
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel boryl complexes and nickel-catalyzed alkyne borylation†
Lukas
Tendera‡
a,
Felipe
Fantuzzi‡
 b,
Todd B.
Marder
ac and
Udo
Radius
*a
b,
Todd B.
Marder
ac and
Udo
Radius
*a
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: u.radius@uni-wuerzburg.de
bSchool of Chemistry and Forensic Science, University of Kent, Park Wood Rd, Canterbury, CT2 7NH, UK
cInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 27th January 2023
Abstract
The first nickel bis-boryl complexes cis-[Ni(iPr2ImMe)2(Bcat)2], cis-[Ni(iPr2ImMe)2(Bpin)2] and cis-[Ni(iPr2ImMe)2(Beg)2] are reported, which were prepared via the reaction of a source of [Ni(iPr2ImMe)2] with the diboron(4) compounds B2cat2, B2pin2 and B2eg2 (iPr2ImMe = 1,3-di-iso-propyl-4,5-dimethylimidazolin-2-ylidene; B2cat2 = bis(catecholato)diboron; B2pin2 = bis(pinacolato)diboron; B2eg2 = bis(ethylene glycolato)diboron). X-ray diffraction and DFT calculations strongly suggest that a delocalized, multicenter bonding scheme dictates the bonding situation of the NiB2 moiety in these square planar complexes, reminiscent of the bonding situation of “non-classical” H2 complexes. [Ni(iPr2ImMe)2] also efficiently catalyzes the diboration of alkynes using B2cat2 as the boron source under mild conditions. In contrast to the known platinum-catalyzed diboration, the nickel system follows a different mechanistic pathway, which not only provides the 1,2-borylation product in excellent yields, but also provides an efficient approach to other products such as C–C coupled borylation products or rare tetra-borylated compounds. The mechanism of the nickel-catalyzed alkyne borylation was examined by means of stoichiometric reactions and DFT calculations. Oxidative addition of the diboron reagent to nickel is not dominant; the first steps of the catalytic cycle are coordination of the alkyne to [Ni(iPr2ImMe)2] and subsequent borylation at the coordinated and, thus, activated alkyne to yield complexes of the type [Ni(NHC)2(η2-cis-(Bcat)(R)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R)(Bcat))], exemplified by the isolation and structural characterization of [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] and [Ni(iPr2ImMe)2(η2-cis-(Bcat)(H7C3)CC(C3H7)(Bcat))].
C(R)(Bcat))], exemplified by the isolation and structural characterization of [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] and [Ni(iPr2ImMe)2(η2-cis-(Bcat)(H7C3)CC(C3H7)(Bcat))].
Introduction
Numerous homogeneous catalytic borylation reactions have been developed over the past decades,1 which include the Suzuki–Miyaura borylation of aryl and alkyl halides,2 catalytic addition reactions to unsaturated organic molecules such as alkenes, alkynes, allenes, α,β-unsaturated compounds, and carbonyl compounds via hydroboration, diboration, β-borylation or carboboration,3,4 or the direct functionalization of C–H bonds.5 In all of these transformations, transition metal boryl complexes6 play a pivotal role and are key intermediates.7 Thus, research on transition metal boryl complexes [LnM-BX2], in general, is attractive due to their interesting properties and their utility in catalysis, in which poly-boryl complexes often play a dominant role. Among the most important transition metal poly-boryl complexes employed in catalysis are iridium tris-boryl or rhodium bis-boryl complexes, initially synthesized by Marder and Baker et al. in 1993 (Fig. 1: compounds I and III),8 and nowadays frequently employed for C–H borylations of arenes, alkenes, and alkanes.5,9,10 Complexes such as [Ir(dtbpy)(COE)(Bpin)3] II (Fig. 1, dtbpy = di-tert-butylbipyridine, COE = cyclooctene; pin = pinacolato)9a are key catalytic intermediates in iridium-catalyzed C–H borylation reactions. Another well studied class of poly-boryl complexes are platinum bis-boryl complexes such as cis-[Pt(PPh3)2(Bcat)2] IV (cat = catecholato),11 pre-catalysts for the addition of diborane(4) compounds to alkynes, reported independently by the groups of Suzuki and Miyaura,12a,b Smith III,12c and Marder and Norman.13 The platinum-catalyzed insertion of alkynes into the B–B bond of a diborane(4) reagent is of interest as it provides the most atom economical route for the stereoselective synthesis of tri- and tetra-substituted alkenes.4 The resulting 1,2-diboryl alkenes are important building blocks in organic synthesis and materials science.14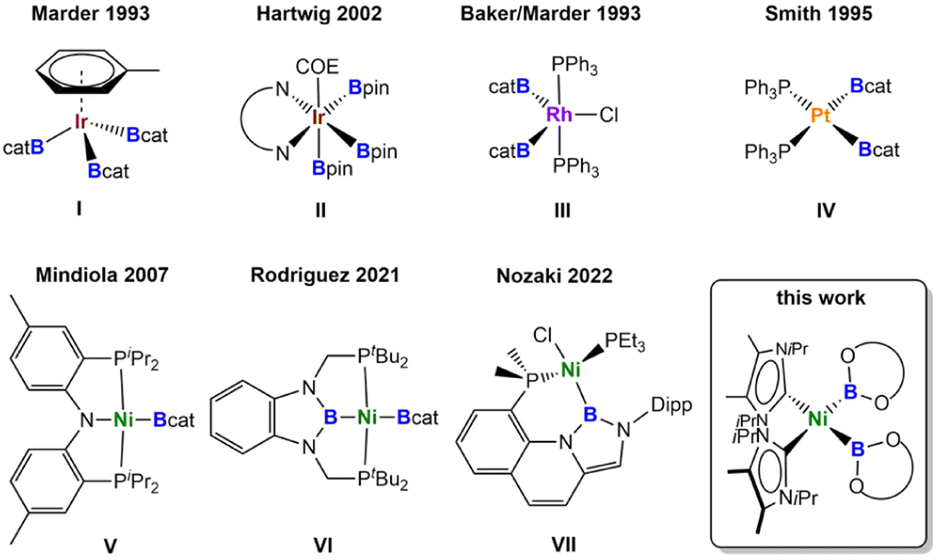 | ||
| Fig. 1 Selected examples of transition-metal boryl complexes. | ||
Most of the transition metal-catalyzed borylation reactions developed initially employed precious metals as the catalyst precursors. As first-row d-block metals are less toxic, less expensive, Earth-abundant, and environmentally benign, they are very attractive alternatives to these expensive noble metals. Recently developed borylations employing 3d-metal catalysts show excellent reactivity and selectivity and often facilitate unique transformations previously unobserved in traditional precious metal-catalyzed processes.1h Good examples for outstanding reactivity are copper(I) boryl complexes, using a diverse range of ligands with phosphines and NHCs (N-heterocyclic carbenes) being the most commonly employed. These reagents are attractive for different transformations, featuring mild reaction conditions, good functional group tolerance, and low cost of the metal catalyst.1h,15 For example, in situ generated copper boryl complexes of the type [LCu(Bpin)] (L = phosphine or NHC) have been employed successfully in the borylation of aryl or alkyl chlorides, bromides, and iodides.15
We recently investigated the use of NHC nickel complexes for the borylation of aryl chlorides, aryl fluorides, and indoles.16 For each of our nickel-catalyzed borylation reactions, a nickel boryl complex was proposed as a key intermediate, but has never been fully characterized in situ or isolated.17 Nickel boryl complexes are generally considered to be elusive, in contrast to other 3d-metals such as iron,18 cobalt,19 or copper.15,20 Only a few structurally characterized nickel boryl complexes have been isolated thus far, all of them bearing large, rigid pincer ligands (Fig. 1). In 2007, Mindiola et al.21a,b reported the synthesis of the first nickel mono-boryl complex [(PNP)Ni(Bcat)] V (PNP = N[2-P(CHMe2)2-4-methylphenyl]2). In 2014, Peters et al.21c and Rodriguez et al.21d independently reported several boryl complexes of PBP pincer ligands [(tBuPBP)NiL] (L = H, Cl, Br, OTf, OC(H)O, Me; tBuPBP = C6H4{N(CH2PtBu2)}2B), in which the boryl moiety is embedded in the pincer system. They also introduced a dimeric nickel(I) complex [(PhPBP)Ni]2 with bridging boryl ligands (PhPBP = C6H4{N(CH2PPh2)}2B) and the first bis-boryl complex [(tBuPBP)Ni(Bcat)] VI.21c,e Very recently, Nozaki et al. reported the phosphine/boryl chelating complex VII (Fig. 1), synthesized via dehydrochloroborylation of a nickel(0) σ-borane precursor.21f
Results and discussion
Synthesis and characterization of nickel boryl complexes
We recently investigated, in detail, the differences in the reactivity of NHC nickel complexes of the type [Ni(NHC)2] dependent upon the stereo-electronic features of the NHC ligands,22 which was the key to the success of the present study. In earlier work, we found that reactions of synthetic equivalents of the complexes [Ni(iPr2Im)2], [Ni(Cy2Im)2], and [Ni(Mes2Im)2] (R2Im = 1,3-di-organyl-imidazolin-2-ylidene; Cy = cyclohexyl; Mes = mesityl; iPr = iso-propyl) with B2cat2, B2pin2, or B2eg2 (=bis(ethylene glycolato)diboron) did not lead to isolable nickel boryl complexes. For the smaller NHCs, decomposition with formation of nickel black and NHC diborane adducts typically occurred. The NHC diborane adducts often underwent subsequent NHC ring expansion reactions, which destroyed the core structure of the NHC and made the process irreversible.23 Furthermore, in the course of our work on the defluoroborylation of polyfluoroarenes, on the borylation of aryl chlorides, and on the C–H borylation of indoles using [Ni(Mes2Im)2] as a catalyst,16 we postulated nickel boryl complexes as decisive intermediates, but never detected such compounds. Complexes of the type [Ni(Mes2Im)2(ArF)(B{OR}2)] (B{OR}2 = Bcat, Bpin) were not observed in stoichiometric reactions of [Ni(Mes2Im)2(ArF)F] with B2pin2 or B2cat2, as reductive elimination leading to the borylation product ArF-B(OR)2 was rapid, reforming [Ni(Mes2Im)2].16a However, in the course of our work on the borylation of aryl chlorides, a resonance at 44.5 ppm was observed in the 11B{1H} NMR spectrum for the reaction of [Ni(Cy2Im)2(Ar)Cl] with B2pin2, which indicated the formation of a nickel boryl complex.16d Unfortunately, this complex of the N-cyclohexyl-substituted NHC was not stable in solution and defied isolation despite several attempts. Therefore, we reasoned that using an NHC ligand with similar donor properties and only slightly modified steric demand might lead to the successful synthesis of nickel boryl complexes. As it has been demonstrated previously that backbone substitution at the C4 and C5 position of the imidazole framework by methylation effects the sterics of the NHC ligands as repulsion between the C4/C5 methyl group and the N-organyl substituent leads to smaller Ccarbene-N-Csubstituent angles,24 we used synthetic equivalents of the backbone-methylated [Ni(iPr2ImMe)2] for this study.[Ni(iPr2ImMe)2] was provided from a mixture of [Ni2(iPr2ImMe)4(μ-(η2:η2)-COD)] 1 and [Ni(iPr2ImMe)2(η4-COD)] 1a, which can be prepared by the reaction of [Ni(COD)2] with two equivalents of iPr2ImMe, as reported previously.22l The stoichiometric reaction of such a mixture of 1 and 1a with B2cat2 at room temperature cleanly led to the formation of cis-[Ni(iPr2ImMe)2(Bcat)2] 2a (Scheme 1), which is the first cis-nickel bis-boryl complex synthesized and isolated thus far. This complex was isolated as a pale brown solid in 58% yield and was characterized by IR- and NMR-spectroscopy, X-ray diffraction, and elemental analysis (vide infra).
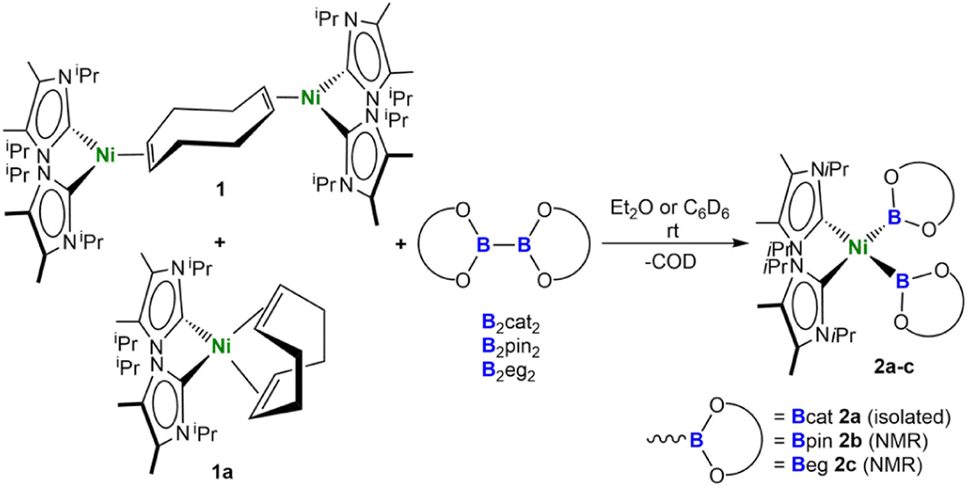 | ||
| Scheme 1 Synthesis of cis-[Ni(iPr2ImMe)2(Bcat)2] 2a, cis-[Ni(iPr2ImMe)2(Bpin)2] 2b, and cis-[Ni(iPr2ImMe)2(Beg)2] 2c. | ||
If the reaction was carried out with either B2pin2 or B2eg2 instead of B2cat2, it did not proceed quantitatively at room temperature, even if a large excess of the diboron(4) reagent was employed. In all cases, the reaction started at approximately 0 °C, but did not proceed at lower temperatures. An increase of the temperature above room temperature rapidly led to a darkening of the reaction mixture with decomposition of the bis-boryl complexes, which is especially rapid for 2b and 2c. This behavior reflects that of copper(I) boryl complexes, which easily decompose upon warming.20d–g The use of modified starting materials, such as the ethylene complex [Ni(iPr2ImMe)2(η2-C2H4)] 1b or the cyclooctene (COE) complex [Ni(iPr2ImMe)2(η2-COE)] 1c (see ESI†), was also unsuccessful for the bulk production of pure 2b and 2c. However, the formation of the bis-boryl complexes cis-[Ni(iPr2ImMe)2(Bpin)2] 2b and cis-[Ni(iPr2ImMe)2(Beg)2] 2c was clearly detected by NMR spectroscopy, and small amounts of these complexes suitable for X-ray diffraction crystallized from these reaction mixtures (Fig. 2). The bis-boryl complexes reveal different stabilities in solution. Whereas cis-[Ni(iPr2ImMe)2(Bpin)2] 2b was still detected in the reaction mixture in a solution kept at room temperature for one month, complexes 2a and 2c completely decompose in C6D6 over a period of 6–14 days with formation of multiple, as yet unidentified, species.
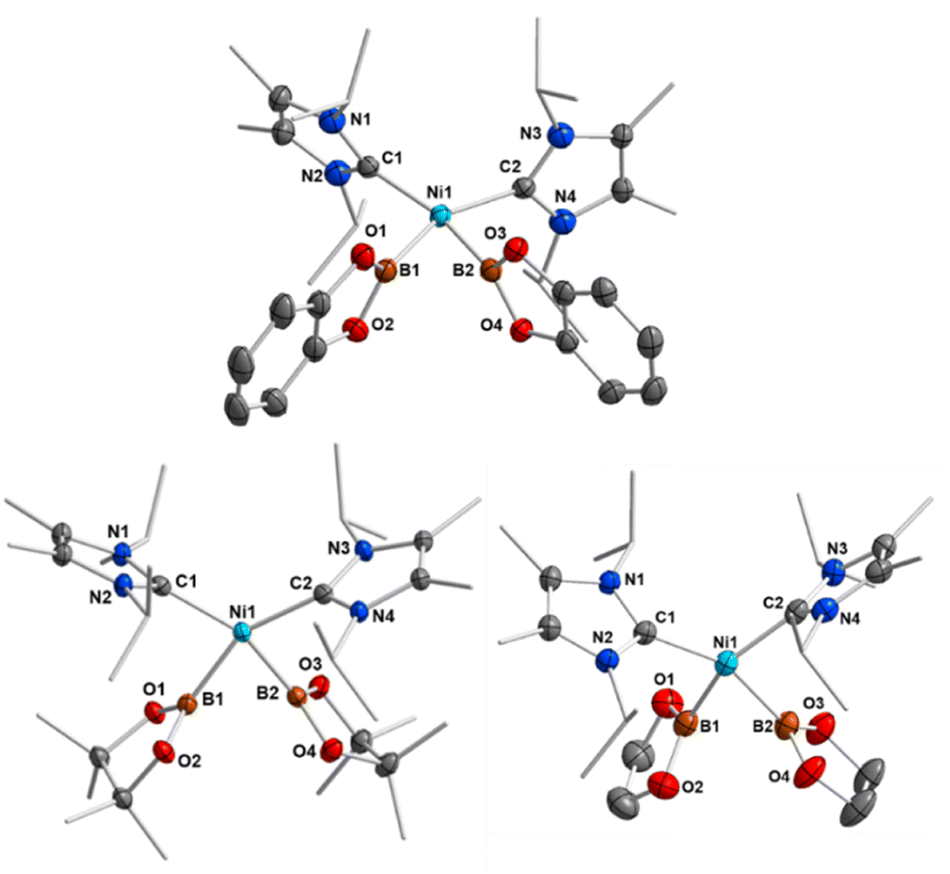 | ||
| Fig. 2 Molecular structures of cis-[Ni(iPr2ImMe)2(Bcat)2] 2a (top), cis-[Ni(iPr2ImMe)2(Bpin)2] 2b (bottom left), and cis-[Ni(iPr2ImMe)2(Beg)2] 2c (bottom right) in the solid state (ellipsoids shown at 50% probability level). Hydrogen atoms are omitted for clarity. For selected bond lengths and angles see Table 1 and ESI Fig. S4–S6.† | ||
Characteristic for complexes 2a–c is a broad resonance at 48.7 ppm (2a), 46.1 ppm (2b), and 46.5 ppm (2c) in the 11B{1H} NMR spectrum (see Table 1), which is the region typically observed for transition metal boryl complexes,6c.f. 47.0 ppm for cis-[Pt(PPh3)2(Bcat)2].13 In the 13C{1H} NMR spectra, the NHC carbene carbon resonances are also significantly shifted compared to those of the starting materials 1 (206.5 ppm) and 1a (205.4 ppm) to 194.3 ppm (2a), 199.4 ppm (2b), and 198.5 ppm (2c). The complexes adopt cis-configurations in solution as their 1H NMR spectra indicate pseudo-C2v species with two resonances for the N-iso-propyl methyl protons (2a: 1.28 ppm and 1.45 ppm, 2b: 1.32 ppm and 1.69 ppm, 2c: 1.28 ppm and 1.58 ppm) and only one signal for the N-iso-propyl methine (2a: 6.05 ppm, 2b: 5.99 ppm, 2c: 6.04 ppm) and for the backbone methyl protons (2a: 1.63 ppm, 2b: 1.84 ppm, 2c: 1.78 ppm).
| Ni–B (Å) | B–B (Å) | Ni–C (Å) | B–Ni–B (°) | δ B B(OR)2 (ppm) |
δ
c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NHC [ppm] NHC [ppm] |
|
|---|---|---|---|---|---|---|
| 2a | 1.9231(19) | 2.156(3) | 1.9393(16) | 68.45(7) | 48.7 | 194.3 |
| 1.9092(18) | 1.9448(15) | |||||
| 2b | 1.936(2) | 2.247(3) | 1.9318(18) | 70.82(8) | 46.1 | 199.4 |
| 1.942(2) | 1.9185(17) | |||||
| 2c | 1.939(2) | 2.189(4) | 1.9180(15) | 68.79(8) | 46.5 | 198.5 |
| 1.9353(19) | 1.9265(17) | |||||
|
V21a |
1.9091(18) | — | — | — | 47.0 | — |
Crystals suitable for X-ray diffraction of 2a–c were obtained by storing the reaction mixtures in diethylether at −30 °C. Complexes 2a–c crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and adopt a distorted square planar geometry with cis-boryl ligands, as observed for platinum bis-boryl complexes cis-[Pt(PR3)2(B{OR}2)2].6,11 The Ni–C and Ni–B distances lie in a narrow range between 1.9092(18) Å and 1.9448(15) Å (see Table 1). We attribute the formation of cis-configured complexes to the strong trans-influence of the boryl ligands25 and a remaining B–B interaction between the two boryl boron atoms (vide infra). This situation is similar to that observed previously for NHC-stabilized bis-silyl and hydro–silyl complexes cis-[Ni(NHC)2(SiR3)2] and cis-[Ni(NHC)2(H)(SiR3)].26 The B–B separations of 2.156(3) Å (2a), 2.247(3) Å (2b), and 2.189(4) Å (2c) (see Table 1) are much smaller than those observed for bis-boryl platinum complexes (2.451–2.667 Å),11–13 consistent with small B1–Ni–B2 angles of 68.45(7)° (2a), 70.82(8)° (2b), and 68.79(8)° (2c).
and adopt a distorted square planar geometry with cis-boryl ligands, as observed for platinum bis-boryl complexes cis-[Pt(PR3)2(B{OR}2)2].6,11 The Ni–C and Ni–B distances lie in a narrow range between 1.9092(18) Å and 1.9448(15) Å (see Table 1). We attribute the formation of cis-configured complexes to the strong trans-influence of the boryl ligands25 and a remaining B–B interaction between the two boryl boron atoms (vide infra). This situation is similar to that observed previously for NHC-stabilized bis-silyl and hydro–silyl complexes cis-[Ni(NHC)2(SiR3)2] and cis-[Ni(NHC)2(H)(SiR3)].26 The B–B separations of 2.156(3) Å (2a), 2.247(3) Å (2b), and 2.189(4) Å (2c) (see Table 1) are much smaller than those observed for bis-boryl platinum complexes (2.451–2.667 Å),11–13 consistent with small B1–Ni–B2 angles of 68.45(7)° (2a), 70.82(8)° (2b), and 68.79(8)° (2c).
Thus, the B–B distances are only 0.478 Å (2a), 0.540 Å (2b), and 0.485 Å (2c) longer than those in the solid state molecular structures of B2cat2 (1.678(3) Å), B2pin2 (1.707(5) Å), and B2eg2 (1.704(3) Å).23d,27 The BO2 planes of both boryl ligands are nearly perpendicular to the NiC2B2 square plane with angles of 87.85(7)° and 86.21(6)° (2a), 88.41(9)° and 88.07(9)° (2b), and 85.85(10)° and 85.30(10)° (2c). Thus, the structures are best represented by valence tautomer B in Scheme 2, which lies in-between the limiting structures of a Ni(II) bis-boryl complex A and a Ni(0) diborane(4) complex C, i.e., incomplete oxidative addition with a residual B⋯B interaction. This is reminiscent of “non-classical” H2 complexes. As the Ni–B distances of 1.9231(19) Å and 1.9092(18) Å (2a), 1.936(2) Å and 1.942(2) Å (2b), and 1.939(2) Å and 1.9353(19) Å are of similar magnitude as those observed in [(PNP)Ni(Bcat)] V (1.9091(18) Å)21a and [(PBP)Ni(Bcat)] VI (1.942(2) Å; 2.015(2) Å),21d which feature 2-center-2-electron Ni–B bonds, the oxidative addition is nearly complete.
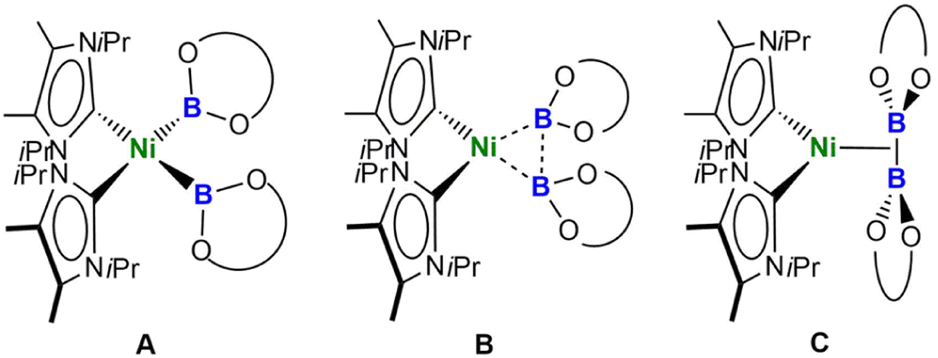 | ||
| Scheme 2 Valence tautomers of complexes cis-[Ni(iPr2ImMe)2(B{OR}2)2] 2a–c. | ||
This situation is closely related to that observed for the paramagnetic cobalt complexes [Co(PMe3)3(Bcat)2] (B–B: 2.185 Å, B–Co–B: 67.9(4)°) and mer-[Co(PMe3)3(Bcat)3] (B–B: 2.1541(5) Å, B–Co–B: 65.78(1) °), which also feature two Bcat ligands with short B–B distances.19a,g DFT calculations on mer-[Co(PMe3)3(Bcat)3] revealed bond critical points at the Co–B vector with substantial electron densities and a bond critical point along the B–B vector, which was characterized by a substantial electron density associated with a much smaller yet positive Laplacian compared to the Co–B bond. It was concluded that mer-[Co(PMe3)3(Bcat)3] maintains a degree of B–B interaction, which is essential for the stabilization of this boryl complex.
The preference for isomer B to describe the bonding situation of the NiB2 motif in 2a–c is also supported by DFT computations on complex 2a. Inspection of the canonical Kohn–Sham molecular orbitals of 2a reveals that the HOMO (Fig. 3a) is mainly composed of a combination of 3d orbitals of nickel that expands across the B–B bonding region. Accordingly, a Mayer bond order (MBO)28 of 0.451 is found for the B–B bond, whereas the corresponding MBOs of the Ni–B bonds are 0.711 each. These findings strongly suggest that a delocalized, multicenter bonding scheme dictates the bonding situation of the NiB2 moiety. This picture is corroborated by further calculations based on the intrinsic bond orbital (IBO) approach.29 Analysis of the IBOs of 2a indicates that two doubly occupied IBOs are participating in the NiB2 bonding. The first orbital (Fig. 3b) is mainly localized at the B–B bonding region, with partial delocalization on the Ni center and across the Ni–B bonds. In contrast, the second orbital (Fig. 3c) is mostly localized across the Ni–B bonds, but with a larger contribution coming from the Ni center. From the IBO point of view, the bonding situation of the NiB2 motif is better described as composed of two three-center two-electron (3c,2e) bonds. Taken together, these results are in accordance with the analysis based on the X-ray structures of 2a–c and support the multicenter bonding situation depicted in isomer B.
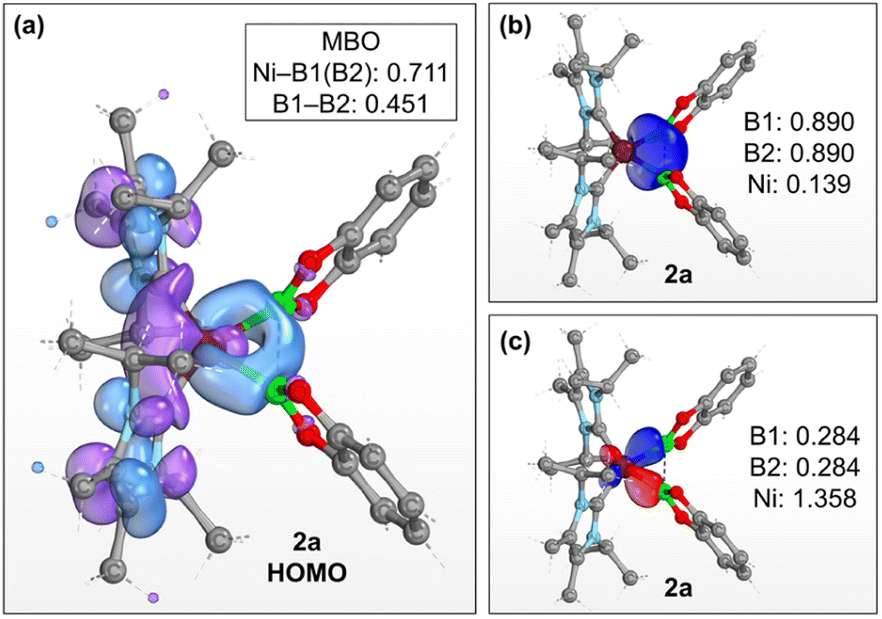 | ||
| Fig. 3 (a) HOMO of 2a (isovalue: 0.03 au). The Ni–B and B–B Mayer bond orders of 2a are shown in the top right box. (b) and (c) Intrinsic bond orbitals of 2a involved in the bonding of the NiB2 motif. Numerical values indicate the fraction of electrons of the doubly occupied orbital assigned to the individual atoms. Level of theory: PBE0-D3(BJ)/def2-SVP/def2-TZVP(Ni).30 Hydrogen atoms are omitted for clarity. | ||
Nickel-catalyzed borylation of alkynes
Bis-boryl complexes are regarded as the key intermediates for the catalytic diboration of alkynes in platinum chemistry.11–13 It has been demonstrated that complexes cis-[Pt(PR3)2(Bcat)2] or synthetic equivalents for [Pt(PR3)2] are highly active catalyst precursors for the cis-stereospecific diboration of alkynes and 1,3-diynes. In the platinum system, phosphine dissociation is a critical step in the catalytic cycle (see ESI Fig. S1†), which includes formation of the bis-boryl complex cis-[Pt(PR3)2(B{OR}2)2] from the catalyst precursor and subsequent phosphine dissociation to give sterically and electronically unsaturated [Pt(PR3)(B{OR}2)2]. This complex can add the alkyne, and insertion of the alkyne into the Pt–B{OR}2 bond and reductive elimination lead to the corresponding cis-alkene-1,2-bis(boronate) ester with regeneration of [Pt(PR3)] or [Pt(PR3)2]. DFT calculations have shown that similar Pd(0) complexes cannot catalyze the alkyne diboration due to differences in the oxidative addition step of the B–B bond of the diborane to [M(PR3)2].12d Although the kinetic barrier is lower, the addition is endothermic for the palladium complex and thus the addition product is not stable due to a very small reverse barrier. For the diboration of alkynes using B2pin2 as the boron source, the optimized phosphine:platinum ratio was later shown to be 1:1, with catalyst activity being strongly dependent on the nature of the phosphine.13b Sterically bulky, strong electron donors, such as PCy3, allowed diborations to be performed at ambient temperatures. Thus, the isolable and stable compound [Pt(PCy3)(η2-C2H4)2] was shown to be an excellent catalyst precursor for alkyne diboration even at room temperature.13b We were thus interested to see whether our nickel complexes are also able to catalyze this reaction.
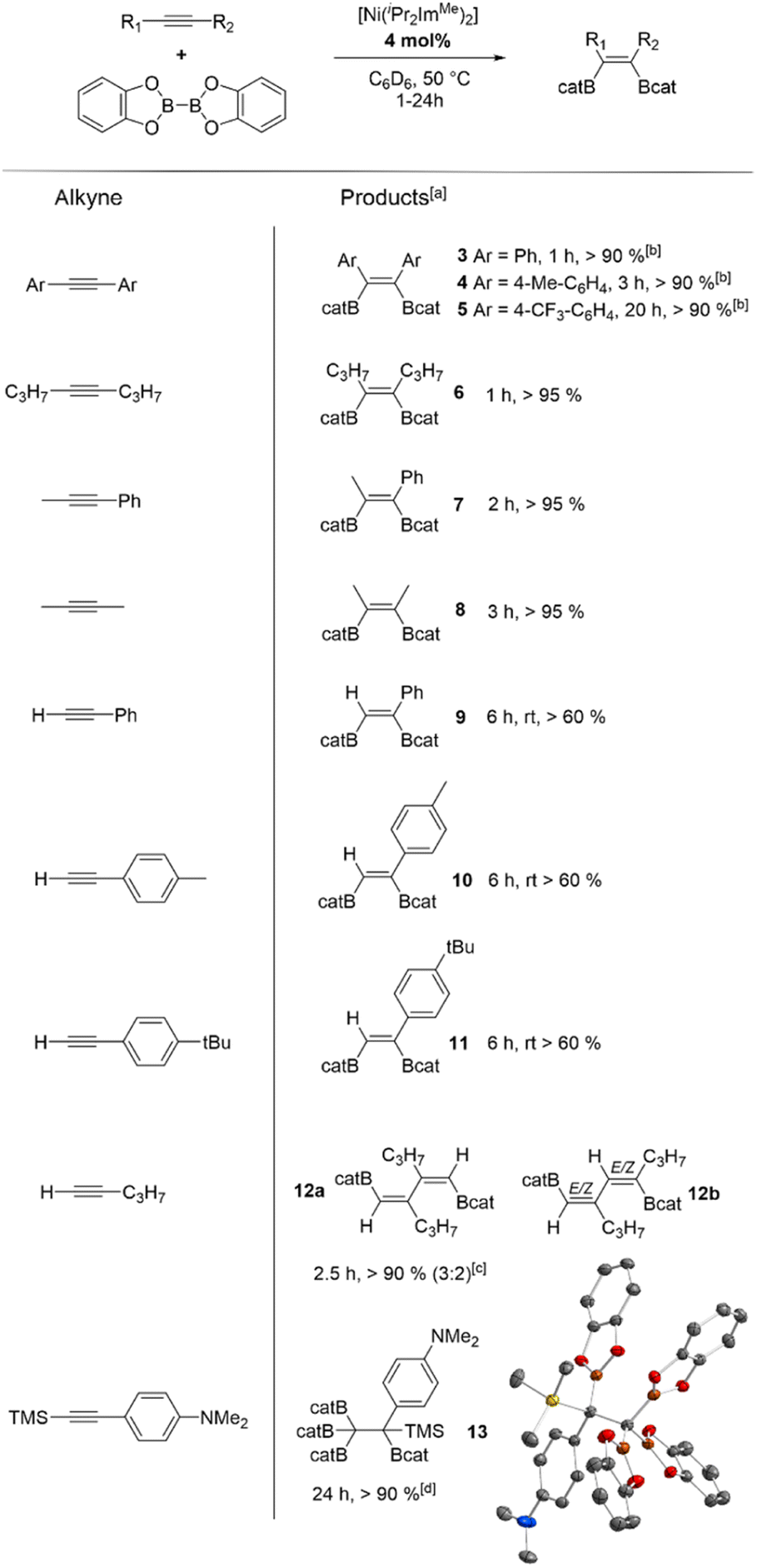
Catalytic reactions were typically carried out in Youngs tap-NMR tubes using different internal and terminal alkynes (see Table 2). As standard reaction conditions, 4 mol% of [Ni(iPr2ImMe)2] (the mixture of 1 and 1a was used directly) and equimolar amounts of alkyne and B2cat2 were reacted using C6D6 as the solvent at 50 °C. Reaction progress was monitored via1H and 11B{1H} NMR spectroscopy. After completion, the resulting products were identified by NMR spectroscopy and GC/MS analysis of the reaction mixture. Internal alkynes led selectively to the quantitative formation of the cis-1,2-diborylalkenes Z-(Bcat)(Ph)CC(Ph)(Bcat) 3, Z-(Bcat)(4-Me-C6H4)CC(4-Me-C6H4)(Bcat) 4, Z-(Bcat)(4-CF3-C6H4)CC(4-CF3-C6H4)(Bcat) 5, Z-(Bcat)(C3H7)CC(C3H7)(Bcat) 6, and Z-(Bcat)(Me)CC(Ph)(Bcat) 7. However, for the synthesis of 3, 4, and 5, a higher catalyst loading of 10 mol% was necessary to reach full conversion as the catalyst is deactivated by transfer of the NHC ligands to the borylation product to yield the corresponding mono NHC-adducts (vide infra). The NHC adduct of compound 4, Z-(Bcat)(4-Me-C6H4)CC(4-Me-C6H4)(Bcat)·(iPr2ImMe) 4NHC, was isolated and characterized separately by the reaction of 4 with one equivalent of iPr2ImMe (see Scheme 3).
| a Reaction conditions: [Ni(iPr2ImMe)2] 1/1a (4 mol%), alkyne (1.0 equiv), B2cat2 (1.0 equiv), C6D6 (0.6 mL), 50 °C (if not otherwise stated). Products after total consumption of the alkynes, monitored by NMR and GC/MS. Yields are combined yields of the products and were estimated by 1H-NMR with respect to the consumption of B2cat2. b [Ni(iPr2ImMe)2] 1/1a (10 mol% needed for completion). c Excess of alkyne (>4 equiv). Products after total consumption of B2cat2. d B2cat2 (2 equiv). |
|---|
|
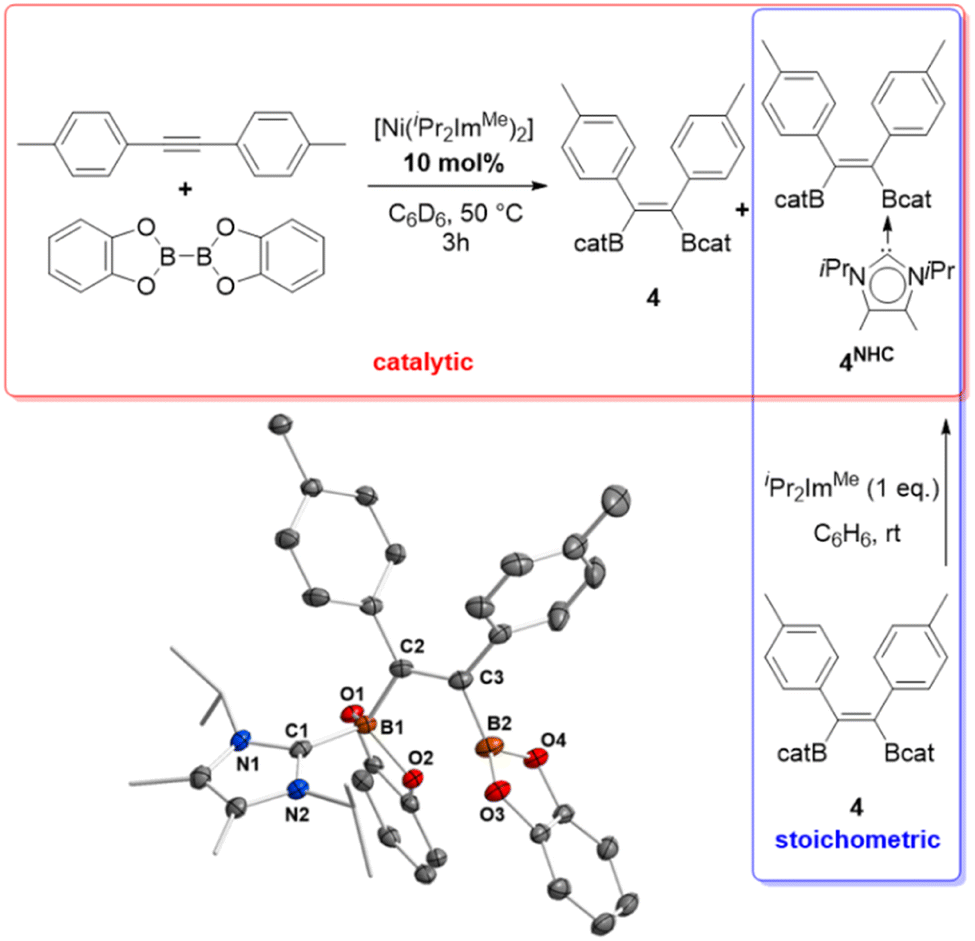 | ||
| Scheme 3 Deactivation of the catalyst and independent synthesis of Z-(Bcat)(4-Me-C6H4)CC(4-Me-C6H4)(Bcat)·(iPr2ImMe) 4NHC. For selected bond lengths and angles in 4NHC see Fig. S17 in the ESI.† | ||
The reaction of aryl substituted terminal alkynes also led to the formation of the cis-1,2-diborylalkenes E-(Bcat)HCC(Ph)(Bcat) 9, E-(Bcat)HCC(4-Me-C6H4)(Bcat) 10 and E-(Bcat)HCC(4-tBu-C6H4)(Bcat) 11, but after consumption of the alkyne, approximately 40% unreacted B2cat2 was always detected besides the 1,2-diborylalkene products. Analysis of the reaction mixtures via high resolution mass spectrometry revealed that alkyne cyclotrimerization products and different partially borylated coupling products were formed as side-products, which are hard to identify via NMR spectroscopy (see ESI, Fig. S72–S83†). Note that the use of more than one equivalent of the alkynes inhibits the borylation, so that no transformation at all was observed when 4 equivalents of the alkynes were used.
Compared to the well-established platinum catalysts,12,13 our nickel complex shows very good activity towards internal alkynes under mild conditions. Only the mono-phosphine platinum complexes reported by Marder et al.13b show a higher efficiency, as they catalyze the diboration at room temperature with a low catalyst loading. For terminal alkynes, the platinum diphosphine complexes and, especially, the palladium NHC complex [Pd(Me2ImMe)2(η2-PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)] reported by Navarro et al., deliver higher yields (79–95%).31 Interestingly, the reactions of alkyl substituted 1-pentyne or TMS-substituted N,N-dimethyl-4-[(trimethylsilyl)-ethynyl]aniline led to new, previously unknown reaction products. The borylation of 1-pentyne selectively afforded the C–C coupled borylation products Z,Z-(Bcat)HCC(C3H7)–(C3H7)CCH(Bcat) 12a (for proof of connectivity see ESI Fig. S18†) and E/Z,E/Z-(Bcat)HCC(C3H7)–HCC(Bcat)(C3H7) 12b in a 3:2 ratio, according to NMR and GC/MS analysis. An excess of 1-pentyne (4 equiv.) was needed to reach full consumption of B2cat2. On the other hand, the borylation of the TMS-substituted alkyne selectively afforded the formation of poly-borylated (4-NMe2-C6H4)(Bcat)(TMS)C–C(Bcat)313. In this case, 2 equivalents of B2cat2 were needed for a full conversion and the TMS-group undergoes a formal 1,2-shift. Insertion of transition metal complexes into C(sp)–SiMe3 bonds of alkynyl silanes were observed previously and are well explored in platinum chemistry.13a,32 C(sp)–Si cleavage of Ar–CC–SiMe3 should result in the formation of TMS–Bcat and Ar–CC–Bcat, which can react together in a transition metal-catalyzed reaction. The resulting product can then be diborated to yield different isomers Ar–C2(Bcat)4(SiMe3), which have been observed for platinum-catalyzed reactions.33 Therefore, the high yield synthesis of 13 is remarkable.
CPh)] reported by Navarro et al., deliver higher yields (79–95%).31 Interestingly, the reactions of alkyl substituted 1-pentyne or TMS-substituted N,N-dimethyl-4-[(trimethylsilyl)-ethynyl]aniline led to new, previously unknown reaction products. The borylation of 1-pentyne selectively afforded the C–C coupled borylation products Z,Z-(Bcat)HCC(C3H7)–(C3H7)CCH(Bcat) 12a (for proof of connectivity see ESI Fig. S18†) and E/Z,E/Z-(Bcat)HCC(C3H7)–HCC(Bcat)(C3H7) 12b in a 3:2 ratio, according to NMR and GC/MS analysis. An excess of 1-pentyne (4 equiv.) was needed to reach full consumption of B2cat2. On the other hand, the borylation of the TMS-substituted alkyne selectively afforded the formation of poly-borylated (4-NMe2-C6H4)(Bcat)(TMS)C–C(Bcat)313. In this case, 2 equivalents of B2cat2 were needed for a full conversion and the TMS-group undergoes a formal 1,2-shift. Insertion of transition metal complexes into C(sp)–SiMe3 bonds of alkynyl silanes were observed previously and are well explored in platinum chemistry.13a,32 C(sp)–Si cleavage of Ar–CC–SiMe3 should result in the formation of TMS–Bcat and Ar–CC–Bcat, which can react together in a transition metal-catalyzed reaction. The resulting product can then be diborated to yield different isomers Ar–C2(Bcat)4(SiMe3), which have been observed for platinum-catalyzed reactions.33 Therefore, the high yield synthesis of 13 is remarkable.
As we observed the formation of coupled and tetra-borylated products 12a/b and 13, we had a closer look at the catalytic borylation reaction of the internal alkyne 2-butyne, which is another special case (Scheme 4).
 | ||
| Scheme 4 Borylation of 2-butyne yielding Z-(Bcat)(Me)CC(Me)(Bcat) 8, (Bcat)2(Me)C–C(Me)(Bcat)28a or E,E-(Bcat)(Me)CC(Me)–(Me)CC(Me)(Bcat) 8b, depending on the stoichiometry used. | ||
The reaction of 2-butyne with B2cat2 and [Ni(iPr2ImMe)2] as a catalyst afforded three different reaction products depending on the reaction conditions used, namely Z-(Bcat)(Me)CC(Me)(Bcat) 8, (Bcat)2(Me)C–C(Me)(Bcat)28a or E,E-(Bcat)(Me)CC(Me)–(Me)CC(Me)(Bcat) 8b (compare Scheme 4). The products obtained were often mixtures which cannot be separated by column chromatography, but the product ratios can be controlled to some extent via the ratio of alkyne to B2cat2 employed. The reaction of one equivalent of 2-butyne with a slight excess of B2cat2 and 4 mol% of [Ni(iPr2ImMe)2] (1 and 1a) in C6D6 was monitored by 1H- and 11B{1H}-NMR spectroscopy, which showed complete consumption of the alkyne and B2cat2 after 3 h at 50 °C. NMR spectroscopy and GC/MS analysis of the final reaction products revealed the selective formation of the bis-borylated product Z-(Bcat)(Me)CC(Me)(Bcat) 8 as the main product and traces of tetra-borylated product (Bcat)2(Me)C–C(Me)(Bcat)28a in a combined quantitative yield. If 2 equivalents of B2cat2 were used, 8a was formed exclusively in quantitative yields. Applying a large excess of 2-butyne (>4 equiv.) led to a mixture of 8, 8a and 8b, with 8b being the main product, after full consumption of the diboron reagent (4d, rt). To our knowledge, the formation of compounds 8a and 13 are the only examples for tetra-borylation of alkynes, beside the Pt-catalyzed tetra-borylation of (Bcat)CC(Bcat) to yield hexa-borylated ethane (Bcat)3C–C(Bcat)3, which was reported by Siebert et al. in 1999.34 Products 8b and 12a/b are very rare examples for a combined one-step coupling and borylation of alkynes, which was first described by Marder et al., who observed small amounts of coupling products (via GC/MS) during the borylation of phenylacetylene with their platinum-catalyst.13a In recent years, Buñuel and Cárdenas et al. reported some closely related nickel-catalyzed borylative cyclization reactions of enynes and allenynes using either HBpin or B2pin2 as the boron source.35
In our case, the use of alternative diboron sources B2pin2, B2eg2 and B2neop2 did not achieve borylation at all or showed large quantities of byproducts from oligomerization reactions. We attribute this lack of reactivity to the fact that B2cat2 is the most Lewis acidic diborane(4) under consideration and therefore the most reactive of the diboron reagents used.36 Furthermore, we have shown recently that bis-NHC adducts of the type (NHC)2·B2(OR)4 are sources of boryl radicals of the type NHC–BR2˙, exemplified by Me2ImMe·Bneop˙ (Me2ImMe = 1,3,4,5-tetramethyl-imidazolin-2-ylidene, neop = neopentylglycolato), which can be used for the transition metal- and additive-free boryl transfer to aryl iodides and bromides giving aryl boronate esters.37a We also reported the related phosphine-catalyzed hydroboration of 1,3-diynes with pinacolborane that affords (E)-1-boryl-1,3-enynes and proceeds with excellent selectivity for boron addition to the external carbon of the 1,3-diyne framework.37b According to our control experiments, the free carbene iPr2ImMe is not a good catalyst for the borylation of alkynes.
Mechanistic investigations – experimental
Interestingly, no major differences in catalytic activity were observed using the nickel alkene complexes 1a–c, [Ni(iPr2ImMe)2(η2-MeCCMe)] 14a, [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] 15a (vide infra) or even the bis-boryl complex 2a as the catalyst precursor, as all of them appear to serve as a source of [Ni(iPr2ImMe)2]. We were also able to isolate analytically pure compounds 4 (60% yield), 7 (65% yield), 8a (38% yield) and 13 (46% yield) from scaled-up reactions, allowing full characterization, including X-ray diffraction. Additionally, crystals of the compounds 3 (structures of 3 and 4 were reported earlier by Marder et al.13a,38), 5, 8, and 8b were obtained by slow evaporation of the reaction mixtures (for details see ESI†).
To study the catalytic reaction of 2-butyne, B2cat2, and [Ni(iPr2ImMe)2] in more detail, we investigated several stoichiometric reactions. Interestingly, the reaction of cis-[Ni(iPr2ImMe)2(Bcat)2] 2a with stoichiometric amounts of 2-butyne did not lead to the cis-alkene-1,2-bis(boronate) ester or to the exchange of the boryl ligands with the alkyne to form [Ni(iPr2ImMe)2(η2-MeCCMe)] 14a.22l Instead, the formation of small amounts of the [Bcat2]− anion, traces of a species which was later identified as [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] 15a, and the slow formation of hexamethylbenzene was detected via NMR spectroscopy. Following the complete consumption of 2-butyne after ca. 20 h, complex 2a began to decompose. Although we did not observe the formation of alkyne complex 14a, the formation of hexamethylbenzene, especially at higher temperatures, suggests that the boryl ligands of 2a are labile via B–B reductive elimination and exchange with the alkyne. However, the reaction of the alkyne complex [Ni(iPr2ImMe)2(η2-MeCCMe)] 14a22l with B2cat2 led to the isolation of the complex of the cis-alkene-1,2-bis(boronate) ester [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] 15a (Scheme 5). This contrasts with the platinum phosphine system, for which Iverson and Smith demonstrated previously that the stoichiometric reaction of [Pt(PPh3)2(η2-H7C3CCC3H7)] with B2cat2 yields the bis-boryl complex [Pt(PPh3)2(Bcat)2] with extrusion of free alkyne (Scheme 5).11a We verified this reactivity by using the octyne complex [Ni(iPr2ImMe)2(η2-H7C3CCC3H7)] 14b, which led to the isolation of [Ni(iPr2ImMe)2(η2-cis-(Bcat)(H7C3)CC(C3H7)(Bcat))] 15b (Scheme 5). These borylation reactions of the alkyne coordinated at nickel are quantitative if performed in an NMR tube.
 | ||
| Scheme 5 Reactivity of NHC nickel alkyne complexes and platinum phosphine alkyne complexes with B2cat2. | ||
Complexes 15a and 15b were isolated as orange to brown solids and were completely characterized using IR- and NMR-spectroscopy, elemental analysis, and X-ray diffraction. The reduction of symmetry on going from 2a (pseudo-C2v) to 15a and 15b (pseudo-Cs) is reflected in the resonances in the 1H and 13C{1H} NMR spectra of these complexes, which are doubled. The olefinic carbon atoms of the alkene ligand were not detected in the 13C{1H} NMR spectra due to the quadrupolar coupling to boron, but were assigned from an HMBC spectrum to be at 40.0 ppm (15a) and at 47.3 ppm (15b). One broad resonance was observed at 33.3 ppm (15a) and 31.9 ppm (15b) for the boryl substituents in the 11B{1H} NMR spectrum, which are clearly distinct from the resonance of 2a at 48.7 ppm.
Crystals of 15a and 15b suitable for X-ray diffraction were obtained from saturated hexane solutions of the compounds at −30 °C (Fig. 4). The complexes crystallize in the monoclinic space groups P21/c (15a) and P21/n (15b). Both complexes adopt a pseudo-trigonal planar structure with Ni–CNHC distances of 1.9454(14)–1.9560(13) Å in a typical range.22 The C3–C4 distances of the coordinated alkene of 1.453(2) Å (15a) and 1.4550(17) Å (15b) are in line with those of coordinated olefins reported previously22a,j and are much larger compared to those of alkyne complexes (c.f.14a: 1.285(2) Å).22l Both olefin ligands are distorted in such a way that one of the electron-deficient boryl substituents can interact with the electron-rich nickel center (see ESI Fig. S15 and S16†), which results in very different Ni⋯B distances of 2.3694(16) Å (Ni1–B2) and 3.0525(19) Å (Ni1–B1) for complex 15a and 2.3376(14) Å (Ni1–B2) and 3.0262(14) Å (Ni1–B1) for complex 15b, respectively. The formation of 15a and 15b indicates that the catalytic bis-borylation of alkynes at d10-[Ni(iPr2ImMe)2] most likely proceeds via a different mechanistic pathway than reported previously for the d10-[PtLn] platinum system. However, the addition of 2-butyne to 15a did not lead to the extrusion of the borylation product and regeneration of the alkyne complex 14a even at higher temperatures, but to formation of hexamethylbenzene.
 | ||
| Fig. 4 Molecular structures of [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] 15a (left) and [Ni(iPr2ImMe)2(η2-cis-(Bcat)(H7C3)CC(C3H7)(Bcat))] 15b (right) in the solid state (ellipsoids shown at 50% probability level). Hydrogen atoms have been omitted for clarity. For selected bond lengths and angles see ESI Fig. S15 and S16.† | ||
Mechanistic investigations – DFT calculations
By combining the results obtained from stoichiometric reactions with additional DFT computations, we were able to rationalize the formation of the borylated products 8, 8a, and 8b from 2-butyne, B2cat2, and [Ni(iPr2ImMe)2]. Our proposed catalytic cycles are shown in Schemes 6 (bis-borylation), 7 (tetra-borylation), and 8 (alkyne-coupling); for a comparison to the well-known platinum catalysis see ESI, Fig. S1.† Initially, [Ni(iPr2ImMe)2] 1 reacts with 2-butyne to form the alkyne complex [Ni(iPr2ImMe)2(η2-MeCCMe)] 14a (ΔG1 = −13.7 kcal mol−1), which is competitive to the reaction with B2cat2 (ΔG1' = −17.5 kcal mol−1).
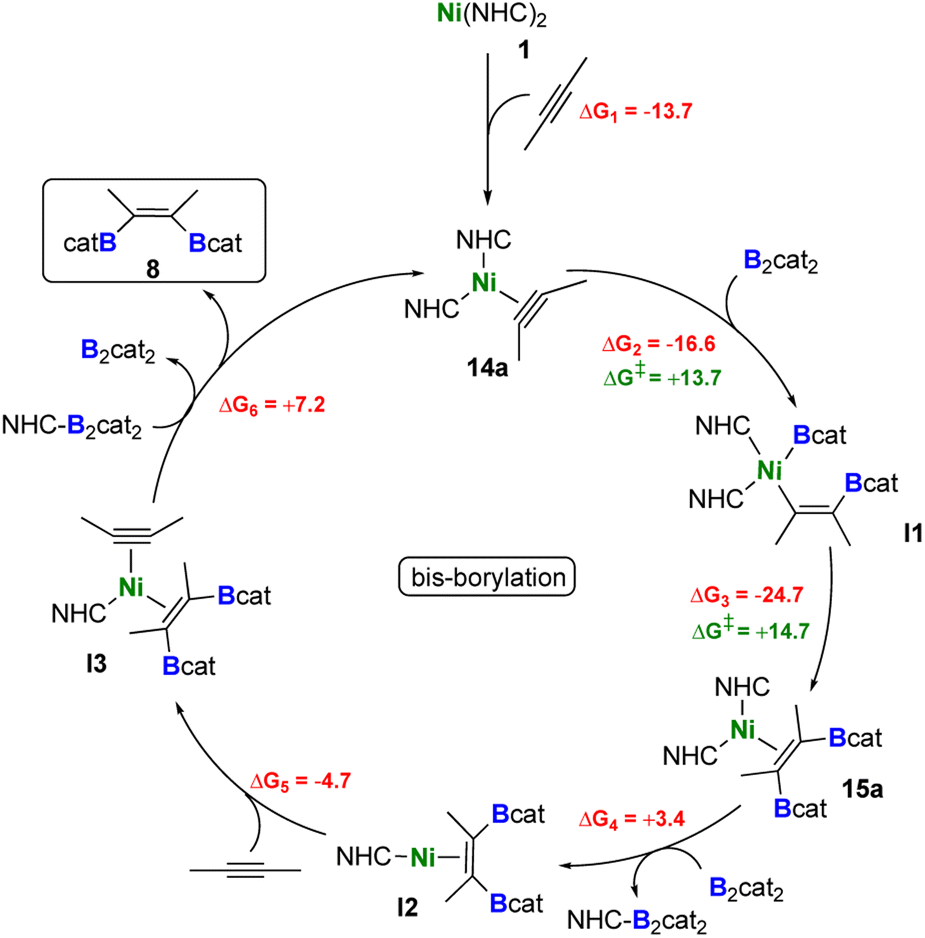 | ||
| Scheme 6 Proposed catalytic cycle for the formation of 8. Reaction free energies (kcal mol−1) calculated at the DFT level are shown in red, while energy barriers are shown in green (see ESI† for more details). | ||
The next steps leading to [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] 15a are crucial to understand the formation of new tetra-borylation and alkyne-coupling products and the different reaction pathways the system can enter. A detailed energy profile of the mechanistic pathway for the addition of B2cat2 to the alkyne complex 14a to yield 15a is therefore presented in Fig. 5. The DFT calculations show that the barrier for the direct addition of B2cat2 to the coordinated alkyne is too high in energy (ΔG‡ = +32.0 kcal mol−1, see TS1′ in Fig. 5). Interestingly, oxidative addition of B2cat2 to the alkyne complex 14 to yield a nickel(IV) intermediate is sterically hindered and thus also out of reach. Alternatively, 15a is formed by insertion of B2cat2 into a Ni–C bond maintaining the B–B bond (ΔG‡ = +13.7 kcal mol−1). This σ-complex assisted methathesis (σ-CAM)-type39 insertion leads to a five-membered NiC2B2 intermediate B that exergonically isomerizes to a relatively stable nickel monoboryl vinyl complex I1 (ΔG = −16.6 kcal mol−1). The next step of the sequence is reductive elimination of the vinyl and the boryl substituent of I1 to yield the complex of the cis-alkene-1,2-bis(boronate) ester [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] 15a. The barrier to pass transition state TS3 yielding the thermodynamically highly favored product 15a is calculated to be ΔG‡ = +14.7 kcal mol−1 and, thus, in accordance with our experimental findings and surmountable under our reaction conditions. The direct release of bis-borylation product 8 from 15a is rather endergonic (ΔG = +19.6 kcal mol−1) and, therefore, we propose that in the next step an NHC is transferred from 15a to another B2cat2 molecule (Scheme 6, ΔG4 = +3.4 kcal mol−1). This leads to the mono-NHC intermediate I2. Addition of an alkyne to I2 leads to I3 and is slightly exergonic (ΔG5 = −4.7 kcal mol−1). The release of 8 from I3 is then mediated by a transfer of the NHC ligand from the ligand-activated B2cat2 species, whose step is endergonic by ΔG6 = +7.2 kcal mol−1 and regenerates 14a and B2cat2.
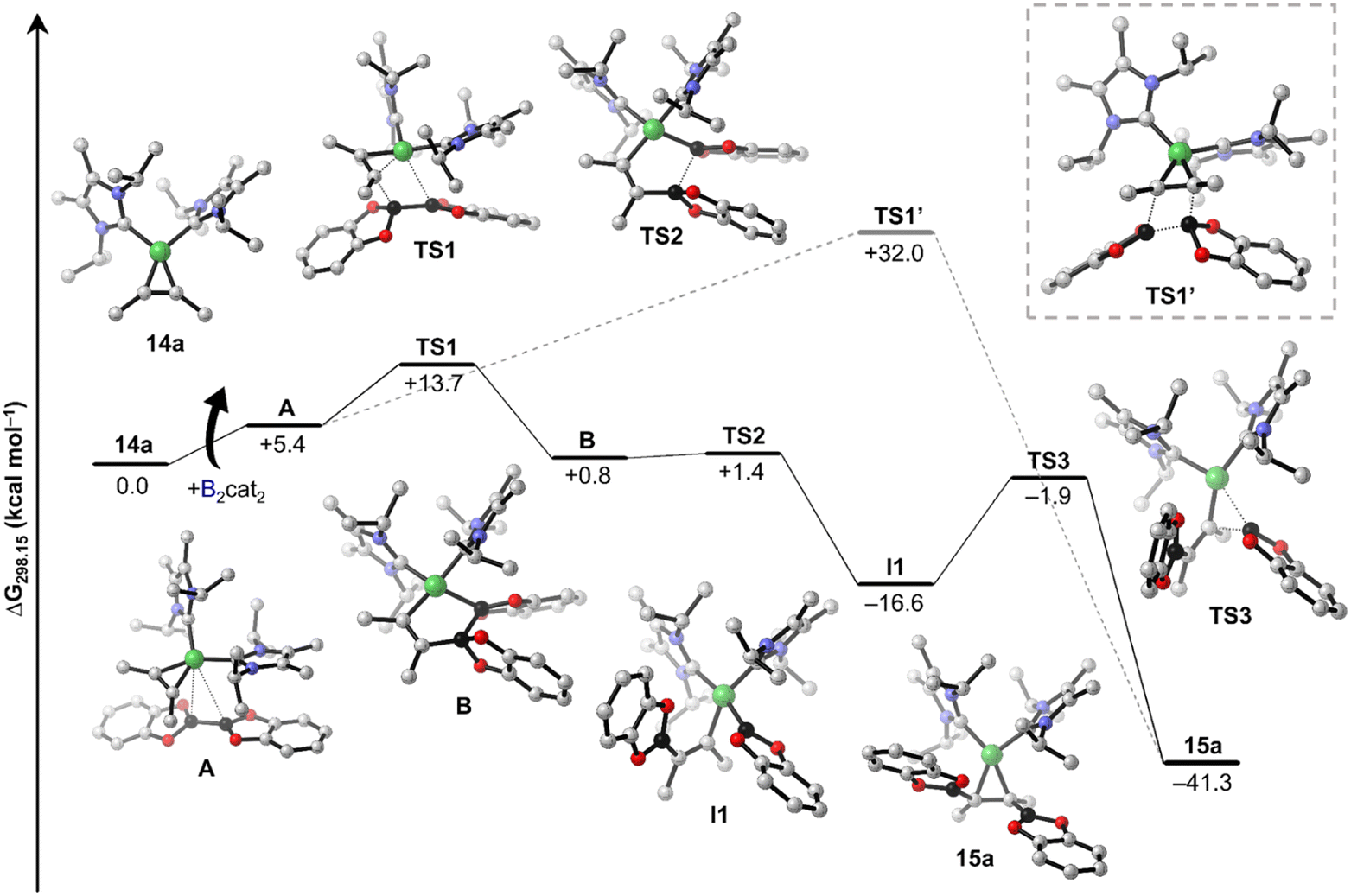 | ||
| Fig. 5 Free energy profile of the proposed mechanistic pathway for the addition of B2cat2 to the alkyne complex [Ni(iPr2ImMe)2(η2-MeCCMe)] 14a to yield [Ni(iPr2ImMe)2(η2-cis-(Bcat)(Me)CC(Me)(Bcat))] 15a. Black lines connect the preferred pathway, while dashed lines indicate a pathway involving a high-energy transition state TS1′. Calculated free energies (kcal mol−1) are at the SMD(benzene)/PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)/def2-SVP/def2-TZVP(Ni) level of theory. Hydrogen atoms are omitted for clarity. | ||
The catalytic cycle leading to the tetra-borylated product 8a is shown in Scheme 7. The mono-NHC intermediate I2 can react with B2cat2 leading exergonically to I4 (ΔG7 = −5.4 kcal mol−1). This species can undergo facile B–B bond dissociation and formation of the nickel monoboryl species I5 (ΔG8 = +5.6 kcal mol−1), where the other boryl group is transferred to the alkene moiety. Although a transition state for this reaction step was not successfully found despite our best efforts, relaxed scan calculations revealed that the energy barrier for this transformation is around 9 kcal mol−1 (see Fig. S91 in the ESI†).
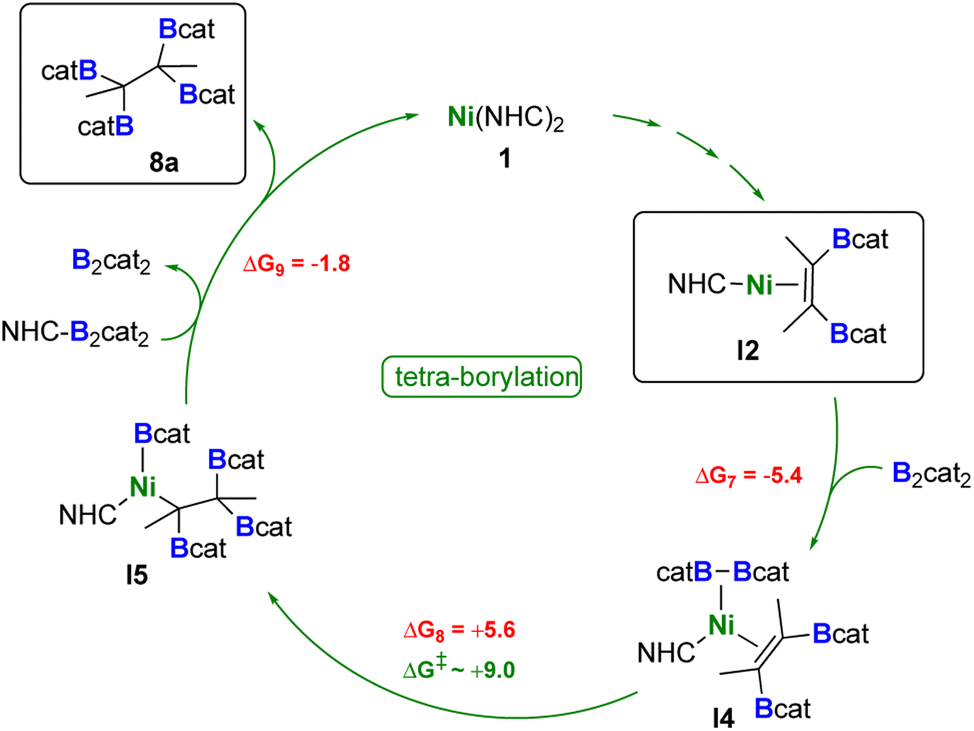 | ||
| Scheme 7 Proposed catalytic cycle for the formation of 8a. Reaction free energies (kcal mol−1) calculated at the DFT level are shown in red, while energy barriers are shown in green (see ESI† for more details). | ||
The release of the tetra-borylated product 8a (ΔG9 = −1.8 kcal mol−1) is then mediated by the NHC-activated B2cat2 species, with further regeneration of B2cat2 and 1.
We found that the formation of the alkyne coupling product 8b can occur via two competitive pathways, starting either from nickel monoboryl vinyl complex I1 (see blue cycle in Scheme 8) or from 15a (see pink cycle in Scheme 8). Assuming that bis-borylation is very fast and all B2cat2 has been consumed, 15a can transfer an NHC to the product 8 (NHC-prod, ΔG10 = +3.7 kcal mol−1), which would again lead to I2. As already discussed, this intermediate can be converted to I3 after addition of an alkyne.
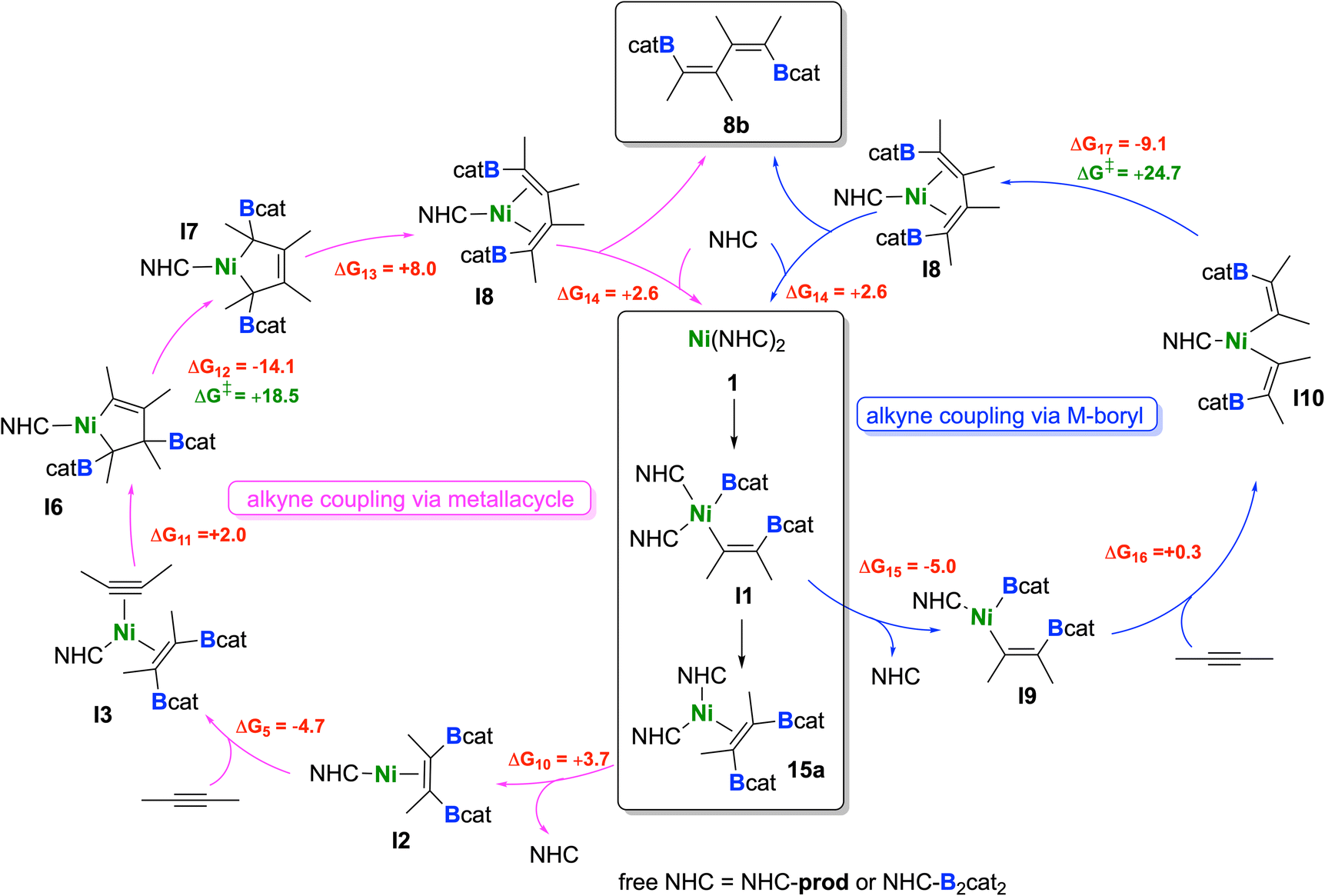 | ||
| Scheme 8 Proposed catalytic cycles for the formation of 8b. Reaction free energies (kcal mol−1) calculated at the DFT level are shown in red, while energy barriers are shown in green (see ESI† for more details). | ||
Details of the alkyne coupling step going from I3 to I8 are shown in Fig. 6. We propose that the alkyne coupling can start from I3 with insertion of 2-butyne into the Ni–C bond of the coordinated cis-alkene-1,2-bis(boronate) ester, which leads to I6 (ΔG11 = +2.0 kcal mol−1), a bis-borylated unsymmetrical metallacyclopentene, energetically close in energy to I3. I6 then undergoes a subsequent 1,3-shift of a boryl group leading to the symmetrical metallacyclopentene complex I7 (ΔG12 = −14.1 kcal mol−1). This concerted 1,3-shift of the boryl substituent viaTS4 is associated with a relatively low barrier (ΔG‡ = +18.5 kcal mol−1). The resulting metallacyclopentene complex I7 rearranges to a 1,4-bis-boryl butadiene complex I8, which is stabilized by a mono-NHC nickel moiety (ΔG13 = +8.0 kcal mol−1). The release of 8b is then facilitated by re-coordination of an NHC to the Ni center (ΔG14 = +2.6 kcal mol−1) regenerating complex 1.
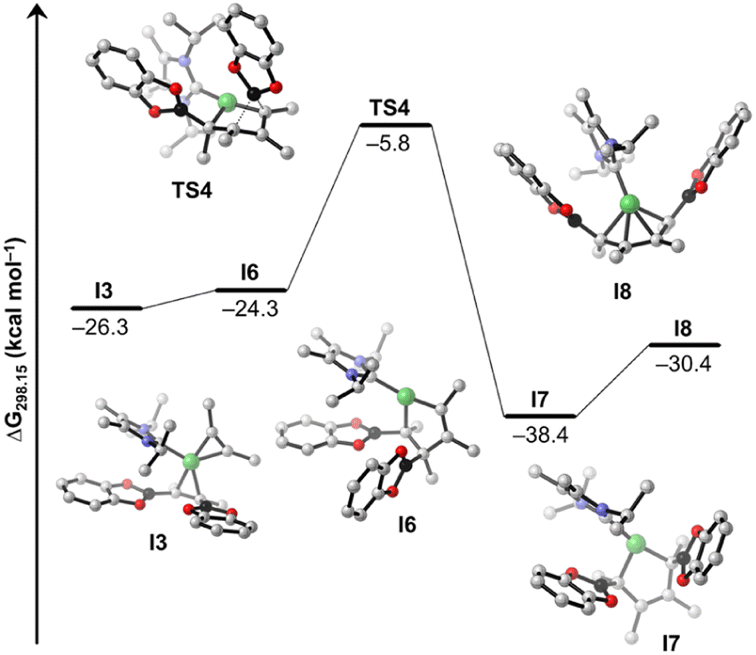 | ||
| Fig. 6 Free energy profile of the proposed mechanistic pathway from I3 to I8. Calculated free energies (kcal mol−1) are at the SMD(benzene)/PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)/def2-SVP/def2-TZVP(Ni) level of theory, and are referenced to the resting state 14a + C2Me2 + B2cat2. Hydrogen atoms are omitted for clarity. | ||
Following the blue reaction path of Scheme 8, dissociation of an NHC from I1 (and/or transfer to any boron compound) leads to the mono-NHC-stabilized boryl species I9 (ΔG15 = −5.0 kcal mol−1 considering NHC dissociation), a step which is well known from platinum chemistry (see ESI Scheme S1†). Surprisingly, formation of I9 from I1 is exergonic even considering just the dissociation of the NHC ligand. This happens because the formally three-coordinate Ni center of I9 is stabilized by interaction with an oxygen atom of the Bcat group (Ni⋯O contact in the optimized structure: 2.056 Å). However, reductive elimination of the cis-alkene-1,2-bis(boronate) ester from I9 is high in energy and interception of this intermediate by another alkyne gives a mixed substituted alkyne boryl vinyl complex. All attempts to optimize this latter structure failed, as the Bcat group at nickel undergoes immediately facile transfer to the vinyl moiety so that reductive elimination to form intermediate I3 takes place. On the other hand, insertion of the coordinated alkyne into the Ni–B bond of I9 yields the nickel bis(vinyl) complex I10 (ΔG16 = +0.3 kcal mol−1). Subsequent C–C reductive elimination from I10viaTS5 (Fig. 7, ΔG‡ = +24.7 kcal mol−1) yields the NHC–Ni butadiene I8 (ΔG18 = −9.1 kcal mol−1), which releases the product 8b, similarly as computed for the other pathway. The results indicate that both pathways are competitive and accessible under the reaction conditions employed, with a preference for the pink cycle due to its lower energy barrier.
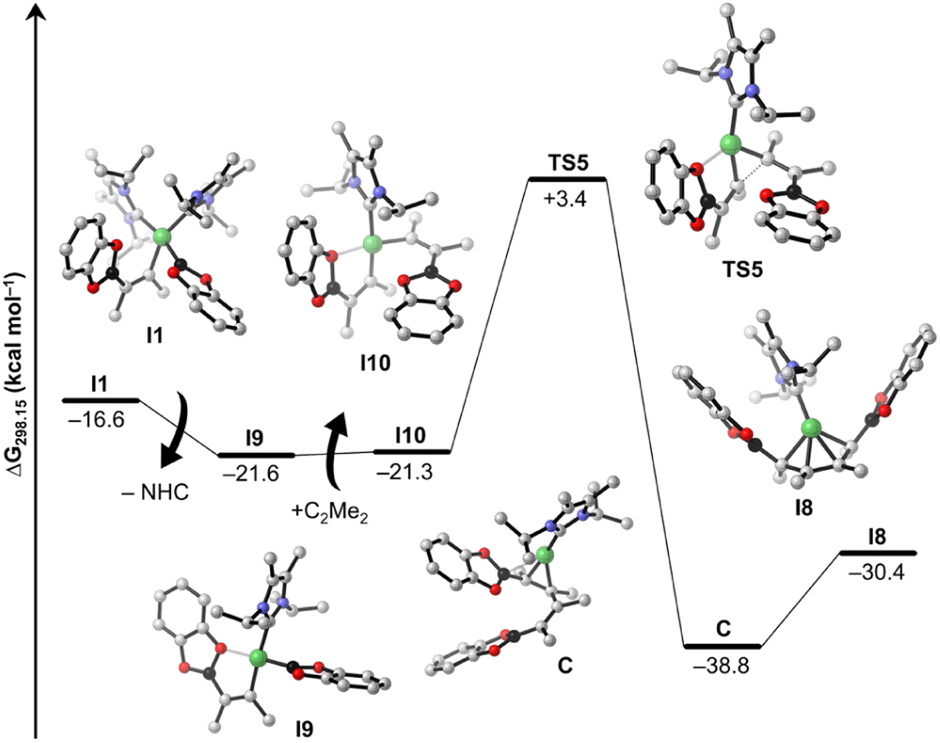 | ||
| Fig. 7 Free energy profile of the proposed mechanistic pathway from I1 to I8. Calculated free energies (kcal mol−1) are at the SMD(benzene)/PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)/def2-SVP/def2-TZVP(Ni) level of theory, and are referenced to the resting state 14a + C2Me2 + B2cat2. Hydrogen atoms are omitted for clarity. | ||
Conclusions
It has been shown over the last decades that transition metal poly-boryl complexes play pivotal roles and are key intermediates in many borylation processes. Such complexes were typically associated with noble metals but, as first-row d-block metals are less toxic, less expensive, Earth-abundant, and environmentally benign, they are very attractive alternatives to these expensive noble metals. Nickel boryl complexes are generally considered to be elusive, in contrast to other 3d-metals such as iron, cobalt, or copper. We report herein the first nickel bis-boryl complexes cis-[Ni(iPr2ImMe)2(Bcat)2] 2a, cis-[Ni(iPr2ImMe)2(Bpin)2] 2b and cis-[Ni(iPr2ImMe)2(Beg)2] 2c, which can be synthesized from the reaction of a source of [Ni(iPr2ImMe)2] with the diboron(4) compounds B2cat2, B2pin2, and B2eg2. Key to the successful synthesis was the choice of the NHC used, showing the right stereo-electronic properties. Whereas cis-[Ni(iPr2ImMe)2(Bcat)2] 2a was isolated as a pale brown solid in 58% yield, the reaction with either B2pin2 or B2eg2 instead of B2cat2 did not proceed quantitatively at room temperature, as observed by NMR spectroscopy. X-ray diffraction studies on 2a–c and DFT calculations on 2a suggest that a delocalized, multicenter bonding scheme best describes the bonding situation of the NiB2 moiety in these complexes, which is reminiscent of the bonding situation in “non-classical” H2 complexes.We also demonstrate that [Ni(iPr2ImMe)2] catalyst precursors provide excellent catalytic activity for the diboration of alkynes under mild conditions, using B2cat2 as the boron source. Beside the well-known cis-alkene-1,2-bis(boronate) esters, the formation of C–C coupled borylation products such as 8b, 12a, and 12b as well as tetra-borylated products such as 8a and 13 were observed or produced as main products of the reaction, which significantly expands the (poly)borylation of alkynes and the scope of accessible boron compounds for further transformations. Therefore, we demonstrated that these 3d metal catalysts provide the potential for new selectivities for the borylation of alkynes compared to the well-established catalysts.
Mechanistic investigations supported by DFT calculations revealed significant differences between our NHC nickel system and the well-established platinum-phosphine chemistry. The formation of borylated alkene π-complexes 15a and 15b as catalytic intermediates is crucial to understand the new catalytic pathway and the formation of new borylation products. Further studies concerning the reactivity of nickel bis-boryl complexes are currently under investigation.
Data availability
Additional data and spectra, crystallographic data, NMR spectra, details on the DFT calculations and Cartesian coordinates of the DFT optimized structures can be found in the ESI.†Author contributions
L. T., U. R., T. B. M., and F. F. conceived the project. L. T. performed all the experiments. F. F. performed the quantum chemical calculations. L. T., F. F., T. B. M., and U. R. wrote the manuscript. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
U.R. and T.B.M. thank the Julius-Maximilians-Universität Würzburg for financial support. F.F. thanks Dr Tim Kinnear and the Centre for Astrophysics and Planetary Science of the University of Kent for providing additional high performance computing resources. We thank AllyChem Co. Ltd. for a generous gift of diboron(4) reagents.References
- (a) H. C. Brown, Science, 1980, 210, 485–492 CrossRef CAS PubMed; (b) N. Miyarura, Top. Curr. Chem., 2002, 219, 11–59 CrossRef; (c) Boron compounds, Science of Syntheses, ed. D. E. Kaufmann and D. S. Matteson, Georg Thieme Verlag, Stuttgart, 2005, vol. 6 Search PubMed; (d) Boronic Acids-Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed; (e) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (f) M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348–7398 CrossRef CAS PubMed; (g) Y.-M. Tian, X.-N. Guo, H. Braunschweig, U. Radius and T. B. Marder, Chem. Rev., 2021, 121, 3561–3597 CrossRef CAS PubMed; (h) S. K. Bose, L. Mao, L. Kuehn, U. Radius, J. Nekvinda, W. L. Santos, S. A. Westcott, P. G. Steel and T. B. Marder, Chem. Rev., 2021, 121, 13238–13341 CrossRef CAS PubMed; (i) J. Hu, M. Ferger, Z. Shi and T. B. Marder, Chem. Soc. Rev., 2021, 50, 13129–13188 RSC; (j) S. Manna, K. K. Das, S. Nandy, D. Aich, S. Paul and S. Panda, Coord. Chem. Rev., 2021, 448, 214165 CrossRef CAS.
- (a) T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS; (b) L. T. Pilarski and K. J. Szabó, Angew. Chem., Int. Ed., 2011, 50, 8230–8232 CrossRef CAS PubMed; (c) M. Murata, Heterocycles, 2012, 85, 1795–1819 CrossRef CAS; (d) C. M. Vogels and S. A. Westcott, ChemCatChem, 2012, 4, 47–49 CrossRef CAS; (e) W. K. Chow, O. Y. Yuen, P. Y. Choy, C. M. So, C. P. Lau, W. T. Wong and F. Y. Kwong, RSC Adv., 2013, 3, 12518–12539 RSC; (f) K. Kubota, H. Iwamoto and H. Ito, Org. Biomol. Chem., 2017, 15, 285–300 RSC; (g) Y. P. Budiman, S. A. Westcott, U. Radius and T. B. Marder, Adv. Synth. Catal., 2021, 363, 2224–2255 CrossRef CAS.
- (a) I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS; (b) V. Lillo, A. Bonet and E. Fernández, Dalton Trans., 2009, 2899–2908 RSC; (c) J. Takaya and N. Iwasawa, ACS Catal., 2012, 2, 1993–2006 CrossRef CAS; (d) H. Wen, G. Liu and Z. Huang, Coord. Chem. Rev., 2019, 386, 138–153 CrossRef CAS; (e) W. Fan, L. Li and G. Zhang, J. Org. Chem., 2019, 84, 5987–5996 CrossRef CAS PubMed; (f) O. Salvadó and E. Fernández, Molecules, 2020, 25, 1758 CrossRef PubMed; (g) X. Wang, Y. Wang, W. Huang, C. Xia and L. Wu, ACS Catal., 2021, 11, 1–18 CrossRef CAS.
- (a) T. B. Marder and N. C. Norman, Top. Catal., 1998, 5, 63–73 CrossRef CAS; (b) R. Barbeyron, E. Benedetti, J. Cossy, J.-J. Vasseur, S. Arseniyadis and M. Smietana, Tetrahedron, 2014, 70, 8431–8452 CrossRef CAS; (c) H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS; (d) E. Buñuel and D. J. Cárdenas, Eur. J. Org. Chem., 2016, 5446–5464 CrossRef; (e) M. B. Ansell, O. Navarro and J. Spencer, Coord. Chem. Rev., 2017, 336, 54–77 CrossRef CAS; (f) F. Zhao, X. Jia, P. Li, J. Zhao, Y. Zhou, J. Wang and H. Liu, Org. Chem. Front., 2017, 4, 2235–2255 RSC; (g) E. Buñuel and D. J. Cárdenas, Chem.–Eur. J., 2018, 24, 11239–11244 CrossRef PubMed; (h) J. Carreras, A. Caballero and P. J. Perez, Chem.–Asian J., 2019, 14, 329–343 CrossRef CAS PubMed.
- (a) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2003, 680, 3–11 CrossRef CAS; (b) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS; (c) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC; (d) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed; (e) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (f) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed; (g) Y. Ping, L. Wang, Q. Ding and Y. Peng, Adv. Synth. Catal., 2017, 359, 3274–3291 CrossRef CAS; (h) T. Gensch, M. J. James, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 2296–2306 CrossRef CAS PubMed; (i) Y. Shi, Q. Gao and S. Xu, Synlett, 2019, 30, 2107–2112 CrossRef CAS; (j) E. Fernandez, Top. Organomet. Chem., 2021, 69, 207–225 CrossRef CAS.
- (a) G. J. Irvine, M. J. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, E. G. Robins, W. R. Roper, G. R. Whittell and L. J. Wright, Chem. Rev., 1998, 98, 2685–2722 CrossRef CAS PubMed; (b) H. Braunschweig, Angew. Chem., Int. Ed., 1998, 37, 1786–1801 CrossRef CAS; (c) H. Braunschweig and M. Colling, Coord. Chem. Rev., 2001, 223, 1–51 CrossRef CAS; (d) S. Aldridge and D. L. Coombs, Coord. Chem. Rev., 2004, 248, 535–559 CrossRef CAS; (e) H. Braunschweig, C. Kollann and D. Rais, Angew. Chem., Int. Ed., 2006, 45, 5254–5274 CrossRef CAS PubMed; (f) D. L. Kays and S. Aldridge, Struct. Bond., 2008, 130, 29–122 CrossRef CAS; (g) U. Kaur, K. Saha, S. Gayen and S. Ghosh, Coord. Chem. Rev., 2021, 446, 214106 CrossRef CAS.
- (a) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2000, 611, 392–402 CrossRef CAS; (b) N. Miyaura, Bull. Chem. Soc. Jpn., 2008, 81, 1535–1553 CrossRef CAS; (c) L. Dang, Z. Lin and T. B. Marder, Chem. Commun., 2009, 3987–3995 RSC; (d) X. Guo, T. Yang, F. K. Sheong and Z. Lin, ACS Catal., 2021, 11, 5061–5068 CrossRef CAS.
- (a) P. Nguyen, H. P. Blom, S. A. Westcott, N. J. Taylor and T. B. Marder, J. Am. Chem. Soc., 1993, 115, 9329–9330 CrossRef CAS; (b) R. T. Baker, J. C. Calabrese, S. A. Westcott, P. Nguyen and T. B. Marder, J. Am. Chem. Soc., 1993, 115, 4367–4368 CrossRef CAS; (c) K. Burgess, W. A. Van der Donk, S. A. Westcott, T. B. Marder, R. T. Baker and J. C. Calabrese, J. Am. Chem. Soc., 1992, 114, 9350–9359 CrossRef CAS.
- (a) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (b) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr and M. R. Smith III, Science, 2002, 295, 305–308 CrossRef CAS PubMed; (c) M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287–4299 CrossRef CAS PubMed.
- (a) A. K. Cook, S. D. Schimler, A. J. Matzger and M. S. Sanford, Science, 2016, 351, 1421–1424 CrossRef CAS PubMed; (b) K. T. Smith, S. Berritt, M. Gonzaĺez-Moreiras, S. Ahn, M. R. Smith, M.-H. Baik and D. J. Mindiola, Science, 2016, 351, 1424–1427 CrossRef CAS PubMed; (c) S. Ahn, D. Sorsche, S. Berritt, M. R. Gau, D. J. Mindiola and M.-H. Baik, ACS Catal., 2018, 8, 10021–10031 CrossRef CAS.
- (a) C. N. Iverson and M. R. Smith III, J. Am. Chem. Soc., 1995, 117, 4403–4404 CrossRef CAS; (b) C. Borner and C. Kleeberg, Eur. J. Inorg. Chem., 2014, 2486–2489 CrossRef CAS.
- (a) T. Ishiyama, N. Matsuda, N. Miyaura and A. Suzuki, J. Am. Chem. Soc., 1993, 115, 11018–11019 CrossRef CAS; (b) T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki and N. Miyaura, Organometallics, 1996, 15, 713–720 CrossRef CAS; (c) C. N. Iverson and M. R. Smith, Organometallics, 1996, 15, 5155–5165 CrossRef CAS; (d) Q. Cui, D. G. Musaev and K. Morokuma, Organometallics, 1998, 17, 742–751 CrossRef CAS.
- (a) G. Lesley, P. Nguyen, N. J. Taylor, T. B. Marder, A. J. Scott, W. Clegg and N. C. Norman, Organometallics, 1996, 15, 5137–5154 CrossRef CAS; (b) R. Ll. Thomas, F. E. S. Souza and T. B. Marder, Dalton Trans., 2001, 1650–1656 RSC.
- (a) T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 4, 271–280 CrossRef PubMed; (b) M. W. Carson, M. W. Giese and M. J. Coghlan, Org. Lett., 2008, 10, 2701–2704 CrossRef CAS PubMed; (c) K. Yavari, S. Moussa, B. Ben Hassine, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2012, 51, 6748–6752 CrossRef CAS PubMed; (d) Q.-X. Lin and T.-L. Ho, Tetrahedron, 2013, 69, 2996–3001 CrossRef CAS; (e) M. W. Carson, J. G. Luz, C. Suen, C. Montrose, R. Zink, X. Ruan, C. Cheng, H. Cole, M. D. Adrian, D. T. Kohlman, T. Mabry, N. Snyder, B. Condon, M. Maletic, D. Clawson, A. Pustilnik and M. J. Coghlan, J. Med. Chem., 2014, 57, 849–860 CrossRef CAS PubMed; (f) J. Yang, M. Chem, J. Ma, W. Huang, H. Zhu, Y. Huang and W. Wang, J. Mater. Chem. C, 2015, 3, 10074–10078 RSC.
- (a) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS PubMed; (b) C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528–532 CrossRef CAS PubMed; (c) S. K. Bose, S. Brand, H. O. Omoregie, M. Haehnel, J. Maier, G. Bringmann and T. B. Marder, ACS Catal., 2016, 6, 8332–8335 CrossRef CAS; (d) D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC; (e) L. Kuehn, M. Huang, U. Radius and T. B. Marder, Org. Biomol. Chem., 2019, 17, 6601–6606 RSC; (f) B. S. Takale, R. R. Thakore, E. Etemadi-Davan and B. H. Lipshutz, Beilstein J. Org. Chem., 2020, 16, 691–737 CrossRef CAS PubMed; (g) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10, 11578–11622 CrossRef CAS.
- (a) J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250–5253 CrossRef CAS PubMed; (b) Y.-M. Tian, X.-N. Guo, M. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen and T. B. Marder, J. Am. Chem. Soc., 2018, 140, 17612–17623 CrossRef CAS PubMed; (c) M. W. Kuntze-Fechner, H. Verplancke, L. Tendera, M. Diefenbach, I. Krummenacher, H. Braunschweig, T. B. Marder, M. C. Holthausen and U. Radius, Chem. Sci., 2020, 11, 11009–11023 RSC; (d) L. Kuehn, D. G. Jammal, K. Lubitz, T. B. Marder and U. Radius, Chem.–Eur. J., 2019, 25, 9514–9521 CrossRef CAS PubMed; (e) Y.-M. Tian, X.-N. Guo, I. Krummenacher, Z. Wu, J. Nitsch, H. Braunschweig, U. Radius and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 18231–18242 CrossRef CAS PubMed; (f) Y.-M. Tian, X.-N. Guo, Z. Wu, A. Friedrich, S. A. Westcott, H. Braunschweig, U. Radius and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 13136–13144 CrossRef CAS PubMed.
- The reaction of [Ni(Cy2Im)2(Ar)Cl] with stoichiometric amounts of B2pin2 at room temperature led to a resonance at 44.5 ppm in the 11B{1H} NMR spectrum of the reaction mixture which indicated the formation of a nickel-boryl complex, see the ESI† of ref. 16 d..
- (a) J. F. Hartwig and S. Huber, J. Am. Chem. Soc., 1993, 115, 4908–4909 CrossRef CAS; (b) X. He and J. F. Hartwig, Organometallics, 1996, 15, 400–407 CrossRef CAS; (c) K. M. Waltz, C. N. Muhoro and J. F. Hartwig, Organometallics, 1999, 18, 3383–3393 CrossRef CAS; (d) S. Aldridge, R. J. Calder, R. E. Baghurst, M. E. Light and M. B. Hursthouse, J. Organomet. Chem., 2002, 649, 9–14 CrossRef CAS; (e) A. Rossin, S. Aldridge and L.-l. Ooi, Appl. Organomet. Chem., 2005, 19, 181–182 CrossRef CAS; (f) T. J. Mazzacano and N. P. Mankad, Chem. Commun., 2015, 51, 5379–5382 RSC; (g) R. B. Bedford, P. B. Brenner, E. Carter, T. Gallagher, D. M. Murphy and D. R. Pye, Organometallics, 2014, 33, 5940–5943 CrossRef CAS; (h) T. Dombray, C. G. Werncke, S. Jiang, M. Grellier, L. Vendier, S. Bontemps, J.-B. Sortais, S. Sabo-Etienne and C. Darcel, J. Am. Chem. Soc., 2015, 137, 4062–4065 CrossRef CAS PubMed; (i) L. Vondung, N. Frank, M. Fritz, L. Alig and R. Langer, Angew. Chem., Int. Ed., 2016, 55, 14450–14454 CrossRef CAS PubMed; (j) K. Nakajima, T. Kato and Y. Nishibayashi, Org. Lett., 2017, 19, 4323–4326 CrossRef CAS PubMed; (k) T. Kato, S. Kuriyama, K. Nakajima and Y. Nishibayashi, Chem.–Asian J., 2019, 14, 2097–2101 CrossRef CAS PubMed.
- (a) C. Dai, G. Stringer, J. F. Corrigan, N. J. Taylor, T. B. Marder and N. C. Norman, J. Organomet. Chem., 1996, 513, 273–275 CrossRef CAS; (b) C. J. Adams, R. A. Baber, A. S. Batsanov, G. Bramham, J. P. Charmant, M. F. Haddow, J. A. K. Howard, W. H. Lam, Z. Lin, T. B. Marder, N. C. Norman and A. G. Orpen, Dalton Trans., 2006, 1370–1373 RSC; (c) J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133–4136 CrossRef CAS PubMed; (d) J. V. Obligacion, S. P. Semproni, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 10645–10653 CrossRef CAS PubMed; (e) S. M. Rummelt, H. Zhong, N. G. Léonard, S. P. Semproni and P. J. Chirik, Organometallics, 2019, 38, 1081–1090 CrossRef CAS PubMed; (f) R. Arevalo, T. P. Pabst and P. J. Chirik, Organometallics, 2020, 39, 2763–2773 CrossRef CAS PubMed; (g) W. Drescher, D. Schmitt-Monreal, C. R. Jacob and C. Kleeberg, Organometallics, 2020, 39, 538–543 CrossRef CAS.
- (a) D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed; (b) K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem.–Eur. J., 2013, 19, 7125–7132 CrossRef CAS PubMed; (c) C. M. Wyss, J. Bitting, J. Bacsa, T. G. Gray and J. P. Sadighi, Organometallics, 2016, 35, 71–74 CrossRef CAS; (d) C. Borner, L. Anders, K. Brandhorst and C. Kleeberg, Organometallics, 2017, 36, 4687–4690 CrossRef CAS; (e) C. Kleeberg and C. Borner, Organometallics, 2018, 37, 4136–4146 CrossRef CAS; (f) W. Drescher and C. Kleeberg, Inorg. Chem., 2019, 58, 8215–8229 CrossRef CAS PubMed; (g) W. Drescher, C. Borner and C. Kleeberg, New J. Chem., 2021, 45, 14957–14964 RSC; (h) P. Ríos, M. S. See, R. C. Handford, S. J. Teat and T. D. Tilley, Chem. Sci., 2022, 13, 6619–6625 RSC; (i) P. M. Rutz, J. Grunenberg and C. Kleeberg, Organometallics, 2022, 41, 3044 CrossRef CAS.
- (a) D. Adhikari, J. C. Huffman and D. J. Mindiola, Chem. Commun., 2007, 4489–4491 RSC; (b) B. L. Tran, D. Adhikari, H. Fan, M. Pink and D. J. Mindiola, Dalton Trans., 2010, 39, 358–360 RSC; (c) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS PubMed; (d) N. Curado, C. Maya, J. López-Serrano and A. Rodríguez, Chem. Commun., 2014, 50, 15718–15721 RSC; (e) P. Ríos, J. Borge, F. Fernández de Córdova, G. Sciortino, A. Lledós and A. Rodríguez, Chem. Sci., 2021, 12, 2540–2548 RSC; (f) F. W. Seidel and K. Nozaki, Angew. Chem., Int. Ed., 2022, 61, e202111691 CrossRef CAS PubMed.
- (a) T. Schaub and U. Radius, Chem.–Eur. J., 2005, 11, 5024–5030 CrossRef CAS PubMed; (b) T. Schaub, M. Backes and U. Radius, Organometallics, 2006, 25, 4196–4206 CrossRef CAS; (c) U. S. D. Paul, C. Sieck, M. Hähnel, K. Hammond, T. B. Marder and U. Radius, Chem.–Eur. J., 2016, 21, 11005–11014 CrossRef PubMed; (d) U. S. D. Paul and U. Radius, Chem.–Eur. J., 2017, 23, 3993–4009 CrossRef CAS PubMed; (e) U. S. D. Paul and U. Radius, Organometallics, 2017, 36, 1398–1407 CrossRef CAS; (f) J. H. J. Berthel, M. W. Kuntze-Fechner and U. Radius, Eur. J. Inorg. Chem., 2019, 2618–2623 CrossRef CAS; (g) J. H. J. Berthel, L. Tendera, M. W. Kuntze-Fechner, L. Kuehn and U. Radius, Eur. J. Inorg. Chem., 2019, 3061–3072 CrossRef CAS; (h) J. H. J. Berthel, M. J. Krahfuss, U. Radius and Z. Anorg, Allg. Chem., 2020, 646, 692–704 CrossRef CAS; (i) M. J. Krahfuss, J. Nitsch, F. M. Bickelhaupt, T. B. Marder and U. Radius, Chem.–Eur. J., 2020, 26, 11276–11292 CrossRef CAS PubMed; (j) L. Tendera, T. Schaub, M. J. Krahfuss, M. W. Kuntze-Fechner and U. Radius, Eur. J. Inorg. Chem., 2020, 3194–3207 CrossRef CAS; (k) S. Sabater, D. Schmidt, H. Schmidt nee Schneider, M. Kuntze-Fechner, T. Zell, C. J. Isaac, H. Grieve, W. J. M. Blackaby, J. P. Lowe, S. A. Macgregor, M. F. Mahon, F. M. Miloserdov, N. Rajabi, U. Radius and M. K. Whittlesey, Chem.–Eur. J., 2021, 27, 13221–13234 CrossRef CAS PubMed; (l) L. Tendera, M. Helm, M. J. Krahfuss, M. W. Kuntze-Fechner and U. Radius, Chem.–Eur. J., 2021, 27, 17849–17861 CrossRef CAS PubMed.
- (a) S. Würtemberger-Pietsch, U. Radius and T. B. Marder, Dalton Trans., 2016, 45, 5880–5895 RSC; (b) S. Pietsch, U. Paul, I. A. Cade, M. J. Ingleson, U. Radius and T. B. Marder, Chem.–Eur. J., 2015, 21, 9018–9021 CrossRef CAS PubMed; (c) S. Würtemberger-Pietsch, H. Schneider, T. B. Marder and U. Radius, Chem.–Eur. J., 2016, 22, 13032–13036 CrossRef PubMed; (d) M. Eck, S. Würtemberger-Pietsch, A. Eichhorn, J. H. J. Berthel, R. Bertermann, U. S. D. Paul, H. Schneider, A. Friedrich, C. Kleeberg, U. Radius and T. B. Marder, Dalton Trans., 2017, 46, 3661–3680 RSC.
- (a) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC; (b) S. Felten, S. F. Marshall, A. J. Groom, R. T. Vanderlinden, R. M. Stolley and J. Louie, Organometallics, 2018, 37, 3687–3697 CrossRef CAS.
- (a) W. Clegg, F. J. Lawlor, G. Lesley, T. B. Marder, N. C. Norman, A. G. Orpen, M. J. Quayle, C. R. Rice, A. J. Scott and F. E. S. Souza, J. Organomet. Chem., 1998, 550, 183–192 CrossRef CAS; (b) J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384–9390 CrossRef CAS PubMed; (c) H. Braunschweig, P. Brenner, A. Müller, K. Radacki, D. Rais and K. Uttinger, Chem.–Eur. J., 2007, 13, 7171–7176 CrossRef CAS PubMed.
- (a) T. Zell, T. Schaub, K. Radacki and U. Radius, Dalton Trans., 2011, 40, 1852–1854 RSC; (b) C. Hauf, J. E. Barquera-Lozada, P. Meixner, G. Eickerling, S. Altmannshofer, D. Stalke, T. Zell, D. Schmidt, U. Radius and W. Scherer, Z. Anorg. Allg. Chem., 2013, 639, 1996–2004 CrossRef CAS; (c) D. Schmidt, T. Zell, T. Schaub and U. Radius, Dalton Trans., 2014, 43, 10816–10827 RSC.
- W. Clegg, M. R. J. Elsegood, F. J. Lawlor, N. C. Norman, N. L. Pickett, E. G. Robins, A. J. Scott, P. Nguyen, N. J. Taylor and T. B. Marder, Inorg. Chem., 1998, 37, 5289–5293 CrossRef CAS.
- (a) I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS; (b) I. Mayer, Int. J. Quantum Chem., 1984, 26, 151–154 CrossRef CAS.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- (a) M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS; (b) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS; (c) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (d) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (e) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. B. Ansell, V. H. Menezes da Silva, G. Heerdt, A. A. C. Braga, J. Spencer and O. Navarro, Catal. Sci. Technol., 2016, 6, 7461–7467 RSC.
- See for example: (a) J. E. Poist and C. S. Kraihanzel, Chem. Commun., 1968, 607–608 RSC; (b) Y. Okuda, Y. Ishiguro, S. Mori, K. Nakajima and Y. Nishihara, Organometallics, 2014, 33, 1878–1889 CrossRef CAS.
- (a) E. G. Robins, X. Liu, N. C. Norman and T. B. Marder, unpublished results; (b) E. G. Robins, PhD Thesis, Bristol University, 1997.
- (a) M. Bluhm, A. Maderna, H. Pritzkow, S. Bethke, R. Gleiter and W. Siebert, Eur. J. Inorg. Chem., 1999, 1693–1700 CrossRef CAS; (b) For a related tetra-borylation of (MeO)CH2-CC-CH2(OMe) see: H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita and K. Takaki, Angew. Chem., Int. Ed., 2012, 51, 235–238 CrossRef CAS PubMed.
- (a) N. Cabrera-Lobera, M. T. Quirós, W. W. Brennessel, M. L. Neidig, E. Buñuel and D. J. Cárdenas, Org. Lett., 2019, 21, 6552–6556 CrossRef CAS PubMed; (b) N. Cabrera-Lobera, M. T. Quirós, E. Buñuel and D. J. Cárdenas, Catal. Sci. Technol., 2019, 9, 1021–1029 RSC; (c) I. Manjón-Mata, M. T. Quirós, E. Velasco-Juárez, E. Buñuel and D. J. Cárdenas, Adv. Synth. Catal., 2022, 364, 1716–1723 CrossRef.
- L. Dang, H. Zhao, Z. Lin and T. B. Marder, Organometallics, 2008, 27, 1178–1186 CrossRef CAS.
- (a) L. Kuehn, L. Zapf, L. Werner, M. Stang, S. Würtemberger-Pietsch, I. Krummenacher, H. Braunschweig, E. Lacôte, T. B. Marder and U. Radius, Chem. Sci., 2022, 13, 8321–8333 RSC; (b) S. Jos, C. Szwetkowski, C. Slebodnick, R. Ricker, K. L. Chan, W. C. Chan, U. Radius, Z. Lin, T. B. Marder and W. L. Santos, Chem.–Eur. J., 2022, 28, e202202349 CrossRef CAS PubMed.
- W. Clegg, A. J. Scott, G. Lesley, T. B. Marder and N. C. Norman, Acta Crystallogr. C, 1996, 52, 1991–1995 CrossRef.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem. Int., Ed., 2007, 46, 2578–2592 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures, experimental section, crystallographic details, NMR spectra, computational details. CCDC 2202537–2202552. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04690c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |