
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCholesterol-coated gold nanorods as an efficient nano-carrier for chemotherapeutic delivery and potential treatment of breast cancer: in vitro studies using the MCF-7 cell line
Nouf N. Mahmoud *a,
Dima A. Sabbaha,
Rana Abu-Dahabb,
Duaa Abuarqoubc,
Maha Abdallahb,
Ameerah (Hasan Ibrahim)a and
Enam A. Khalilb
*a,
Dima A. Sabbaha,
Rana Abu-Dahabb,
Duaa Abuarqoubc,
Maha Abdallahb,
Ameerah (Hasan Ibrahim)a and
Enam A. Khalilb
aFaculty of Pharmacy, Al-Zaytoonah University of Jordan, Amman 11733, Jordan. E-mail: nouf.mahmoud@zuj.edu.jo
bSchool of Pharmacy, The University of Jordan, Amman 11942, Jordan
cCell Therapy Center, The University of Jordan, Amman 11942, Jordan
First published on 24th April 2019
Abstract
Gold nanorods (GNRs) have a recognized role in treatment of cancers as efficient nanocarriers for chemotherapeutic drug delivery. In this study, GNRs modified with cholesterol-PEG were employed as a nanocarrier for a hydrophobic compound having a promising phosphatidylinositol 3-kinase (PI3Kα) inhibitory activity. The acquired nanocomplex was characterized by optical and infra-red (IR) absorption spectroscopies, in addition to hydrodynamic size and zeta potential. Glide docking and superposing of docked poses of the hydrophobic ligand and cholesterol moiety demonstrated that hydrophobic interactions drive the conjugation and attachment of the ligand to the cholesterol moiety of the nanocarrier. In vitro release study under a cellular environment indicates that the presence of cells has enhanced the release and the cellular uptake of the conjugated ligand. Furthermore, the anti-proliferative assay of the nanocomplex revealed potent cytotoxicity over a low concentration range of the nanocomplex against MCF-7 breast cancer cells compared to the free compound or the nanocarrier alone. Analysis of cellular death modality by flow cytometry showed that the nanocomplex has a rapid effect on cell death, as cells went toward the late apoptotic/necrotic stage rapidly and proportionally to the increase of the nanocomplex concentration. The overall results propose that cholesterol-decorated GNRs could be considered as a promising nanocarrier for hydrophobic drugs to achieve efficient delivery and potential therapy against breast cancer cells.
1. Introduction
Breast cancer is one of the major health burdens despite the rapid progress in cancer research and therapy.1 Classical chemotherapy is the first line of cancer treatment using cytotoxic chemotherapeutic agents that interfere with the process of cell division. However, the severe toxicity to normal tissues, such as hair loss, bone marrow toxicity, nausea and vomiting, in addition to poor bioavailability and targeting potentials, often limit the effectiveness of chemotherapy against cancers.2,3 Combined chemotherapy of two or more anticancer drugs has been extensively exploited to treat a variety of cancers. However, the toxicity of such combinations is still unreasonable.Failure of numerous chemotherapeutic agents coupled with growing the understanding of cancer pathology urge the development of novel and targeted therapeutic platforms with minimal toxicity and resistance.
Nanoparticle-based approaches are distinctly attractive for breast cancer therapy, diagnosis and drug delivery owing to their tiny size, capability of surface modification using a variety of small ligands and biomacromolecules, and ability to accumulate into tumors owing to the enhanced permeability and retention effect.4,5
The employment of nanocarriers as drug delivery is a key strategy for treatment of cancers. Several types of nano-systems have been extensively investigated for chemotherapy delivery on a more effective and less toxic fashion, including polymers, liposomes, dendrimers, solid lipid and inorganic nanoparticles.6–11
Utilizing gold nanoparticles (GNPs) as delivery nano-systems is substantially examined in the literature due to ease of surface modifications and unique optical properties.12–14 GNPs of diverse sizes and surface chemistries were used for delivery of several drugs and biomacromolecules.15–17 For example, TAT peptide-functionalized GNPs were used to improve delivery and reduce toxicity of doxorubicin towards breast cancer metastasis to brain.18 Furthermore, conjugation of quercetin to GNPs has induced cell apoptosis in two cell lines of breast cancer.19
Although covalent conjugation of GNPs to thiolated ligands provides a considerable stability of the conjugates and produces a controlled release of the cargo, such a slow release may produce challenges for actual clinical applications.20 Accordingly, non-specific binding between nanocarriers and payloads is becoming a more efficacious approach for competent drug delivery and massive cellular uptake.20
In this work, a simple gold-based nano-platform was designed as a nanocarrier for a hydrophobic ligand into MCF-7 breast cancer cells. GNRs were synthesized using a low concentration of cetyl trimethylammonium bromide (CTAB) and their surfaces were modified and stabilized by a cholesterol moiety linked to a thiolated poly ethylene glycol (PEG) in order to enhance its colloidal stability and aqueous dispersibility. The modified GNRs (Chol-GNRs) were utilized to conjugate a small hydrophobic molecule, as a chemotherapeutic drug model, by non-covalent hydrophobic interactions with the hydrophobic pocket of the nanocarrier (the cholesterol moiety). Such non-covalent binding is supposed to enhance the delivery of the payload upon exposure to cells and ultimately will accelerate its cytotoxicity.
The hydrophobic compound used in this study is N-(4-hydroxyphenyl)4-hydroxy-2-quinolone-3-carboxamide (Compound A), which is one of a new series of N-substituted-4-hydroxy-2-quinolone-3-carboxamides that were synthesized, characterized, and biologically evaluated previously in our lab, and they displayed a promising phosphatidylinositol 3-kinase (PI3Kα) inhibitory activity.21 PI3Kα is responsible for synthesis of triphosphates that induce cell growth and division.22,23 In this study, Chol-GNRs were conjugated to Compound A and the obtained “nanocomplex” was characterized in terms of ultraviolet-visible (UV-vis) and infra-red (IR) absorption spectroscopies, hydrodynamic size, surface charge and in vitro release behavior. The cytotoxicity of the nanocomplex on MCF-7 breast cancer cell line and human dermal fibroblasts was investigated and the cellular death modality of the designed nanocomplex was assessed using flow cytometry assay.
2. Experimental part
2.1 Chemical synthesis of GNRs solution
GNRs solution was synthesized using a mixture of CTAB (Sigma-Aldrich Chemicals, USA) and sodium oleate (NaOL, Sigma-Aldrich Chemicals, USA) following a previous protocol with slight modifications.24 Briefly, seeds were prepared by adding sodium borohydride (NaBH4, 99%), (Sigma-Aldrich Chemicals, USA) to a stirred solution of CTAB. A volume of 18.0 mL (4.0 mM) of silver nitrate (AgNO3, 99%, Sigma-Aldrich Chemicals, USA) was transferred to a growth solution of CTAB and NaOL and kept under stirring for 15 min. Chloroauric acid (250 mL, 1.0 mM) (HAuCl4·3H2O, 99.9%, Sigma-Aldrich Chemicals, USA) was transferred to the growth solution and the pH of the solution was adjusted by HCl. Ascorbic acid (99%, Sigma-Aldrich Chemicals, USA), and the seeds (0.8 mL) were added to the growth solution and the obtained mixture was kept undisturbed at 30 °C for 24 h. GNRs solution was spun down twice by centrifugation (Centrifuge, Labofuge I, Heraeus Christ, Germany) for 10 min at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 580 × g, suspended in Milli-Q water and stored at 4 °C.
580 × g, suspended in Milli-Q water and stored at 4 °C.
2.2 Surface modification of GNRs with cholesterol-polyethylene glycol-thiol (Chol-PEG-SH); Chol-GNRs
A volume of 1.0 mL (25.0 mg mL−1) of Chol-PEG-SH solution (MW ∼ 2000 g mol−1, Nanosoft Polymers, USA) was added to a solution of GNRs (10.0 mL) and kept under stirring overnight. The obtained modified GNRs solution was spun down by centrifugation twice for 10 min at 11510 × g and were stored at 4 °C.
2.3 Surface modification of GNRs with methoxy-polyethylene glycol-thiol (m-PEG-SH); PEG-GNRs
A volume of 1.0 mL of thiolated m-PEG solution (10.0 mg mL−1) (MW ∼ 2000 g mol−1, Sigma-Aldrich Chemicals, USA) was added to a solution of GNRs (10.0 mL) and kept under stirring overnight. The functionalized GNRs solution was spun down by centrifugation twice for 10 min at 11510 × g and were stored at 4 °C.
2.4 Conjugation of Compound A into Chol-GNRs and PEG-GNRs to obtain Chol-A-GNRs and PEG-A-GNRs, respectively
Compound A was synthesized in-house and fully characterized according to a previous published protocol.21 For conjugation of Compound A to the hydrophobic moiety of Chol-GNRs or PEG-GNRs, 10 μL of Compound A stock solution (50 mg mL−1 of dimethyl sulfoxide (DMSO)) was added to each 1.0 mL of Chol-GNRs or PEG-GNRs (2.0 nM), and the solution was kept under stirring for 4 h at room temperature. The nanocomplex solution was then spun down by centrifugation twice at 11510 × g for 8 min to remove the unbound Compound A, and was suspended in phosphate buffer saline (PBS, pH 7.4).
2.5 Characterization of GNRs, PEG-GNRs and cholesterol-coated GNRs; Chol-GNRs
Chol-GNRs were characterized by optical absorption spectroscopy (UV-1800 spectrophotometer, Shimadzu, Japan), dynamic light scattering (DLS) and zeta potential (Nicomp Nano Z3000 zeta potential/particle size analyzer, CA, USA), in addition to transmission electron microscope (TEM) imaging (FEI Morgani 268, operating voltage of 60 kV, Holland).2.6 Characterization of the nanocomplexes; Chol-A-GNRs and PEG-A-GNRs
Chol-A-GNRs were characterized by optical absorption spectroscopy, DLS, effective surface charge and by Fourier-transform infrared (FTIR) spectroscopy (Shimadzu FTIR Affinity spectrophotometer, Japan). Potassium bromide (Acros, Belgium) was used to prepare the sample discs for FTIR spectroscopy. PEG-A-GNRs were characterized by optical absorption spectroscopy, DLS and effective surface charge.The amount of the conjugated Compound A (μg) was estimated using a well validated UV-vis absorption spectroscopy method. A standard calibration curve of Compound A was obtained by measuring the UV-vis absorbance of known concentrations of Compound A in PBS (pH 7.4) at 297 nm.
2.7 Glide docking of Compound A and cholesterol moiety
To explore the interaction of cholesterol moiety and Compound A, we performed Glide docking approach25–27 against homology modeled28 structure of phosphoinositide-3 kinase (PI3Kα) (PDB ID: 2RD0).29 The structure was treated and energy minimized using the Protein Preparation and Macro Model wizards in Schrödinger27 software to maximize H-bond interactions and reduce the steric clashes, respectively. Additionally, 3D models of cholesterol and Compound A were built using MAESTRO27 Build module and energetically minimized by the Macro Model panel recruiting the OPLS2005 force field.2.8 In vitro release study of Compound A from the nanocomplex (Chol-A-GNRs) in PBS or tissue culture medium
A volume of 1.0 mL of the nanocomplex (80 μg mL−1 of gold) were transferred into a dialysis bag (MWCO 12–14 kD, Spectrum Lab, USA) which was placed in a glass vial containing 50.0 mL of PBS (pH 7.4) or tissue culture medium; Roswell Park Memorial Institute medium (RPMI) 1640 medium (Euroclone, Europe) in 0.1% Tween 80 to facilitate the solubility of the compound. Then, the vials were placed into a shaking water bath (37 °C, 120 rpm) (GFL 1083, Burgwedel, Germany). The same experiment was conducted for the nanocarrier alone (Chol-GNRs) to exclude any interference.A volume of 0.5 mL of the release media was removed at specific time intervals (0.5, 1, 2, 3, 4, 5, 6, 24, 48 and 72 h) and replaced with a fresh PBS (pH 7.4) or tissue culture medium to keep the sink condition. The concentration (μg mL−1) and subsequently the amount (μg) of Compound A in the removed media was estimated by measuring the optical absorbance at each time point at 297 nm against a standard calibration curve of Compound A in PBS (pH 7.4) or tissue culture medium containing Tween 80 (0.1%). The release profile of Compound A was presented as percentage of released cumulative amount vs. time. The results represent the average of four independent experiments.
2.9 In vitro release study of Compound A from the nanocomplex (Chol-A-GNRs) under cellular environment
In order to predict the effect of cells on the release pattern of Compound A from the nanocomplex, the release study was performed under cellular environment. A density of 2 × 105 of MCF-7 cells were seeded into 6-well plates in 3.0 mL of tissue culture medium (RPMI) for each well and kept attaching for 48 h (in three replicates). Then, the freshly prepared dilution of Chol-A-GNRs in the cell culture medium without FBS to appropriate concentrations of the nanocomplex (20.0 μg mL−1 of gold) was immediately added to the cells. Untreated cells were considered as control in each experiment.The concentration of Compound A (μg mL−1) in the supernatant of each treatment vs. control (untreated cells) at different time intervals (60, 120 and 240 min) was estimated by measuring the optical absorbance of the supernatants at 297 nm after centrifuging the media for 8 min at 11510 × g. The amount of Compound A released into the tissue culture medium under cellular environment was estimated against a standard calibration curve of the compound.
2.10 Anti-proliferative activity against MCF-7 and human dermal fibroblasts cell lines
2.11 Quantification of the cellular uptake of Chol-GNRs and PEG-GNRs into MCF-7 breast cancer cell line and human dermal fibroblasts by ICP-OES
A density of 2 × 106 cells of MCF-7 were seeded into 25 cm2 flasks in 12.0 mL tissue culture media and kept attaching for 48 hours. Then, the freshly prepared dilutions of PEG-GNRs or Chol-GNRs in tissue culture medium (20.0 μg mL−1 of gold) without FBS were added to the cells and incubated for 3 h. After two washing steps with PBS, the cells were trypsinized and collected after centrifugation for 30 min at 4 °C. The collected pellets were digested with fresh aqua regia (HNO3 and HCl; 1:3) and kept in a water bath (60–70 °C) for 3 h, then, diluted with Milli-Q water up to 4.0 mL, filtered by Teflon syringe filter (0.22 μm) and analyzed by ICP-OES (Optima 2000 DV, ICP-OES, PerkinElmer, USA). Untreated cell were presented as control in each experiment. The same previous cellular uptake experiment of Chol-GNRs and PEG-GNRs was performed using human dermal fibroblasts. The experiments were done in triplicate.
Gold was quantified at 242.795 nm and the concentration of gold was estimated against a standard calibration curve of gold standard for ICP (1000 ppm, Sigma-Aldrich Chemicals, USA) (0.1–10.0 ppm). The measurements were performed in triplicate.
2.12 Cell death analysis by flow cytometry
MCF-7 cells (4 × 105 cells) were seeded into 6-well plates, then they were treated with Chol-A-GNRs (4.0 and 8.0 μg mL−1 of gold). After 24 h, the cells were collected by trypsin EDTA 0.25%, washed with PBS, and the obtained pellets were stained with annexin V/PI dye according to the manufacturer's instructions. Doxorubicin (1.0 μM, Doxil) was used as a positive control. Cell death modality, either apoptosis or necrosis, was analyzed by fluorescein activated sorter FACS Canto II, Becton Dickinson (BD) Biosciences, USA.
2.13 Statistical analysis
Statistical analysis was conducted by unpaired t-test by GraphPad Prism version 7.0. Results are considered significant when p < 0.05.3. Results and discussion
3.1 Chemical synthesis and characterization of GNRs and Chol-GNRs
In this study, GNRs were synthesized, characterized and modified with a cholesterol moiety linked to a thiolated-PEG moiety (Fig. 1A and C). Such gold-based nanocarrier was employed as a template to conjugate a hydrophobic chemical molecule, as a hydrophobic chemotherapeutic drug model (Compound A) (Fig. 1B) into the hydrophobic pocket of cholesterol-decorated gold nanocarrier.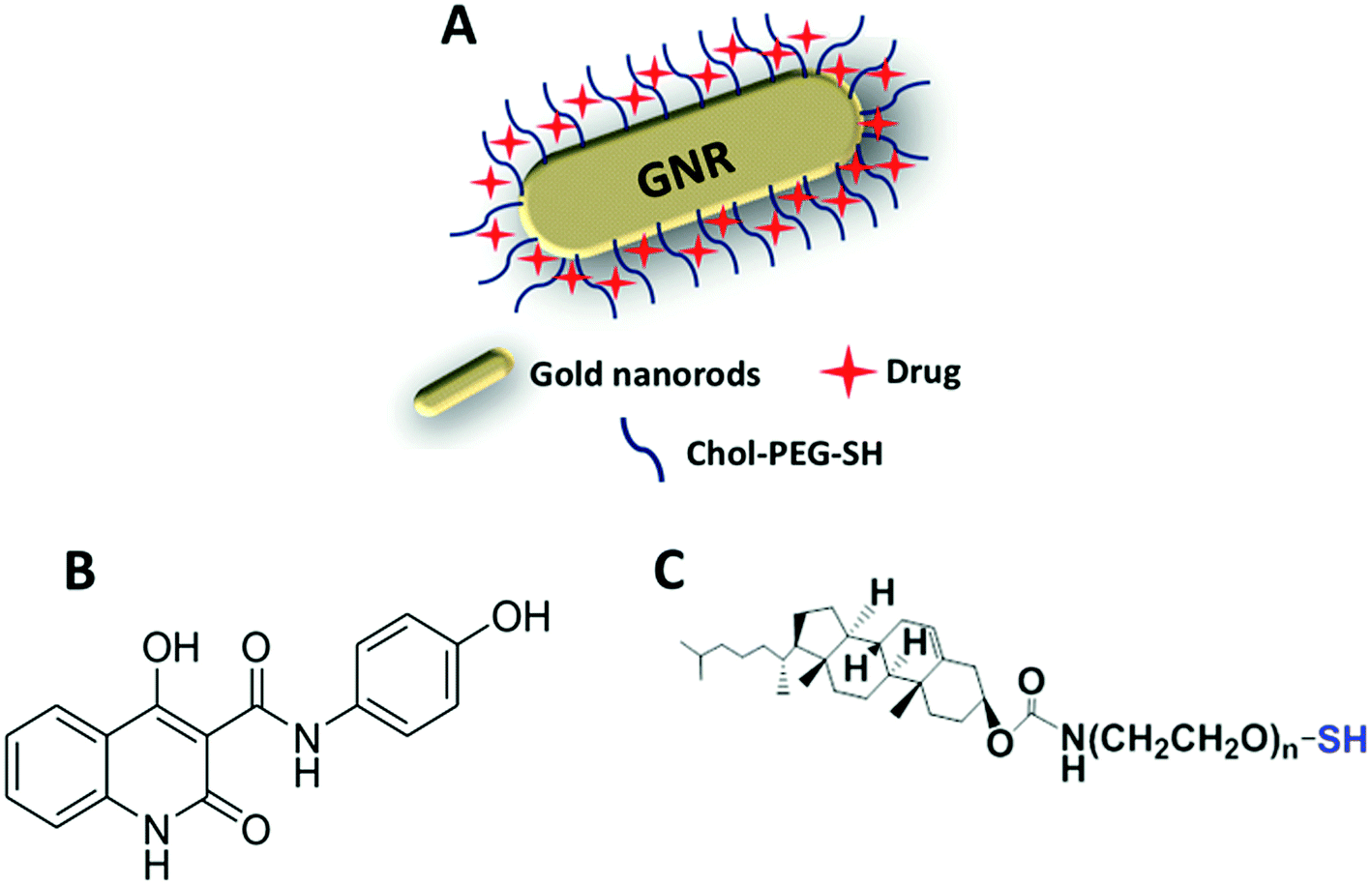 | ||
| Fig. 1 Illustration demonstrates the nanocomplex (A) of Chol-GNRs conjugated with Compound A (B) by non-covalent hydrophobic interactions Compound A and the cholesterol moiety that linked to a thiolated PEG (C). | ||
The method used in this study to prepare GNRs was based on utilizing a mixture of CTAB and oleic acid instead of CTAB alone to acquire GNRs with an aspect ratio (AR) of ∼4. The prepared GNRs has a low concentration of CTAB which has a well-documented cytotoxicity.30,31 The concentration of CTAB that bound to the surface of GNRs was further reduced by effective surface modification with a cholesterol moiety that was linked to a thiolated PEG. PEG has a pivotal role in enhancing the colloidal stability of the nanoparticles upon cellular exposure since it retards the non-specific adsorption of biomacromolecules by steric repulsion in addition to enhancing the aqueous dispersibility of Chol-PEG-SH in water.32 Besides, the PEG moiety extends blood circulation time, and ultimately increases specific tumor accumulation via the EPR effect.33 On the other hand, the presence of thiol in the molecule facilitated the effective displacement of the surface bound CTAB by cholesterol moiety via a strong covalent bond between thiol and gold (S–Au).34
The synthesized GNRs using a mixture of CTAB and oleic acid displayed a typical optical absorption profile with a small transverse peak and an intense longitudinal peak at ∼530 nm and ∼850 nm, respectively (Fig. 2A). After surface modification of GNRs with PEG-SH or Chol-PEG-SH moiety, a small shift of the longitudinal peak was observed, however, no significant tailing or broadening of the plasmon peaks was reported, indicating a good colloidal stability of the cholesterol-modified GNRs (Fig. 2A). Furthermore, an extra optical absorption peak was observed for GNRs functionalized with cholesterol moiety at ∼250 nm, that is corresponding to the optical absorption peak of Chol-PEG-SH dispersed in water (Fig. 2A). Moreover, the hydrodynamic size of GNRs upon surface modification with Chol-PEG-SH was increased to ∼64 nm and the effective surface charge was reduced to an almost neutral for both PEGylated and cholesterol-modified GNRs which confirm the successful replacement of the surface-bound CTAB with PEG or cholesterol-PEG-SH moieties (Fig. 2C). The shape and size of the synthesized GNRs and Chol-GNRs were further verified by TEM imaging; Fig. 2D shows a representative TEM image of GNRs having a rod-like shape nanoparticles of average length and width of ∼52.1 and ∼12.5 nm, respectively and an average corresponding aspect ratio (AR) of ∼4. The shape and size of GNRs upon conjugation with Chol-PEG-SH were verified by TEM imaging which revealed a rod-like shape nanoparticles having average length and width of ∼54.1 nm and ∼12.4 nm, respectively, and an average corresponding aspect ratio (AR) of ∼4 (Fig. 2E).
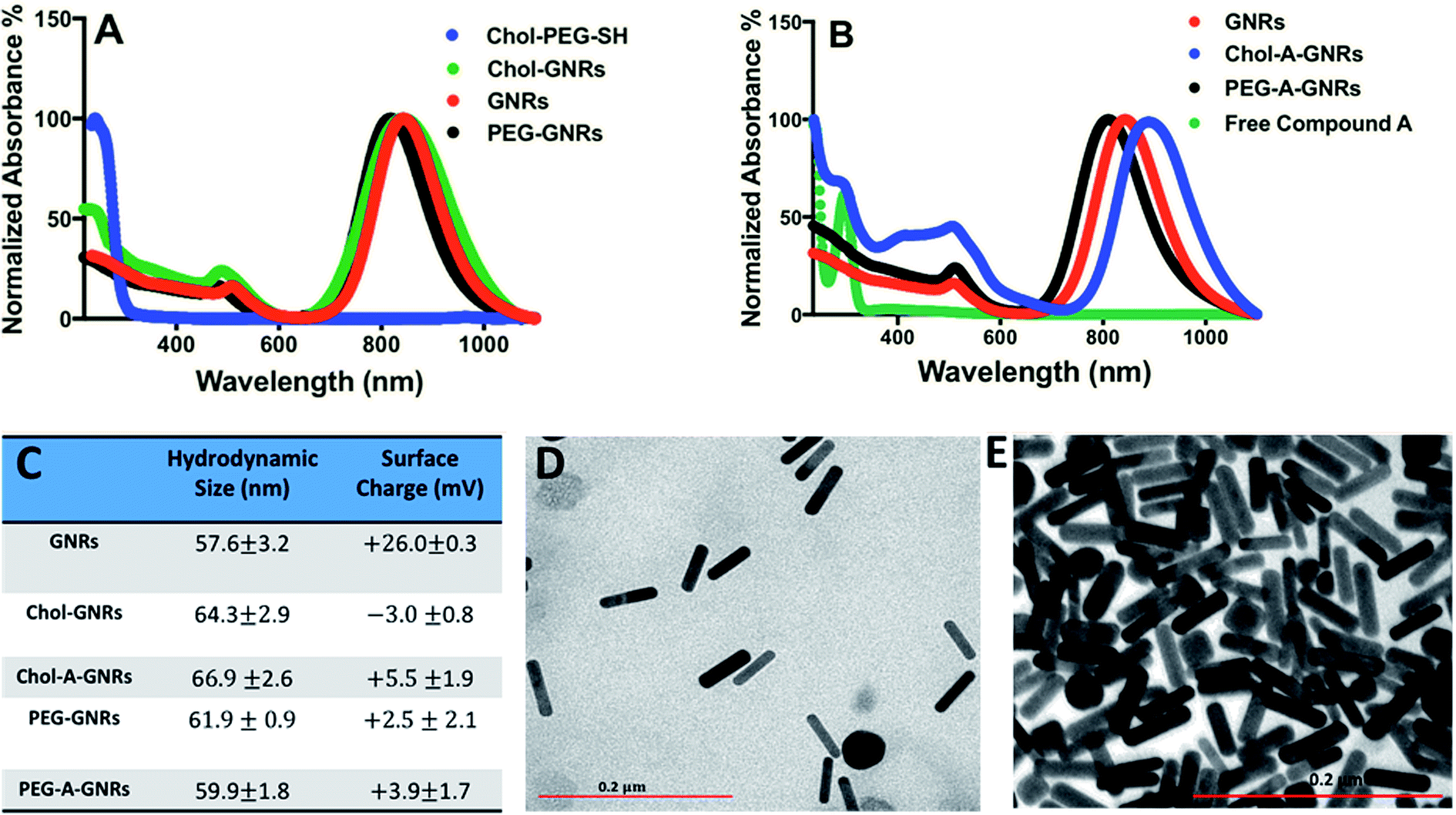 | ||
| Fig. 2 Characterization of GNRs, Chol-GNRs, PEG-GNRs, Chol-A-GNRs and PEG-A-GNRs. (A) Optical absorption spectra of GNRs, Chol-GNRs, PEG-GNRs and Chol-PEG-SH dispersed in water. An extra peak was observed in the optical spectrum of Chol-GNRs at ∼250 nm which confirms the successful surface modification. (B) Optical absorption spectra of GNRs, Compound A in PBS, Chol-A-GNRs and PEG-A-GNRs. An extra absorption peak was observed at ∼297 for Chol-A-GNRs which confirms the successful conjugation of Compound A to Chol-GNRs. (C) Hydrodynamic sizes and effective surface charges of GNRs, Chol-GNRs, Chol-A-GNRs, PEG-GNRs and PEG-A-GNRs. The size of Chol-GNRs was increased due to functionalization with Chol-PEG-SH, however, no significant change in the size and charge of Chol-A-GNRs upon conjugation with Compound A. (D) TEM image of GNRs that confirms a rod-shape of the nanoparticles having average length and width of ∼52.1 nm and ∼12.5 nm, respectively, and an AR of ∼4. (E) TEM image of Chol-GNRs that confirms the rod-shape of the nanoparticles having average length and width of ∼54 nm and ∼12.5 nm, respectively, and an AR of ∼4. | ||
3.2 Conjugation of Compound A to the surface of Chol-GNRs or PEG-GNRs and characterization of the “nanocomplexes”; Chol-A-GNRs and PEG-A-GNRs
Hydrophobic drugs are the most commercially available drugs. However, loading hydrophobic drugs to aqueous nano-systems is considered as a real challenge. Conjugation of a hydrophobic molecule into the hydrophobic pocket of nano-system mediates the proper release of the loaded compound through hydrophobic interactions of the compound with the hydrophobic sites of the cellular membrane.20In the current study, the synthesized Chol-GNRs were used as a nanocarrier of a small hydrophobic molecule; Compound A, that was synthesized previously by our research group by the structure-based drug design as a targeted agent for treatment of cancers such as colon and breast cancers by inhibiting PI3Kα. This study presents a facile and simple method for efficient conjugation of Compound A into the hydrophobic pocket of cholesterol-modified GNRs. Compound A has an intense optical absorption peak at ∼297 nm (Fig. 2B). Chol-GNRs conjugated to Compound A demonstrated typical transverse and longitudinal optical absorption peaks without significant broadening or tailing of the peaks, in addition to another peak at ∼297 nm that is corresponding to the conjugated compound (Fig. 2B).
PEG-A-GNRs showed no characteristic optical absorption peak at ∼297 nm, suggesting its poor ability to conjugate the hydrophobic drug (Fig. 2B).
The obtained nanocomplex (Chol-A-GNRs) was characterized by their hydrodynamic size and effective surface charge. Conjugation of Compound A to Chol-GNRs was resulted in slight change in hydrodynamic size and similar surface charge compared to that of Chol-GNRs (Fig. 2C). However, PEG-A-GNRs demonstrated no significant change in the hydrodynamic diameter upon conjugation with Compound A (Fig. 2C).
The amount of Compound A conjugated to Chol-GNRs was estimated using a validated optical absorption spectroscopy method against a standard calibration curve of known concentrations of Compound A in PBS. We found that for each 1.0 mL of Chol-GNRs (2.0 nM), ∼202 μg ± 4.2 μg of Compound A was conjugated to the hydrophobic region of Chol-GNRs and the drug loading percentage on Chol-GNRs was around 41% relative to the initial amount of the drug.
On the other hand, ∼60 μg ± 9.2 μg of Compound A were conjugated into PEG-GNRs, and the drug loading percentage was around 12% relative to the initial amount of drug. These results indicate the crucial role of cholesterol moiety in conjugating the hydrophobic drug into GNRs by hydrophobic interactions between their similar hydrophobic core structures.
Furthermore, the successful surface modification of GNRs with cholesterol moiety and Compound A was evaluated by comparing the FTIR spectra of the free Compound A and Chol-GNRs to that of the nanocomplex (Chol-A-GNRs). The detailed description of FTIR spectrum of the Compound A was described previously.21 Briefly, FTIR spectrum of Compound A showed the following characteristic bands: 3340 (–OH stretching), 3078 cm−1 (–CH stretching), 2870–2816 cm−1 (–NH stretching), 1913 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching), 1658 cm−1 (–CO amide in ring stretching), 1612 cm−1 (–CO external amide stretching), 1404 cm−1 (–C–O stretching), 1234 cm−1 (–C–C stretching) (Fig. 3). The nanocarrier alone (Chol-GNRs) demonstrated the following characteristic bands: 3325 cm−1 (–CH stretching), 2870 cm−1 (–NH stretching), 1714 cm−1 (–CO ester stretching), 1110 cm−1 (–C–O stretching) (Fig. 3). On the other hand, the following characteristic bands of the nanocomplex (Chol-A-GNRs) clearly confirm the successful decoration of GNRs with both cholesterol moiety and Compound A: 3336 cm−1 (–OH stretching), 2860–2920 cm−1 (–NH stretching), 1690 cm−1 (–CO ester stretching), 1650 cm−1 (–CO amide in ring stretching), 1608 cm−1 (–CO external amide stretching) and 1221 cm−1 (–C–O stretching) (Fig. 3). FTIR spectra of Compound A, Chol-GNRs and Chol-A-GNRs revealed similarities in the absorption peaks with slight shifts suggesting successful conjugation of Compound A to the cholesterol moiety of GNRs by hydrophobic interactions occurred between their structural backbones (Fig. 3).
C stretching), 1658 cm−1 (–CO amide in ring stretching), 1612 cm−1 (–CO external amide stretching), 1404 cm−1 (–C–O stretching), 1234 cm−1 (–C–C stretching) (Fig. 3). The nanocarrier alone (Chol-GNRs) demonstrated the following characteristic bands: 3325 cm−1 (–CH stretching), 2870 cm−1 (–NH stretching), 1714 cm−1 (–CO ester stretching), 1110 cm−1 (–C–O stretching) (Fig. 3). On the other hand, the following characteristic bands of the nanocomplex (Chol-A-GNRs) clearly confirm the successful decoration of GNRs with both cholesterol moiety and Compound A: 3336 cm−1 (–OH stretching), 2860–2920 cm−1 (–NH stretching), 1690 cm−1 (–CO ester stretching), 1650 cm−1 (–CO amide in ring stretching), 1608 cm−1 (–CO external amide stretching) and 1221 cm−1 (–C–O stretching) (Fig. 3). FTIR spectra of Compound A, Chol-GNRs and Chol-A-GNRs revealed similarities in the absorption peaks with slight shifts suggesting successful conjugation of Compound A to the cholesterol moiety of GNRs by hydrophobic interactions occurred between their structural backbones (Fig. 3).
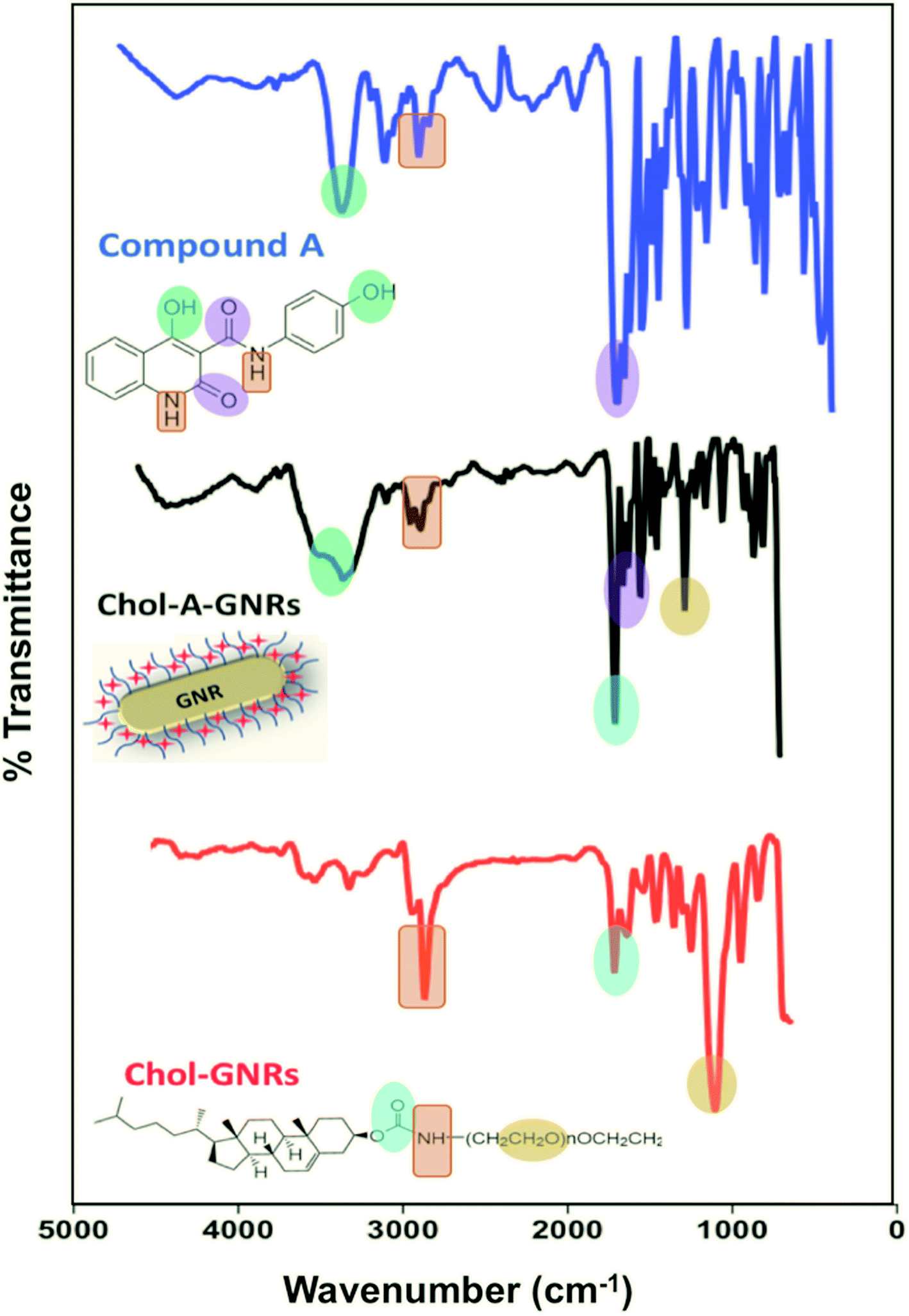 | ||
| Fig. 3 Infra-red spectra of Compound A, Chol-GNRs and Chol-A-GNRs revealed similarities in the core structures' peaks with slight shifts suggesting successful conjugation of Compound A to the cholesterol moiety of GNRs by hydrophobic interactions occurred between their structural backbones. | ||
3.3 Glide docking and superposing of docked poses of Compound A and cholesterol moiety
In order to understand the prospective interaction between Compound A and the cholesterol moiety, we carried out a Glide docking approach25–27 against the active domain of phosphoinositide-3-kinase α (PI3Kα) (PDB ID: 2RD0) (Fig. 4A and B) and superposed the docked poses of cholesterol and Compound A (Fig. 5A–C). The superposing tactic illustrates that hydrophobic interaction drives the attachment of Compound A to cholesterol. Fig. 5A–C show overlapping of hydrophobic motifs of both compounds in different positions. The values of root mean square distance of superposed atoms in both compounds range from 2.803–2.820 Å; these values infer that overlaying of similar core structures might presides hydrophobic interaction, and the hydrophobic backbones of both cholesterol and Compound A enhance their binding interaction.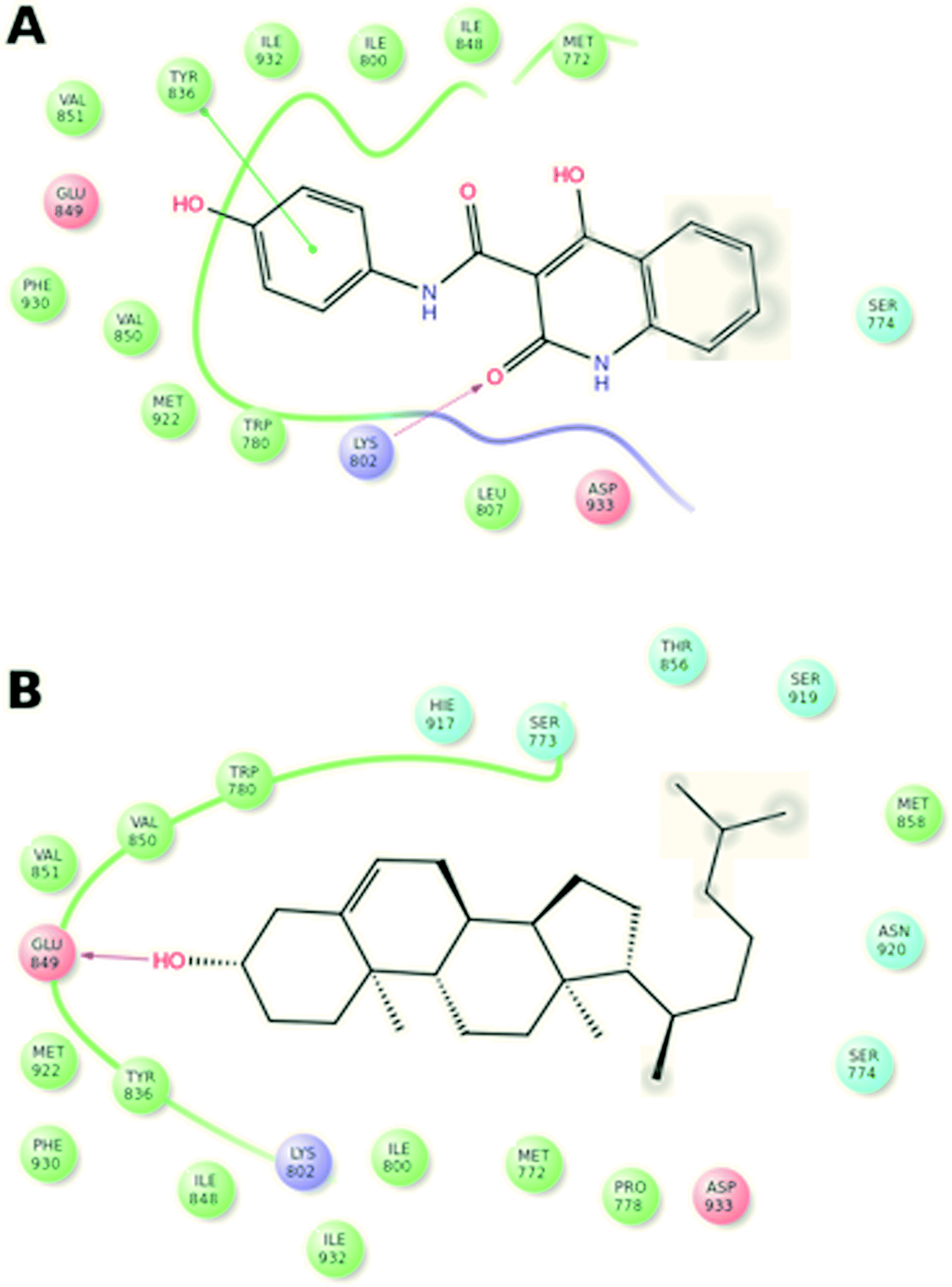 | ||
| Fig. 4 Glide docking and complex formation of Compound A/PI3Kα (A) and cholesterol/PI3Kα (B). | ||
 | ||
| Fig. 5 Superposing of docked poses of cholesterol and Compound A in 3D models. The best docked poses of Compound A and cholesterol are extracted and overlaid to each other. The hydrophobic interaction drives the attachment of Compound A to cholesterol moiety where hydrophobic carbon backbone motifs match their peers in each compound due to their similar hydrophobic core structures. Cholesterol is depicted in pink (A), yellow (B) and white color (C). Compound A carbon atoms are represented in green color, blue color for nitrogen atoms (N), and red color for oxygen (O). | ||
3.4 In vitro release of Compound A from the nanocomplex; Chol-A-GNRs in PBS, tissue culture medium and under cellular environment
The release pattern of Compound A that was conjugated to Chol-GNRs nano-system was studied in both PBS (pH 4.7) and the tissue culture medium (RPMI 1640). As shown in Fig. 6A, no burst release of the drug was observed and less than 45% of the drug was released over the first 6 h. However, the release was much slow afterward and reached an almost complete release after 72 h (Fig. 6A). On other hand, the release pattern of Compound A in the tissue culture medium was similar to that in PBS, however, it was slightly faster and reached ∼100% after 48 h (Fig. 6A). The release behavior of the drug in both PBS and the tissue culture medium reflects the strength of the hydrophobic interaction between Compound A and the Chol-GNRs carrier.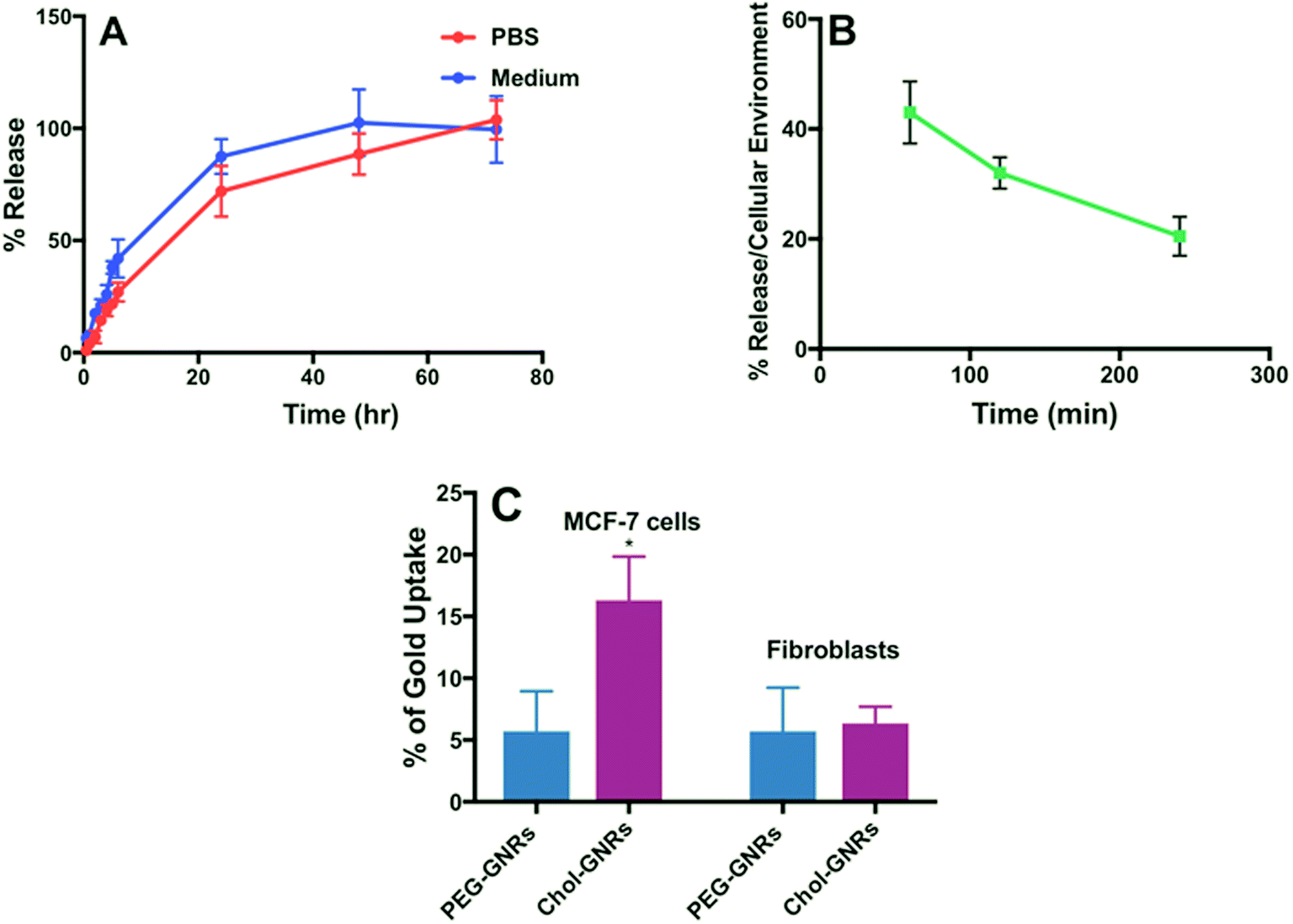 | ||
| Fig. 6 In vitro release profile of Compound A from the nanocomplex (Chol-A-GNRs) in PBS and in tissue culture medium (A). Release profile of Compound A from the nanocomplex (Chol-A-GNRs) under cellular environment (B). Percentages of cellular uptake of Chol-GNRs and PEG-GNRs into MCF-7 breast cancer cells and human dermal fibroblasts (C). Data are presented as mean ± SD, n = 3. Unpaired t-test was used to assess the differences; * represent p < 0.05. | ||
Since the release of the compound in PBS or the medium does not exactly resemble the real situation, the release of Compound A was examined upon exposure to cells under cellular environment. Generally, hydrophobic drugs are preferentially up-taken into lipids and hydrophobic sites such as cellular lipid membrane which maximize their cellular uptake.20
Thus, we further examined the compound release profile when the nanocomplex was exposed to cancer cells under cellular environment. The MCF-7 cells were incubated with Chol-A-GNRs, and at predetermined time intervals from 30 min to 240 min, the amount of the released Compound A was recorded by measuring the optical absorbance at 297 nm of the supernatant after centrifuging the medium. The release of the compound was ∼43% after 1 h of incubation and it was reduced over time and reached ∼20% after 4 h of incubation under cellular environment (Fig. 6B). Clearly, the presence of cells enhanced the compound release from the nanocomplex and might accelerate the cellular uptake of the compound, which may be due to the lipophilicity of the cell membrane.20 Similarly, Ren et al. have found that the release of paclitaxel from GNRs stabilized with PEG linked to 11-mercaptoundecanoic acid was accelerated in the presence of cancer cells compared to drug release in PBS or serum due to the lipophilicity of the cell membrane which enhances the uptake of hydrophobic drugs.35
Obviously, the release of Compound A from the nanocomplex was slightly fast in tissue culture medium compared to PBS, and the release was accelerated in the presence of cells compared to PBS or tissue culture medium.
3.5 Cellular uptake of Chol-GNRs and PEG-GNRs into MCF-7 breast cancer cells and human dermal fibroblasts
The cellular uptake of gold into the breast cancer cells was quantified using a well validated ICP-OES method. Fig. 6C shows an average of ∼15% of the applied dose of GNRs were penetrated into the cells compared to PEGylated nanorods which demonstrated an average penetration of 5% after 3 h of incubation with the cells. Besides the principal role of Chol-A-GNRs in the effective delivery of the hydrophobic ligand into cancer cells, the detected cellular uptake of GNRs may be responsible for the possible synergistic cytotoxicity for the Compound A and the nanocarrier (Chol-GNRs). Kim et al. have designed gold-based nanoparticles having a hydrophobic pocket and a hydrophilic shell containing a zwitterionic head group for delivery of three different hydrophobic ligands into MCF-7 cells. They have reported that such a nano-system has demonstrated an efficient cellular uptake of the drug into the cells with a little uptake into the cancer cells.20On the other hand, the cellular uptake of Chol-GNRs or PEG-GNRs into the human dermal fibroblasts was less than 7% compared to the uptake of Chol-GNRs into the breast cancer cells (∼15%).
3.6 Anti-proliferative activity of Compound A, Chol-GNRs and nanocomplex (Chol-A-GNRs) against MCF-7 breast cancer cell line and human dermal fibroblasts
The cytotoxicity of Compound A as free drug was investigated against MCF-7 breast cancer cells and human dermal fibroblasts. MCF-7 is an estrogen receptor positive cell line. The biological effects of estrogen are mediated through either the alpha or the beta receptor. It has been demonstrated that PI3K is necessary for the estrogen signaling, in addition, PI3K is one of the key enzymes which regulates phosphoinositide metabolism and is responsible for generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3) via phosphorylation of D-3 position of the inositol ring of (PIP2). The activation of PI3K results in PIP3-mediated activation of serine–threonine kinase Akt by phosphorylation. The Akt protein in turn modulates the function of many substrates involved in the regulation of cell functions including proliferation and cell cycle progression.36 That is why MCF-7 cell line was selected to investigate the cytotoxicity effect of the PI3K inhibitor (Compound A) alone and upon conjugation with GNRs decorated with cholesterol-PEG moiety (the nanocomplex).The IC50 of Compound A (235.9 μg mL−1) was determined over the concentration range of 0.97–500 μg mL−1 against MCF-7 breast cancer cells (Fig. 7).
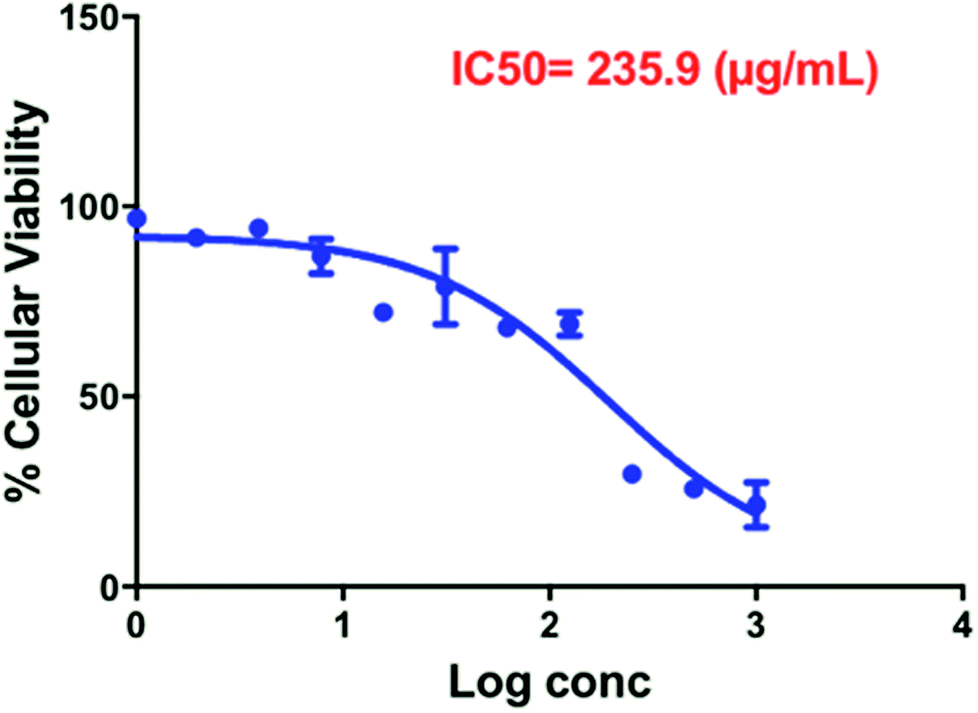 | ||
| Fig. 7 Estimation of IC 50 of free Compound A over the concentration range of 0.97–500 μg mL−1 against MCF-7 breast cancer cells (results present the average and standard deviation of three determinations). | ||
Fig. 8A shows the percentage of cellular viability of MCF-7 upon treatment with the nanocomplex (Chol-A-GNRs), free Compound A and the nanocarrier (Chol-GNRs). Interestingly, the nanocomplex revealed potent cytotoxicity over very low range of concentrations (10.0/12.5, 2.5/3.125 and 0.625/0.75 μg mL−1 of Chol-GNRs/Compound A). For example, at concentration of ∼3.125 μg mL−1 of Compound A in Chol-A-GNRs, the nanocomplex exhibited cellular viability of only about 20% compared to around 70% for the nanocarrier alone (∼3.5 folds), and around 100% for the free compound at the same concentration (5 folds). Interestingly, the effective concentrations of Compound A in the nanocomplex were significantly less than the IC50 of Compound A alone against MCF-7 breast cancer cells.
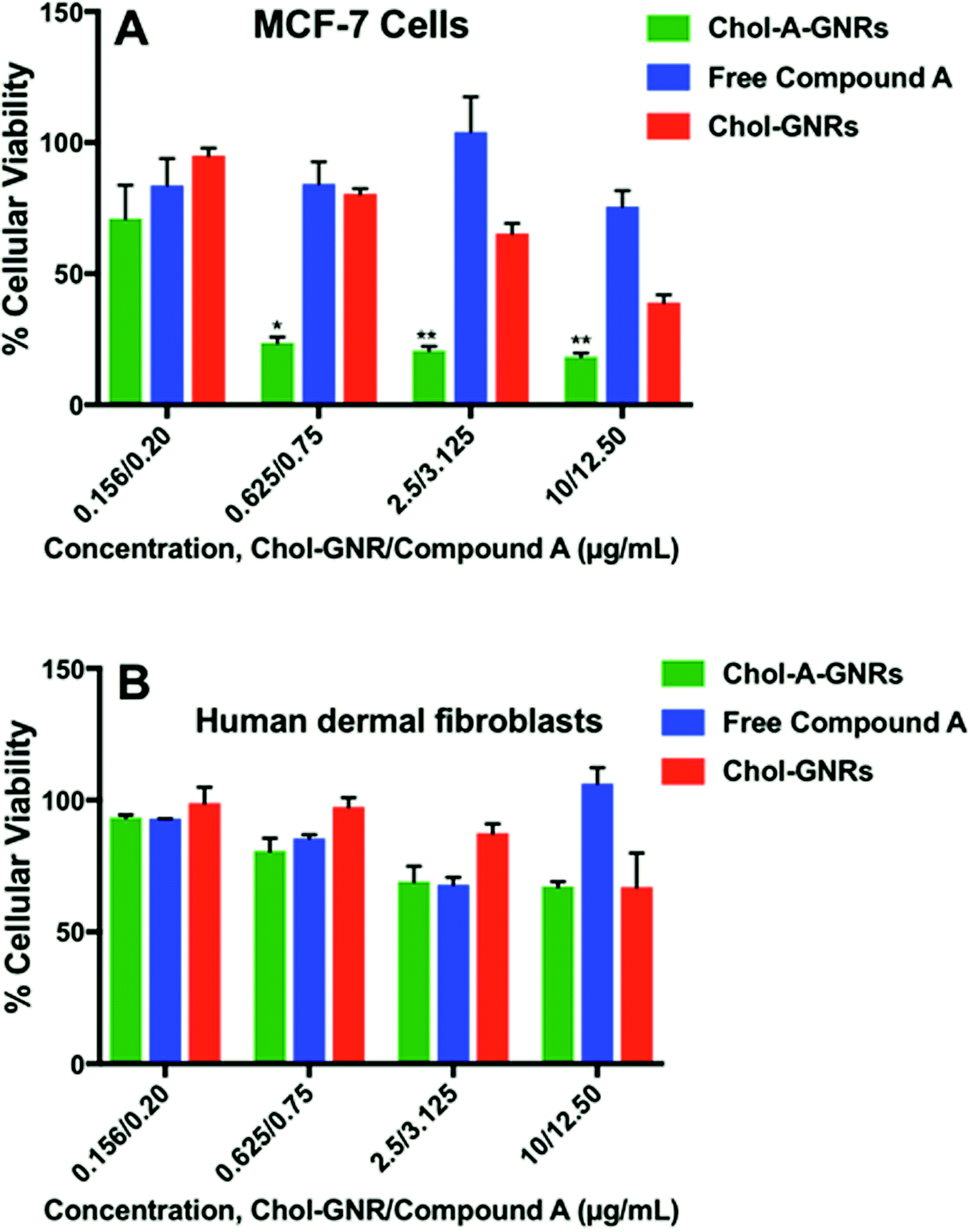 | ||
| Fig. 8 Anti-proliferative activity of the nanocomplex (Chol-A-GNRs), free Compound A, and the nanocarrier (Chol-GNRs) against MCF-7 breast cancer cells (A) and human dermal fibroblasts (B). The nanocomplex demonstrates potent anti-proliferative activity against MCF-7 cells over low range of concentrations compared to human dermal fibroblasts. Data are given as mean ± SD, n = 3. Unpaired t-test was used to assess the differences; * represent p < 0.05, ** represents p < 0.01. | ||
The cellular viability of the nanocarrier (Chol-GNRs) against MCF-7 cells was around 40% at the highest concentration used, which may be due to the detected cellular uptake of Chol-GNRs (∼15%) into MCF-7 cells in particular at high concentration.
Its worth's to note that the nanocomplex at high concentration (40.0/50 μg mL−1 of Chol-GNRs/Compound A) demonstrated no significant antiproliferative activity (data not shown). This could be attributed to the poor colloidal stability of the nanocomplex upon mixing with the tissue culture medium at high concentration compared to low concentrations (as indicated by the obvious aggregation of the nanocomplex in the medium). Nanoparticles in solution generally have a high tendency to increase in size due to rapid collision of particles, van der Waals bonding and water bridging.37
Interestingly, no appreciable cytotoxicity was observed for the free compound, free nanocarrier nor the nanocomplex against normal human dermal fibroblasts over the same concentration range used against the cancer cells (Fig. 8B). As shown in Fig. 6C, the cellular uptake of the nanocarrier (Chol-GNRs) into the human dermal fibroblasts (healthy cells) was significantly less than that observed for the cancerous cells (7% vs. 15%) which might explain their observed safety profile upon application to the normal cells.
Cancer cells in general have fundamental differences in the composition, amount and arrangement of phospholipids and cholesterol in the cell membrane compared to the normal cells.38 Cholesterol (cholest-5-en-3-ol) is a rigid planar molecule with a steroid skeleton of four fused rings, three six-membered and one five-membered, and is one of the most important structural components of the cell membrane.39 Furthermore, the presence of cholesterol in the cell membrane is abundant, and it plays a key role in the membrane integrity and fluidity,40 however, it has been reported that malignant cells have elevated levels of cholesterol precursors.41 Breast and prostate cancer cell lines have elevated amount of lipid rafts and they are more sensitive to cell death caused by cholesterol depletion compared to the healthy cells.42
We suppose that the nanocarrier (Chol-GNRs) which composed of a cholesterol moiety may enhanced the adsorption of the nanocomplex into the lipid cell membrane of the breast cancer cells compared to normal cells, and subsequently enhanced the uptake of the loaded hydrophobic ligand. Besides, the observed cellular uptake of the nanocarriers themselves may also contribute to the detected massive uptake of Compound A into the cells. This also could explain the little toxicity of the nanocomplex against human dermal fibroblasts and their preferential cellular uptake into the cancer cells. Moreover, Compound A was designated as PI3Kα inhibitory agent for a targeted therapy of cancers that demonstrated an elevated activation of PI3Kα. Breast cancer was found to be highly associated with a pathologic activation of PI3Kα pathway compared to healthy cells.43
Employment of cholesterol and lipids for drug delivery was demonstrated in the literature. Among various lipid-based formulations, liposomes have demonstrated efficiency in several biomedical applications.44 Furthermore, lipid conjugated to a polymer possess several characteristics that make them highly suitable for drug delivery for diverse of hydrophobic anti-cancer drugs.45 Moreover, cholesterol-coupled to poly(D,L-lactide)-micelles were used for efficient delivery of curcumin into melanoma and breast cancer cell.46
3.7 Analysis of cell death modality of MCF-7 cells upon treatment with Chol-A-GNRs by flow cytometry
The results indicate that the nanocomplex (Chol-A-GNRs) significantly reduced the cell viability of MCF-7 cells compared to the control untreated cells. Interestingly, both 4.0 and 8.0 μg mL−1 of the nanocomplex showed similar decrease in the percentage of viable cells; ∼31.47% and ∼32.14%, respectively, compared to the control untreated cells; ∼58.72% (P < 0.05) (Fig. 9A–D). On the other hand, 8.0 μg mL−1 of the treatment was able to increase the percentage of necrotic population statistically by 3.35 folds compared to the untreated cells (∼58.77% vs. ∼17.5% respectively, P < 0.05), while 4.0 μg mL−1 of the treatment showed only 1.5 folds change compared to the untreated cells (∼25.94% vs. ∼17.5%, respectively) (Fig. 9A–D).
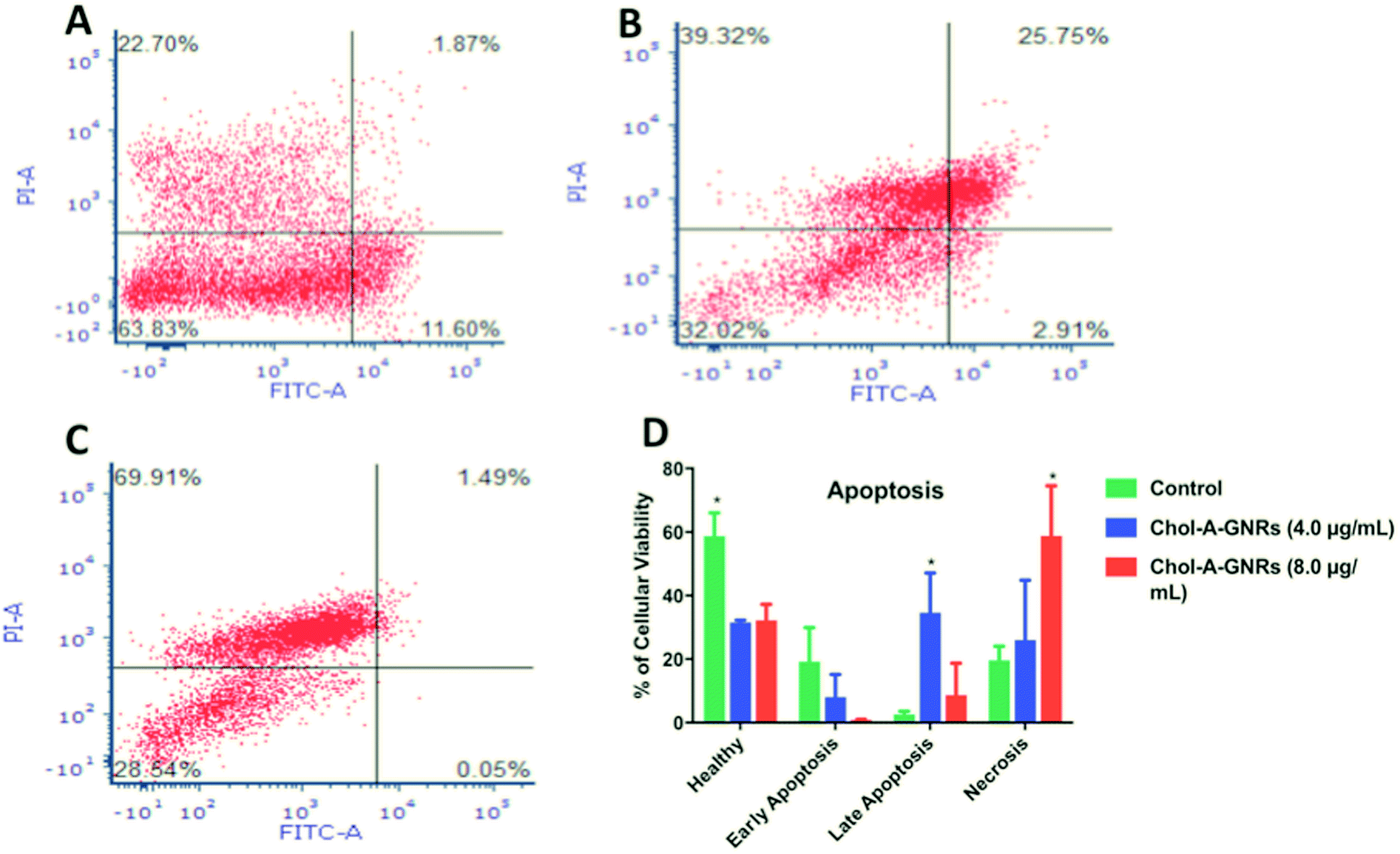 | ||
| Fig. 9 Flow cytometry assay of apoptosis of the control untreated MCF-7 cells (A); MCF-7 cells incubated for 24 h in the presence of 4.0 μg mL−1 (B) and 8.0 μg mL−1 (C) of the nanocomplex and stained with FITC-conjugated annexin V and PI-stained. The dot plot for each sample was divided into four quadrants to indicate viable cells (lower left quadrant), early apoptotic cells (lower right quadrant), necrotic cells (upper left quadrant), and late apoptotic cells (upper right quadrant). A significant difference was observed in the percentage of necrotic and late apoptotic cells upon treating the MCF-7 cells with 8.0 μg mL−1 and 4.0 μg mL−1 of the nanocomplex, respectively compared to the untreated cells (P < 0.05). Data are presented as mean ± SD, n = 3 (D). Unpaired t-test was used to assess the differences; * represent p < 0.05. (A)–(C) Plots are examples of one run of the analysis. | ||
On the contrary, the percentage of late apoptotic population was high in the cells treated with 4.0 μg mL−1 of the nanocomplex, as the percentage was increased by 13 folds compared to the control (∼34.15% vs. ∼2.56%, respectively, P < 0.05). On the other hand, the nanocomplex of 8.0 μg mL−1 showed only 3.37 folds change compared to the untreated cells (∼8.630% vs. average of ∼2.56%, respectively) (Fig. 9A–D).
Quite the opposite of late stages; early apoptotic population was larger in the control untreated cells (average of ∼19.18%) compared to the cells treated with the nanocomplex. The cells treated with 4.0 or 8.0 μg mL−1 of the nanocomplex showed low percentages of early apoptotic cells (average of ∼8% and ∼0.455%, respectively) (Fig. 9A–D). This is clearly explained by the rapid effect of the nanocomplex on cell death, as cells went through early apoptotic stage toward late apoptotic/necrotic stage rapidly and proportionally to the increase of the nanocomplex concentration.
Consequently, we concluded that the nanocomplex has direct impact on cell death modality and this effect is a concentration dependent.
Cell cycle parameters of cells treated with 8.0 μg mL−1 of the nanocomplex were compared to the control (untreated cells). As shown in Fig. 10, our results interestingly show a significant difference in the proportion of cells in G0/G1, S and G2/M phases when cells were treated with 8.0 μg mL−1 of the nanocomplex compared to the untreated cells (P < 0.05). As cells treated with the aforementioned treatment showed 100% arrest at G0/G1 phases and were unable to go through other cell cycle phases (S and G2/M phases) (Fig. 10A). While untreated cells did not show any perturbations in the cell cycle phases (Fig. 10B). Thus, this arrest might be resulted in the activation of different pathways that lead to the programmed cell death.47,48
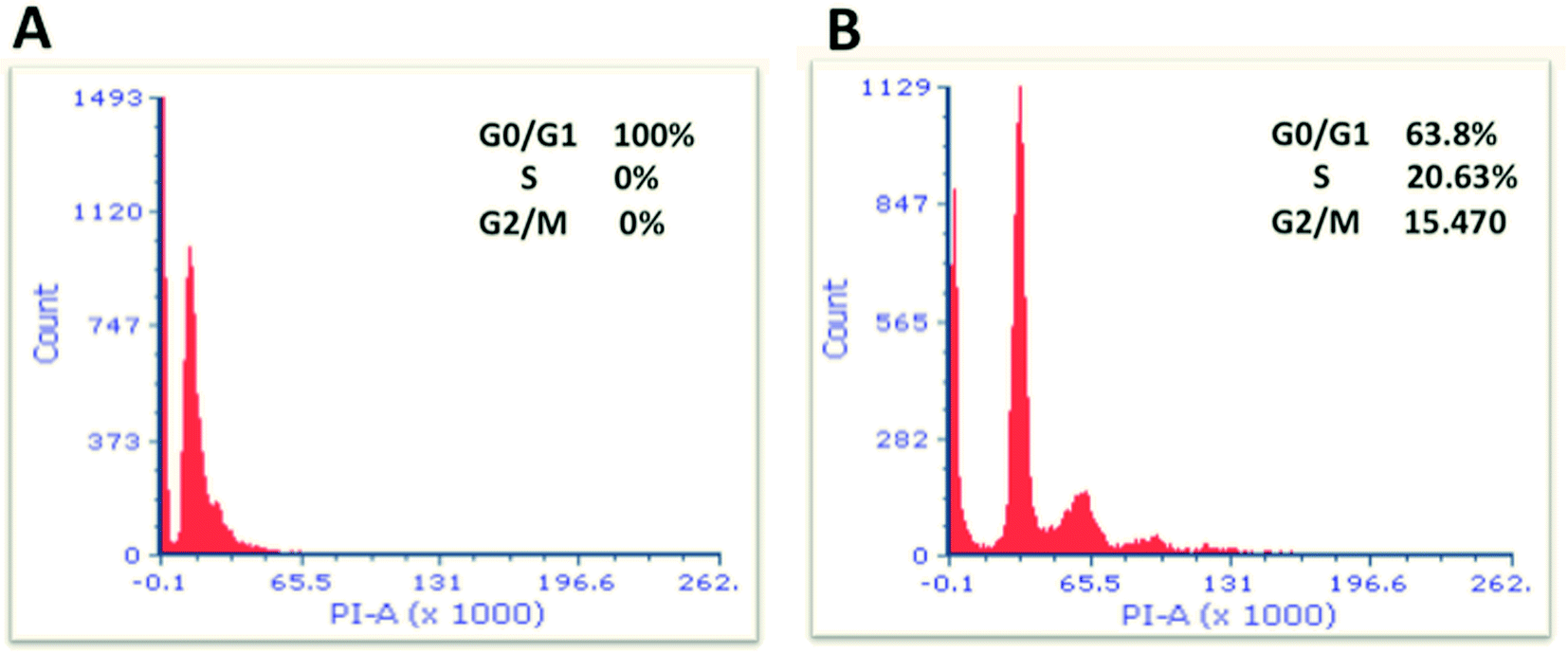 | ||
| Fig. 10 Flow cytometric analysis of cell cycle parameters. MCF-7 cells were incubated for 24 h; in the presence of 8.0 μg mL−1 of the nanocomplex (A) and without additive of treatment (untreated) (B). Cells were stained with propidium iodide for analysis by flow cytometry. The cells treated with the nanocomplex showed 100% arrest at G0/G1 phases. A statistical difference was observed between MCF-7 cells treated with the nanocomplex and their control untreated cells (P < 0.05). Data are given as means, n = 3, and unpaired t-test was used to assess the difference. | ||
As a result, we indicate that the nanocomplex is considered as a strong inhibitor for cell cycle progression.
4. Conclusions
Effective delivery of hydrophobic chemotherapeutic agents is considered as a real challenge in cancer therapy. Gold-based nanocarriers have a distinguished role in competent and efficient drug delivery. In this study, a facile and simple GNRs decorated with cholesterol moiety was utilized as a promising nanocarrier to conjugate a small hydrophobic ligand, having a PI3Kα inhibitory activity, into the hydrophobic pocket of the nanocarrier. The acquired nanocomplex possess an accelerated release of the ligand under cellular environment and exhibited potent anti-proliferative activity over low range of concentrations. Furthermore, the nanocomplex demonstrated a potent inhibition of the cell cycle and hastened the cellular apoptotic/necrotic events. Further investigations are needed to understand the specific pathways responsible for such observed potent cytotoxic features of the nanocomplex.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the Deanship of Scientific Research and Graduate Studies at Al-Zaytoonah University of Jordan Grant (2017-2016/64/04), and The Scientific Research Support Fund (MPH/1/15/2015), Amman, Jordan, for financial funding.Notes and references
- Z. Anastasiadi, G. D. Lianos, E. Ignatiadou, H. V. Harissis and M. Mitsis, Updates in surgery, 2017, 69, 313–317 CrossRef PubMed.
- K. Sak, Chemother. Res. Pract., 2012, 2012, 282570 Search PubMed.
- A. H. Partridge, H. J. Burstein and E. P. Winer, J. Natl. Cancer Inst. Monogr., 2001, 135–142 CrossRef CAS.
- Q. Mu, H. Wang and M. Zhang, Expert Opin. Drug Delivery, 2017, 14, 123–136 CrossRef CAS PubMed.
- S. K. Singh, S. Singh, J. W. Lillard Jr and R. Singh, Int. J. Nanomed., 2017, 12, 6205–6218 CrossRef CAS PubMed.
- S. S. Qi, J. H. Sun, H. H. Yu and S. Q. Yu, Drug Delivery, 2017, 24, 1909–1926 CrossRef CAS PubMed.
- R. X. Zhang, H. L. Wong, H. Y. Xue, J. Y. Eoh and X. Y. Wu, J. Controlled Release, 2016, 240, 489–503 CrossRef CAS PubMed.
- E. J. Feldman, J. E. Lancet, J. E. Kolitz, E. K. Ritchie, G. J. Roboz, A. F. List, S. L. Allen, E. Asatiani, L. D. Mayer, C. Swenson and A. C. Louie, J. Clin. Oncol., 2011, 29, 979–985 CrossRef CAS PubMed.
- J. R. Hasenstein, H. C. Shin, K. Kasmerchak, D. Buehler, G. S. Kwon and K. R. Kozak, Mol. Cancer Ther., 2012, 11, 2233–2242 CrossRef CAS PubMed.
- H. C. Shin, A. W. Alani, H. Cho, Y. Bae, J. M. Kolesar and G. S. Kwon, Mol. Pharmaceutics, 2011, 8, 1257–1265 CrossRef CAS PubMed.
- H. C. Shin, H. Cho, T. C. Lai, K. R. Kozak, J. M. Kolesar and G. S. Kwon, J. Controlled Release, 2012, 163, 93–99 CrossRef CAS PubMed.
- S. Anniebell and S. C. B. Gopinath, Curr. Med. Chem., 2018, 25, 1433–1445 CrossRef CAS PubMed.
- H. Daraee, A. Eatemadi, E. Abbasi, S. Fekri Aval, M. Kouhi and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 410–422 CrossRef CAS PubMed.
- J. Lee, D. K. Chatterjee, M. H. Lee and S. Krishnan, Cancer Lett., 2014, 347, 46–53 CrossRef CAS PubMed.
- A. O. Elzoghby, A. L. Hemasa and M. S. Freag, J. Controlled Release, 2016, 243, 303–322 CrossRef CAS PubMed.
- Y. Ding, Z. Jiang, K. Saha, C. S. Kim, S. T. Kim, R. F. Landis and V. M. Rotello, Mol. Ther., 2014, 22, 1075–1083 CrossRef CAS PubMed.
- V. S. Marangoni, J. Cancino-Bernardi and V. Zucolotto, J. Biomed. Nanotechnol., 2016, 12, 1136–1158 CrossRef CAS PubMed.
- R. A. Morshed, M. E. Muroski, Q. Dai, M. L. Wegscheid, B. Auffinger, D. Yu, Y. Han, L. Zhang, M. Wu, Y. Cheng and M. S. Lesniak, Mol. Pharmaceutics, 2016, 13, 1843–1854 CrossRef CAS PubMed.
- S. Balakrishnan, S. Mukherjee, S. Das, F. A. Bhat, P. Raja Singh, C. R. Patra and J. Arunakaran, Cell Biochem. Funct., 2017, 35, 217–231 CrossRef CAS PubMed.
- C. K. Kim, P. Ghosh, C. Pagliuca, Z. J. Zhu, S. Menichetti and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 1360–1361 CrossRef CAS PubMed.
- D. A. Sabbah, B. Hishmah, K. Sweidan, S. Bardaweel, M. AlDamen, H. A. Zhong, R. Abu Khalaf, A. Hasan Ibrahim, T. Al-Qirim, G. Abu Sheikha and M. S. Mubarak, Anti-Cancer Agents Med. Chem., 2018, 18, 263–276 CrossRef CAS PubMed.
- B. Vanhaesebroeck and M. D. Waterfield, Exp. Cell Res., 1999, 253, 239–254 CrossRef CAS PubMed.
- B. Vanhaesebroeck, J. Guillermet-Guibert, M. Graupera and B. Bilanges, Nat. Rev. Mol. Cell Biol., 2010, 11, 329–341 CrossRef CAS PubMed.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Nano Lett., 2013, 13, 765–771 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- Protein Preparation Wizard, Maestro, Macromodel, and QPLD-dock, Schrödinger, LLC, Portland, OR, USA, 97204, 2016 Search PubMed.
- D. A. Sabbah, J. L. Vennerstrom and H. A. Zhong, J. Chem. Inf. Model., 2012, 52, 3213–3224 CrossRef CAS PubMed.
- C.-H. Huang, D. Mandelker, O. Schmidt-Kittler, Y. Samuels, V. E. Velculescu, K. W. Kinzler, B. Vogelstein, S. B. Gabelli and L. M. Amzel, Science, 2007, 318, 1744–1748 CrossRef CAS PubMed.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- K. Nishida and H. Kawasaki, RSC Adv., 2017, 7, 18041–18045 RSC.
- A. S. Karakoti, S. Das, S. Thevuthasan and S. Seal, Angew. Chem., Int. Ed. Engl., 2011, 50, 1980–1994 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- H. Hinterwirth, S. Kappel, T. Waitz, T. Prohaska, W. Lindner and M. Lämmerhofer, ACS Nano, 2013, 7, 1129–1136 CrossRef CAS PubMed.
- F. Ren, S. Bhana, D. D. Norman, J. Johnson, L. Xu, D. L. Baker, A. L. Parrill and X. Huang, Bioconjugate Chem., 2013, 24, 376–386 CrossRef CAS PubMed.
- Y. R. Lee, J. Park, H. N. Yu, J. S. Kim, H. J. Youn and S. H. Jung, Biochem. Biophys. Res. Commun., 2005, 336, 1221–1226 CrossRef CAS PubMed.
- C. P. Tso, C. M. Zhung, Y. H. Shih, Y. M. Tseng, S. C. Wu and R. A. Doong, Water Sci. Technol., 2010, 61, 127–133 CrossRef CAS PubMed.
- C. R. Santos and A. Schulze, FEBS J., 2012, 279, 2610–2623 CrossRef CAS PubMed.
- N. M. Cerqueira, E. F. Oliveira, D. S. Gesto, D. Santos-Martins, C. Moreira, H. N. Moorthy, M. J. Ramos and P. A. Fernandes, Biochemistry, 2016, 55, 5483–5506 CrossRef CAS PubMed.
- J. M. Crane and L. K. Tamm, Biophys. J., 2004, 86, 2965–2979 CrossRef CAS PubMed.
- R. E. Duncan, A. El-Sohemy and M. C. Archer, J. Biol. Chem., 2004, 279, 33079–33084 CrossRef CAS PubMed.
- Y. C. Li, M. J. Park, S. K. Ye, C. W. Kim and Y. N. Kim, Am. J. Pathol., 2006, 168, 1107–1118 CrossRef CAS PubMed.
- S. X. Yang, E. Polley and S. Lipkowitz, Cancer Treat. Rev., 2016, 45, 87–96 CrossRef CAS PubMed.
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Front. Pharmacol., 2015, 6, 286 Search PubMed.
- J. H. Kang and Y. T. Ko, Int. J. Nanomed., 2015, 10(spec iss), 33–45 CAS.
- P. Kumari, O. S. Muddineti, S. V. Rompicharla, P. Ghanta, A. K. B B N, B. Ghosh and S. Biswas, Drug Delivery, 2017, 24, 209–223 CrossRef CAS PubMed.
- J. A. Pietenpol and Z. A. Stewart, Toxicology, 2002, 181–182, 475–481 CrossRef CAS PubMed.
- B. Pucci, M. Kasten and A. Giordano, Neoplasia, 2000, 2, 291–299 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |