
Tailoring functionality through synthetic strategy in heterolanthanide assemblies
Flavia
Artizzu
*ab,
Francesco
Quochi
b,
Angela
Serpe
a,
Elisa
Sessini
a and
Paola
Deplano
ab
aDipartimento di Scienze Chimiche e Geologiche, University of Cagliari, SS 554 Bivio per Sestu, I-09042, Monserrato-Cagliari, Italy. E-mail: f.artizzu@unica.it
bDipartimento di Fisica, University of Cagliari, SS 554 Bivio per Sestu, I-09042, Monserrato-Cagliari, Italy
First published on 15th January 2015
Abstract
An overview of the different strategies proposed for the preparation of heterolanthanide assemblies suitable to work as (multi-) functional materials, highlighting their structure/property relationship, is provided. Three classes of compounds are selected: multi-dimensional coordination frameworks, polynuclear discrete molecules and flexible large molecules formed by two or more coordinating units connected by a linker. Synthetic approaches and potential functionalities are discussed on the basis of lanthanide ion discrimination and the structural arrangement of the heterometallic assembly.
Introduction
Lanthanide (Ln) ions nowadays belong to the class of “critical metals”, materials whose availability is essential for high-technology, green and defense applications, but vulnerable to politically or economically driven fluctuations in supply.1 Their very peculiar luminescent and magnetic properties, related to the nature of their 4f core-like electrons, are in fact exploited in several fields ranging from high-tech devices to biomedical applications. Correspondingly, lanthanide compounds have been widely studied in view of their numerous potential applications, depending on the nature of the lanthanide ion, such as, for example, MRI contrast agents (Gd3+), catalysts, magnetic materials with large anisotropy and reduced quantum tunneling of the magnetisation (Dy3+, Tb3+), luminescent materials for solar energy conversion, optical imaging, bioprobes, sensors, visible emitters for display and lighting technology (Eu3+,Tb3+, Sm3+) and near-infrared (NIR) emitters for lasers, optical fibers and amplifiers (Er3+, Nd3+, Yb3+).2,3In this context, the most challenging frontier is represented by heterometallic assemblies containing two or more different lanthanide cations as carriers of distinctive functionalities, where different physical properties can coexist or be in interplay thanks to intermetallic communication. These materials can include, for example: multiple emitters, directional light converters, double-function compounds acting as MRI contrast agents and luminescent probes, and systems with simultaneously activated catalytic sites.4,5
However, the preparation of well-defined and easily reproducible polymetallic lanthanide compounds is often a difficult task, and fully characterized mixed-lanthanide coordination compounds still represent very rare examples in the literature. Nonetheless, the extraordinary versatility of lanthanide coordination chemistry, which obeys completely different rules from those common to transition metal ions, is basically related to the lack of orbital directionality, the strong Lewis acid character and the tendency to reach high coordination numbers of the Ln ions. These factors can lead to the isolation of a wide variety of coordination compounds, ranging from multi-dimensional networks to polynuclear discrete assemblies, through judicious selection of the coordinating ligands or even simply through the variation of the reaction conditions. This offers a unique opportunity to mix and play with different lanthanide ions to create diverse architectures and to achieve tunability of their physical properties through composition control and rational synthetic strategies. In fact, depending on the desired resulting functionality and potential applications of the material, tailored synthetic approaches, leading to specific coordination frameworks, can be selected.
The aim of this article is therefore to provide an overview, covering the most significant literature on the different strategies that have been proposed for the preparation of heterolanthanide assemblies, shedding light on the structure/property relationship of these materials.
Discussion
A. General considerations
Coordination chemistry can provide several benefits for the the use of Ln ions in functional materials. Among them, the following are worth highlighting: (i) luminescent Ln ions, exhibiting intrinsically weak absorptivity, can take advantage of indirect optical pumping through resonance energy transfer (sensitisation) from a suitable organic ligand acting as an antenna chromophore (see Fig. 1); (ii) magnetic anisotropy of some lanthanide ions can be tuned by changing the coordination symmetry,4a,6a and the magnetic coupling between paramagnetic centers can be mediated by covalent or noncovalent interactions of organic moieties;6b (iii) metal encapsulation with organic ligands improves Ln solubility into host matrices, providing processing versatility. The electronic, optical and magnetic properties of Ln ions are significantly affected by the structural constraints of the metal ion site (geometry, symmetry, nature of the first and second coordination spheres, nature of the metal ions, and site accessibility). Consequently, careful design of the coordination architecture for the selective incorporation of Ln ions is crucial for achieving programmed functionalities.4 Moreover, for application purposes, synthesised compounds should be: (i) fully characterised, for a detailed understanding of the structure/property relationship; (ii) highly reproducible; (iii) kinetically inert and thermodynamically stable; and (iv) processable without alterations. | ||
| Fig. 1 A Jablonski diagram depicting the sensitisation mechanism of a Ln ion through resonance energy transfer from an organic ligand. The ligand is photoexcited to the singlet level S1, then intersystem-crossing (ISC) occurs to lower-lying excited triplet states (T1, T2) that are suitable to feed the Ln3+ upper levels by direct energy transfer (ET). Alternatively, ligand-to-metal ET can occur directly from the S1 state. FS = fluorescence; PL = photoluminescence; NR = non-radiative decay; PS = phosphorescence. | ||
Basically, two considerations should be taken into account for the design of functional heterolanthanide systems: (i) whether different physical properties are intended to be cooperative or simply superimposed and (ii) if functionality has to be achieved in the bulk materials or at a molecular level.
When dealing with the first point, one has to bear in mind that, for efficient intermetallic communication (magnetic coupling, electron transfer, energy transfer), the distance between the interacting Ln ions must be reduced below 3.5–4 Å for mechanisms requiring orbital overlap (Dexter energy transfer, magnetic exchange) or below 10 Å for efficient dipolar through-space interactions (Förster resonance energy transfer, magnetic dipolar interactions), see Fig. 2.4a,7,8
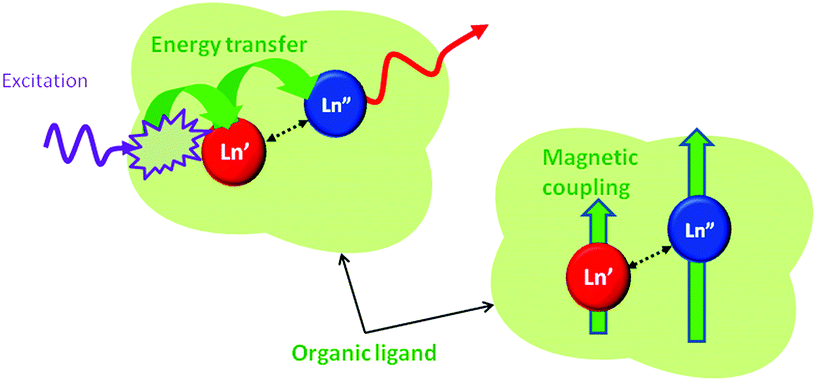 | ||
| Fig. 2 A schematic representation of the intermetallic communication by energy transfer or magnetic coupling between close-contact lanthanide pairs embedded in an organic framework. | ||
Intermetallic separation control can be achieved through the use of bridging ligands/atoms, multi-compartmental flexible receptors or by programmed supramolecular interactions, such as hydrogen bonding, π-stacking between adjacent coordinated ligands, or halogen bonding. This is definitely a crucial factor to be considered, as undesired and uncontrolled interactions between Ln ions, especially energy transfer, may severely alter the properties exhibited by the system and hamper their interpretation.
The second point that has to be addressed more generally concerns the intended final application purpose for these multifunctional materials. To give some practical examples, while several technological applications (e.g. in photonic devices), take general advantage of the properties of a material processed in bulk as long as sample homogeneity is ensured, on the other hand, exploitation of single-molecule properties is clearly compulsory for certain uses (e.g. as bioassays and catalysts), i.e. when simultaneous independent functionalities of active centres interacting with an external matrix are sought. This brings to attention one of the biggest challenges in the design of heterolanthanide polynuclear arrays, i.e. the achievement of metal composition and speciation control at the molecular level aimed at obtaining purely heterometallic compounds. In fact, the discrimination of trivalent Ln cations is severely hampered by the close similarity of their chemical behaviour and ionic radii along the series. Therefore, statistical metal distribution over available alike coordination sites inevitably governs the synthesis of heterolanthanide compounds. While this can certainly be advantageous in some cases, especially when dealing with materials intended to be used as solid state or processed samples, it represents a severe limitation when single-molecule properties are sought. Some synthetic approaches have been proposed in this regard in recent years, essentially based on two main concepts: self assembly by size selectivity or multi-step sequential metallation. However, very rare examples of purely heterometallic molecules have been isolated so far, leaving large room for improvement and for the further development of methods of general validity.
In light of these considerations, three main classes of heterolanthanide assemblies can be discussed: multi-dimensional frameworks such as metal-organic-frameworks (MOFs) and chain polymers; polynuclear discrete molecules; and large molecules formed by two or more coordinating units connected by a linker.
B. Multi-dimensional frameworks
Lanthanide-containing coordination polymers (CPs) have been investigated in the last decade in view of their potential applications in gas storage, sensing, heterogeneous catalysis, chemical separation and drug delivery.9 These materials offer the opportunity to mix lanthanide ions in a framework consisting of repeating equivalent coordinating units, so that the metal properties are independent to the occupied coordination sites. This makes heterolanthanide CPs interesting in view of a random statistical distribution of Ln ions over the coordination framework that provides homogeneity of the structural features and the physical properties across the bulk material. Moreover, these architectures allow the modulation of intermetallic distances through an appropriate selection of the organic linker, so that cooperative or superimposed independent functionalities between active metal centres can, in principle, be achieved.CPs are usually prepared through one-step reactions of the selected Ln salts in a desired stoichiometric amount and an organic linker (by reaction in solution, diffusion into gels, or by molten salt or solvothermal syntheses). However, post-synthetic modifications have also been used, in which Ln ions are incorporated into a preformed organic framework, to prevent structural alterations.9f,10
Heterolanthanide multi-dimensional networks, have so far been studied mainly in view of their luminescence properties. Recently, heterolanthanide MOFs have been smartly proposed as NIR or Vis luminescent barcodes, taking advantage of the narrow, non-overlapping and easily distinguishable emission lines of Ln ions (Fig. 3).10–13
 | ||
| Fig. 3 A schematic representation of a Ln-based barcoded material. Reproduced with permission from ref. 11. Copyright 2009 American Chemical Society. | ||
In these materials, organic antenna ligands can be introduced, enabling the simultaneous emission of different lanthanide ions upon single-wavelength excitation. A wide variety of barcodes can thus be obtained simply by varying the metal stoichiometry, so long as the optically active Ln centres are homogeneously distributed across the framework and kept far enough apart (e.g. by using large organic linkers) to avoid undesired and uncontrolled intermetallic energy transfers that can significantly affect the emission properties. In this regard, the non-luminescent Gd3+ ion has been used as a codopant in 2∞[Gd2−x−yEuxTbyCl6(bipy)3]·2bipy MOF, to dilute the two communicating Tb3+ and Eu3+ ions, separated by a short bipy ligand (Fig. 4), allowing the modulation of the emission colour, resulting from the overlap of the green Tb3+ emission (5D4→7FJ, J = 6–3) and the red Eu3+ one (5D0→7FJ, J = 0–4), on varying the metal content.13 Interestingly, it has also been observed that Gd3+ codoping can enhance the luminescence of Ln3+:Y2WO6 (Ln = Sm, Eu, Dy) microstructures.14
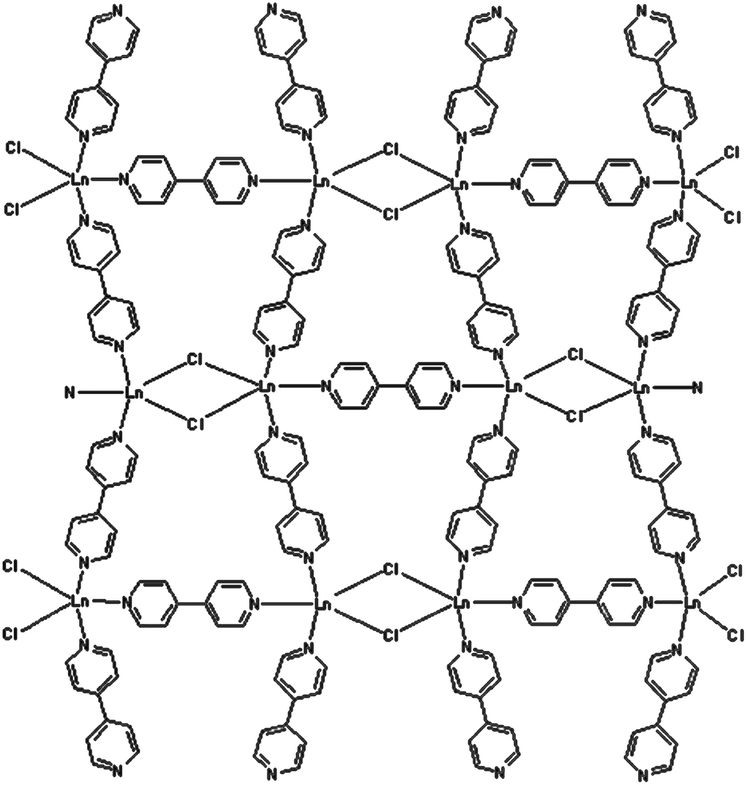 | ||
| Fig. 4 The structure of 2∞[Gd2−x−yEuxTbyCl6(bipy)3]·2bipy. Reproduced from ref. 13. | ||
Another proposed application for mixed Ln CPs relies on their use as luminescent thermometric probes. In fact, it is well known that luminescence intensity decreases when temperature increases as a consequence of the thermal deactivation of excited states. In the heterolanthanide ∞[Ln(L)2(NO3)2]·Cl·2H2O (Ln = Eu, Tb, and Gd; L = 1,4-bis(pyridinil-4-carboxylato)-l,4-dimethylbenzene) CP (Fig. 5a), temperature sensitivity in the luminescence is observed as the Tb → Eu energy transfer efficiency is thermally enhanced (from 22.64% at 25 K to 34.41% at 200 K).
 | ||
| Fig. 5 Above. Structure of the heterometallic Eu–Tb ∞[Ln(L)2(NO3)2]·Cl·2H2O CPs: (a) Ring-like structure unit. (b) One dimensional chain structure and the simplified diagram. (c) Two dimensional supramolecular layer. All the hydrogen atoms, chlorine atoms, and water molecules have been omitted for clarity. Blue dot line: hydrogen bond. Below. Luminescent response to the temperature for Eu (red) and Tb (green) compared with the trends for the corresponding homometallic CPs (in the inset). Adapted from ref. 15. | ||
The emission intensity ratio ITb/IEu (5D4→7F5, Tb3+, 544 nm to 5D0→7F2, Eu3+, 613 nm), can therefore be used as a self referenced signal probe for temperature (Fig. 5).15
In light of these few examples, it is clear that heterolanthanide CPs are promising for several applications in view of the functionalities attainable in these materials. Among the numerous figures of merit, such as their structural porosity (for uses as host matrices), they can also incorporate a high density of active metal centres homogeneously dispersed across the bulk material. However, little control can be achieved in the Ln distribution, so mixed Ln CPs can best perform as “diluted” materials where independent physical properties carried by different Ln3+ ions are superimposed. The numerous possible distance-dependent Ln–Ln interactions can in fact be difficult to control so that recognition and quantification of the resulting properties may be complicated.
C. Discrete polynuclear molecules
Discrete polynuclear complexes can offer several advantages for the development of functional molecular materials for application purposes. In fact, they can often be easily processed through convenient, easy-to-handle, solution methods or by techniques commonly used for the fabrication of technological devices, such as vacuum deposition, without altering their properties.16 In addition, unlike CPs, control of the separation between the lanthanide ions and their relative positions, is, in principle, feasible through judicious chemical design. This means that it would be possible to achieve controlled efficient energy transfer or magnetic coupling between different Ln ions sitting at short distances in rigid, preorganized positions in the same molecular architecture. This can be accomplished through the use of small bridging coordinating units such as CN−, oxalate,4a ligands having suitable bridging atoms,7,17–19 multi-compartmental macrocycles,4,20 and flexible multidentate ligands.8,21–24 Some significant examples of ligands used to prepare polynuclear heterolanthanide complexes with short intermetallic separation and their coordination modes are reported in Chart 1.4,7,17–24 | ||
| Chart 1 | ||
Generally, the easiest and least time/resources-consuming synthetic approach for heterolanthanide polynuclear assemblies, consists of a one-pot reaction between the selected metal salts and the ligand(s) in suitable ratios, taking advantage of the very similar reactivity of the Ln ions across the series. The corresponding downside is that, in the absence of some metal selectivity restrictions, statistical mixtures of homo- and heterolanthanide complexes are invariably obtained. In general, for a bimetallic complex with ligand L:4,7,18–23
| Ln′3+ + Ln′′3+ + xL → Ln′Ln′′Lx + Ln′Ln′Lx + Ln′′Ln′′Lx | (1) |
Relative abundances of 50% for the heterometallic, and 25% for the homometallic species, are predicted for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Ln′:Ln′′ molar ratio, while this statistical distribution can be tuned on varying metal composition. Following this reaction scheme, heterobimetallic compounds such as [ErxYb3−x(Q)9],7 (ligand I, Chart 1), [Er1.4Yb0.6(Ba)6(Phen)2] and [ErYb(PBa)6(Phen)2],19 (ligand III, Chart 1), have been obtained. [La1−xLn′′x(L2)(NO3)4]·1.2CH3OH (Ln′′ = Tb, Eu) complexes with the macrocyclic ligand L2 (IV, Chart 1), have been prepared in a similar procedure with the La:Tb and La:Eu ratios in the reaction mixtures being 8:2 and 9:1, respectively.20a Likewise, a series of mixed lanthanide compounds [Ln′1−xLn′′x(L7) (NO3)]·nCH3OH (Ln′ = Gd or Lu; Ln′′ = Eu or Tb) have been isolated with the acyclic Schiff base ligand L7 (V, Chart 1).21a A slight modification of the aforementioned synthetic pathway (1) allows the use of Ln acetylacetonate complexes, [Ln(acac)3]·H2O as precursor materials. Direct reaction of Ln′ and Ln′′ precursors in a 1:1 molar ratio in solution yields dimeric species [Ln′Ln′′(acac)6] (Ln′ = Tb, Ln′′ = Eu), linked by oxo-bridges (ligand II, Chart 1),18 whereas when using the salen ligand (VI, Chart 1) as a unit assembler, [Ln′Ln′′(salen)2(acac)2] (Ln′ = Er, Ln′′ = Yb) is formed.21b
:1 Ln′:Ln′′ molar ratio, while this statistical distribution can be tuned on varying metal composition. Following this reaction scheme, heterobimetallic compounds such as [ErxYb3−x(Q)9],7 (ligand I, Chart 1), [Er1.4Yb0.6(Ba)6(Phen)2] and [ErYb(PBa)6(Phen)2],19 (ligand III, Chart 1), have been obtained. [La1−xLn′′x(L2)(NO3)4]·1.2CH3OH (Ln′′ = Tb, Eu) complexes with the macrocyclic ligand L2 (IV, Chart 1), have been prepared in a similar procedure with the La:Tb and La:Eu ratios in the reaction mixtures being 8:2 and 9:1, respectively.20a Likewise, a series of mixed lanthanide compounds [Ln′1−xLn′′x(L7) (NO3)]·nCH3OH (Ln′ = Gd or Lu; Ln′′ = Eu or Tb) have been isolated with the acyclic Schiff base ligand L7 (V, Chart 1).21a A slight modification of the aforementioned synthetic pathway (1) allows the use of Ln acetylacetonate complexes, [Ln(acac)3]·H2O as precursor materials. Direct reaction of Ln′ and Ln′′ precursors in a 1:1 molar ratio in solution yields dimeric species [Ln′Ln′′(acac)6] (Ln′ = Tb, Ln′′ = Eu), linked by oxo-bridges (ligand II, Chart 1),18 whereas when using the salen ligand (VI, Chart 1) as a unit assembler, [Ln′Ln′′(salen)2(acac)2] (Ln′ = Er, Ln′′ = Yb) is formed.21b
Small deviations in the statistical abundances of the molecular species have occasionally been observed as a consequence of the differences in their reactivities upon ligand coordination to the Ln ions belonging to the first (La–Eu), or the second (Gd–Lu) half of the series.25a Instead, for the mixed lanthanide compounds [Ln′xLn′′2−x(L8) (DMF)5]·nDMF (Ln′ = Eu, Tb; Ln′′ = Nd, Tb, Gd, Ho) with the macrocyclic p-tert-butylcalix[8]arene ligand L8 (VII, Chart 1), ICP-AES determination of the Ln3+ content shows a clear selectivity of the ligand for ions in the middle of the lanthanide series.20b Lanthanide ions can, in fact, be solely discriminated on the basis of a continuous and smooth decrease in their ionic radii (lanthanide contraction) that barely drops, about 15%, on going from La3+ to Lu3+, with a ∼1% variation between two successive lanthanides with a relative maximum (cusp) corresponding to the half-filled 4f7 shell of Gd3+ (Fig. 6).26 To address this point, the most reasonable route relies on polynuclear architectures where coordination sites are inequivalent, i.e., by tailoring binding sites for the specific recognition of Ln ions. This task can be particularly complicated in view of the flexible coordination behavior of the Ln ions, for which coordination numbers raging from 6 to 12 are likely to be observed.
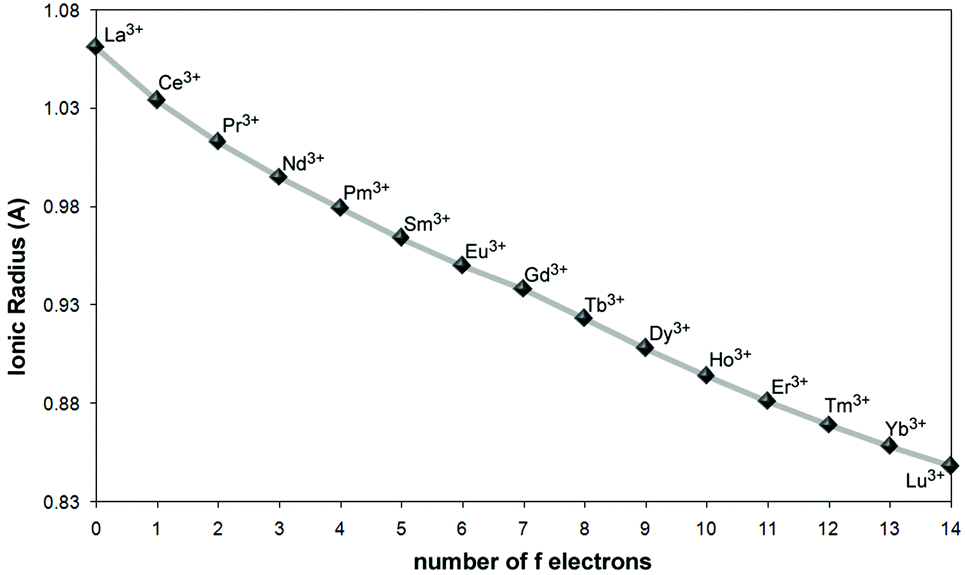 | ||
| Fig. 6 The ionic radii of trivalent lanthanide ions with a coordination number of six.26 | ||
This has so far limited the strive toward pure discrete heterolanthanide complexes through simple one-pot reactions, and only very few examples are to be mentioned in this regard. In particular, Bünzli and coworkers have extensively investigated bimetallic helicates with flexible multidentate ligands as selective dinucleating agents.22 Specifically, ligand LAB (VIII, Chart 1), has been elegantly designed to host two Ln ions in two different cavities formed after self-assembly of a triple-stranded helicate [Ln′Ln′′(LAB)3]6+. This synthetic approach takes advantage of the templating effect arising from the N2O coordination moiety, favoring the complexation of small lanthanides, and the more rigid N3 binding unit, which, on the other hand, is better suited for large lanthanides (La–Gd) due to the stabilisation of the ligand wrapping around the ion (see Fig. 7). Accurate molecular speciation studies in solution by means of mass spectrometry and NMR have evidenced the remarkably high yield (90%) of the thermodynamic formation of the heterometallic complex in the case of a La–Lu pair, having the largest ionic radius difference (ΔLa–Lu = 0.18 Å). For other lanthanide pairs, the size selectivity significantly decreases as the difference in ionic radii decreases. However, this size discriminating effect still remains the most relevant example reported in the literature so far in the case of one-pot syntheses. Unfortunately, ligand conformational flexibility, giving rise to head-to-head-to-head (HHH) and symmetrical head-to-head-to-tail (HHT) isomers, has hampered further investigations of these systems. Similar results have been more recently predicted by Piguet and his group, for the [LaLu(L9)]6+ complex with a tripodal ligand IX in Chart 1.
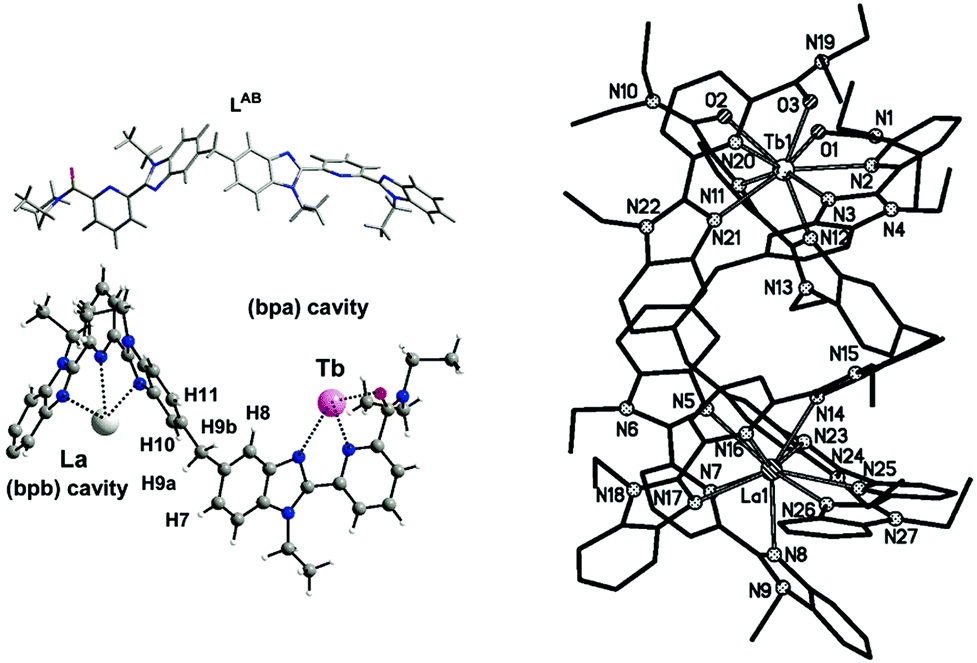 | ||
| Fig. 7 Left, the LAB ligand structure and coordination modes and right, the crystal structure of the [LaTb(LAB)3]6+ complex. Adapted with permission from ref. 22b. Copyright 2004. American Chemical Society. | ||
Very recently, relevant ion-size-driven control of molecular speciation has been reported in the case of the trimetallic [NdyErxYb3−(x+y)(Q)9] compound which shows multiple NIR emissions upon single-wavelength excitation of the Q ligand. Results obtained from XRD structural studies, EDX and ICP-mass analyses and ESI-mass spectrometry, have evidenced that the lighter and larger Nd3+ ion can be chemically discriminated from the heavier and almost “vicariant” Er3+ and Yb3+ ions and helps to control molecular speciation, leading to an approximately 90% formation of the heterometallic species [NdLn2(Q)9] (Ln = Er, Yb).7b
A second synthetic approach makes use of preformed lanthanide complexes with peripheral binding arms (see ligands X and XI, Chart 1) that are able to react with a second lanthanide ion:8,17,24
| Ln′Lx + Ln′′3+ → Ln′Lx ⊂ Ln′′ | (2) |
Following this route, a few examples of pure heterometallic species have been isolated and structurally characterised, such as the [Lu ⊂ (EuLT2)6](Otf)9 complex, with the terpyridine derivative ligand X (Chart 1) and the [(YbLa)LS(NO3)2]2·[La(NO3)5(H2O)] complex with the multidentate acyclic Schiff base ligand LS (XI, Chart 1).8a,17,24 However, an excess of the Ln′′3+ salt is often necessary to ensure product formation, leading to cocrystallisation of multiple species. Moreover, due to complex lability, dissociation and scrambling effects, mixtures of homo-and heterometallic species are likely to occur, and accurate studies of molecular speciation in solution (by NMR and mass spectrometry) are needed to ensure that the considered product is the stable thermodynamic one rather than a kinetic intermediate.4a,24
The evident limit of both of the described synthetic strategies is represented by the mandatory requirement of pairing lanthanide ions with a relevant difference between their ionic radii to reach for high selectivity. Unfortunately, the very rare examples available in the literature of highly discriminated, structurally characterised, heterometallic complexes, usually comprise optically and magnetically silent lanthanide ions such as La3+ and Lu3+, to take advantage of the extreme limits of ionic radii along the series (La3+ is the largest, first term of the Ln series whereas Lu3+ places right at the end being the heaviest and smallest lanthanide ion). This points out the stringent need to further develop suitable synthetic methods and ligands for achieving a better tunability of the selectivity properties with the aim to couple selected optically/magnetically active lanthanide ions in purely heterometallic assemblies that show programmed functionalities.
However, the investigation of the magnetic properties of heterolanthanide pairs embedded in the same coordination framework has so far been limited, whereas the intermetallic resonance energy transfer (RET) in luminescent compounds has attracted more attention in past years for the development of directional molecular light converters. In this regard, it has to be pointed out that, since energy transfer requires energy matching of excited levels, only selected couples of lanthanide ions can give rise to efficient interplay of optical properties. Among them, Tb → Eu and Yb → Er RET mechanisms have been studied the most, respectively in visible (Eu3+ 5D0→7FJ, 615 nm) and near-infrared (Er3+ 4I13/2→4I15/2, 1540 nm) luminescent materials (Fig. 8). This phenomenon can, in fact, be of particular importance for the enhanced excitation of poorly ligand-sensitised Ln ions or as a diagnostic tool for assessing intermetallic distances, for example for the investigation of protein structures.27
 | ||
| Fig. 8 Simplified resonance energy transfer mechanisms in the Tb → Eu and Yb → Er pairs. Dashed arrows represent non-radiative processes. | ||
The RET from a donor (Ln′) to an acceptor (Ln′′) is in fact a dipolar distance-dependent through space mechanism (Förster RET), whose effectiveness, ηRET, is mainly regulated through the interlanthanide separation R and the Förster radius R0 (which can be determined spectroscopically28 and can be calculated through use of the following equations:
 | (3) |
 | (4) |
It must be pointed out that, for an accurate assessment of interlanthanide RET efficiency, a close investigation of molecular speciation is crucial to determine the τRET value for purely heterometallic molecules. Among the few relevant examples reported in the literature, a remarkable Tb → Eu ηRET = 76% value was found for a triple-stranded helicate with a symmetrical derivative of ligand VIII in Chart 1, where the distance between the two lanthanide ions lies around 9 Å.22c Recently, a fully efficient Yb → Er RET at the molecular level has been demonstrated in trinuclear Yb2ErQ9 (Q = quinolinolate, ligand I, Chart 1), where the intermetallic separation is below 3.5 Å.7a The Er3+ sensitisation is in this case the result of a two step process involving ligand photoexcitation followed by efficient RET to Yb and then to Er. This latter example is of particular significance as lanthanide quinolinolates have been studied for a long time as luminescent active centers in optoelectronic devices and their processing potential is therefore well known.16b–d,25b See Fig. 9.
 | ||
| Fig. 9 Schematic representations of the light conversion in a Tb–Eu triple-stranded helicate (above) and in a trinuclear Yb2ErQ9 complex. Adapted with permission from ref. 22c and 7. Copyright 1993, 2013. American Chemical Society. | ||
As already pointed out in this paper, full disclosure of the optical or magnetic properties resulting in functional/multi-functional lanthanide systems requires the knowledge of their structural arrangement. Moreover, it is crystal clear that the final properties of the product, obtained following the above described synthetic approaches, will result from the independent contribution of the different species of which it consists, i.e. a superposition of the properties of hetero- and homometallic molecules weighted to their respective amounts in the sample.7b While purely heterometallic compounds are certainly advantageous for practical applications to ensure homogeneity and control of the emission signal at the molecular level, nonetheless programmed speciation of hetero- and homometallic mixtures (e.g. by varying reactant stoichiometry in the synthetic procedure) can be useful for achieving tunability of the different Ln emission intensities in broadband emitters. This latter approach can therefore be parallel to that described for multi-dimensional coordination polymers, with the additional advantage of the higher processability of small molecules.
D. Preformed coordinated units connected with a linker
The most effective strategy for affording purely heterometallic assemblies relies on multi-step synthetic procedures involving preformed coordinating units (Cn) connected with a covalent linker (link), according to the following proposed reaction pathways:29–31| Ln′C1 + linkC2 → (Ln′C1)linkC2 + Ln′′ → (Ln′C1)link(C2Ln′′) | (5) |
| (Ln′C1)link + (Ln′′C2) → (Ln′C1)link(C2Ln′′) | (6) |
The downside of these smart synthetic approaches, however, is that the completeness of the reaction leading to the formation of covalent bonding between the organic moieties cannot be effectively controlled. Therefore, a mixture of the desired heterometallic complex and the starting precursors is likely to result,31 and additional separation steps (e.g. by chromatography) are required to isolate the pure product. A variation of reaction schemes (5) and (6) is represented by sequential metallation/selective demetallation/metallation steps:32
| C1linkC2 + Ln′ → (Ln′C1)link(C2Ln′) → (Ln′C1)linkC2 + Ln′′ → (Ln′C1)link(C2Ln′′) | (7) |
This reaction pathway takes advantage of the decomplexation behaviour of the C2 unit under specific conditions, in that it is efficient enough to leave a coordination site free to accommodate a second Ln′′ cation, with a high yield.32
A few examples of these quite large and flexible assemblies prepared according to the above mentioned synthetic pathways are reported in Chart 2. Unfortunately, no X-ray diffraction studies are available so far, likely as a consequence of ligand fluxionality, so intermetallic distances can be only assessed through molecular structure modelling. In particular, interesting conclusions can be drawn from the structural arrangement of the Tb–Eu complex IV, Chart 2, on the basis of photophysical studies. In fact, estimation of the Tb → Eu RET efficiency (66%) from the experimental determination of the Tb3+ emission lifetime in the hetero- and in the corresponding homodimetallic complex (τRET and τ0, respectively, see eqn (3)), is in better accordance (eqn (4)) with a pseudo-endo molecular configuration (Fig. 10b) rather than a protracted arrangement (Fig. 10a), where the estimated intermetallic distance is much higher (7.8 Å and 17.8 Å for the two configurations, respectively).
 | ||
| Chart 2 | ||
 | ||
| Fig. 10 Structural models for complex IV, Chart 2 in protracted (a) and pseudo-endo (b) configurations. Reproduced with permission from ref. 31. Copyright 2011 American Chemical Society. | ||
In the case of the Tb–Eu complex III in Chart 2, an interesting emission spectral change upon the variation of solvent polarity is observed, i.e. enhancement of the Eu3+ luminescence intensity and the corresponding decrease of the Tb3+ emission when passing from water to alcohols. A possible explanation for this phenomenon may rely on molecule fluxionality and configurational changes leading to a shorter intermetallic separation in less polar solvents and consequently an enhanced Tb → Eu RET. This makes complex III a potential ratiometric probe for solvent polarity.
These flexible polynuclear molecules bearing specific coordination sites are therefore suitable for achieving, in principle, purely heterometallic assemblies through accurate ligand design and rational synthesis. However, the lack of the structural control over intermetallic distances and the coordination environments in these systems makes them ideal candidates when independent active sites in a single molecule are sought, for example for multiple emissive bioprobes, double-function MRI contrast agents and luminescent sensors, and catalysts with multi-active sites.
Conclusions
Heterolanthanide coordination compounds are extremely promising materials, with the potentiality of showing multi-functionality depending on the nature of the Ln ions and optical or/and magnetic properties, and are of interest for several applications. The rich and versatile chemistry of the f-elements allows the preparation of a variety of coordination edifices through ligand design and rational synthetic strategies. Three main classes of compounds have been selected on the basis of their structural arrangement, which determines the interplay or coexistence of the physical properties (related to the entity of metal separation), and their application as bulk materials or single-molecules.Multi-dimensional frameworks, where Ln ions are randomly distributed over the available coordination sites can best perform as “diluted” bulk materials where functionality results from a superposition of different properties related to non-interacting active metal centres. These kind of edifices have been successfully proposed as luminescent barcodes where tunability of emission lines can be achieved by varying the metal content.
Conversely, discrete and rigid heterometallic molecules offer the potential advantage of controlling intermetallic communication (magnetic coupling, resonance energy transfer) between Ln ions sitting at short distances and in fixed positions in the same, solution-processable, molecular architecture. A variety of magnetically active materials or directional light converters can be obtained in this way. However, in this context, the big chemical challenge is represented by severe limits in the discrimination of Ln ions, and synthetic approaches that often lead to the isolation of statistical mixtures of hetero- and homometallic species. Whereas these multi-speciated compounds can be nonetheless of interest for applications as bulk materials (e.g. as tunable broadband emitters in photonic devices), a step toward the isolation of purely heterometallic molecules can be made by taking into account a third class of coordination assemblies. These are usually large and flexible architectures consisting of preformed coordinating units, hosting different Ln ions, connected with an organic linker. Efficient discrimination of Ln ions can in principle be achieved through programmed synthesis and ligand design. However, the flexibility and fluxionality of these systems hamper the control of intermetallic communication. Therefore these assemblies can be proposed for applications where specific functionalities of independent active sites embedded in a single-molecule, can be exploited.
It is evident that the potentialities of heterometallic assemblies look well beyond the examples described in this paper, and a wide variety of (multi-) functional materials can be explored. This should stimulate the strive for a deeper understanding and further development of the complex coordination chemistry of the lanthanides. For this reason, a precise study, corroborated by robust experimental analyses on solid state and solution samples, of all of the equilibria involved in the selected reaction pathway, to reach a detailed picture of molecular speciation and therefore the reactivity of the different Ln ions, must not be neglected. Despite this point generally seeming to be only barely addressed in the literature, it is, nonetheless, of basic importance, not only to achieve fully characterised functional materials for application purposes, but also to provide a theoretical and experimental background for improving methodologies for the recovery and separation of these metals. This is of crucial importance to exploit the recycling of Ln-based end-life devices as a secondary source for these elements in view of their current “critical raw materials”1 status.
Acknowledgements
The Regione Autonoma della Sardegna (CRP-17571) and Banco di Sardegna Foundation are gratefully acknowledged for financial support.Notes and references
-
European Commission, Report on critical raw materials, May 2014
.
-
(a)
Rare Earth Coordination Chemistry, Fundamentals and Applications, ed. C. Huang, Wiley, 2010 Search PubMed
-
(a) M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496 CrossRef CAS PubMed
-
(a) C. Piguet and J.-C. G. Bünzli, Chem. Rev., 2002, 102, 1897 CrossRef PubMed
-
(a) A. Tsubouchi and T. C. Bruice, J. Am. Chem. Soc., 1994, 116, 11614 CrossRef CAS
-
(a) S. Floquet, N. Ouali, B. Bocquet, G. Bernardinelli, D. Imbert, J.-C. G. Bünzli, G. Hopfgartner and C. Piguet, Chem. – Eur. J., 2003, 9, 1860 CrossRef CAS PubMed
-
(a) F. Artizzu, F. Quochi, L. Marchiò, E. Sessini, M. Saba, A. Serpe, A. Mura, M. L. Mercuri, G. Bongiovanni and P. Deplano, J. Phys. Chem. Lett., 2013, 4, 3062 CrossRef CAS
-
(a) J.-P. Costes, F. Dahan, A. Dupuis, S. Lagrave and J.-P. Laurent, Inorg. Chem., 1998, 37, 153 CrossRef CAS
-
(a) R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703 CrossRef CAS PubMed
- Y. Lu and B. Yan, J. Mater. Chem. C, 2014, 2, 7411 RSC
- K. A. White, D. A. Chengelis, K. A. Gogick, J. Stehman, N. L. Rosi and S. Petoud, J. Am. Chem. Soc., 2009, 131, 18069 CrossRef CAS PubMed
- X. Fan, S. Freslon, C. Daiguebonne, G. Calvez, L. Le Pollès, K. Bernot and O. Guillou, J. Mater. Chem. C, 2014, 2, 5510 RSC
- P. R. Matthes, C. J. Höller, M. Mai, J. Heck, S. J. Sedlmaier, S. Schmiechen, C. Feldmann, W. Schnick and K. Müller-Buschbaum, J. Mater. Chem., 2012, 22, 10179 RSC
- A. M. Kaczmarek, K. Van Hecke and R. Van Deun, Inorg. Chem., 2014, 53, 9498 CrossRef CAS PubMed
- X. Meng, S.-Y. Song, X.-Z. Song, M. Zhu, S.-N. Zhao, L.-L. Wu and H.-J. Zhang, Inorg. Chem. Front., 2014 10.1039/x0xx00000x
-
(a) L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487 CrossRef CAS PubMed
- H. B. Xu, J. G. Deng, L. Y. Zhang and Z. N. Chen, Cryst. Growth Des., 2013, 13, 849 CAS
- F. Tanaka and T. Ishibashi, J. Chem. Soc., Faraday Trans., 1996, 92, 1105 RSC
-
(a) L. Song, Q. Wang, D. Tang, X. Liu and Z. Zhen, New J. Chem., 2007, 31, 506 RSC
-
(a) K. D. Matthews, I. A. Kahwa and D. J. Williams, Inorg. Chem., 1994, 33, 1382 CrossRef CAS
-
(a) R. C. Howell, K. V. N. Spence, I. A. Kahwa, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 1996, 961 RSC
-
(a) N. André, R. Scopelliti, G. Hopfgartner, C. Piguet and J.-C. G. Bünzli, Chem. Commun., 2002, 214 RSC
-
(a) P. E. Ryan, G. Canard, S. Koeller, B. Bocquet and C. Piguet, Inorg. Chem., 2012, 51, 10012 CrossRef CAS PubMed
- X.-Y. Chen, Y. Bretonnière, J. Pécaut, D. Imbert, J.-C. G. Bünzli and M. Mazzanti, Inorg. Chem., 2007, 46, 625 CrossRef CAS PubMed
-
(a) S. Petoud, J.-C. G. Bünzli, F. Renaud, C. Piguet, K. J. Schenk and G. Hopfgartner, Inorg. Chem., 1997, 36, 5750 CrossRef CAS PubMed
-
F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, 5th edn, Wiley, N.Y., 1988, ch. 20, p. 21 Search PubMed
- D. Chaudhuri, W. e W. Horrocks Jr., J. C. Amburgey and D. J. Weber, Biochemistry, 1997, 36, 9674 CrossRef CAS PubMed
- Experimental R0 values reported in the literature are usually in the range 7–10 Å.
- S. Faulkner and S. J. A. Pope, J. Am. Chem. Soc., 2003, 125, 10526 CrossRef CAS PubMed
- M. P. Placidi, A. J. L. Villaraza, L. S. Natrajan, D. Sykes, A. M. Kenwright and S. Faulkner, J. Am. Chem. Soc., 2009, 131, 9916 CrossRef CAS PubMed
- D. J. Lewis, P. B. Glover, M. C. Solomons and Z. Pikramenou, J. Am. Chem. Soc., 2011, 133, 1033 CrossRef CAS PubMed
- M. S. Tremblay and D. Sames, Chem. Commun., 2006, 4116 RSC
| This journal is © the Partner Organisations 2015 |