
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe spatial distribution of cobalt phthalocyanine and copper nanocubes controls the selectivity towards C2 products in tandem electrocatalytic CO2 reduction†
Min
Wang
a,
Anna
Loiudice
b,
Valery
Okatenko
a,
Ian D.
Sharp
 b and
Raffaella
Buonsanti
*a
b and
Raffaella
Buonsanti
*a
aLaboratory of Nanochemistry for Energy (LNCE), Institute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne, CH-1950 Sion, Switzerland. E-mail: raffaella.buonsanti@epfl.ch
bWalter Schottky Institute and Physics Department, Technische Universität München, Am Coulombwall 4, 85748 Garching, Germany
First published on 4th January 2023
Abstract
The coupling of CO-generating molecular catalysts with copper electrodes in tandem schemes is a promising strategy to boost the formation of multi-carbon products in the electrocatalytic reduction of CO2. While the spatial distribution of the two components is important, this aspect remains underexplored for molecular-based tandem systems. Herein, we address this knowledge gap by studying tandem catalysts comprising Co-phthalocyanine (CoPc) and Cu nanocubes (Cucub). In particular, we identify the importance of the relative spatial distribution of the two components on the performance of the tandem catalyst by preparing CoPc-Cucub/C, wherein the CoPc and Cucub share an interface, and CoPc-C/Cucub, wherein the CoPc is loaded first on carbon black (C) before mixing with the Cucub. The electrocatalytic measurements of these two catalysts show that the faradaic efficiency towards C2 products almost doubles for the CoPc-Cucub/C, whereas it decreases by half for the CoPc-C/Cucub, compared to the Cucub/C. Our results highlight the importance of a direct contact between the CO-generating molecular catalyst and the Cu to promote C–C coupling, which hints at a surface transport mechanism of the CO intermediate between the two components of the tandem catalyst instead of a transfer via CO diffusion in the electrolyte followed by re-adsorption.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) is an attractive route for CO2 recycling, wherein value-added chemicals and fuels are produced from CO2 while also storing electricity generated from renewable energy sources.1,2 Among all catalysts, Cu-based materials provide suitable intermediate binding energies to drive CO2RR towards highly reduced products (e.g. methane, ethylene, ethanol), which are of interest due to their high energy densities.3,4 To date, tremendous effort has been dedicated to increasing the selectivity towards targeted products of the reaction, which produces over sixteen different products in various yields when performed over a polycrystalline Cu foil.5Cu-based tandem schemes have emerged as a valid strategy to enhance the selectivity of CO2RR towards multicarbon products (C2+) by decoupling the CO2 to CO and the CO to C2+ reduction steps.6–9 Previous work has demonstrated that increasing the local concentration of CO and/or the surface coverage of adsorbed CO (*CO) decreases the energetic barrier for C–C coupling, which is the rate-determining step towards C2+ products on Cu surfaces.10–17 In addition to their intrinsic catalytic properties (i.e. turnover frequency, selectivity, overpotential), the relative spatial arrangement of the CO-producing component and of the Cu catalyst plays an important role in defining the efficiency of such tandem systems.9,18–20 For example, researchers have synthesized reverse core–shell structures, with the CO-producing catalyst (Ag) in the core and the Cu as a shell, which was found to maximize CO utilization and enhance the C2+ activity compared to core–shell structures with Cu in the core.19 In an alternative approach, similar benefit was obtained by rationally segmenting the Ag and Cu components in gas-diffusion electrodes.20 In both cases, the spatial distribution of the two catalysts impacts the modality of CO transport to the active sites, which can occur either via surface diffusion or via sequential adsorption following transport through the gas phase or the electrolyte. Thus, optimization of the tandem catalyst configuration emerges as an important parameter to modulate the CO utilization efficiency.
Most studies on the design of tandem systems have so far focused on the coupling of Cu with a second metallic domain (Zn,21,22 Au23 or Ag24–27) as the CO-generating catalyst. Molecular catalysts, such as cobalt phthalocyanine28 and iron porphyrin29,30 have also been successfully employed as the CO-generating catalyst in the context of tandem schemes. Compared to metals, molecular catalysts offer additional versatility for varying the local CO concentration and production rate thanks to the superior chemical tunability via synthetic modifications of molecules versus materials.30 Despite their promise, no experimental work has investigated yet the influence of the spatial arrangement of the components on the catalytic performance of molecular-Cu based tandem systems.
In this work, we investigate the impact of a direct interface between the molecular catalyst and the Cu electrode on the C–C coupling efficiency. We use colloidally dispersible Cu nanocubes (Cucub) enclosed by (100) facets as model catalysts. Their high surface to volume ratio, which is ideal for exploring surface phenomena, as well as their intrinsic selectivity towards ethylene production, justify this choice.31–33 To work alongside Cucub, we select cobalt phthalocyanine (CoPc) as the CO-producing molecular catalyst. In addition to being highly active and stable when immobilized on conductive substrates for CO2RR, the reduction potential window matches the applied potentials of Cucub in aqueous solutions.34–37 Additionally, CoPc readily adsorbs on different surfaces, including carbon and metallic Cu, via electrostatic interactions, thanks to the Pc planar structure.38–40 Finally, both catalysts (i.e. the CoPc and the Cucub) can be produced in the form of inks, which facilitates the manipulation of their surface interactions. We exploit these intrinsic characteristics of the CuCub and of the CoPc to create tandem catalysts comprising CoPc-Cucub/C, where the CoPc shares a direct interface with the Cucub, and CoPc-C/Cucub, where the CoPc is first adsorbed on carbon black and then mixed with the Cucub. We find that the CoPc-Cucub/C catalyst exhibits a faradaic efficiency (FE) and partial current density for C2 products which are double the values measured for pristine Cucub/C without the molecular component present. In contrast, the FE for C2 products decrease by half for the CoPc-C/Cucub compared to the Cucub/C. These results demonstrate that CO utilization is much more efficient in the CoPc-Cucub/C, where a close contact between the two components of the tandem catalyst exists. Thus, we propose that surface transport, which requires a direct interface, dominates over electrolyte-mediated diffusion-readsorption and is the favorable pathway to maximize the CO utilization efficiency and C2 product formation in these molecular-based tandem catalysts.
Results and discussion
The CoPc-Cucub/C catalyst was obtained by first mixing the CoPc molecules with the Cucub in dimethylformamide (DMF), followed by the addition of carbon black into CoPc-Cucub. Prior to mixing, the native ligands of the Cucub were removed via solvent washing, which allowed them to become dispersible in DMF. The molecular catalyst was added in excess to the washed Cucub in order to maximize the Cu surface coverage (see Experimental section in ESI† for details). The amount of the adsorbed molecular catalyst (∼1 nmol per cm2 per working electrode) was quantified by inductively coupled plasma-optical emission spectrometry (ICP-OES) (Table S1†). The CoPc-C/Cucub catalyst was prepared by adsorbing the same total amount of CoPc on the carbon black prior to mixing with the Cucub. For reference, carbon black was also added to the individual Cucub and CoPc systems (Cucub/C and CoPc/C) to enable a direct comparison among each of the different systems.
Fig. 1 reports the transmission electron microscopy (TEM) and Fourier transform infrared spectroscopy (FTIR) characterization of the as-obtained catalysts. TEM reveals the intact cubic shape of the Cucub in both CoPc-Cucub/C (Fig. 1A) and CoPc-C/Cucub (Fig. 1B), which proves that the synthesis procedure does not impact the Cucub morphology. The low contrast material in both images is the carbon black. The FTIR data presented in Fig. 1C show that, first of all, the washed Cucub (Cucub–ligand stripped) has ligand-free surfaces. Indeed, the intensity of the –CH– stretching at 2915 cm−1 is negligible compared to the as-synthesized sample and the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration is completely absent. Both vibrational modes are representative of the native trioctylphosphine oxide ligands of the Cucub (area shaded in blue), confirming their removal from the Cu nanoparticles. The characteristic peaks of CoPc (area shaded in yellow: with Pc ring at 753 cm−1, pyrrole C–N asymetric stretch at approximately 1086 cm−1, and the isoindole and pyrrole stretch at 1200–1500 cm−1) are present in the CoPc-Cucub sample even after thoroughly washing, proving that the CoPc is strongly adsorbed on the Cucub surface, likely due to the expected electrostatic interactions. Similarly, the characteristic IR peaks of CoPc are present for the CoPc-C/Cucub catalyst. The background signal in the region from 700 cm−1 to 2000 cm−1, is attributed to the carbon black (Fig. S1†).
O vibration is completely absent. Both vibrational modes are representative of the native trioctylphosphine oxide ligands of the Cucub (area shaded in blue), confirming their removal from the Cu nanoparticles. The characteristic peaks of CoPc (area shaded in yellow: with Pc ring at 753 cm−1, pyrrole C–N asymetric stretch at approximately 1086 cm−1, and the isoindole and pyrrole stretch at 1200–1500 cm−1) are present in the CoPc-Cucub sample even after thoroughly washing, proving that the CoPc is strongly adsorbed on the Cucub surface, likely due to the expected electrostatic interactions. Similarly, the characteristic IR peaks of CoPc are present for the CoPc-C/Cucub catalyst. The background signal in the region from 700 cm−1 to 2000 cm−1, is attributed to the carbon black (Fig. S1†).
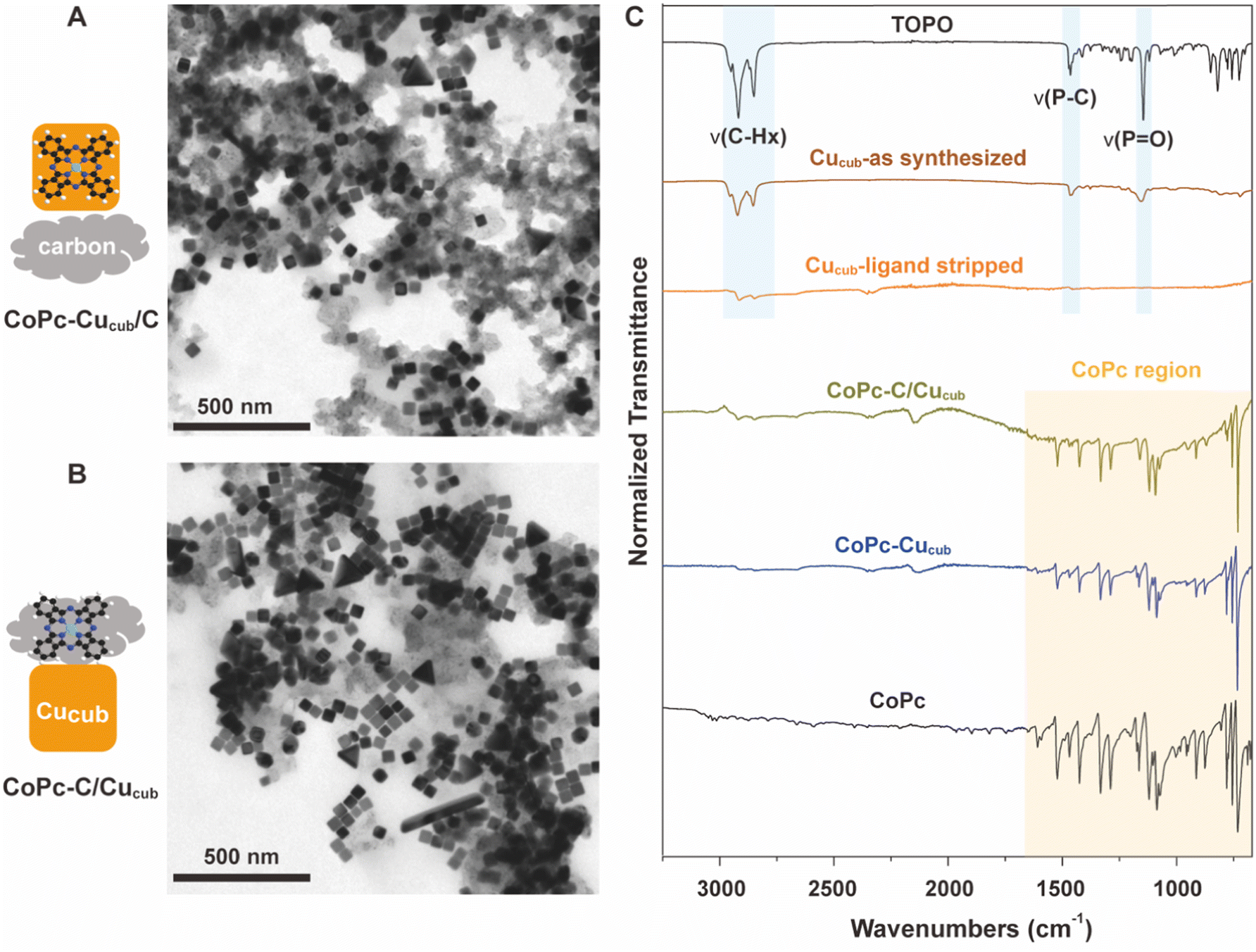 | ||
| Fig. 1 (A and B) Bright field TEM images of CoPc-Cucub/C (A) and CoPc-C/Cucub (B), confirming that the structure of Cucub is retained in both cases following catalyst preparation. The synthesized tandem catalyst structures are schematically indicated on the left. (C) FTIR spectra of TOPO ligand, the as-synthesized Cucub, ligand-stripped Cucub, CoPc-Cucub, CoPc-C/Cucub and CoPc. These results demonstrate the successful removal of TOPO ligand from Cucub and the retention of CoPc within both catalyst assemblies. | ||
We further characterized the catalysts by high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) coupled with energy-dispersive X-ray (EDX) spectroscopy to learn more about the spatial distributions of the different components (Fig. 2). For the case of the CoPc-Cucub catalyst, the Cu and Co signals are co-localized on the Cucub and no free CoPc molecules are present on the grid (Fig. 2A and B). By contrast, for the case of CoPc-C/Cucub catalyst, the CoPc is present only on the carbon black and not on the Cucub (Fig. 2C and D). These results confirm the successful assembly of two different catalyst motifs according to the designed structures depicted by the schematic illustrations in Fig. 1A.
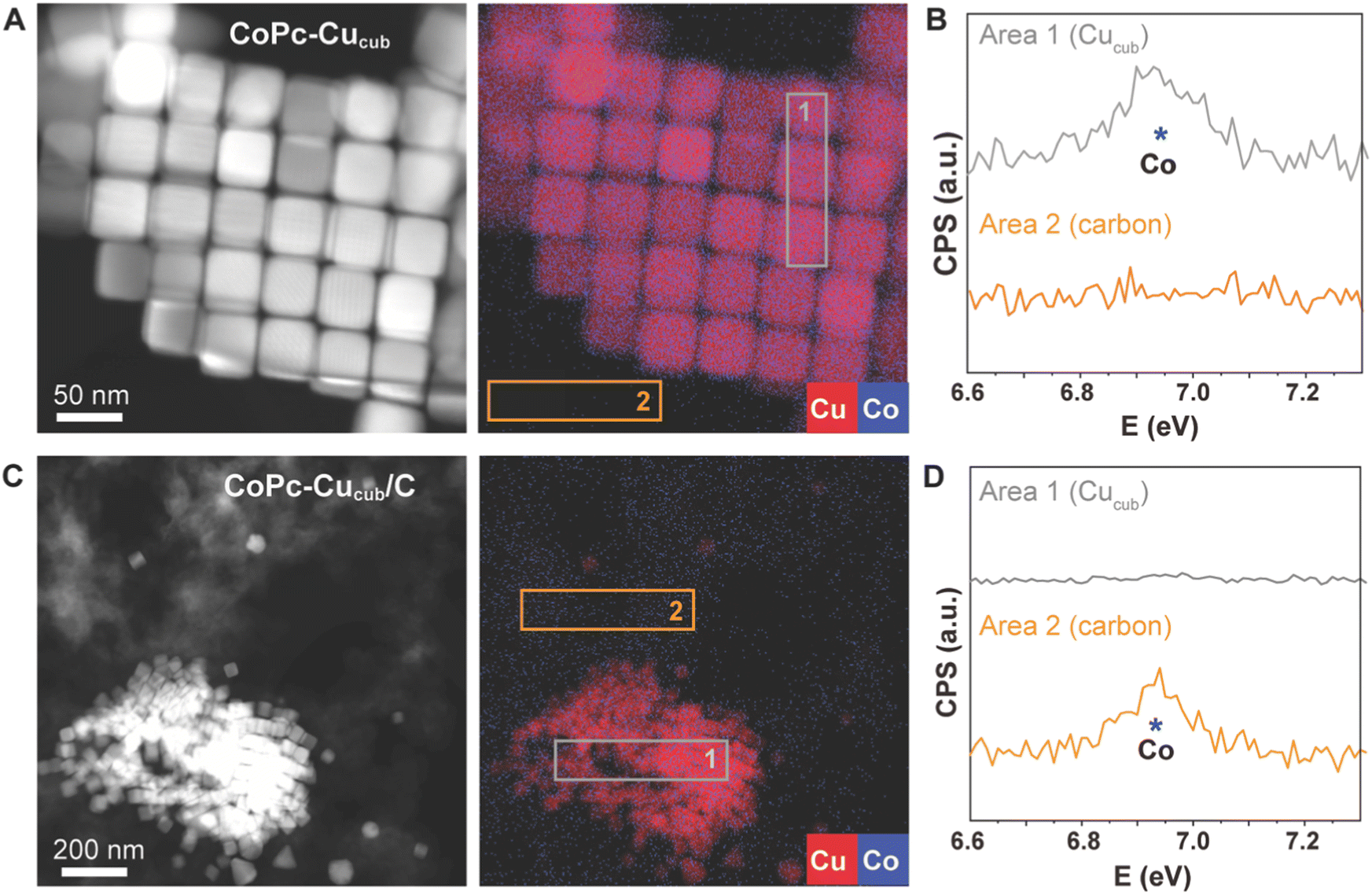 | ||
| Fig. 2 (A and C) HAADF-STEM images and corresponding EDX elemental maps, along with (B and D) EDX spectra for CoPc-Cucub/C (A and B) and CoPc-C/Cucub (C and D). The EDX maps show the spatial distributions of Cu (red) and Co (blue). The carbon is the low contrast material in (C) and is not colored for the sake of clarity. The EDX spectra report the intensity of the Co signals in two different areas of the map, which correspond to the Cucub (area 1) and to the carbon (area 2), as indicated on the colored maps. | ||
Having completed the characterization of the two catalysts, we evaluated the behavior of CoPc-Cucub/C and CoPc-C/Cucub towards CO2RR in a H-cell system, using CO2-saturated 0.1 M aqueous KHCO3 as the supporting electrolyte (Fig. 3). We chose an electrochemical potential of −1.0 V vs. RHE (reversible hydrogen electrode) for initial comparison based on the previous knowledge that Cucub exhibits a significant selectivity for C2H4 and that CoPc possesses maximum activity for CO near this working potential. For reference, the product distributions for the tandem catalyst assemblies are also compared to those possessing only the individual components, Cucub/C and CoPc/C. All reported current densities are normalized by the electrochemically active surface area (ECSA) (Fig. S2†).
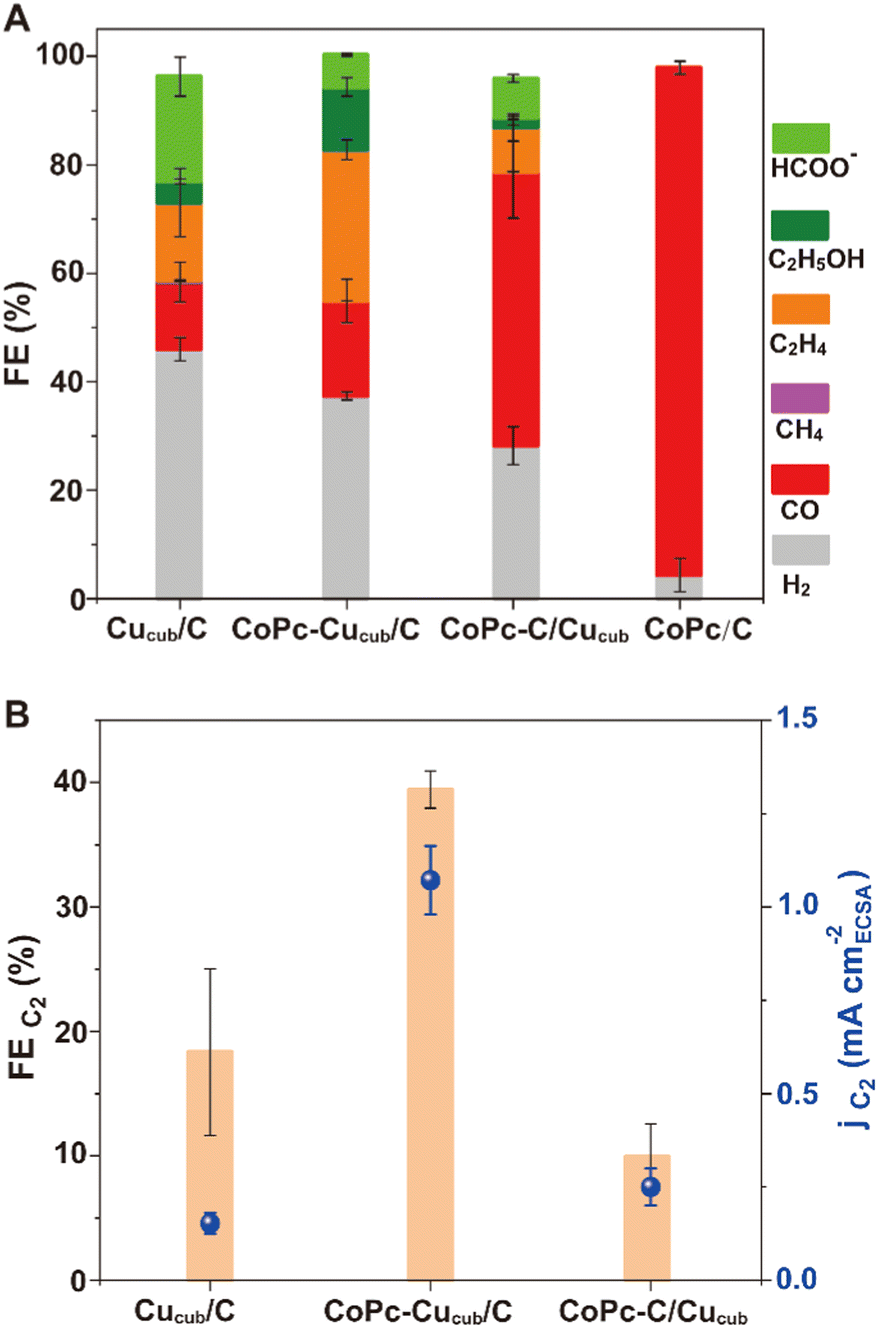 | ||
| Fig. 3 (A) FEs for all gaseous products (i.e., H2, CO, CH4, C2H4) and the main liquid products (i.e., formate, C2H5OH), (B) FEC2 and corresponding partial current density jC2 normalized by the ECSA for the Cucub/C, CoPc-Cucub/C, CoPc-C/Cucub and CoPc/C. The loading of Cucub was 20 μg cm−2 and the loading of CoPc anchored on the Cucub and on the carbon black was 1.1 nmol cm−2 and 0.9 nmol cm−2, respectively. These data were collected in an H-cell configuration using 0.1 M KHCO3 as the electrolyte with the working electrode poised at −1.0 V vs. RHE for 1 h. The reported values are an average of three independent experiments with error bars indicating the standard deviations. Compared to the reference Cucub/C, a significant increase of FEC2 is observed for CoPc-Cucub/C, while a decrease is observed for CoPc-C/Cucub. | ||
The product distribution analysis (Fig. 3A), which is represented by the FE values, reveals that the major products observed for Cucub/C are still present for the CoPc-Cucub/C, but with a clear increase of the FE for C2 products (i.e. ethylene + ethanol). However, for the CoPc-C/Cucub, CO production notably increases while the other products are suppressed. Importantly, FEH2 is suppressed for both CoPc-Cucub/C and CoPc-C/Cucub compared to the Cucub/C. As expected, the major product for CoPc/C is CO, with minor production of H2, indicating that the activity of the heterogenized molecular component is preserved.
To facilitate the comparison of C2 products, Fig. 3B reports the FEC2 and the corresponding partial current densities (jC2) of all Cucub-based systems. The CoPc-Cucub/C clearly shows an enhancement of C2 products with the highest FE of 39.4% and jC2 of 1.07 mA cm−2. In contrast, the FEC2 for CoPc-C/Cucub is only 10%, much lower than that of the Cucub/C, which is 18.3%. The jC2 are instead comparable and equal to 0.2 mA cm−2 for CoPc-C/Cucub and 0.16 mA cm−2 for the Cucub.
The high FECO and the suppressed FEC2 for CoPc-C/Cucub suggests that CO released from the molecule diffuses away rather than being utilized by the Cucub. Longer reaction times up to 10 hours do eventually result in a slight increase of FEC2 and jC2 for CoPc-C/Cucub (Fig. S3†), which might result from the overall increasing concentration of CO in the electrolyte. However, the performance of CoPc-C/Cucub remain always lower than CoPc-Cucub/C and Cucub/C. Decreasing the loading of the CoPc does decrease the FECO but does not improve the FEC2 compared to CoPc-Cucub/C with same CoPc loading and to Cucub/C (Fig. S4†), which indicates that the CO transfer from CoPc-C to Cucub does not efficiently occur in these catalysts.
To further investigate the mechanism behind the observed catalytic activity differences, we investigated the CoPc-Cucub/C and CoPc-C/Cucub CO2RR performance over a wide range of applied potentials and compared them to those of Cucub/C and CoPc/C (Fig. 4). We note that the CoPc stability decreases and the selectivity for methane increases at more negative voltages (Fig. S5†). Consistent with the data in Fig. 3, CoPc-Cucub/C exhibits a similar potential-dependent product distribution compared to Cucub/C, but with an increased C2 product yield and suppressed hydrogen generation across the entire potential range (Fig. 4A). In particular, the FEC2 and the corresponding partial current density of the CoPc-Cucub/C are consistently higher than the values measured for the Cucub/C and increase at more cathodic potentials (Fig. 4B). The best-performing CoPc-Cucub/C possess a FEC2 of 48% with a partial current density of 1.5 mA cm−2 at −1.05 V vs. RHE, which is 1.7 times the FEC2 of the Cucub/C. In contrast, for the CoPc-C/Cucub assembly, CO is the major product and the FEC2 is reduced over the whole potential range compared to Cucub/C (Fig. 4D). Furthermore, the partial current density of the C2 products in CoPc-C/Cucub and Cucub/C is always similar, which indicates that the Cucub intrinsic selectivity is unaffected by the CoPc molecules (Fig. 4D). We also note that the total current densities of the tandem catalysts are higher compared to those of either the Cucub or the CoPc alone, which indicates that both components are active (Fig. S8†). Furthermore, the overall C2 production rate is higher in the tandem catalysts compared to the Cucub alone (Fig. S8†), which is similar to what has been reported for some of the metallic/Cu tandem catalysts.41,42 We highlight that morphology and composition of all catalysts were maintained after CO2RR at −1.05 V vs. RHE for 1 hour (Fig. S6†).
 | ||
| Fig. 4 (A and D) FEs for all gaseous products (i.e., H2, CO, CH4, C2H4) and the main liquid products (i.e., formate, C2H5OH), (B and E) FEs and partial current densities of C2 products (i.e. C2H4 + C2H5OH), jC2, normalized by the ECSA and (C and F) FEs for CO as a function of potential for Cucub/C, CoPc-Cucub/C, CoPc-C/Cucub and CoPc/C. The loading of Cucub was 20 μg cm−2 and the loading of CoPc anchored on the Cucub and on the carbon black was 1.1 nmol cm−2 and 0.9 nmol cm−2, respectively. These data were collected in a H-cell system using 0.1 M KHCO3 as the electrolyte for 1 h. The reported values are an average of three independent experiments with error bars indicating the standard deviations. | ||
To understand whether the C–C coupling enhancement in the CoPc-Cucub/C catalyst is consistent with a tandem mechanism, we plotted the FECO of the different systems at various electrochemical potentials (Fig. 4C and F). The FECO for the CoPc-Cucub/C decreases as the potential becomes more cathodic (Fig. 4C), concomitant with the C2 production increase. This correlation is consistent with a tandem mechanism in which the CO formed on CoPc is consumed by Cucub. By contrast, the FECO of the CoPc-C/Cucub catalyst remains approximately constant and is similar to that of CoPc/C across the entire potential range (Fig. 4F), proving that the CO formed on CoPc is not consumed by Cucub and the sequential tandem reaction does not occur.
To further validate the conclusion that the obtained results are due to a tandem effect, we performed control experiments to investigate whether other factors could be involved. XPS revealed that the electronic properties of the catalysts are not impacted by interactions between the different components and are, therefore, not responsible for the observed catalytic differences (Fig. S8 and Table S2†). In addition, we verified that neither the phthalocyanine ligand nor the Co alone on the Cu surface can provide the enhanced C–C coupling observed for CoPc-Cucub/C (Fig. S9–S11†). Finally, no major changes in hydrophobicity, which could justify hydrogen suppression, were observed upon adsorption of the CoPc on the Cucub (Fig. S12†). Overall, the tandem effect emerges as the main reason underlying the C–C coupling enhancement observed in the CoPc-Cucub/C catalysts. Finally, encouraged by the results measured in a H-cell configuration, we included the spatial configurated systems into a gas-fed flow cell, comprising the CoPc-Cucub/C and the CoPc-C/Cucub supported on a gas diffusion electrode as the cathode, in order to realize CO2 to C2 conversion at high current density (Fig. S13†). In agreement with the data discussed above, the CoPc-Cucub/C catalyst demonstrated enhanced C2 production compared to the Cucub/C reaching a FE for C2 of 62% with a partial current density of 125 mA cm−2 in 1 M KHCO3.
Fig. 5 summarizes the results of this study and the proposed mechanism responsible for the observed electrocatalytic activity of molecule-copper tandem catalysts. We find that the close vicinity and direct interface between the CO-generating molecules and the Cu surface is essential to promote C–C coupling via the tandem effect. When the molecular component is spatially separated from the copper, the sequential tandem process does not occur and the overall multicarbon production is suppressed. These results suggest that the tandem mechanism in these catalysts is enabled by surface diffusion of the CO rather than CO diffusion in the electrolyte and sequential reabsorption.
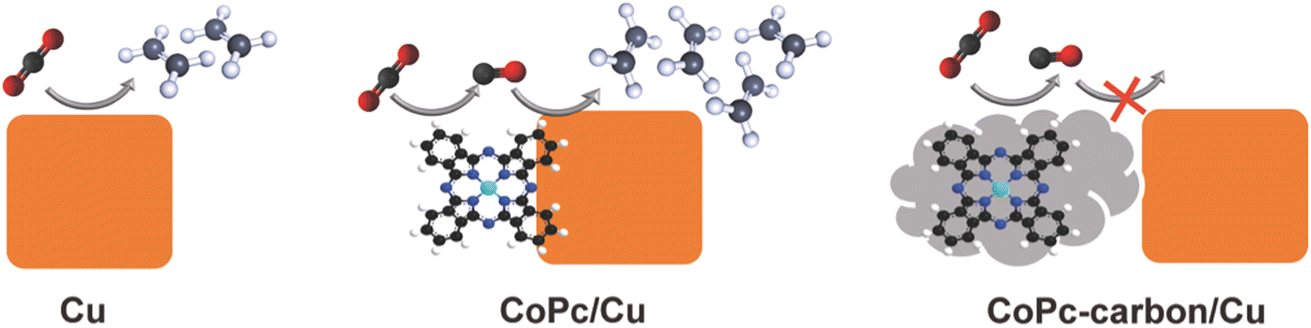 | ||
| Fig. 5 Schematic illustration summarizing the effect of the spatial distribution of CO-producing molecular catalysts and Cu catalysts in tandem systems. The close spatial coupling and direct interface between the two components is crucial for enhancing C–C coupling via enhanced CO generation in the vicinity of Cu. | ||
Conclusions
In summary, we have demonstrated that tandem catalysts must be carefully engineered to promote efficient C–C coupling. In particular, our results reveal that a direct interface between the CO-generating molecular catalyst and the Cu is a key factor for efficient CO utilization by the copper. Indeed, we have reported a CO2-to-C2 conversion with FE of 48% at −1.05 V vs. RHE for CoPc-Cucub/C catalysts, which is 1.7 times higher than that of the Cucub/C when tested under the same conditions. In contrast, the CoPc-C/Cucub assembly, in which the Cucub and the CoPc were physically separated, exhibited a dramatically reduced FEC2 compared to the Cucub. Overall, these findings advance our fundamental understanding of the performance-driving parameters necessary for constructing more efficient hybrid molecule/nanocrystal tandem catalysts in the future. For example, the discovery that a close spatial coupling is crucial for efficient tandem catalysts suggests that their integration into gas diffusion electrodes operating at high current density may benefit from functional groups to anchor the molecular catalysts on the surface of the nanocatalysts.Data availability
Data for this paper, including TEM images, FT-IR, HAADF-STEM images and corresponding EDX and electrochemical characterization, are available at Zenodo at: https://doi.org/10.5281/zenodo.7327628.Author contributions
M. W. carried out the synthesis of the tandem catalysts and electrocatalytic testing for CO2RR. A. L. performed X-ray photoelectron spectroscopy, HAADF-STEM images and corresponding EDX and contributed to daily discussions. V. O. and I. D. S. contributed to discussions. R. B. supervised this work and coordinated the project. All the authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support from EU-funded LICROX FET project (Grant agreement 951843).References
- G. Centi and S. Perathoner, Opportunities and prospects in the chemical recycling of carbon dioxide to fuels, Catal. Today, 2009, 148(3–4), 191–205 CrossRef CAS.
- D. T. Whipple and P. J. A. Kenis, Prospects of CO2 utilization via direct heterogeneous electrochemical reduction, J. Phys. Chem. Lett., 2010, 1(24), 3451–3458 CrossRef CAS.
- H.-R. M. Jhong, S. Ma and P. J. A. Kenis, Electrochemical conversion of CO2 to useful chemicals: Current status, remaining challenges, and future opportunities, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4(9), 732–745 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Y. Lum and J. W. Ager, Sequential catalysis controls selectivity in electrochemical CO2 reduction on Cu, Energy Environ. Sci., 2018, 11(10), 2935–2944 RSC.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon and D. Sinton, et al., Cascade CO2 electroreduction enables efficient carbonate-free production of ethylene, Joule, 2021, 5(3), 706–719 CrossRef CAS.
- Y. Zhu, X. Cui, H. Liu, Z. Guo, Y. Dang, Z. Fan and W. Hu, et al., Tandem catalysis in electrochemical CO2 reduction reaction, Nano Res., 2021, 14, 4471–4486 CrossRef CAS.
- B. Cao, F. Z. Li and J. Gu, Designing Cu-Based Tandem Catalysts for CO2 Electroreduction Based on Mass Transport of CO Intermediate, ACS Catal., 2022, 12, 9735–9752 CrossRef CAS.
- X. Wang, J. F. de Araújo, W. Ju and P. Strasser, et al., Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2-CO co-feeds on Cu and Cu-tandem electrocatalysts, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- A. J. Garza, A. T. Bell and M. Head-Gordon, Mechanism of CO2 reduction at copper surfaces: pathways to C2 products, ACS Catal., 2018, 8(2), 1490–1499 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, Mechanistic understanding of CO2 reduction reaction (CO2RR) toward multicarbon products by heterogeneous copper-based catalysts, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang and D. Sinton, et al., Constraining CO coverage on copper promotes high-efficiency ethylene electroproduction, Nat. Catal., 2019, 2(12), 1124–1131 CrossRef CAS.
- S. Louisia, D. Kim, Y. Li, M. Gao, S. Yu, I. Roh and P. Yang, The presence and role of the intermediary CO reservoir in heterogeneous electroreduction of CO2, Proc. Natl. Acad. Sci., 2022, 119(18), e2201922119 CrossRef CAS PubMed.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, Electrochemical Reduction of CO2 Using Copper Single-Crystal Surfaces: Effects of CO* Coverage on the Selective Formation of Ethylene, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, Selective C-C coupling in carbon dioxide electroreduction via efficient spillover of intermediates as supported by operando Raman spectroscopy, J. Am. Chem. Soc., 2019, 141(47), 18704–18714 CrossRef CAS PubMed.
- P. B. O'Mara, P. Wilde, T. M. Benedetti, C. Andronescu, S. Cheong, J. J. Gooding, R. D. Tilley and W. Schuhmann, Cascade Reactions in Nanozymes: Spatially Separated Active Sites inside Ag-Core–Porous-Cu-Shell Nanoparticles for Multistep Carbon Dioxide Reduction to Higher Organic Molecules, J. Am. Chem. Soc., 2019, 141(36), 14093–14097 CrossRef PubMed.
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen and Y. Peng, et al., Geometric Modulation of Local CO Flux in Ag@Cu2O Nanoreactors for Steering the CO2RR Pathway toward High-Efficacy Methane Production, Adv. Mater., 2021, 33(32), 2101741 CrossRef CAS PubMed.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Highly selective and productive reduction of carbon dioxide to multicarbon products via in situ CO management using segmented tandem electrodes, Nat. Catal., 2022, 5(3), 202–211 CrossRef CAS.
- S. B. Varandili, D. Stoian, J. Vavra, K. Rossi, J. R. Pankhurst, Y. T. Guntern, L. Núria and R. Buonsanti, Elucidating the structure-dependent selectivity of CuZn towards methane and ethanol in CO2 electroreduction using tailored Cu/ZnO precatalysts, Chem. Sci., 2021, 12(43), 14484–14493 RSC.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Atomic Layer Deposition of ZnO on CuO Enables Selective and Efficient Electroreduction of Carbon Dioxide to Liquid Fuels, Angew. Chem., Int. Ed., 2019, 131, 15178–15182 CrossRef.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Improved CO2 reduction activity towards C2+ alcohols on a tandem gold on copper electrocatalyst, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, Binding site diversity promotes CO2 electroreduction to ethanol, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Cu-Ag Tandem Catalysts for High-Rate CO2 Electrolysis toward Multicarbons, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- P. Iyengar, M. J. Kolb, J. R. Pankhurst, F. Calle-Vallejo and R. Buonsanti, Elucidating the facet-dependent selectivity for CO2 electroreduction to ethanol of Cu-Ag tandem catalysts, ACS Catal., 2021, 11(8), 4456–4463 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, Structural sensitivities in bimetallic catalysts for electrochemical CO2 reduction revealed by Ag-Cu nanodimers, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- X. Kong, J. Zhao, J. Ke, C. Wang, S. Li, R. Si, B. Liu, J. Zeng and Z. Geng, Understanding the Effect of *CO Coverage on C–C Coupling toward CO2 Electroreduction, Nano Lett., 2022, 22(9), 3801–3808 CrossRef CAS PubMed.
- F. Li, Y. C. Li, Z. Wang, J. Li, D. H. Nam, Y. Lum and E. H. Sargent, Cooperative CO2-to-ethanol conversion via enriched intermediates at molecule–metal catalyst interfaces, Nat. Catal., 2020, 3(1), 75–82 CrossRef CAS.
- M. Wang, V. Nikolaou, A. Loiudice, I. D. Sharp, A. Llobet and R. Buonsanti, Tandem electrocatalytic CO2 reduction with Fe-porphyrins and Cu nanocubes enhances ethylene production, Chem. Sci., 2022, 13, 12673–12680 RSC.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Tailoring copper nanocrystals towards C2 products in electrochemical CO2 reduction, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, Facet-dependent selectivity of Cu catalysts in electrochemical CO2 reduction at commercially viable current densities, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, Plasma-activated copper nanocube catalysts for efficient carbon dioxide electroreduction to hydrocarbons and alcohols, ACS Nano, 2017, 11(5), 4825–4831 CrossRef CAS PubMed.
- Y. Wu, Y. Liang and H. Wang, Heterogeneous Molecular catalysts of metal phthalocyanines for electrochemical CO2 reduction reactions, Acc. Chem. Res., 2021, 54(16), 3149–3159 CrossRef CAS PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu and H. Wang, et al., Highly selective and active CO2 reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures, Nat. Commun., 2017, 8(1), 1–8 CrossRef PubMed.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell, Science, 2019, 365(6451), 367–369 CrossRef CAS PubMed.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, C. P. Berlinguette and M. Robert, et al., CO2 electrochemical catalytic reduction with a highly active cobalt phthalocyanine, Nat. Commun., 2019, 10(1), 1–8 CrossRef PubMed.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Porphyrins at interfaces, Nat. Chem., 2015, 7(2), 105–120 CrossRef PubMed.
- K. Shen, B. Narsu, G. Ji, H. Sun, J. Hu, Z. Liang and F. Song, et al., On-surface manipulation of atom substitution between cobalt phthalocyanine and the Cu (111) substrate, RSC Adv., 2017, 7(23), 13827–13835 RSC.
- A. Belser, K. Greulich, P. Grüninger, R. Karstens, R. Ovsyannikov, E. Giangrisostomi and H. Peisert, et al., Perfluorinated Phthalocyanines on Cu(110) and Cu(110)-(2×1)O: The Special Role of the Central Cobalt Atom, J. Phys. Chem. C, 2021, 125(16), 8803–8814 CrossRef CAS.
- T. T. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel and A. A. Gewirth, et al., Nanoporous copper–silver alloys by additive-controlled electrodeposition for the selective electroreduction of CO2 to ethylene and ethanol, J. Am. Chem. Soc., 2018, 140(17), 5791–5797 CrossRef CAS PubMed.
- L. R. L. Ting, O. Pique, S. Y. Lim, M. Tanhaei, F. Calle-Vallejo and B. S. Yeo, Enhancing CO2 electroreduction to ethanol on copper–silver composites by opening an alternative catalytic pathway, ACS Catal., 2020, 10(7), 4059–4069 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06359j |
| This journal is © The Royal Society of Chemistry 2023 |