
Fluorescence visualization of cucurbit[8]uril-triggered dynamic host–guest assemblies†
Xiaodong
Zhang
,
Tao
Sun
and
Xin-Long
Ni
 *
*
Key Laboratory of Macrocyclic and Supramolecular Chemistry of Guizhou Province, Guizhou University, Guiyang, 550025, China. E-mail: longni333@163.com
First published on 9th June 2020
Abstract
Cucurbit[8]uril (Q[8] or CB[8]), as a synthetic macrocyclic host, plays an important role in constructing various water soluble dynamic supramolecular polymers and materials. Here we report host–guest interactions of Q[8] with π-conjugated bispyridinium guests in aqueous solution that resulted in a series of unprecedented dynamic 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 2:3, and 1:2 assemblies (guest/Q[8]). Most importantly, these subtly different host–guest binding modes could be distinguished by naked-eye observation of the resulting quantitative fluorescence emissions. Further characterization suggested that the Q[8] host-stabilized charge-transfer interactions could be dissociated and controlled by the N-substituted alkyl chains on the cationic guests via the formation of a 2:3 quinary complex or a U-shaped conformation, which led to the discovery of new possible binding modes for the Q[8] host.
:2, 2:3, and 1:2 assemblies (guest/Q[8]). Most importantly, these subtly different host–guest binding modes could be distinguished by naked-eye observation of the resulting quantitative fluorescence emissions. Further characterization suggested that the Q[8] host-stabilized charge-transfer interactions could be dissociated and controlled by the N-substituted alkyl chains on the cationic guests via the formation of a 2:3 quinary complex or a U-shaped conformation, which led to the discovery of new possible binding modes for the Q[8] host.
Introduction
Cucurbit[8]uril (Q[8] or CB[8]),1,2 a large homologue of the cucurbituril family, is unique because of its ability to form ternary complexes by encapsulating two hetero- or homo-guests in its rigid macrocyclic cavity. In particular, host-stabilized charge-transfer (HSCT),3 as the dominant interaction force, has resulted in the wide use of Q[8] to construct various water soluble dynamic materials in the past decade.4–15 Meanwhile, it was also found that Q[8] is able to act as molecular handcuffs to simultaneously include two chromophore dyes in its cavity to form a dimer assembly. This not only increases the π–π stacking of the aromatic moieties,16 but also enhances intra/inter-molecular charge transfer (ICT) of the inclusion guest dyes.17Recently, it has been discovered that π-conjugated bispyridinium derivatives could serve as both electron-deficient and electron-rich groups that could dimerize inside the Q[8] cavity to form highly stable Q[8]-enhanced ternary complexes18–20 and supramolecular polymers21–23 with enhanced light emission efficiency. More recently, Scherman's group,24–26 Schalley's group,27 and Ma's group28 revealed that complexation of diaryl viologens bearing electron-donating groups with Q[8] could be used to achieve a series of unique dimer emissions in the 2:2 binding mode. These studies indicated the potential of Q[8] as a host in the fabrication of smart tunable luminescent materials. However, to our knowledge, less attention has been paid to Q[8] encapsulation-triggered fluorescence signals for monitoring of the accompanying dynamic host–guest interactions.
Fluorescence has been widely used to explore dynamic processes, such as self-assembly,29,30 molecular motion31 and conformational changes.32 The high sensitivity of fluorescence methods allows tracking of molecular interactions even at nanomolar concentrations. In particular, it is possible to visualize directly and in real time monitor processes in situ. For example, Tang et al. exploited aggregation-induced emission fluorescence imaging to successfully demonstrate the transition processes of surfactant micelles and microemulsion droplets.33 Li and co-workers directly observed the distribution of gelatin in calcium carbonate crystals by super-resolution fluorescence microscopy.34 Yang et al. found that the process and dynamics of coordination-driven self-assembly could be tracked in real-time by fluorescence-resonance energy transfer.35
In this work, a series of different length alkyl chains were introduced into π-conjugated bispyridinium species as guest molecules (Scheme 1). We found that the alkyl chains as the terminal moieties played a crucial role in driving the Q[8] host to form various dynamic host–guest assemblies including 2:2, 2:3, and 1:2 assemblies. In particular, the multiple dynamic Q[8]-based host–guest interactions could be distinguished by naked-eye observation of the resulting quantitative fluorescence emissions. Further studies revealed that the Q[8]-based HSCT interaction could be dissociated or controlled by the alkyl chain lengths and a slight modification of the π-conjugated aromatic core of the guests.
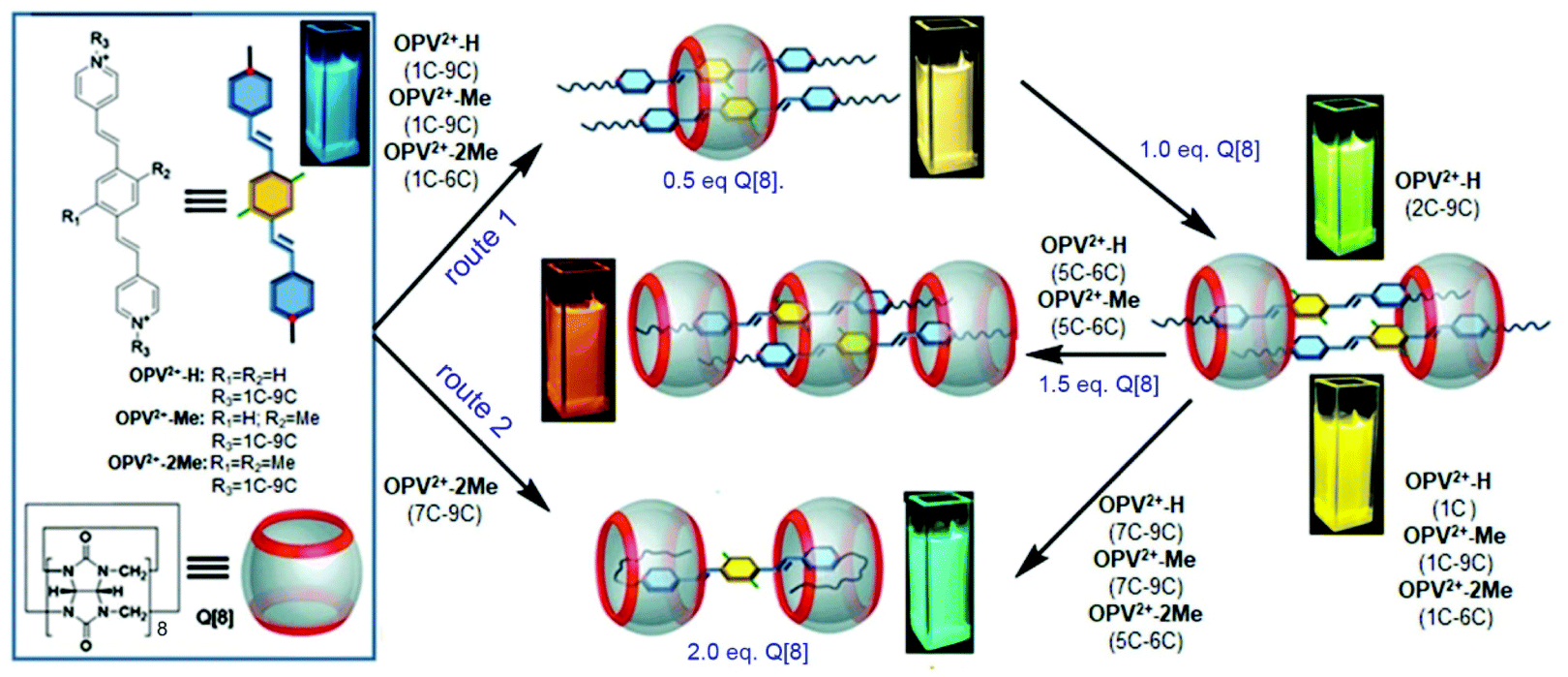 | ||
| Scheme 1 Chemical structures of OPV2+ derivatives and Q[8], and representation of Q[8]-based dynamic host–guest assemblies with multiple tunable fluorescence signals. Route 1: Q[8] concentration dependent host–guest interactions of 2:1, 2:2, 2:3, and 1:2 (guest/Q[8]); route 2: the formation of a U-shaped conformation of Q[8] with OPV2+-2Me (7C–9C). | ||
Results and discussion
Our previous work revealed that the photophysical properties of oligo(p-phenylenevinylene) (OPV2+)-based bispyridinium guest molecules could be sensitively switched by the Q[8] host.21 Here, to conveniently monitor multimodular host–guest assemblies of Q[8] with the alkyl chain-appended guests, the π-conjugated OPV2+ core was selected as the fluorescent reporting moiety. In order to capture and distinguish dynamic host–guest interactions, such as whether the Q[8] host could enclose the phenyl part of the guest, larger barrier groups, such as one or two methyl (Me) groups, were appended to the phenyl moiety to serve as a “brake.” The molecular structures of guests together with plausible fluorescent visible host–guest assemblies are shown in Scheme 1. The guests were OPV2+-H, OPV2+-Me, and OPV2+-2Me with different alkyl chains (containing carbon atoms from 1C to 9C), in which I− acted as counter anions for 1C appended guests, and Br− as counter anions for other guests. The related guests are hereafter abbreviated as OPV2+-(H, Me, 2Me) (1C to 9C).The fluorescence and UV-vis spectra of the guests showed similar emission peaks around 480 nm and absorption peaks around 390 nm in aqueous solutions, respectively, indicating that the appended alkyl chains did not affect the optical properties of the OPV2+ core. In the presence of the Q[8] host, OPV2+-H and OPV2+-Me derived guests showed similar optical responses, while some different fluorescence response behaviour was observed in the case of OPV2+-2Me based guests (Fig. 1, Fig. S1–S3 and Table S1†). For example, an enhanced bathochromic fluorescence emission (yellow colour) shift was observed for all of the guest solutions when the concentration of Q[8] was fixed at 0.5 eq., except for the OPV2+-2Me guests (7C–9C), where slight red-shifts were noted (Fig. 1f). When the concentration of Q[8] was increased to 1.0 eq., considerably enhanced emission around 550 nm was observed (Fig. 1a, d, and Fig. S1†), except for guests OPV2+-H (2C–9C), where a tunable enhanced blue-shift emission was observed (Fig. S1 and Table S1†).
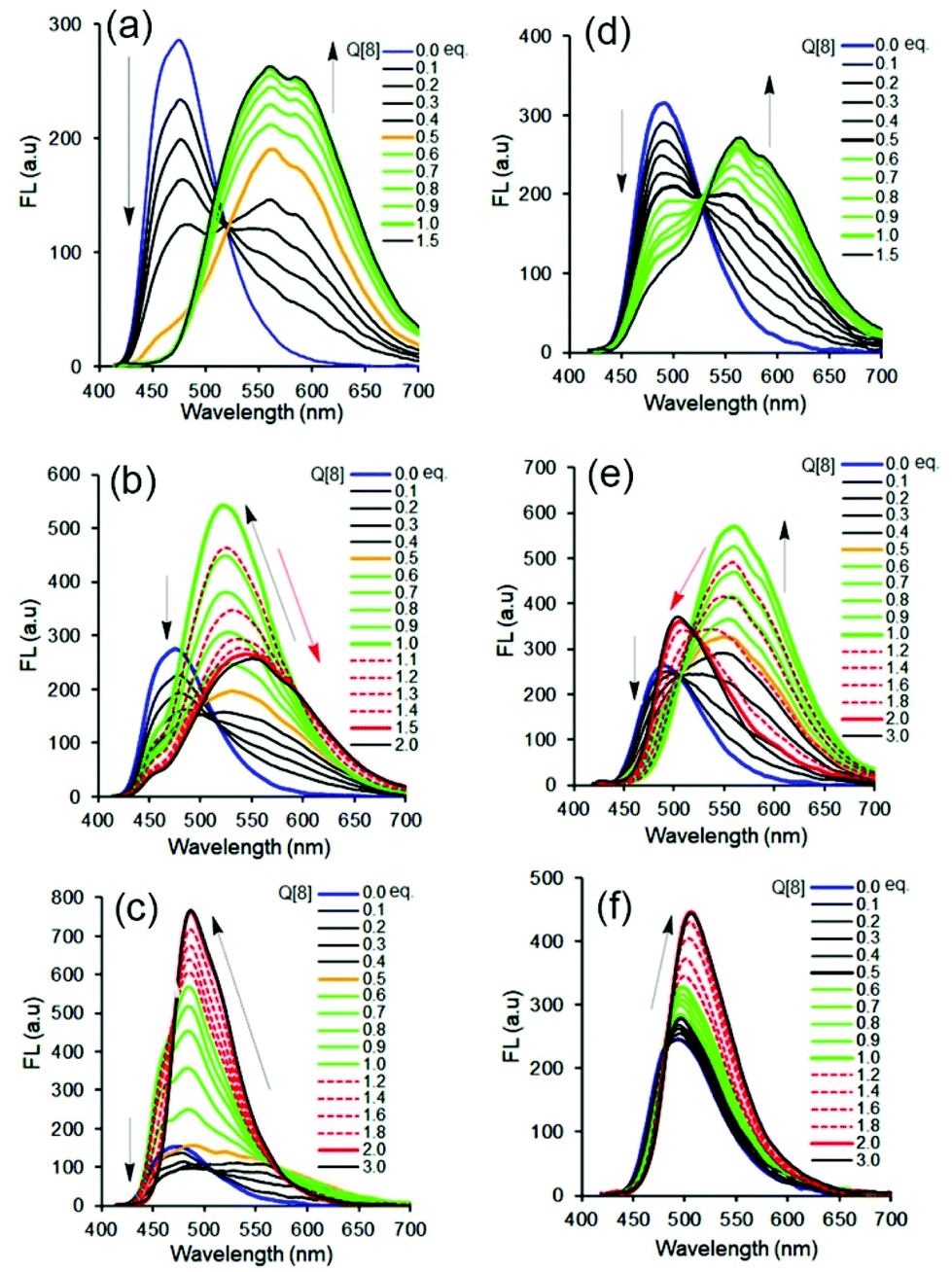 | ||
| Fig. 1 Fluorescence spectrum changes of OPV2+ core based guests (a–c) OPV2+-H (1C, 5C, and 9C), (d–f) OPV2+-2Me (1C, 5C, and 9C) (each of 10.0 μM in aqueous solution, pH 7.2) in the presence of increasing concentrations of the Q[8] host, respectively (λex = 400 nm). | ||
When the concentration of Q[8] was increased to 1.5 eq., no changes in the fluorescence spectra were observed for any of the 1C–4C appended guests (Fig. S1 and Table S1†). However, OPV2+-H guests (5C–6C) (Fig. 1b) and OPV2+-Me (5C–6C) underwent significant red-shift emissions (Table S1†), and a blue-shift emission change was observed in the OPV2+-2Me (5C–6C) solution (Fig. 1e).
Interestingly, OPV2+-H guests (7C–9C) and OPV2+-Me (7C–9C) exhibited a blue-shift with enhanced emission intensity (Fig. 1c and Table S1†), and a slight red-shift of OPV2+-2Me (7C–9C) in the case of the increase of concentrations of Q[8] from 0.5 eq. to 2.0 eq., respectively (Fig. 1f and Table S1†). Essentially, the different fluorescence responses of the same alkyl-attached guests when Q[8] was added are attributed to the modification by the Me group on the OPV2+ core (Scheme 1).
Compared to the multiple fluorescence signals triggered by different concentrations of the Q[8] host, similar changes in the UV-vis absorption spectra were observed (Fig. S2 and S3†). At first glance, all of the maximum absorbance peaks of the guests at 390 nm decreased remarkably, with a bathochromic shift of the band at 400–470 nm in the presence of the Q[8] host. Close inspection revealed considerably enhanced red-shifted absorbance peaks for OPV2+-H (5C–6C) and OPV2+-Me (5C–6C) guests with two isoabsorption points around 413 and 432 nm, and enhanced blue-shifted absorbance peaks (but still red-shifted compared to the free guest) with isoabsorption points around 428 and 441 nm for OPV2+-2Me (5C–6C) guests.
Generally, guest complexation within the cavity of Q[8] typically results in an upfield shift of the encapsulated protons (shielding effect of the hydrophobic cavity) and a downfield shift of protons in close proximity to the portals of the host (deshielding effect of the carbonyl-rimmed portal). For example, upon titration of 0.5 eq. Q[8] into a guest OPV2+-H (1C) solution (neutral D2O), nearly all of the protons on the parent OPV2+core underwent upfield shifts (Fig. 2a). Particular protons on the ethylene moieties and the phenyl group, including the methyl groups on the OPV2+-Me and OPV2+-2Me guests (Fig. S4–S7,† apart from OPV2+-2Me (7C–9C), which will be discussed later on), were upfield shifted significantly. In contrast, no obvious proton chemical shift changes were observed for the N-substituted alkyl chains. These results indicated that the phenyl aromatic and ethylene moieties were encapsulated in the Q[8] cavity to form 2:1 host–guest complexes (guest/Q[8]).21 It is believed that the Q[8] host is threaded onto the guests such that it can move back and forth along the aromatic OPV2+ core of the guests. In particular, ion dipole interactions between the positively charged nitrogen atoms of the guests and the carbonyl oxygen atoms of the Q[8] host prevent dissociation. This shuttling motion is relatively fast on the NMR time scale; so all of the signals for the protons on the aromatic core are shifted upfield in the spectrum, and no proton chemical shift changes are observed for the N-substituted alkyl chains. Similar proton signal changes were apparent in the host–guest complexes of OPV2+-Me (Fig. S5†) and OPV2+-2Me (Fig. S6†) systems, but less upfield shift was observed for the protons on the OPV2+ core (Fig. S7†), suggesting that the barrier Me and 2Me groups on the phenyl moiety as “brakes” slowed the rate of the Q[8] shuttling motion.
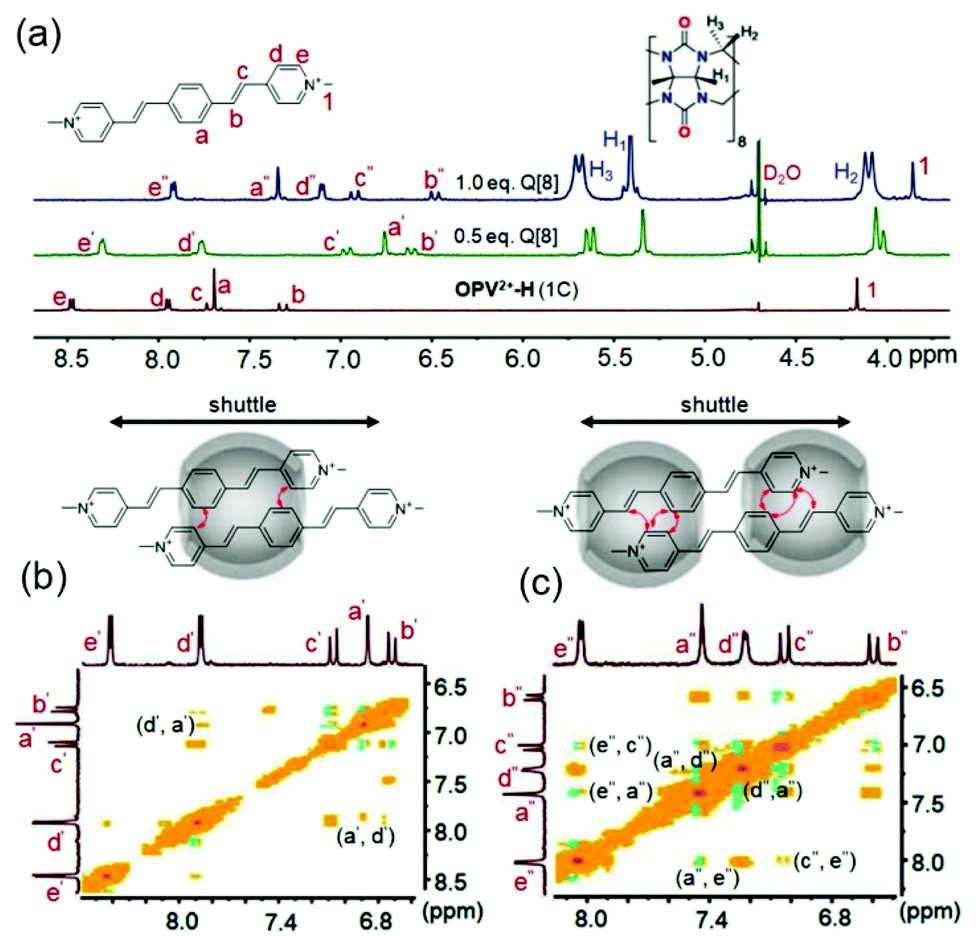 | ||
| Fig. 2 (a) 1H NMR spectra of OPV2+-H (1C) (1.0 mM in D2O) with different concentrations of Q[8] (0–1.0 mM); 1H–1H NOESY of OPV2+-H (1C)/Q[8] at molar ratios of 2:1 (b) and 2:2 (c). | ||
As an example, 2D NMR spectra such as 1H–1H NOESY (nuclear Overhauser effect spectroscopy) of Q[8] with guest OPV2+-H (1C) showed NOE correlation of Hd′–Ha′, implying the formation of head-to-tail dimer complexes (Fig. 2b). Therefore, complexation inside the confined space of the Q[8] cavity results in distinct π–π stacking (substantial electron-delocalization due to dimeric stacking along the long molecular axis), which leads to the significant red-shift fluorescence emission (yellow colour) as mentioned in Fig. 1. Compared to the shorter fluorescence life time (around 0.13 ns) and lower quantum yield (around 5%) of the free guests, all of the Q[8]-encapsulated guest assemblies in solution had a long lifetime of 11–15 ns (Table S2†) and an enhanced quantum yield (Table S3†), apart from OPV2+-H (7C–9C), where the fluorescence lifetime was 6.7–8.6 ns. This may be attributed to the long alkyl chain placing the π-conjugated OPV2+ fluorophore near the Q[8] carbonyl portals, which was confirmed by the lower upfield chemical shift of the phenyl protons of OPV2+-H (7C–9C) (Fig. S7a†).
As the concentration of Q[8] was increased to ∼1.0 eq., for OPV2+-H guests, the signals for all guest protons, including those of the N-substituted alkyl chains, were located in the upfield area (Fig. 2 and S4†). Protons on the pyridinium moiety (Hd and He) appeared to be shifted further upfield, whereas the ethylene protons were still in the same position and phenyl protons were shifted downfield (still upfield shifted compared to the free guest). The NOE correlations between He′′–Hc′′, He′′–Ha′′, and Ha′′–Hd′′, (Fig. 2c) indicated that a dynamic 2:2 complex was formed in this process. Interestingly, closer inspection revealed that the Hd and He protons exhibited a gradual downfield shift as the length of the N-substituted alkyl chains increased (Fig. 3). In particular, for OPV2+-H (7C–9C) guests, it was observed that the signal for Hd underwent a downfield shift compared to 0.5 eq. of Q[8] (Hd was even downfield shifted, to 7.98 ppm from 7.90 ppm in the free state, suggesting that Hd was in close proximity to the Q[8] portal). This result indicated that the longer alkyl chains might play an important role in switching the dynamic 2:2 host–guest complex from head-to-tail geometries to head-to-head arrangements, which is also supported by the fluorescence spectral changes (weak intensity around 550 nm) and shorter life time (Table S2†). Similar NMR and fluorescence spectral changes were observed in the OPV2+-Me and OPV2+-2Me systems, suggesting similar host–guest assembly modes. However, the significant proton downfield shift of the Me group and strong fluorescence intensity around 555 nm for OPV2+-2Me (1C–6C) implied that the double Me moieties can control and retard the shuttling rate of Q[8] on the guest and favor the head-to-tail complex (Fig. S7c†).
 | ||
| Fig. 3
1H NMR spectra of Q[8]/OPV2+-H (1C–9C) at a molar ratio of 2:2. | ||
Upon increasing the concentration of Q[8] up to 1.5 eq., no further chemical shift changes of the protons were observed in any of the 1C–4C-appended host–guest complexes. However, the proton peaks of the ethylene (Hb and Hc) and phenyl (Ha) moieties and the alkyl chains on the 5C–6C-based OPV2+-H and OPV2+-Me guests, including the monomer Me group on OPV2+-Me, exhibited significant upfield shifts (Fig. 4a and S4, S5†). The signals of the other protons, such as Hd and He, exhibited slight downfield shifts compared to 1.0 eq. Q[8], but were still upfield relative to their original positions in the free state. There were no further proton peak shifts when the concentration of Q[8] was increased above 1.5 eq. The 2D NOE correlations for OPV2+-H (5C) (He′–Hc′, Hd′–Ha′, He′–H5′, He′–H4′, Hd′–H2′, Hd′–H1′, H1′–H4′, H2′–H5′, and H2′–H4′) suggested a unique 2:3 assembly of the guests with Q[8]36 (Fig. 4b and S9, 10†). From a structural viewpoint, this binding mode would strengthen the acceptor–donor–acceptor (A–D–A) electronic interaction of the π-conjugated OPV2+-derived cationic guest. In particular, the central Q[8] encapsulation of the dimeric guest not only increases the π-surface of the OPV2+ core, but also greatly enhances the electron-donor group promoting the ICT effect of the guest. Therefore, a further red-shifted emission was observed in that host–guest system.
 | ||
| Fig. 4 (a) 1H NMR spectra of OPV2+-H (5C) (1.0 mM in D2O) with different concentrations of Q[8] (0–1.5 mM); (b) 1H–1H NOESY of OPV2+-H (5C)/Q[8] at a molar ratio of 2:3. | ||
In contrast, in the case of OPV2+-2Me (5C–6C), it was found that the signals for protons on the ethylene and phenyl groups (including the two Me groups on the phenyl parent) underwent downfield shifts when compared to their original proton peaks in the free guest (Fig. S6†), suggesting that the moieties were located at the carbonyl portals of the Q[8] host. The proton signals of the pyridinium moiety and alkyl chains exhibited remarkable upfield shifts, implying that these groups were deep in the Q[8] cavity. The detailed 1H NMR titration experiment results revealed that a 1:2 host–guest interaction (Q[8]/guest) occurred in this system and with all of the 7C–9C appended guests (Fig. S4–S6†).
Interestingly, the NOE correlations (Ha′–H9′, He′–H5′, He′–H4′, He′–H3′, and Hb′–H9′) from the 1:2 complex of OPV2+-H (9C) (Fig. 5) provided evidence that the N-substituted alkyl chain was folded into the cavity of Q[8] to form a U-shaped conformation together with partial inclusion of the pyridinium moiety. This is slightly different from the observation by Kim,37 where the alkyl chains directly adopted the U-shape themselves in the Q[8] cavity. In addition, for OPV2+-2Me (7C–9C) guests (Fig. S6†), the NMR titration results demonstrated that all the signals of the guest protons were upfield shifted from the initial concentration of Q[8] (below 1.0 eq.). The proton peaks for the Me and parent phenyl moieties were downfield shifted when the concentration of Q[8] was increased from 1.0 to 2.0 eq. In particular, the continued upfield shifts of the proton signals for the alkyl chains and the pyridinium groups in the NMR spectra suggested that no ternary complexes were formed throughout the Q[8] titration. This result implied that there were no π–π interactions between the OPV2+-2Me (7C–9C) guests in the Q[8] cavity (Fig. S12†), which is consistent with the observation from the fluorescence emission (Fig. 1f). Evidently, the two Me groups on the phenyl group of these guests was the key factor in controlling such molecular assembly modes.
 | ||
| Fig. 5 (a) 1H NMR spectra of OPV2+-H (9C) (0.7 mM in D2O) with different concentrations of Q[8] (0–1.4 mM); (b) 1H–1H NOESY of OPV2+-H (9C)/Q[8] at a molar ratio of 1:2. | ||
Recently, Scherman and co-workers reported pioneering work exploring the properties of Q[8]-based complexes by isothermal titration calorimetry (ITC). Their studies revealed that the enthalpies of 2:2 quaternary complexes of Q[8] with guests were typically below −50 kJ mol−1.24 The large binding enthalpies can be attributed to the occupation of the whole Q[8] cavity by the guest to release the cavity-bound high energy water.38 In an effort to gain more detailed information on the binding interactions between Q[8] and the OPV2+-derived guests, ITC studies were also carried out in this work. As shown in Table S4,† the obtained enthalpy data from representative guests were found to range from −95 kJ mol−1 to −50 kJ mol−1, suggesting that the Q[8] cavities were fully occupied by guests, except for OPV2+-Me(1C)/Q[8] and OPV2+-2Me(1C)/Q[8] complexes, where the enthalpies were below −40 kJ mol−1. This can be attributed to the Me group and shorter alkyl chain making the guest less included in the Q[8] cavity and leading to less displacement of high energy water molecules from the cavity. Similarly, the much lower binding enthalpies of OPV2+-H (5C)/Q[8] (−95.12 kJ mol−1) and OPV2+-Me (5C)/Q[8] (−88.75 kJ mol−1) indicated that a larger number of cavity-bound water molecules were displaced by these novel host–guest binding modes. Therefore, the 2:2, 2:3, and U-shaped 1:2 binding modes of guests with Q[8] were reasonably confirmed by the ITC data and they are in agreement with the related 1H NMR titration experiment results. Additionally, the binding constants (Ka) for the guests and Q[8] complexes were found to be between 1.0 × 105 M−1 and 9.0 × 106 M−1, indicating strong host–guest binding.
Fluorescence lifetime data systematically obtained by time-correlated single photon counting (TCSPC) was fully in line with the above observations. As shown in Table S2,† after addition of Q[8] to the relevant guest solution, the lifetime was significantly increased (in the range of 7 to 15 ns), except in the case of the 1:2 complex (guest/Q[8]) and all host–guest interactions of OPV2+-2Me (7C–9C) with shorter lifetimes were observed. The longest fluorescence lifetimes were observed for the 2:3 complexes of Q[8] with the 5C–6C-appended OPV2+-H and OPV2+-Me guests. Essentially, longer lifetimes imply that the π-conjugated OPV2+ core (fluorophore) is deeply encapsulated in the Q[8] cavity, while shorter lifetimes indicate that the fluorophore is less deeply buried or is farther from the Q[8] cavity.39 Most importantly, the fluorescence lifetime changes were fully in line with the host–guest assembly modes confirmed by the fluorescence and NMR spectra, suggesting that fluorescence decay times may have potential in the characterization of Q[8]-chromophore system binding modes. In particular, the solubility limit of the Q[8] host and the intrinsic hydrophobic effect of the chromophore in water sometimes hinder the performance of the NMR titration experiment. For example, in the present work, the poor solubility of OPV2+-2Me (7C–9C) in aqueous solution meant that the NMR signal changes were not clear enough to detail the host–guest interactions. The shorter fluorescence lifetimes from these complexes in dilute solutions, however, gave strong supplementary evidence to support the host–guest binding modes.
In addition to the above, the Q[8]-derived dynamic host–guest assemblies and the fluorescence signals, such as those of the OPV2+-Me (5C)/Q[8] complex, can be used as competitive fluorescent indicator displacement chemosensors for the discrimination of spermine and spermidine from other species such as cations, anions and amino acids in pure aqueous solutions (Fig. S14†). This suggests that Q[8]-based dynamic assemblies and their resulting fluorescence signals have further applications.
Conclusions
In summary, we have successfully prepared a series of Q[8]-derived multiple dynamic host–guest assemblies, including 2:2, 2:3, and 1:2 complexes, which exhibited quantitative fluorescence. These signals were not only sensitively captured in the fluorescence spectra, but could also be visualized directly with the naked eye under UV light. The host–guest binding modes were further characterized with their 1D and 2D NMR spectra. Interestingly, the systematically determined fluorescence lifetimes obtained from the Q[8] complexes indicated that they could be used to help characterize the host–guest binding modes. Most importantly, the Q[8]-based dynamic assemblies induced by N-substituted alkyl chains and subtle modifications by Me groups on the OPV2+ core had different binding properties according to their HSCT interactions. These dynamic assemblies including the 2:3 quinary complex and the 1:2 interaction-triggered U-shaped conformation have not been reported previously. We expect that the Q[8]-based new binding modes and the resulting photophysical properties will help extend the application of Q[8] as a macrocyclic host in supramolecular polymers and functional materials.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21871063) and Guizhou University (20165656, 20175788, and 20177261).Notes and references
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, New Cucurbituril Homologues: Syntheses, Isolation, Characterization, and X-ray Crystal Structures of Cucurbit[n]uril (n=5, 7, and 8), J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, Controlling Factors in the Synthesis of Cucurbituril and Its Homologues, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS PubMed.
- Y. H. Ko, E. Kim, I. Hwang and K. Kim, Supramolecular assemblies built with host-stabilized charge-transfer interactions, Chem. Commun., 2007, 1305–1315 RSC.
- U. Rauwald and O. A. Scherman, Supramolecular Block Copolymers with Cucurbit[8]uril in Water, Angew. Chem., Int. Ed., 2008, 47, 3950–3953 CrossRef CAS PubMed.
- J. J. Reczek, A. A. Kennedy, B. T. Halbert and A. R. Urbach, Multivalent Recognition of Peptides by Modular Self-Assembled Receptors, J. Am. Chem. Soc., 2009, 131, 2408–2415 CrossRef CAS PubMed.
- Y. Liu, Y. Yu, J. Gao, Z. Wang and X. Zhang, Water-Soluble Supramolecular Polymerization Driven by Multiple Host-Stabilized Charge-Transfer Interactions, Angew. Chem., Int. Ed., 2010, 49, 6576–6579 CrossRef CAS PubMed.
- Y. Wang, D. Li, H. Wang, Y. Chen, H. Han, Q. Jin and J. Ji, pH responsive supramolecular prodrug micelles based on cucurbit[8]uril for intracellular drug delivery, Chem. Commun., 2014, 50, 9390–9392 RSC.
- C. Hu, N. Ma, F. Li, Y. Fang, Y. Liu, L. Zhao, S. Qiao, X. Li, X. Jiang, T. Li, F. Shen, Y. Huang, Q. Luo and J. Liu, Cucurbit[8]uril-Based Giant Supramolecular Vesicles: Highly Stable, Versatile Carriers for Photoresponsive and Targeted Drug Delivery, ACS Appl. Mater. Interfaces, 2018, 10, 4603–4613 CrossRef CAS PubMed.
- P. Dowari, S. Das, B. Pramanik and D. Das, pH clock instructed transient supramolecular peptide amphiphile and its vesicular assembly, Chem. Commun., 2019, 55, 14119–14122 RSC.
- C. Gao, Q. Huang, Q. Lan, Y. Feng, F. Tang, M. P. M. Hoi, J. Zhang, S. M. Y. Lee and R. Wang, A user-friendly herbicide derived from photo-responsive supramolecular vesicles, Nat. Commun., 2018, 9, 2967 CrossRef PubMed.
- J. Liu, Y. Lan, Z. Yu, C. S. Y. Tan, R. M. Parker, C. Abell and O. A. Scherman, Cucurbit[n]uril-Based Microcapsules Self-Assembled within Microfluidic Droplets: A Versatile Approach for Supramolecular Architectures and Materials, Acc. Chem. Res., 2017, 50, 208–217 CrossRef CAS PubMed.
- C. Li, M. J. Rowland, Y. Shao, T. Cao, C. Chen, H. Jia, X. Zhou, Z. Yang, O. A. Scherman and D. Liu, Responsive Double Network Hydrogels of Interpenetrating DNA and CB[8] Host–Guest Supramolecular Systems, Adv. Mater., 2015, 27, 3298–3304 CrossRef CAS PubMed.
- E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. García, Cucurbit[8]uril (CB[8])-Based Supramolecular Switches, Angew. Chem., Int. Ed., 2019, 58, 403–416 CrossRef CAS PubMed.
- K.-D. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C.-H. Sue, T.-Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Toward a Single-Layer Two-Dimensional Honeycomb Supramolecular Organic Framework in Water, J. Am. Chem. Soc., 2013, 135, 17913–17918 CrossRef CAS PubMed.
- M. Yan, X.-B. Liu, Z.-Z. Gao, Y.-P. Wu, J.-L. Hou, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, A pore-expanded supramolecular organic framework and its enrichment of photosensitizers and catalysts for visible-light-induced hydrogen production, Org. Chem. Front., 2019, 6, 1698–1704 RSC.
- N. Barooah, J. Mohanty and A. C. Bhasikuttan, Cucurbit[8]uril-templated H and J dimers of bichromophoric coumarin dyes: origin of contrasting emission, Chem. Commun., 2015, 51, 13225–13228 RSC.
- Y. Si, Q. Zhang, W. Jin, S. Yang, Z. Wang and D. Qu, Aqueous highly emissive host-guest systems by host enhanced intramolecular charge transfer, Dyes Pigm., 2020, 173, 107919 CrossRef.
- T. Jiang, X. Wang, J. Wang, G. Hu and X. Ma, Humidity- and Temperature-Tunable Multicolor Luminescence of Cucurbit[8]uril-Based Supramolecular Assembly, ACS Appl. Mater. Interfaces, 2019, 11, 14399–14407 CrossRef CAS PubMed.
- B. Tang, W.-L. Li, Y. Chang, B. Yuan, Y. Wu, M.-T. Zhang, J.-F. Xu, J. Li and X. Zhang, Supramolecular Radical Dimer: High-Efficiency NIR-II Photothermal Conversion and Therapy, Angew. Chem., Int. Ed., 2019, 58, 15526–15531 CrossRef CAS PubMed.
- Y. Xia, S. Chen and X.-L. Ni, White Light Emission from Cucurbituril-Based Host–Guest Interaction in the Solid State: New Function of the Macrocyclic Host, ACS Appl. Mater. Interfaces, 2018, 10, 13048–13052 CrossRef CAS PubMed.
- X.-L. Ni, S. Chen, Y. Yang and Z. Tao, Facile Cucurbit[8]uril-Based Supramolecular Approach To Fabricate Tunable Luminescent Materials in Aqueous Solution, J. Am. Chem. Soc., 2016, 138, 6177–6183 CrossRef CAS PubMed.
- H.-J. Kim, P. C. Nandajan, J. Gierschner and S. Y. Park, Light-Harvesting Fluorescent Supramolecular Block Copolymers Based on Cyanostilbene Derivatives and Cucurbit[8]urils in Aqueous Solution, Adv. Funct. Mater., 2018, 28, 1705141 CrossRef.
- H. Wu, Y. Chen, X. Dai, P. Li, J. F. Stoddart and Y. Liu, In Situ Photoconversion of Multicolor Luminescence and Pure White Light Emission Based on Carbon Dot-Supported Supramolecular Assembly, J. Am. Chem. Soc., 2019, 141, 6583–6591 CrossRef CAS PubMed.
- G. Wu, M. Olesińska, Y. Wu, D. Matak-Vinkovic and O. A. Scherman, Mining 2:2 Complexes from 1:1 Stoichiometry: Formation of Cucurbit[8]uril–Diarylviologen Quaternary Complexes Favored by Electron-Donating Substituents, J. Am. Chem. Soc., 2017, 139, 3202–3208 CrossRef CAS PubMed.
- M. Olesińska, G. Wu, S. Gómez-Coca, D. Antón-García, I. Szabó, E. Rosta and O. A. Scherman, Modular supramolecular dimerization of optically tunable extended aryl viologens, Chem. Sci., 2019, 10, 8806–8811 RSC.
- G. Wu, Y. J. Bae, M. Olesińska, D. Antón-García, I. Szabó, E. Rosta, M. R. Wasielewski and O. A. Scherman, Controlling the structure and photophysics of fluorophore dimers using multiple cucurbit[8]uril clampings, Chem. Sci., 2020, 11, 812–825 RSC.
- S. Schoder, H. V. Schröder, L. Cera, R. Puttreddy, A. Güttler, U. Resch-Genger, K. Rissanen and C. A. Schalley, Strong Emission Enhancement in pH-Responsive 2:2 Cucurbit[8]uril Complexes, Chem. – Eur. J., 2019, 25, 3257–3261 CAS.
- J. Wang, Z. Huang, X. Ma and H. Tian, Visible-Light-Excited Room-Temperature Phosphorescence in Water by Cucurbit[8]uril-Mediated Supramolecular Assembly, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, Supramolecular Block Copolymers by Seeded Living Polymerization of Perylene Bisimides, J. Am. Chem. Soc., 2019, 141, 12044–12054 CrossRef CAS PubMed.
- X.-T. Liu, K. Wang, Z. Chang, Y.-H. Zhang, J. Xu, Y. Zhao and X.-H. Bu, Engineering Donor–Acceptor Heterostructure Metal–Organic Framework Crystals for Photonic Logic Computation, Angew. Chem., Int. Ed., 2019, 58, 13890–13896 CrossRef CAS PubMed.
- V. N. Vukotic, K. Zhu, G. Baggi and S. J. Loeb, Optical Distinction Between ′′Slow′′ And ′′Fast′′ Translational Motion In Degenerate Molecular Shuttles, Angew. Chem., Int. Ed., 2017, 56, 6136–6141 CrossRef CAS PubMed.
- X. Ye, Y. Liu, Y. Lv, G. Liu, X. Zheng, Q. Han, K. A. Jackson and X. Tao, In Situ Microscopic Observation Of The Crystallization Process Of Molecular Microparticles By Fluorescence Switching, Angew. Chem., Int. Ed., 2015, 54, 7976–7980 CrossRef CAS PubMed.
- W. Guan, W. Zhou, C. Lu and B. Z. Tang, Synthesis and Design Of Aggregation-Induced Emission Surfactants: Direct Observation Of Micelle Transitions And Microemulsion Droplets, Angew. Chem., Int. Ed., 2015, 54, 15160–15164 CrossRef CAS PubMed.
- M. Fu, A. Wang, X. Zhang, L. Dai and J. Li, Direct Observation of the Distribution of Gelatin In Calcium Carbonate Crystals by Super-Resolution Fluorescence Microscopy, Angew. Chem., Int. Ed., 2016, 55, 908–911 CrossRef CAS PubMed.
- C. B. Huang, L. Xu, J. L. Zhu, Y. X. Wang, B. Sun, X. Li and H. B. Yang, Real-Time Monitoring the Dynamics of Coordination-Driven Self-Assembly By Fluorescence-Resonance Energy Transfer, J. Am. Chem. Soc., 2017, 139, 9459–9462 CrossRef CAS PubMed.
- G. Wu, I. Szabó, E. Rosta and O. A. Scherman, Cucurbit[8]uril-mediated pseudo[2,3]rotaxanes, Chem. Commun., 2019, 55, 13227–13230 RSC.
- Y. H. Ko, H. Kim, Y. Kim and K. Kim, U-shaped Conformation of Alkyl chains Bound to a Synthetic Host, Angew. Chem., Int. Ed., 2008, 47, 4106–4109 CrossRef CAS PubMed.
- F. Biedermann, M. Vendruscolo, O. A. Scherman, A. D. Simone and W. M. Nau, Cucurbit[8]uril and Blue-Box: High-Energy Water Release Overwhelms Electrostatic Interactions, J. Am. Chem. Soc., 2013, 135, 14879–14888 CrossRef CAS PubMed.
- R. N. Dsouza, U. Pischel and W. M. Nau, Fluorescent Dyes and Their Supramolecular Host/Guest Complexes with Macrocycles in Aqueous Solution, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qo00649a |
| This journal is © the Partner Organisations 2021 |