 519 |

|
| H/H | 527 |

|
| H/Me | 534 |

|
| F/OMe | 542 |

|
| H/OMe | 546 |

|
| Me/OMe | 550 |

|
| OMe/OMe | 556 |

|
| Br/OMe | 557 |

|
| OMe/diOMe | 558 |

|
| COOMe/OMe | 570 |

|
| OMe/triOMe | 572 |

|
| diOMe/triOMe | 574 |

|
| diOMe/diOMe | 575 |

|
| CN/OMe | 579 |

|
| Ac/OMe | 588 |

|
| triOMe/triOMe | 599 |

|
| SMe/OMe | 602 |

|
| OMe/diOMe-m | 611 |

|
| diOMe-m/diOMe-m | 635 |

|
| NO2/OMe | —a |

|
Moreover, a strong linear correlation emerged when the maximum fluorescence wavelength, obtained from the solid-state fluorescence spectra (Fig. 3a), was plotted as a function of theoretically obtained fluorescence wavelengths, which was obtained from time-dependent (TD) DFT calculations (UM06-2X/6-311+G(d,p)) (Fig. 3b). To investigate the dependence of the obtained results on the functional, we performed the same calculations using different functionals (UCAM-B3LYP/6-311+G(d,p) and UωB97X-D/6-311+G(d,p)). Although slight systematic errors were observed, as in the case of the UM06-2X/6-311+G(d,p) level, a strong linear dependence was obtained (Fig. S60†). These results thus indicate that the fluorescence wavelength of the DAAN radicals can be estimated theoretically with high accuracy, and that it can be expected to be possible to design radicals with a target fluorescence wavelength using TD-DFT calculations.
 | ||
| Fig. 3 (a) Fluorescence spectra (λex = 365 nm) of mixtures of polystyrene and DAAN derivatives after ball-milling. (b) Relationship between theoretical wavelength and experimental wavelength. (c) Molecular orbital level of D0 and D1 states and Jablonski diagram for the DAAN radicals. | ||
Generally speaking, radical species can undergo two possible electronic transitions from the ground state to the excited state, i.e., (1) from the β-HOMO to the β-SOMO and (2) from the α-SOMO to the α-LUMO (Fig. 3c). Similarly, two types of electronic transitions from the excited state to the ground state are possible. In order to investigate which transition is more dominant in the DAAN derivatives, the electronic transitions for absorption and emission were evaluated based on the results of the TD-DFT calculations. The obtained results indicate that for almost all derivatives, except for NO2/OMe, the HOMO–SOMO electronic transition is dominant in both the ground-to-excited-state and the excited-to-ground-state transitions (Tables S2 and S3†). They also confirm that this trend increases with the strength of the electron-donating properties of the substituents. This reflects the fact that the introduction of electron-withdrawing substituents decreases the LUMO energy, which decreases the SOMO–LUMO gap (Table S4, Fig. S61–S80†). In addition, the β-HOMO to β-SOMO and the α-SOMO to α-LUMO transitions could not be confirmed for NO2/OMe (Tables S2 and S3†), which suggests that the electronic transition between the optimized D1 state and the D0 state is a forbidden transition for NO2/OMe.
To clarify the difference between fluorescent and non-fluorescent DAAN radicals, we further analyzed the results of the DFT and TD-DFT calculations. In general, the luminescence quantum yield of radical species (ϕ) is expressed using the rate constant:33
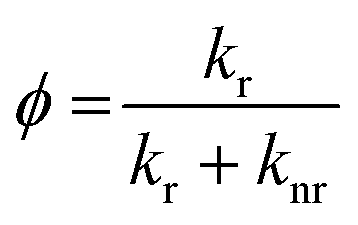 | (1) |
The molecular structures of the DAAN derivatives in the optimized D0 state and the optimized D1 state were obtained from TD-DFT calculations in order to clarify the characteristics of the radiative and non-radiative processes. Specifically, after the structural optimization, the lengths of all the carbon–carbon bonds in the diphenylmethane moiety and the dihedral angle between the two aromatic rings of each DAAN derivative were calculated (Fig. 4a). In the cases of the non-symmetric DAAN derivatives R1/R2, the carbon atoms were numbered with the ipso-position of R1 as C1. The results indicate that the bond lengths between the central carbon atom and the aryl moiety (C6–C7–C8) and between the carbon atoms at the ortho- and meta-positions (C2–C4, C3–C5, C9–C11, C10–C12) of the optimized D1 state are shorter than those in the optimized D0 state, except in the case of NO2/OMe (Table S5†). These results thus also indicate an increased quinoid character for the aryl groups in the optimized D1 state, except in the case of NO2/OMe.34 Moreover, the dihedral angle between the aromatic rings decreases from the optimized D0 state to the optimized D1 state (except in the case of NO2/OMe), confirming the improved planarity of the molecules (Table S6†). The decrease in the dihedral angle should promote stronger π-conjugation between the two aromatic rings (Fig. 4b), and the enhanced π-conjugation should suppress the thermal fluctuation and vibration of molecules, which would lead to non-radiative decay. These findings provide some evidence that the knr of NO2/OMe is significantly higher than that of the other DAAN derivatives.
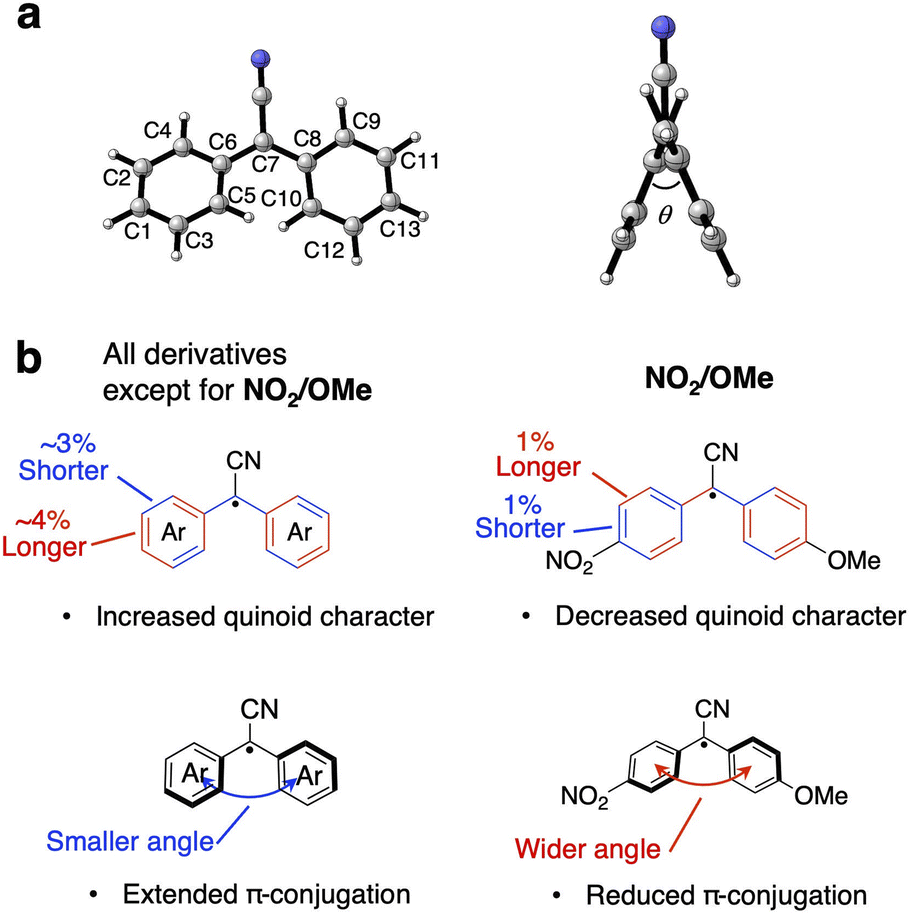 | ||
| Fig. 4 (a) Carbon numbering scheme and dihedral angle in the DAAN radicals. (b) Schematic diagram of the structural changes in the bond lengths and dihedral angles of the DAAN radicals upon transitioning from the optimized D0 state to the optimized D1 state. | ||
The transition dipole moment between the optimized D1 and D0 states (μ01) is related to kr, and molecules with a large μ01 value show high luminescence efficiency.35,36 The μ01 value was calculated based on the results of the TD-DFT calculations, and a positive correlation was observed between the experimental fluorescence quantum yields and μ01 (Table S2, Fig S81†). These results suggest that it is possible to use TD-DFT calculations in order to estimate whether a radical species can exhibit fluorescence. In addition, NO2/OMe showed extremely low values compared to the other DAAN derivatives (Table S7†). This result suggests that NO2/OMe is a non-luminescent radical.
Substituent effects of the DAAN derivatives on the radical generation via polymer mechanochemistry
The following two points are considered to be important for the generation of radicals from DAAN derivatives (Fig. 5). (1) The hydrogen atom at the benzylic position of the DAAN derivatives is easily abstracted by polymeric mechanoradicals. (2) The generated DAAN radicals are more stable than other mechanoradicals due to their aromaticity and electron-withdrawing cyano group.37–42 To confirm these hypotheses, the thermodynamic parameters of each DAAN derivative were determined using DFT calculations, and the amount of DAAN radicals generated upon ball-milling mixed samples of polystyrene and DAAN derivatives was estimated based on EPR measurements by comparing the integral area of each observed spectrum with that of a solution of 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL) as a standard. Unless otherwise noted, DFT calculations were carried out at the UM06-2X/6-311+G(d,p) level given that this level has been reported to be appropriate for fluorescent radicals.4,30,43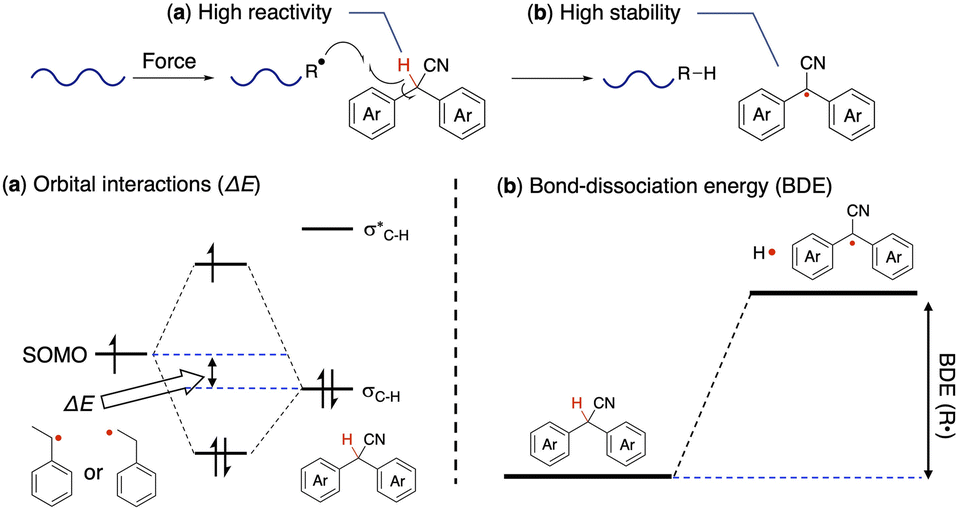 | ||
| Fig. 5 Schematic illustration of (a) orbital interactions and the (b) bond-dissociation energy (BDE) in DAAN derivatives. | ||
The unpaired electron of the mechanoradical generated by the ball-milling of polystyrene is accommodated in the α-SOMO. To abstract the hydrogen atom from the DAAN derivative for the mechanoradical, the α-SOMO of the mechanoradical must interact with the C(sp3)–H bonding orbital (σC–H) of the DAAN derivatives (Fig. 5a). Accordingly, the orbital energy of the α-SOMO of the 1-phenylethyl radical and 2-phenylethyl radical, which was used as the model structure for the mechanoradical generated by the polymer-chain scission of polystyrene, and the σC–H energy of the DAAN derivatives were calculated using DFT calculations. The results of the calculations suggested that the σC–H energy level of the DAAN derivatives and the reactivity with the α-SOMO of the mechanoradical increase with increasing electron-donating properties of the substituents on the DAAN derivatives because the energy difference between α-SOMO of mechanoradical and σC–H decreases (Table S8, Fig. S82–S101†).
We also examined the stability of the DAAN radicals generated when the benzylic hydrogen of the DAAN derivative is abstracted by the mechanoradical. It is generally accepted that the thermodynamic stability from spin delocalization and kinetic stability from steric protection are the two main factors that account for the overall stability of long-lived radicals. On the other hand, the bond-dissociation energy (BDE) is a parameter that is often used to evaluate the thermodynamic stability of radicals.12,44 As shown in eqn (2), the BDE is the energy required for cleaving the hydrogen adduct (R–H) of the radical (R˙) into R˙ and H˙ (Fig. 5b); the smaller this value, the more readily homolysis occurs and the greater the amount of thermodynamically stable radicals generated.
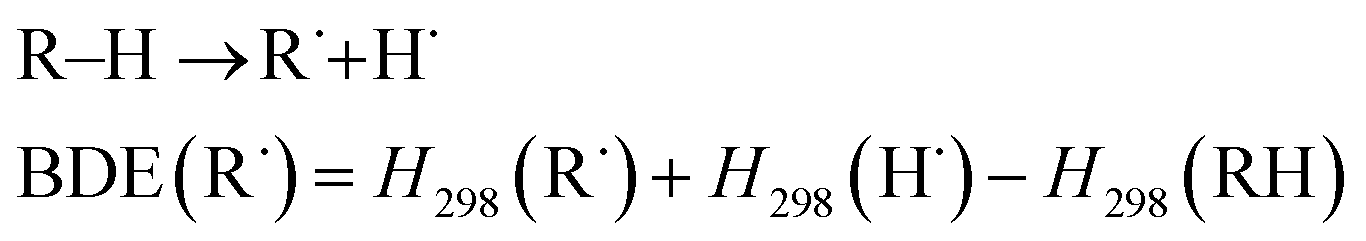 | (2) |
Many recent studies have shown that the thermodynamic stability of such radicals can be predicted with high accuracy using DFT calculations.45–48 In this study, we performed DFT calculations to evaluate the thermodynamic stability of radicals based on the BDE (for the calculation results, see Table S9†). The obtained results suggest that the BDE increases and that the radical species become thermodynamically unstable when there are a total of three or more electron-donating methoxy groups on the aromatic rings, or, conversely, when there are no electron-donating groups. On the other hand, when the aromatic rings have one or two electron-donating groups or one electron-donating group and one electron-withdrawing group, the BDE decreases, suggesting that the radical species become thermodynamically stable. The results of these calculations thus reflect the captodative effect.28,29 In addition, the BDE tends to increase when there is a methoxy group at the ortho-position. This result implies that the DAAN derivatives with substituents at the ortho-position show deviation from ideal molecular planarity due to steric repulsion, commensurate with lower thermodynamic stabilization compared to those without substituents at the ortho-position.
DFT-derived prediction of the conversion efficiency to DAAN radicals from precursors
Fig. 6a and b show the results of our DFT calculations. Higher σC–H energy levels and smaller BDE values (the compounds are plotted in the bottom-right corner) correspond to a higher estimated conversion efficiency of the to DAAN radicals based on thermodynamic considerations.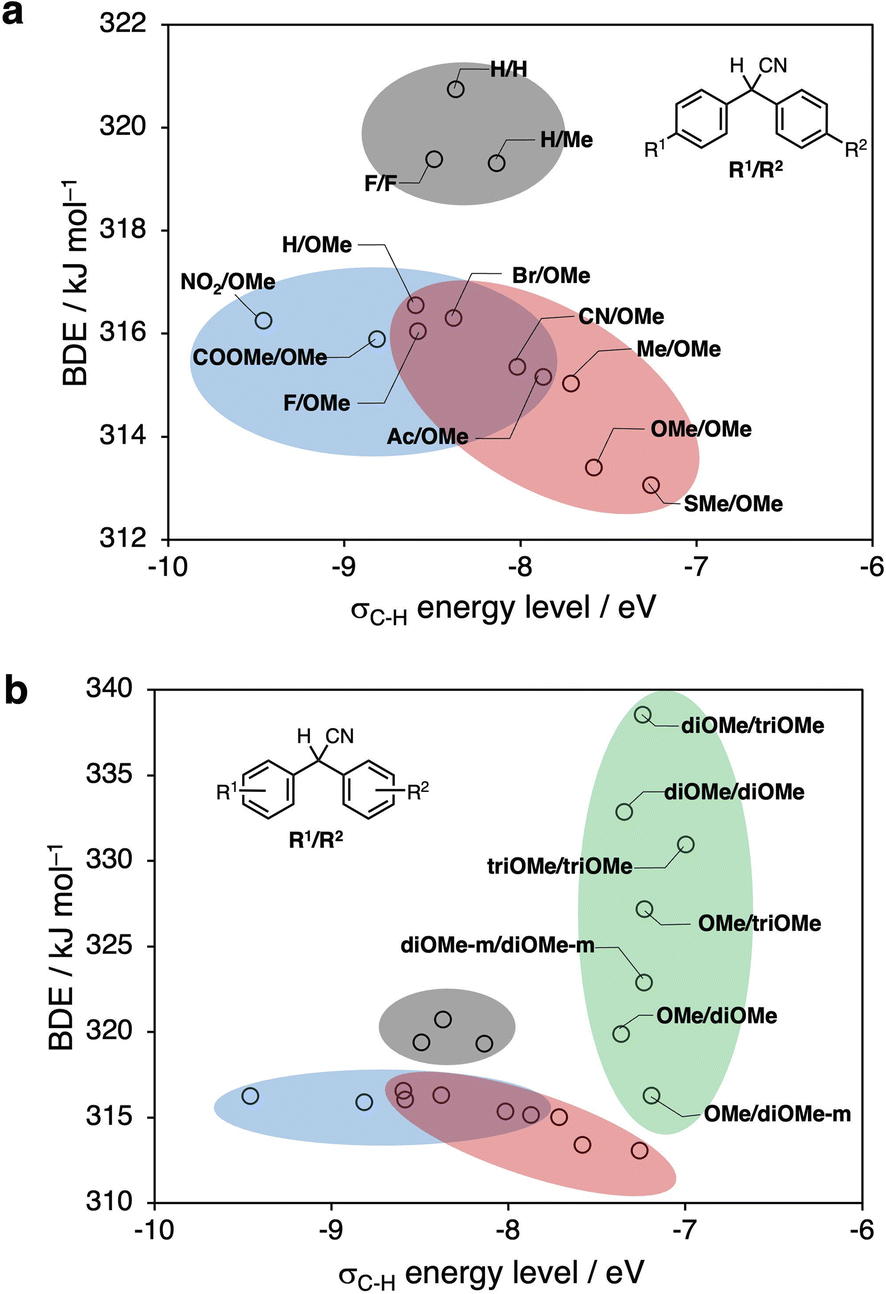 | ||
| Fig. 6 (a) BDE values and σC–H energy levels of DAAN derivatives obtained from DFT calculations (para-substituent derivatives). (b) BDE values and σC–H energy levels of DAAN derivatives obtained from DFT calculations (all derivatives). | ||
Fig. 6a, which summarizes the calculation results for DAAN derivatives with substituents at only the para-position, predicts the highest conversion efficiency to DAAN radicals for SMe/OMe. Moreover, the calculations predict that derivatives from the blue and red groups can be expected to have a higher conversion efficiency to DAAN radicals than the derivatives from the gray group. Furthermore, Fig. 6b shows that, compared to the derivatives with substituents at only the para-position, the derivatives in the green group have higher BDE values and generate radical species with lower thermodynamic stability, albeit that they have a higher σC–H level and higher reactivity with mechanoradicals.
Evaluation of the amount of DAAN radicals
After the ball-milling of the mixtures of polystyrene (Mn = 66.0 kg mol−1; Mw/Mn = 1.46, 100 mg) and the DAAN derivatives (50 μmol), the amount of generated DAAN radicals relative to the corresponding DAAN derivative was evaluated using EPR measurements. The observed DAAN radicals are shown in Fig. 7a (for the EPR spectra, see Fig. S102–S121†). Among the DAAN derivatives with substituents at only the para-position, H/H, F/F, and H/Me furnished only a small amount of DAAN radicals, while SMe/OMe afforded a large amount of DAAN radicals. This result is consistent with the DFT-based prediction that the conversion efficiency to DAAN radicals increases with decreasing BDE value and increasing σC–H energy level. However, the amount of DAAN radicals with methyl groups was smaller than that predicted based on its BDE. This is probably due to side reactions, such as the reaction of the methyl group with the mechanoradicals to produce benzyl radicals (Fig. S122†). Furthermore, in the DFT calculations, the thermodynamic stability of the DAAN radicals was predicted to be lower when substituents were present at the ortho-position. Nevertheless, with increasing number of substituents at the ortho-position, the amount of observed DAAN radicals also increased (Fig. S123†). This result should most likely be attributed to the protection of the central (or α) carbon atom, which is the atom with the highest spin density, due to the introduction of a bulky methoxy group at the ortho-position. In other words, the low thermodynamic stability of the ortho-substituted DAAN derivatives is counterbalanced by their kinetic stability. To evaluate the steric hindrance of the DAAN radicals, their percentages of buried volume (% VBur) were calculated.49 For this purpose, a sphere was set up with the carbon-centred radical at the origin and a 6 Å radius; these dimensions are the most suitable to consider the spatial steric protection of the radical by the surrounding atoms.50 The buried volumes and steric maps presented in this work were obtained using the SambVca 2 Web application.51 According to the topography (Fig. S124†), the methoxy groups at the ortho-position of the DAAN radicals protect the carbon-centred radicals well (Fig. 7b). These results suggest that small BDE values and high σC–H energy levels are important for improving the amount of generated DAAN radicals. In addition, the steric protection of the carbon-centred radicals overcomes the low thermodynamic stability of radicals and increases the amount of generated DAAN radicals.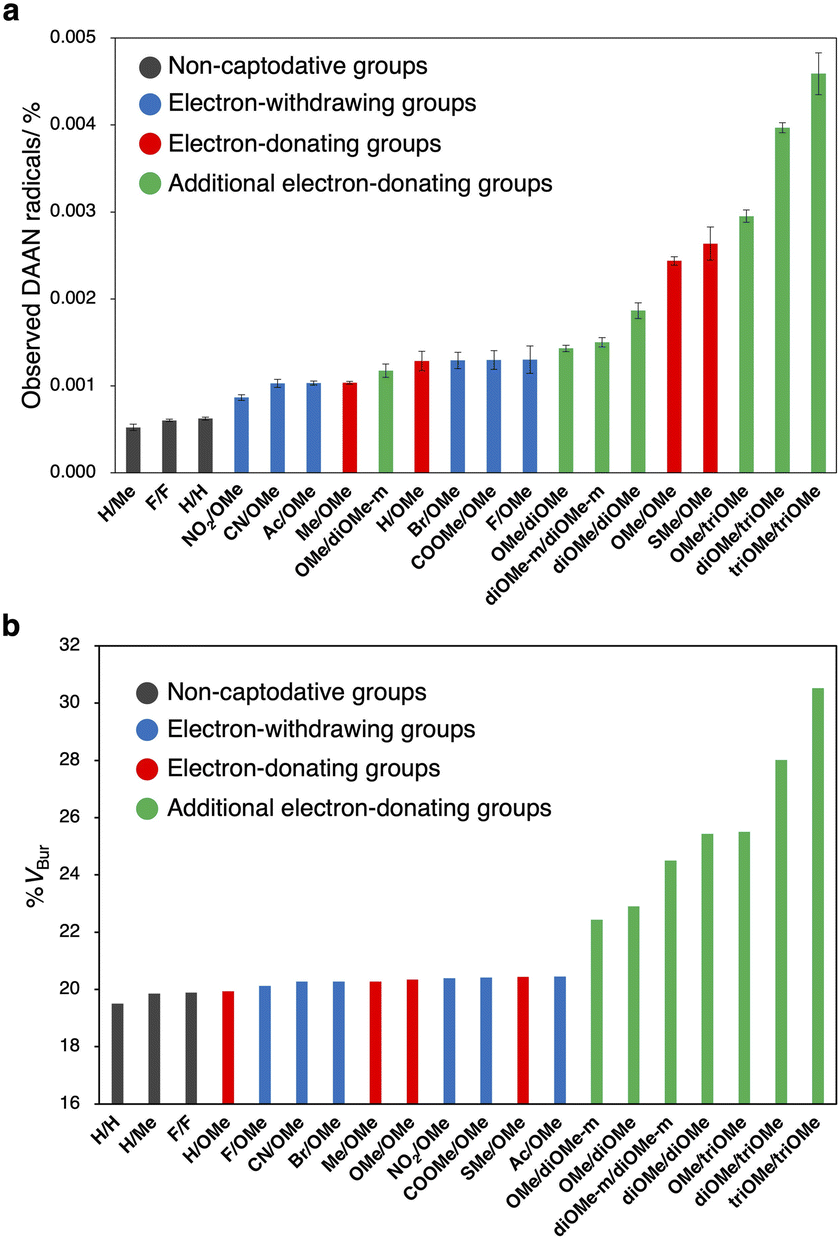 | ||
| Fig. 7 (a) Observed DAAN radicals using EPR measurements and (b) % VBur of DAAN derivatives. | ||
Conclusion
In this study, we conducted a comprehensive study on metastable DAAN radicals with various substituents, that are generated by polymeric mechanochemical reactions. We discovered a significant change in fluorescence wavelength depending on the nature and location of these substituents, which is a phenomenon that has not previously been observed for the extensively studied fluorescent triaryl methyl radicals. The amount of generated DAAN radicals evaluated by EPR is greater for smaller BDE values and higher σC–H energy levels when the substituent are located exclusively at the para-position. When a substituent is introduced at the ortho-position, the thermodynamic stability of the radical is reduced, albeit that the kinetic effect of the steric bulk of the radical centre is outweighed, and the amount of DAAN radicals increases. We thus expect that the present study will inspire the development of unexplored fluorescent radicals and their applications in advanced chemosensors, bioimaging, and organic light-emitting diodes.Author contributions
T. Y., D. A., and H. O. conceived the concept and designed the experiments. T. Y. performed the experiments and analysed the data. K. M. advised the theoretical calculation study. T. Y., D. A., K. M., and H. O. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by KAKENHI grants 21H04689 (to H. O.) from the Japan Society for the Promotion of Science (JSPS), JSPS Research fellowships for Young Scientists 22J21853 (to T. Y.), the ANRI Fellowship (to T. Y.), and JST CREST grant JPMJCR1991 (to H. O.). The numerical calculations were carried out on the TSUBAME3.0 supercomputer at Tokyo Institute of Technology supported by the MEXT project ‘Tokyo Tech Academy for Convergence of Materials and Informatics (TAC-MI).Notes and references
- J. Luo, X.-F. Rong, Y.-Y. Ye, W.-Z. Li, X.-Q. Wang and W. Wang, Molecules, 2022, 27, 1632 CrossRef CAS PubMed.
- P. Murto and H. Bronstein, J. Mater. Chem. C, 2022, 10, 7368–7403 RSC.
- D. Velasco, S. Castellanos, M. López, F. López-Calahorra, E. Brillas and L. Juliá, J. Org. Chem., 2007, 72, 7523–7532 CrossRef CAS PubMed.
- X. Ai, Y. Chen, Y. Feng and F. Li, Angew. Chem., Int. Ed., 2018, 57, 2869–2873 CrossRef CAS PubMed.
- R. Matsuoka, S. Kimura and T. Kusamoto, ChemPhotoChem, 2021, 5, 669–673 CrossRef CAS.
- Y. Mu, Y. Liu, H. Tian, D. Ou, L. Gong, J. Zhao, Y. Zhang, Y. Huo, Z. Yang and Z. Chi, Angew. Chem., Int. Ed., 2021, 60, 6367–6371 CrossRef CAS PubMed.
- H. Cho, S. Kimura, N. C. Greenham, Y. Tani, R. Matsuoka, H. Nishihara, R. H. Friend, T. Kusamoto and E. W. Evans, Adv. Opt. Mater., 2022, 2200628 CrossRef CAS.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 127, 7197–7201 CrossRef.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- J. M. Hudson, T. J. H. Hele and E. W. Evans, J. Appl. Phys., 2021, 129, 180901 CrossRef CAS.
- W. Zheng, X. Li, G. V. Baryshnikov, X. Shan, F. Siddique, C. Qian, S. Zhao and H. Wu, Angew. Chem., Int. Ed., 2023, 62, e202305925 CrossRef CAS PubMed.
- D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed.
- K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 CrossRef CAS PubMed.
- N. Soh, Y. Katayama and M. Maeda, Analyst, 2001, 126, 564–566 RSC.
- A. Jiménez, A. Selga, J. L. Torres and L. Julià, Org. Lett., 2004, 6, 4583–4586 CrossRef PubMed.
- V. Gamero, D. Velasco, S. Latorre, F. López-Calahorra, E. Brillas and L. Juliá, Tetrahedron Lett., 2006, 47, 2305–2309 CrossRef CAS.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2014, 53, 11845–11848 CrossRef CAS PubMed.
- H. Guo, Q. Peng, X. K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J. L. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed.
- P. Mayorga Burrezo, V. G. Jiménez, D. Blasi, I. Ratera, A. G. Campaña and J. Veciana, Angew. Chem., Int. Ed., 2019, 58, 16282–16288 CrossRef CAS PubMed.
- X. Li, Y.-L. Wang, C. Chen, Y.-Y. Ren and Y.-F. Han, Nat. Commun., 2022, 13, 5367 CrossRef CAS PubMed.
- Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CAS.
- T. Yamamoto and H. Otsuka, Polym. Chem., 2023, 14, 2464–2468 RSC.
- T. Yamamoto, D. Aoki and H. Otsuka, ACS Macro Lett., 2021, 10, 744–748 CrossRef CAS PubMed.
- T. Yamamoto, S. Kato, D. Aoki and H. Otsuka, Angew. Chem., Int. Ed., 2021, 60, 2680–2683 CrossRef CAS PubMed.
- D. K. Singh, S. S. Prasad, J. Kim and I. Kim, Org. Chem. Front., 2019, 6, 669–673 RSC.
- X. Hou, Z. Ge, T. Wang, W. Guo, J. Wu, J. Cui, C. Lai and R. Li, Arch. Pharm., 2011, 344, 320–332 CrossRef CAS PubMed.
- L. Hu, M. I. Hussain, Q. Deng, Q. Liu, Y. Feng, X. Zhang and Y. Xiong, Tetrahedron, 2019, 75, 308–314 CrossRef CAS.
- F. G. Bordwell and T. Y. Lynch, J. Am. Chem. Soc., 1989, 111, 7558–7562 CrossRef CAS.
- H. Tanaka, Prog. Polym. Sci., 2003, 28, 1171–1203 CrossRef CAS.
- Y. Hattori, S. Tsubaki, R. Matsuoka, T. Kusamoto, H. Nishihara and K. Uchida, Chem.–Asian J., 2021, 16, 2538–2544 CrossRef CAS PubMed.
- K. Matsuda, R. Xiaotian, K. Nakamura, M. Furukori, T. Hosokai, K. Anraku, K. Nakao and K. Albrecht, Chem. Commun., 2022, 58, 13443–13446 RSC.
- R. Xiaotian, W. Ota, T. Sato, M. Furukori, Y. Nakayama, T. Hosokai, E. Hisamura, K. Nakamura, K. Matsuda, K. Nakao, A. P. Monkman and K. Albrecht, Angew. Chem., Int. Ed., 2023, 62, e202302550 CrossRef CAS PubMed.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2015, 54, 3731–3734 CrossRef CAS PubMed.
- S. Kimura, A. Tanushi, T. Kusamoto, S. Kochi, T. Sato and H. Nishihara, Chem. Sci., 2018, 9, 1996–2007 RSC.
- Z. Cui, A. Abdurahman, X. Ai and F. Li, CCS Chem., 2020, 2, 1129–1145 CrossRef CAS.
- Z. Zhou, C. Qiao, J. Yao, Y. Yan and Y. S. Zhao, J. Mater. Chem. C, 2022, 10, 2551–2555 RSC.
- S. Kato, S. Furukawa, D. Aoki, R. Goseki, K. Oikawa, K. Tsuchiya, N. Shimada, A. Maruyama, K. Numata and H. Otsuka, Nat. Commun., 2021, 12, 126 CrossRef CAS PubMed.
- T. Sumi, R. Goseki and H. Otsuka, Chem. Commun., 2017, 53, 11885–11888 RSC.
- J. Kida, D. Aoki and H. Otsuka, Aggregate, 2021, 2, e50 CrossRef CAS.
- S. Kato, K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1087–1091 CrossRef CAS PubMed.
- M. van Galen, J. P. Kaniraj, B. Albada and J. Sprakel, J. Phys. Chem. C, 2022, 126, 1215–1221 CrossRef CAS PubMed.
- Z. Liu, H. K. Bisoyi, Y. Huang, M. Wang, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202115755 CrossRef CAS PubMed.
- S. Sang, F. Chen and C. Zhang, Int. J. Quant. Chem., 2021, 121, 1–11 CrossRef.
- J. Hioe and H. Zipse, Org. Biomol. Chem., 2010, 8, 3609–3617 RSC.
- T. Kubo, Y. Katada, A. Shimizu, Y. Hirao, K. Sato, T. Takui, M. Uruichi, K. Yakushi and R. C. Haddon, J. Am. Chem. Soc., 2011, 133, 14240–14243 CrossRef CAS PubMed.
- L. Lin and J. Zhu, J. Org. Chem., 2021, 86, 15558–15567 CrossRef CAS PubMed.
- P. C. St. John, Y. Guan, Y. Kim, S. Kim and R. S. Paton, Nat. Commun., 2020, 11, 1–18 CrossRef PubMed.
- Q. Xiang, J. Guo, J. Xu, S. Ding, Z. Li, G. Li, H. Phan, Y. Gu, Y. Dang, Z. Xu, Z. Gong, W. Hu, Z. Zeng, J. Wu and Z. Sun, J. Am. Chem. Soc., 2020, 142, 11022–11031 CrossRef CAS PubMed.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- X. Wang, P. Xue, C. Zhou, Y. Zhang, P. Li and R. Chen, J. Mater. Chem. C, 2022, 10, 18343–18350 RSC.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mr00031a |
| This journal is © The Royal Society of Chemistry 2024 |