
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the existence of low-valent magnesium–calcium complexes†‡
Jonathan
Mai
 ,
Bastian
Rösch
,
Neha
Patel
,
Jens
Langer
and
Sjoerd
Harder
*
,
Bastian
Rösch
,
Neha
Patel
,
Jens
Langer
and
Sjoerd
Harder
*
Inorganic and Organometallic Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 1, Erlangen 91058, Germany. E-mail: sjoerd.harder@fau.de
First published on 13th April 2023
Abstract
DFT-Calculations predict that a low-valent complex (BDI)Mg–Ca(BDI) with bulky β-diketiminate (BDI) ligands is thermodynamically stable. It was attempted to isolate such a complex by salt-metathesis between [(DIPePBDI*)Mg−Na+]2 and [(DIPePBDI)CaI]2 (DIPePBDI = HC[C(Me)N-DIPeP]2; DIPePBDI* = HC[C(tBu)N-DIPeP]2; DIPeP = 2,6-CH(Et)2-phenyl). Whereas in alkane solvents no reaction was observed, salt-metathesis in C6H6 led to immediate C–H activation of benzene to give (DIPePBDI*)MgPh and (DIPePBDI)CaH, the latter crystallizing as a THF-solvated dimer [(DIPePBDI)CaH·THF]2. Calculations suggest reduction and insertion of benzene in the Mg–Ca bond. The activation enthalpy for the subsequent decomposition of C6H62− into Ph− and H− is only 14.4 kcal mol−1. Repeating this reaction in the presence of naphthalene or anthracene led to heterobimetallic complexes in which naphthalene2− or anthracene2− anions are sandwiched between (DIPePBDI*)Mg+ and (DIPePBDI)Ca+ cations. These complexes slowly decompose to their homometallic counterparts and further decomposition products. Complexes in which naphthalene2− or anthracene2− anions are sandwiched between two (DIPePBDI)Ca+ cations were isolated. The low-valent complex (DIPePBDI*)Mg–Ca(DIPePBDI) could not be isolated due to its high reactivity. There is, however, strong evidence that this heterobimetallic compound is a fleeting intermediate.
Introduction
Low-valent MgI complexes like I (Scheme 1) are kinetically stabilized towards disproportionation (2 MgI → MgII + Mg0) by a bulky β-diketiminate ligand (BDI). Ever since this first milestone discovery by Jones in 2007,1 there has been quite some interest to isolate other alkaline-earth metals (Ae) in unusually low oxidation states.2–5 Examples of this activity are the CaI inverse sandwich complex II by Westerhausen and coworkers,6 or the Be0 (III) and BeI (IV) complexes by the Braunschweig and Gilliard groups, respectively.7,8 Since the oxidation state of the Ca metals in II depends on the electronic state and charge on the triphenyl-benzene anion, there is some controversy about the Ca valency in this complex.9 On a similar note, also the oxidation states in CAAC complexes III and IV are the subject of discussion.10–13 Early attempts to isolate (BDI)Ca–Ca(BDI) complexes in which the +I oxidation state on the metal is unambiguous hitherto failed.14 We and others therefore designed BDI ligands with either very large substituents or electron-donating arms aiming to stabilize the larger CaI centres.15–18 Our approach to introduce a ligand without acidic protons in direct vicinity of the metal led to the superbulky DIPePBDI ligand (DIPePBDI = HC[C(Me)N-DIPeP]2; DIPeP = 2,6-CH(Et)2-phenyl).19 The highly dynamic (Et)2CH-substituents in this ligand are relatively inert to deprotonation by strong bases and increase the solubility of its metal complexes in inert (unreactive) solvents like alkanes. Alkane solubility is an important requirement for the isolation of highly reducing low-valent Ae metal complexes.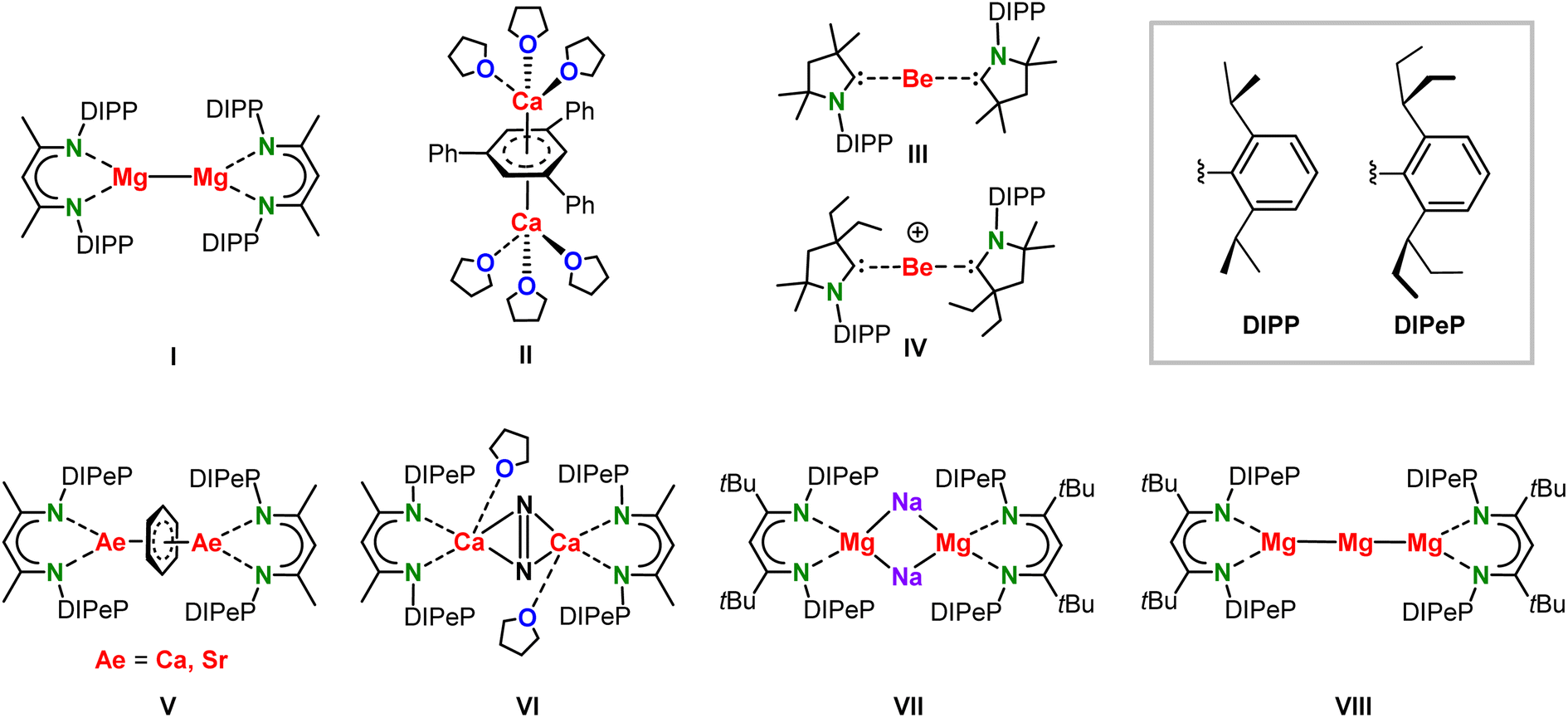 | ||
| Scheme 1 Complexes I–VIII. | ||
Previous attempts to prepare (BDI)Ca–Ca(BDI) or (BDI)Sr–Sr(BDI) complexes in aromatic solvents led to solvent reduction and formation of complexes with bridging C6H62− dianions (V).15,20 The extreme reducing power of (BDI)Ca–Ca(BDI) complexes was further demonstrated by isolation of the N2-activated complex VI when alkanes were used as the solvent and N2 as an “inert” gas.15 Similar N2 reduction to N22−was found in an attempt to isolate a mixed-metal CaI/KI complex.17 Attempting the syntheses of (BDI)Ca–Ca(BDI) complexes under an Ar atmosphere led to intractable mixtures of decomposition products.15,16 Although even larger BDI ligands like DIPePBDI* (HC[C(tBu)N-DIPeP]2) so far did not unlock the challenging isolation of (BDI)Ca–Ca(BDI) complexes, this superbulky ligand was successful in the isolation of the first Mg0 complexes (VII and VIII).21
The fair stability of the Mg–Mg bond and the very high reactivity of proposed but so far never isolated (BDI)Ca–Ca(BDI) complexes, begged the question whether mixed-metal (BDI)Mg–Ca(BDI) complexes may be stable. The slight difference in the Mg and Ca electronegativities (Mg 1.31, Ca 1.00)22 would formally make this a Mg0/CaII combination but large electron densities should be expected on both metals. Although there is rare theoretical work on fictitious species with mixed-metal Mg–Be or Mg–Ca bonds,23,24 the chemistry of mixed low-valent Mg/Ca complexes is hitherto no-man's-land. We here report our theoretical and experimental journey regarding low-valent complexes featuring a Mg–Ca bond.
Results and discussion
DFT-Calculations show that the isolation of low-valent mixed-metal Mg/Ca complexes may be feasible (Scheme 2). Starting from the radicals (DIPPBDI)Mg˙ and (DIPPBDI)Ca˙, the energies for formation of the homo- and hetero-metallic low-valent complexes were calculated (DIPPBDI = HC[C(Me)N-DIPP]2; DIPP = 2,6-CH(Me)2-phenyl). Using the B3PW91/def2TZVP//def2SVP level of theory, structures were optimized with and without consideration of dispersion using Grimme's third dispersion correction with Becke–Johnson damping (GD3BJ) (see ESI‡ for details).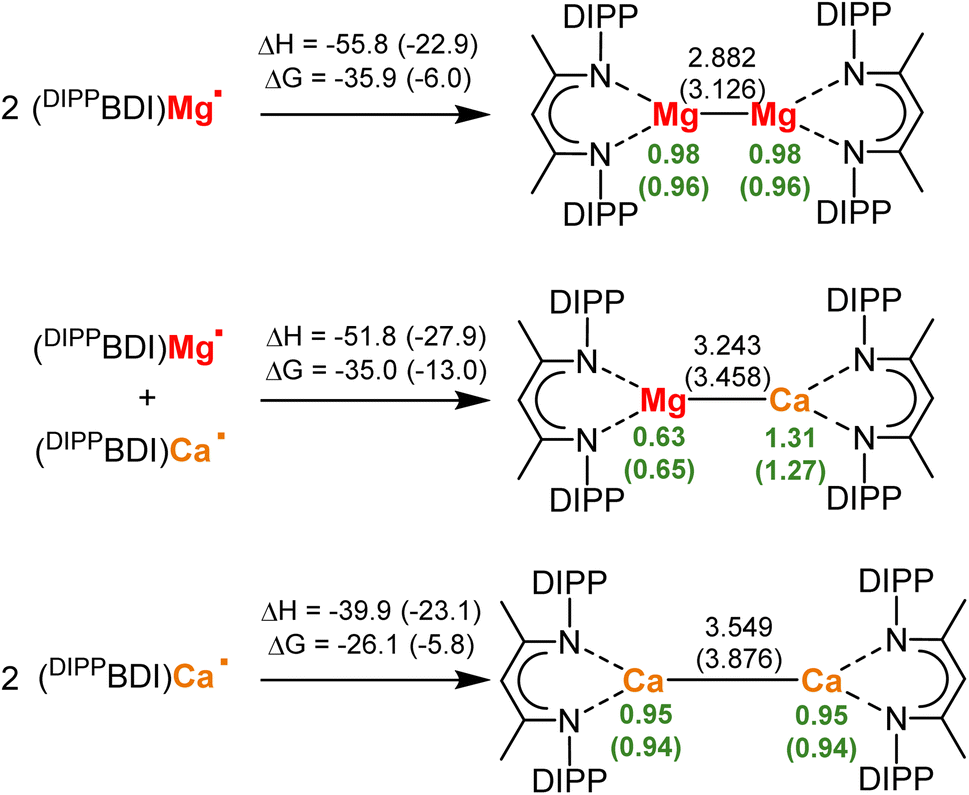 | ||
| Scheme 2 Formation of homo- and hetero-metallic (DIPPBDI)Ae–Ae(DIPPBDI) complexes (Ae = Mg or Ca). Enthalpies and Gibbs free energies at 298 K in kcal mol−1, Ae–Ae distances in Å and NPA charges in green; B3PW91/def2TZVP//def2SVP with dispersion correction (GD3BJ). Values in brackets relate to calculations without dispersion corrections. | ||
Note that correction for attractive dispersive forces between the DIPPBDI ligands has a considerable effect on the metal–metal bond distances, resulting in shorter and stronger Ae–Ae bonds. Use of dispersion calculates a Mg–Mg bond length (2.882 Å) that is close to the experimentally found value (2.846(1) Å).1 The calculated energies of formation (ΔH = −55.8 kcal mol−1, ΔG(298 K) = −35.9 kcal mol−1) are in agreement with previously reported Mg–Mg bond energies varying between 36–45 kcal mol−1.25,26 Unique for the Mg–Mg bond is the occurrence of a Non-Nuclear-Attractor (NNA) in the bond center.27,28 Experimental electron count in this local maximum of electron density shows a basin with 0.81e. This fits very well with the calculated value of 0.80e when dispersion correction was used; See Fig. S58‡ for an Atoms-In-Molecules analysis (AIM). Without correction for dispersion a significantly smaller NNA basin of 0.62e was calculated (Fig. S59‡). This is in agreement with the observation that the NNA is sensitive to the Mg–Mg distance.27 Our calculations also show why it is problematic to isolate (BDI)Ca–Ca(BDI) complexes. Even with inclusion of dispersion the Ca–Ca bond is long (3.549 Å) and the bond formation energies are small (ΔH = −39.9 kcal mol−1, ΔG(298 K) = −26.1 kcal mol−1). AIM analysis of (DIPPBDI)Ca–Ca(DIPPBDI) shows a regular Ca–Ca bond path with a bond-critical-point in the center. A NNA could not be located. Interestingly, the much longer Ca–Ca bond (3.876 Å) optimized without correction for dispersion, shows a NNA but only with a rather small basin of 0.33e (Fig. S61‡). As expected, the Mg–Ca distance in (DIPPBDI)Mg–Ca(DIPPBDI) fits nicely between the values for the Mg–Mg and Ca–Ca bonds. However, surprising is the fact that the Mg–Ca bond in this heterometallic complex is calculated to be nearly as stable as the Mg–Mg bond. Using dispersion, the following Mg–Ca bond formation energies have been calculated: ΔH = −51.8 kcal mol−1, ΔG(298 K) = −35.0 kcal mol−1, values that are close to those for a Mg–Mg bond. AIM analysis on this mixed metal compound does not show the existence of a NNA on the Mg–Ca axis (Fig. S62 and S63‡). Note that metal exchange between the homometallic MgI and CaI dimers to give heterometallic (DIPPBDI)Mg–Ca(DIPPBDI) is exothermic and exergonic (ΔH = −7.9 kcal mol−1, ΔG(298 K) = −8.0 kcal mol−1). This distinct thermodynamic preference for the mixed Mg–Ca complex may originate from polarization of the Mg–Ca bond electron density, resulting in a more favorable electrostatic interaction. Indeed, the NPA charges on Mg (+0.63) and Ca (+1.31) clearly show strong polarization towards a Mg0/CaII complex, however, there is still a considerable electron density on Ca (NPA charges on Ca in regular CaII complexes are circa +1.75).
The recently reported [(DIPePBDI*)Mg−Na+]2 complex (VII) could be the key to the synthesis of such heterobimetallic complexes. Being a sodium salt of a magnesyl anion, the question arose whether a simple salt-metathesis reaction with the previously reported11 dimer [(DIPePBDI)CaI]2 (IX) may give access to a mixed-metal low-valent species (DIPePBDI*)Mg–Ca(DIPePBDI) featuring a Mg–Ca bond. Although two different metals (Mg and Ca) and two different ligands (DIPePBDI and DIPePBDI*) could result in complications due to metal–ligand scrambling, we have chosen to use the somewhat smaller DIPePBDI ligand at Ca for steric reasons. The structure of a recently reported MgI complex with two superbulky ligands, (DIPePBDI*)Mg–Mg(DIPePBDI*), is overcrowded causing one of the BDI ligands to be only monodentate.22
Salt-metathesis between VII and X in methylcyclohexane did not lead to NaI precipitation and the expected product (DIPePBDI*)Mg–Ca(DIPePBDI) (1); see Scheme 3. 1H NMR in this solvent showed that there is essential no interaction between both dimers. We believe that this is due to complete steric saturation of the metal centers. This is the reason why the vast majority of salt-metathesis reactions in s-block metal chemistry are performed in ethereal solvents (preferably THF) that break down aggregates, allowing an encounter between the metal centers of both adducts.
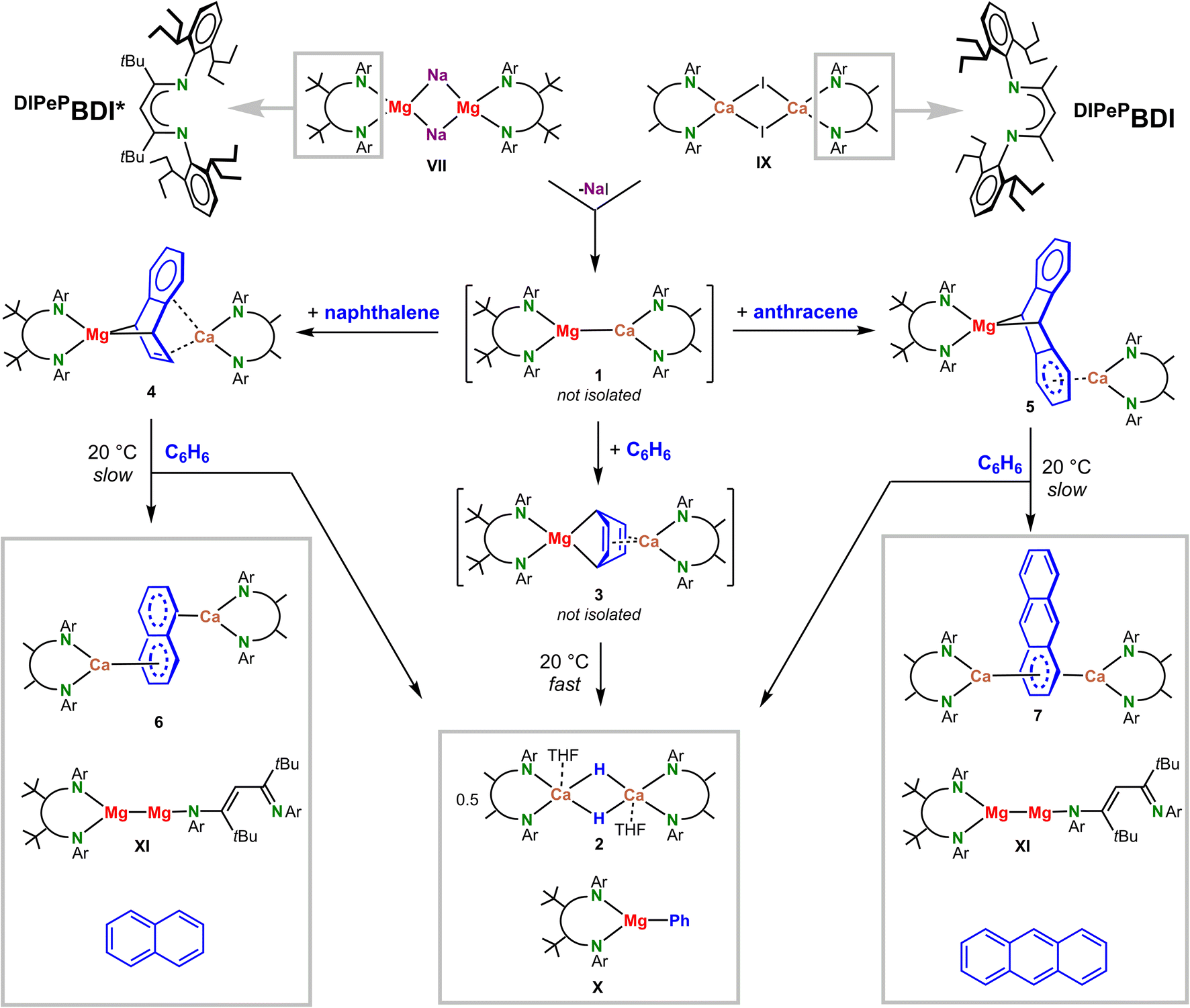 | ||
| Scheme 3 In situ formation, decomposition and reactivity of a low-valent heterobimetallic (BDI)Mg–Ca(BDI) intermediate. | ||
Unfortunately, the sodium magnesyl complex VII is not stable in ethers.21 Recently, successful solid-state salt-metathesis reactions in a ball-mill have been reported.29 A solvent-free reaction between VII and X in the ball-mill gave conversion, however, a mixture of various undefined products was formed. Salt-metathesis in the slightly more polar solvent toluene also gave various intractable products but reaction in benzene was selective. A benzene solution of [(DIPePBDI*)Mg−Na+]2 (VII) and [(DIPePBDI)CaI]2 (IX) slowly changed color from dark-brown to yellow and over a two-day period a precipitate of NaI was formed. 1H NMR of the crude product showed selective formation of two main species: (DIPePBDI*)MgPh (X), which we previously fully characterized,21 and (DIPePBDI)CaH, which after addition of small amounts of THF crystallized as the dimer [(DIPePBDI)CaH·(THF)]2 (2) in 37% yield. The crystal structure of this dimer (Fig. S46‡) resembles the previously reported tetrahydropyran complex.15 The benzene solvent is the source of the Ph anion in X and the hydride in 2: reaction in C6D6 led to the same products with C6D5− and D− anions (Fig. S30–S32‡).
Cleavage of the C–H bond in benzene has also been observed for previously reported (BDI)Mg(μ2,μ4-C6H6)Mg(BDI) complexes (formed by reduction of (BDI)MgI in the presence of TMEDA or by ball-milling),19,30 however, this process needs high temperatures and very long reaction times. Although homometallic (BDI)Mg–Mg(BDI) complexes do not react with benzene, activation of the Mg–Mg bond by UV-light31 or by a Pd0 catalyst32 also led to benzene C–H bond cleavage. In contrast, recently isolated Ca and Sr complexes with a μ6-bridging C6H62− ion (V) reacted with benzene by dehydrogenative C–C coupling to give a biphenyl2− ion.20 For the Mg/Ca combination, we found no indication of biphenyl formation.
These observations demonstrate the importance of solvent effects in salt-metathesis reactions. Although there is currently no proof that benzene lowers the aggregation number of VII and X (DOSY measurements were inconclusive), it is assumed that aromatic (co)solvents influence dimer-monomer equilibria by coordination to the s-block metal. Strong interactions between Ae metal cations and π-systems have been reported33–37 and are also supported by DFT calculations (Fig. S52‡). Minor quantities of monomeric adducts would enable salt-metathesis between VII and X to give (DIPePBDI*)Mg–Ca(DIPePBDI) (1).
Although we found no direct proof for Mg–Ca bond formation, the fleeting existence of such an intermediate seems reasonable. The alternative, formation of (BDI)Mg˙ and (BDI)Ca˙ radicals, can be excluded. The (BDI)Mg˙ radical may react with benzene26 but would also couple to unreactive dimers, whereas a (BDI)Ca˙ radical reacts with benzene to a stable complex with a bridging C6H62− dianion (V).15 Both products could not be observed. Instead, selective cleavage of the benzene C–H bond was achieved.
We propose a mechanism in which the expected heterobimetallic intermediate [(DIPePBDI*)Mg–Ca(DIPePBDI)] (1) reduces benzene to give a complex with a Mg(μ2,μ4-C6H6)Ca bridge (3). DFT-calculations on a model system with DIPP-substituents show that reduction of benzene to give 3 is exothermic by −13.2 kcal mol−1 (Scheme 4a).
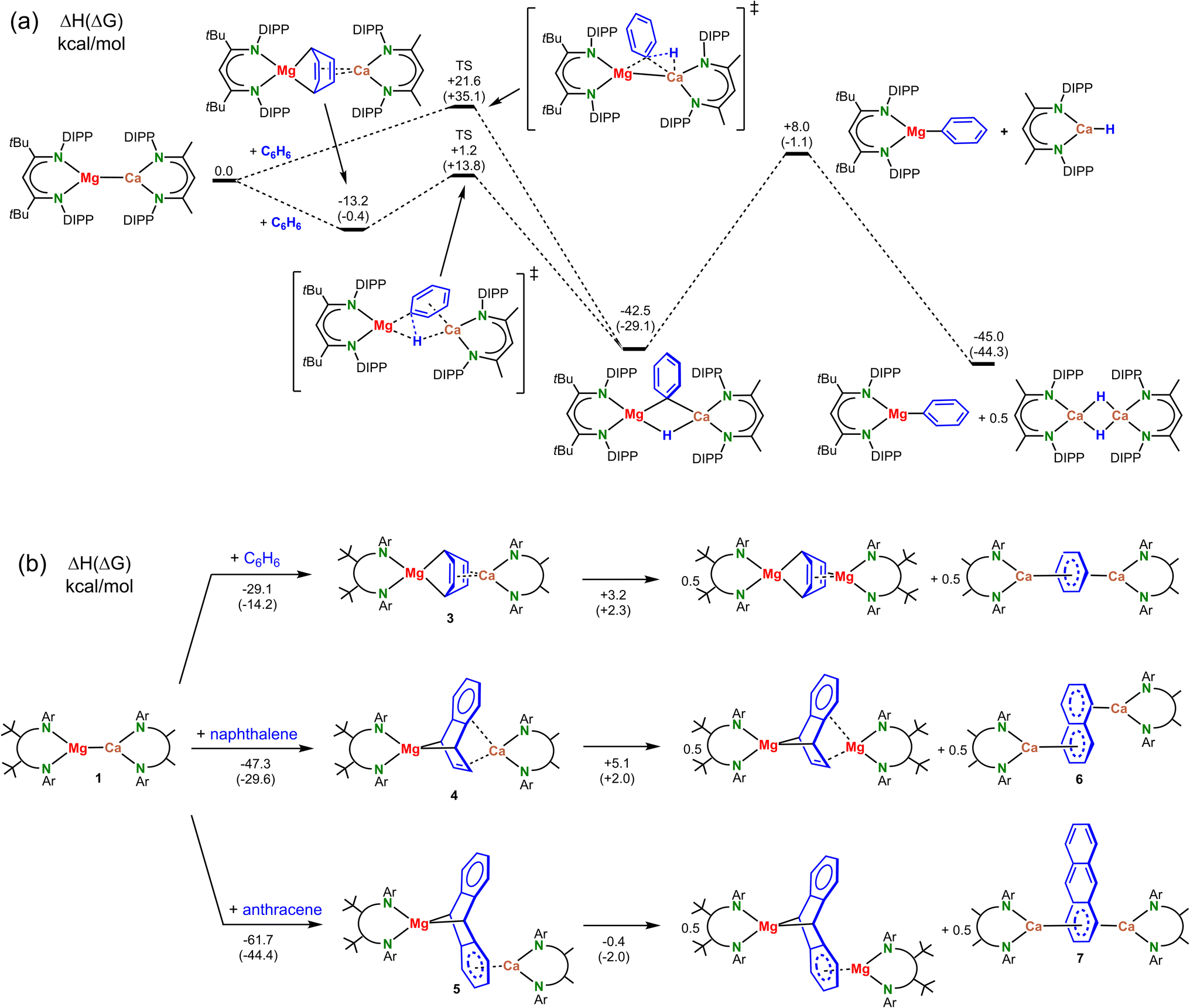 | ||
| Scheme 4 (a) Calculated mechanism for benzene C–H activation at a low-valent heterobimetallic (BDI)Mg–Ca(BDI) intermediate; B3PW91/def2TZVP//def2SVP with dispersion correction (GD3BJ). (b) Enthalpies and free energies (298 K) for the reaction of (DIPePBDI*)Mg–Ca(DIPePBDI) (1) with benzene, naphthalene or anthracene and energies for metal exchange to homometallic complexes. ΔH and ΔG (between parentheses) are given in kcal mol−1. | ||
The smaller Mg2+ cation bridges two C atoms in para-positions and forms strongly polar Mg–C bonds to the non-aromatic ring. The larger more electropositive Ca2+ ion is electrostatically bound to localized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Exchanging Mg and Ca in 3 is unfavorable: a structure with two Ca–C bonds and Mg2+-alkene interactions is 16.1 kcal mol−1 higher in energy (Fig. S51‡). Starting from the most stable μ2,μ4-bridged benzene complex, the transition state for subsequent C–H bond cleavage is only +14.4 kcal higher in energy. Alternatively, benzene could directly react with the polar Mg–Ca bond but this transition state is more than 20 kcal mol−1 higher in energy. Starting from the heterometallic Mg–Ca complex, cleavage of the benzene C–H bond to monomeric (BDI)MgPh and the dimeric Ca hydride complex, is quite exothermic (−45.0 kcal mol−1). Combined with a low activation energy for this process, a complex with a Mg–Ca bond is clearly not stable in benzene.
C bonds. Exchanging Mg and Ca in 3 is unfavorable: a structure with two Ca–C bonds and Mg2+-alkene interactions is 16.1 kcal mol−1 higher in energy (Fig. S51‡). Starting from the most stable μ2,μ4-bridged benzene complex, the transition state for subsequent C–H bond cleavage is only +14.4 kcal higher in energy. Alternatively, benzene could directly react with the polar Mg–Ca bond but this transition state is more than 20 kcal mol−1 higher in energy. Starting from the heterometallic Mg–Ca complex, cleavage of the benzene C–H bond to monomeric (BDI)MgPh and the dimeric Ca hydride complex, is quite exothermic (−45.0 kcal mol−1). Combined with a low activation energy for this process, a complex with a Mg–Ca bond is clearly not stable in benzene.
Since we were unable to isolate the proposed intermediates [(DIPePBDI*)Mg–Ca(DIPePBDI)] (1) or [(DIPePBDI*)Mg(μ2,μ4-C6H6)Ca(DIPePBDI)] (3), we attempted to prepare the more stable naphthalene and anthracene complexes. DFT calculations (Scheme 4b) predict that the reduction of arenes by a Mg–Ca model complex is increasingly more exergonic along the row benzene (ΔG = −14.2 kcal mol−1) < naphthalene (ΔG = −29.6 kcal mol−1) < anthracene (ΔG = −44.4 kcal mol−1).
Repeating the salt-metathesis reaction between [(DIPePBDI*)Mg−Na+]2 (VII) and [(DIPePBDI)CaI]2 (IX) in either methylcyclohexane or benzene in the presence of two equivalents of naphthalene led to a deep purple solution. The crude product is an essentially pure complex (Fig. S33 and S34‡), which after solvent removal and crystallization from pentane at −20 °C, could be isolated in form of orange crystals. The low yield of 29% is due to its very high solubility in pentane. Crystal structure determination shows a heterobimetallic inverse Mg/Ca sandwich with a bridging, non-planar, naphthalene dianion (DIPePBDI*)Mg(μ2,μ4-naphthalene)Ca(DIPePBDI) (4); Fig. 1a and Table 1. The smaller Mg2+ cation bridges two C atoms in para-positions and forms covalent but strongly polar Mg–C bonds to the non-aromatic ring. The larger more electropositive Ca2+ ion is ionically bound to a localized CC bond and the ring π-system. The Mg2+ ion is encapsulated by DIPePBDI* whereas the Ca2+ ion resides in the pocket of the DIPePBDI ligand. There is no indication of metal or ligand exchange. There is also no sign of metal disorder in the crystal structure, illustrating that the Mg and Ca metals are strongly bound in the BDI-ligand pockets. Calculated NPA charges are in agreement with an inverse sandwich consisting of a naphthalene2− ion and Mg2+/Ca2+ ions (Fig. 1a).
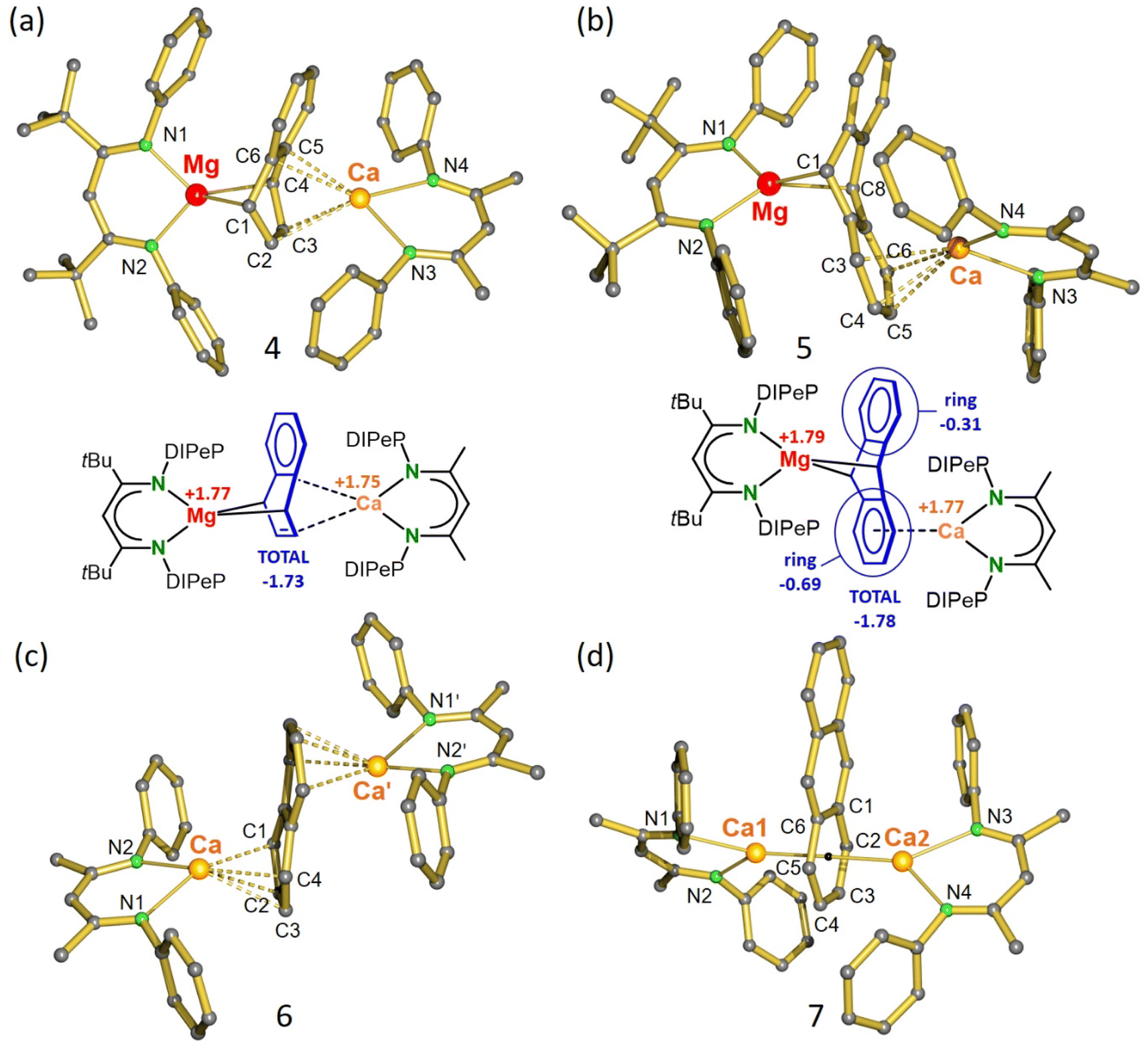 | ||
| Fig. 1 (a) Crystal structure and NPA charges for (DIPePBDI*)Mg(μ2,μ4-naphthalene)Ca(DIPePBDI) (4). (b) Crystal structure and NPA charges for (DIPePBDI*)Mg(μ2,μ4-anthracene)Ca(DIPePBDI) (5). (c) Crystal structure of (DIPePBDI)Ca(μ4,μ4-naphthalene)Ca(DIPePBDI) (6). (d) Crystal structure of (DIPePBDI)Ca(μ6,μ6-anthracene)Ca(DIPePBDI) (7). All Et2CH-substituents and H atoms have been omitted for clarity. Selected bond distances are summarized in Table 1. | ||
| Complex 4 | Complex 5 | Complex 6 | Complex 7 | |
|---|---|---|---|---|
| Mg–N1 2.112(1) | Mg–N1 2.109(3) | Ca–N1 2.330(1) | Ca1–N1 2.345(1) | Ca2–N3 2.348(1) |
| Mg–N2 2.095(1) | Mg–N2 2.128(3) | Ca–N2 2.365(1) | Ca1–N2 2.338(1) | Ca2–N4 2.338(1) |
| Mg–C1 2.344(1) | Mg–C1 2.374(4) | Ca–C1 2.642(1) | Ca1–C1 2.776(2) | Ca2–C1 2.706(1) |
| Mg–C4 2.376(1) | Mg–C8 2.426(4) | Ca–C2 2.654(1) | Ca1–C2 2.715(1) | Ca2–C2 2.708(2) |
| Ca–N3 2.345(1) | Ca–N3 2.341(3) | Ca–C3 2.673(1) | Ca1–C3 2.704(1) | Ca2–C3 2.726(2) |
| Ca–N4 2.357(1) | Ca–N4 2.338(3) | Ca–C4 2.674(1) | Ca1–C4 2.696(1) | Ca2–C4 2.717(2) |
| Ca–C2 2.839(2) | Ca–C3 2.778(4) | Ca1–C5 2.681(2) | Ca2–C5 2.702(1) | |
| Ca–C3 2.732(2) | Ca–C4 2.724(4) | Ca1–C6 2.746(2) | Ca2–C6 2.715(1) | |
| Ca–C5 2.676(1) | Ca–C5 2.744(4) | |||
| Ca–C6 2.825(1) | Ca–C6 2.821(4) | |||
In a similar procedure, salt-metathesis between VII and IX in the presence of anthracene yielded red crystals of (DIPePBDI*)Mg(μ2,μ4-anthracene)Ca(DIPePBDI) (5) in 45% yield. Also in this case, the raw product of the reaction is nearly pure (Fig. S35 and S36‡). In contrast to 4, Mg and Ca in 5 are bound to different rings (Fig. 1b). The Mg2+ ion bridges the 9,10-positions of anthracene in a similar fashion as found in Mg(anthracene)·(THF)3, however, the Mg–C distances in 5 (2.374(4) Å and 2.426(4) Å) are somewhat longer (cf. 2.25(1)-2.33(1) Å) due the bulk of the BDI ligands.38 In both Mg-anthracene complexes, the anthracene2− ion is strongly deformed from planarity illustrating dearomatization of the central ring. Loss of aromatic character is also indicated by the C–C distances and AIM analysis (Fig. S55, S65 and S66‡). The much larger Ca2+ ion is coordinating to the π-system of one of the outer aromatic rings. This leads to asymmetry and considerable polarization of negative charge to the Ca-bound ring (see Fig. 1b for NPA charges). Delocalization of negative charge is indicated by significantly shorter (Mg)C–C(Ar) bonds in the Ca-bound ring (1.442(5) and 1.444(5) Å) in comparison to those in the free ring (1.470(6) and 1.474(6) Å). A similar coordination mode was also found by the Jones group for the anthracene dianion sandwiched between (MesBDI)Mg+ cations (Mes = mesityl).39 It should, however, be noted that the potential energy surface for Ae2+ coordination to naphthalene or anthracene dianions is rather shallow and enthalpy differences between different isomers only vary between 2 and 15 kcal mol−1 (Fig. S51‡). This is in agreement with the 1H NMR spectrum of the anthracene complex 5 (Fig. S12‡) which shows one signal for the bridgehead Mg–CH protons (3.43 ppm) and two broad signals for the aromatic ring protons (5.13/5.72 ppm), indicating fast exchange of Ca between the two aromatic rings. Cooling to −70 °C led to slow exchange and decoalescence, resulting in five signals for the anthracene2− ion (Fig. S41‡), showing that also in solution an asymmetric structure is favored.
The naphthalene and anthracene complexes 4 and 5 both decompose slowly in solution at room temperature. A purple C6D6 solution of the naphthalene complex (4) turned blue and was after two weeks fully decomposed. The 1H NMR spectra show formation of several species (Fig. S37 and S38‡). After 13 days, the main decomposition product was characterized as the naphthalene dianion sandwiched between two (DIPePBDI)Ca+ ions (6 and Fig. 1c). This complex could also be obtained by an independent synthetic procedure in the form of blue crystals (vide infra). Its formation suggests scrambling of the heterobimetallic Mg/Ca complex 4 to give the homometallic Mg/Mg and Ca/Ca analogues. Although the homometallic complex (DIPePBDI)Ca(μ4,μ4-naphthalene)Ca(DIPePBDI) (6) is the main decomposition product, the fate of Mg is found to be different. Apart from the previously reported MgI dimer (DIPePBDI*)Mg–Mg(DIPePBDI*) (XI),26 also free naphthalene and typical benzene C–H bond activation products like (DIPePBDI*)MgPh (X) and the Ca hydride complex 2 could be observed by 1H NMR (Fig. S37 and S38‡). This indicates that the naphthalene2− ion in 4 can partially be replaced by the benzene solvent to form 3 and its decomposition products.
A similar decomposition pathway was observed for the anthracene complex 5 (Fig. S39 and S40‡). The main decomposition product was found to be homometallic (DIPePBDI)Ca(μ6,μ6-anthracene)Ca(DIPePBDI) (7 and Fig. 1d) which could also be obtained by an independent synthetic procedure in the form of dark-blue crystals (vide infra). Other decomposition products are (DIPePBDI*)Mg–Mg(DIPePBDI*) (XI), free anthracene and typical benzene C–H bond activation products like (DIPePBDI*)MgPh (X) and the Ca hydride complex 2.
Full decomposition of the heterobimetallic Mg/Ca complexes 4 and 5 in the homobimetallic Ca/Ca complexes 6 and 7 suggests that the mixed-metal combinations is less stable than the homometallic combinations. Calculations show that the energy differences between hetero- and homobimetallic complexes are small and therefore statistical Mg/Ca scrambling could be expected (Scheme 4b). The driving force for metal exchange is therefore dictated by formation of other side products, e.g. the cleavage of the benzene C–H bond which according to Scheme 4a is highly exothermic.
Alternatively, the bimetallic Ca complexes of naphthalene (6) and anthracene (7) could be prepared by reduction of [(DIPePBDI)CaI]2 (IX) with KC8 in the presence of either naphthalene or anthracene to give the products as intensely blue crystals in reasonable isolated yields (6: 46%, 7: 48%). Their crystal structures differ from those of the heterometallic Mg/Ca complexes 4 and 5 by changes in coordination of the central dianion.
The naphthalene dianion in the centrosymmetric structure of 6 is sandwiched between two Ca2+ ions coordinating to different rings (Fig. 1c). Bonding is mainly ionic and the naphthalene dianion is only slightly distorted from its flat aromatic structure. Each six-membered ring is deformed to a butterfly-type structure with an interplanar angle of 14.92(5)°. A similar geometry was reported for the dilithium salt of naphthalene: (C8H8)Li2·(TMEDA)2.40
The crystal structure of anthracene complex 7 is close to being C2-symmetric but has no crystallographic symmetry (Fig. 1d). The Ca atoms bind in η6-fashion to a common anthracene outer-ring with Ca–C bond distances in a narrow range of 2.681(2)–2.776(2) Å. DFT calculations show that the structure in which the two Ca atoms bind to opposite outer-rings is 13.2 kcal mol−1 higher in energy (Fig. S51‡). While preference for 7 is correctly predicted by DFT theory, the crystal structure of the naphthalene complex 6 is not in agreement with the lowest energy structure. DFT calculations predict that also in this case coordination to a common ring is favorable by ΔH = 14.7 kcal mol−1 (Fig. S51‡). We believe that coordination to a common naphthalene or anthracene ring is favored due to very strong polarizability of these extended π-systems. Indeed, in 7 the electron density on the anthracene dianion is mainly on the ring sandwiched by the two Ca2+ ions (NPA charge: −1.40) and not on the free outer-ring (NPA charge: −0.20). This results in increased electrostatic attraction between Ca2+ and the anthracene dianion. The high electron density on the Ca-bound ring is also evident from the considerably longer C–C bonds (average: 1.441 Å) vs. the C–C bonds in the uncoordinated ring (average: 1.399 Å). This originates from partial occupation of anti-bonding π*-orbitals in the Ca-bound ring. In contrast to the Mg/Ca anthracene complex 5, the relatively shallow potential energy surfaces for Ca/Ca coordination do not allow to freeze out the asymmetric coordination geometry in solution. 1H NMR spectra of 6 and 7 in toluene show down to −70 °C fast exchange between coordination geometries.
Conclusion
Although isolation and structural characterization of a low-valent Ae metal complex with a direct Mg–Ca bond remains a challenge, there are strong indications to assume that such a species has been formed as a fleeting intermediate.(1) The experimental approach, i.e. ion exchange between Mg−–Na+ and Ca+–I− bonds, implies formation of a Mg–Ca bond and NaI salt which is the driving force of this salt-metathesis reaction. The existence of the free (BDI)Mg˙ and (BDI)Ca˙ radicals rather than a Mg–Ca bound species can be ruled out, as they would react differently with benzene or other arenes.
(2) DFT-Calculations underscore that the Mg–Ca bond strength is comparable to that of the Mg–Mg bond for which there are numerous verifiable examples.2–5 These calculations also show that a heterobimetallic (BDI)Mg–Ca(BDI) complex is thermodynamically favored over its homometallic Mg/Mg and Ca/Ca counterparts. Polarization of the Mg–Ca bond, however, makes it highly reactive and kinetically unstable. The herein observed reduction of benzene to C6H62− and subsequent C–H bond cleavage can be achieved at room temperature, giving selectively (BDI)Mg–Ph and (BDI)Ca–H complexes. This reaction has been calculated to be highly exothermic (ΔH = −45.0 kcal mol−1) and has a kinetic barrier of only +14.4 kcal mol−1.
(3) Although the predicted intermediate, (BDI)Mg–(C6H6)–Ca(BDI), could not be isolated, similar but more stable complexes with naphthalene2− and anthracene2− ions were obtained by salt-metathesis in the presence of easily reducible naphthalene or anthracene. The naphthalene and anthracene complexes are exclusively formed as mixed-metal Mg/Ca species (4 and 5) which over time decompose to more stable homometallic Ca/Ca species (6 and 7) and low-valent (BDI)Mg–Mg(BDI). This is indirect proof for prior formation of a low-valent intermediate with two different metals which may feature a Mg–Ca bond.
Reduction of naphthalene or anthracene by a combination of free (BDI)Mg˙ and (BDI)Ca˙ radicals would at most lead to statistical Mg/Mg, Ca/Ca and Mg/Ca distributions or directly to the more stable homometallic Ca/Ca species (6 and 7) and low-valent (BDI)Mg–Mg(BDI). This is indeed the case. Reduction of an equimolar mixture of (DIPePBDI*)MgI and (DIPePBDI)CaI with KC8 in the presence of naphthalene led only to small amounts of the heterobimetallic Mg/Ca complex 4. Instead, major quantities of the homometallic Ca/Ca species 6 and low-valent (DIPePBDI*)Mg–Mg(DIPePBDI*) (XI) were formed (Fig. S45‡). In contrast, salt-metathesis between [(DIPePBDI*)Mg−Na+]2 (VII) and [(DIPePBDI)CaI]2 (IX) in the presence of naphthalene gave clean formation of the heterobimetallic Mg/Ca complex 4.
Arguably, 4 may also be formed by a two-step mechanism: (a) reduction of naphthalene by [(DIPePBDI*)Mg−Na+]2 (VII) to give (DIPePBDI*)Mg(naphthalene)Na and (b) subsequent salt-metathesis with [(DIPePBDI)CaI]2 (IX) yielding the heterobimetallic Mg/Ca complex 4 and NaI. Indeed, [(DIPePBDI*)Mg−Na+]2 (VII) reacts with naphthalene to give a dark-orange solution but so far no products could be isolated. Although this order of reactions may be an alternative pathway for naphthalene or anthracene reduction, it cannot be the pathway for benzene reduction: VII is not able to reduce benzene. Instead, Na+ is slowly reduced to Na0 under simultaneous formation of a trinuclear (BDI)Mg–Mg–Mg(BDI) complex (VIII).21 The cleavage of the C–H bond in benzene to give complexes 2 and X must therefore most likely proceed through the low-valent mixed Mg–Ca species (DIPePBDI*)Mg–Ca(DIPePBDI), formed by the salt-metathesis reaction of [(DIPePBDI*)Mg−Na+]2 (VII) and [(DIPePBDI)CaI]2 (IX) (Scheme 3). This is further supported by a conceivable calculated mechanism for benzene C–H bond activation (Scheme 4a).
Although a first example of a (BDI)Mg–Ca(BDI) complex could not be isolated, the existence of a highly reactive intermediate with a Mg–Ca bond is plausible. After first reactivity studies with benzene, naphthalene and anthracene, we are currently extending our explorations in heterobimetallic low-valent Mg–Ca chemistry.
Experimental section
General considerations
All experiments were conducted in dry glassware under an inert nitrogen atmosphere by applying standard Schlenk techniques or gloveboxes (MBraun) using freshly dried and degassed solvents. All solvents were degassed with nitrogen, dried over activated aluminum oxide (Innovative Technology, Pure Solv 400-4-MD, Solvent Purification System), and then stored under inert atmosphere over molecular sieves (3 Å) unless noted otherwise. Deuterated benzene (C6D6), toluene-d8, cyclohexane-d12 and methylcyclohexane-d14 were purchased from Sigma Aldrich or Deutero GmbH, degassed and dried over molecular sieves (3 Å). The following compounds were synthesized according to literature procedures: [(DIPePBDI*)MgNa]2,21 [(DIPePBDI)CaI]2,15 [(DIPePBDI)CaH]2 (ref. 15) and [(DIPePBDI)CaN(SiMe3)]2·(THF).41 PhSiH3 (Alfa Aesar, >99%) was obtained commercially as well as, naphthalene (Sigma Aldrich, 99%) and anthracene (Sigma Aldrich, 97%) which were sublimed under reduced pressure and stored under an N2 atmosphere. NMR spectra were measured on Bruker Avance III HD 400 MHz and Bruker Avance III HD 600 MHz spectrometers. Chemical shifts (δ) are denoted in ppm (parts per million), coupling constants in Hz (Hertz). For describing signal multiplicities common abbreviations are used: s (singlet), d (doublet), t (triplet), q (quartet), p (quintet), sept (septet), m (multiplet) and br (broad). Spectra were referenced to the solvent residual signal. Assignments of resonance signals in the 1H and 13C{1H} NMR spectra were made based on two-dimensional NMR correlation (HSQC, HMBC, COSY) experiments. Elemental analysis was performed with an Hekatech Eurovector EA3000 analyzer. All crystal structures have been measured on a SuperNova (Agilent) diffractometer with dual Cu and Mo microfocus sources and an Atlas S2 detector. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers: CCDC 2240705 [(DIPePBDI)CaH·(THF)]2 (2), 2240706 (DIPePBDI*)Mg(μ2,μ4-naphthalene)Ca(DIPePBDI) (4), 2240707 (DIPePBDI*)Mg(μ2,μ4-anthracene)Ca(DIPePBDI) (5), 2240708 (DIPePBDI)Ca(μ4,μ4-naphthalene)Ca(DIPePBDI) (6), 2240709 (DIPePBDI)Ca(μ6,μ6-anthracene)Ca(DIPePBDI) (7).Data availability
Crystallographic data has been deposited with the Cambridge Structural Database.Author contributions
J. Mai: conceptualization, investigation, validation, formal analysis, writing – original draft, visualization. B. Rösch: investigation, validation, formal analysis. N. Patel: investigation, validation, formal analysis. J. Langer: formal analysis, validation. Sjoerd Harder: conceptualization, writing – original draft – review and editing, visualization, validation, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Mrs A. Roth (University of Erlangen-Nürnberg) for CHN analyses and J. Schmidt and Dr C. Färber (University of Erlangen-Nürnberg) for assistance with the NMR analyses. We acknowledge the Deutsche Forschungsgemeinschaft (DFG) for funding this project (HA 3218/11-1). Dr Neha Patel is grateful for funding from the European Union's Horizon 2020 research and innovation programme as a Marie Skłodowska-Curie post-doctoral fellow on the NITRO-EARTH project (101064276).References
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- S. Krieck, L. Yu, M. Reiher and M. Westerhausen, Eur. J. Inorg. Chem., 2010, 197–216 CrossRef CAS.
- A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659–5672 RSC.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- L. A. Freeman, J. E. Walley and R. J. Gilliard Jr, Nat. Synth., 2022, 1, 439–448 CrossRef.
- S. Krieck, H. Görls, L. Yu, M. Reiher and M. Westerhausen, J. Am. Chem. Soc., 2009, 131, 2977–2985 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed.
- G. Wang, J. E. Walley, D. A. Dickie, S. Pan, G. Frenking and R. J. Gilliard, J. Am. Chem. Soc., 2020, 142, 4560–4564 CrossRef CAS PubMed.
- W. Huang and P. L. Diaconescu, Dalton Trans., 2015, 44, 15360–15371 RSC.
- D. Jędrzkiewicz, J. Mai, J. Langer, Z. Mathe, N. Patel, S. DeBeer and S. Harder, Angew. Chem., Int. Ed., 2022, 61, e202200511 CrossRef PubMed.
- M. Gimferrer, S. Danes, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2022, 13, 6583–6591 RSC.
- S. Pan and G. Frenking, Chem. Sci., 2023, 14, 379–383 RSC.
- M. Gimferrer, S. Danes, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2023, 14, 384–392 RSC.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem.–Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef PubMed.
- B. Maitland, A. Stasch and C. Jones, Aust. J. Chem., 2022, 75, 543–548 CrossRef CAS.
- R. Mondal, K. Yuvaraj, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2022, 58, 12665–12668 RSC.
- Y. Liu, K. Zhu, L. Chen, S. Liu and W. Ren, Inorg. Chem., 2022, 61, 20373–20384 CrossRef CAS PubMed.
- T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 607–611 CrossRef CAS PubMed.
- J. Mai, M. Morasch, D. Jędrzkiewicz, J. Langer, B. Rösch and S. Harder, Angew. Chem., Int. Ed., 2023, 62, e202212463 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–721 CrossRef PubMed.
- L. R. Murphy, T. L. Meek, A. L. Allred and L. C. Allen, J. Phys. Chem. A, 2000, 104, 5867–5871 CrossRef CAS.
- T. Clark, J. Am. Chem. Soc., 1988, 110, 1672–1678 CrossRef CAS.
- P. Banerjee and P. K. Nandi, Phys. Chem. Chem. Phys., 2016, 18, 12505–12520 RSC.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079–9083 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Eyselein, A. Friedrich, J. Langer and S. Harder, Chem. Commun., 2020, 56, 11402–11405 RSC.
- J. A. Platts, J. Overgaard, C. Jones, B. B. Iversen and A. Stasch, J. Phys. Chem. A, 2011, 115, 194–200 CrossRef CAS PubMed.
- J. Overgaard, C. Jones, A. Stasch and B. B. Iversen, J. Am. Chem. Soc., 2009, 131, 4208–4209 CrossRef CAS PubMed.
- I. R. Speight, S. C. Chmely, T. P. Hanusa and A. L. Rheingold, Chem. Commun., 2019, 55, 2202–2205 RSC.
- D. Jędrzkiewicz, J. Langer and S. Harder, Z. Anorg. Allg. Chem., 2022, 648, e202200138 CrossRef.
- D. D. L. Jones, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 7087–7092 CrossRef CAS PubMed.
- M. Garçon, A. J. P. White and M. R. Crimmin, Chem. Commun., 2018, 54, 12326–12328 RSC.
- J. Pahl, S. Brand, H. Elsen and S. Harder, Chem. Commun., 2018, 54, 8685–8688 RSC.
- L. Garcia, M. D. Anker, M. F. Mahon, L. Maron and M. S. Hill, Dalton Trans., 2018, 47, 12684–12693 RSC.
- J. Pahl, A. Friedrich, H. Elsen and S. Harder, Organometallics, 2018, 37, 2901–2909 CrossRef CAS.
- K. Thum, J. Martin, H. Elsen, J. Eyselein, L. Stiegler, J. Langer and S. Harder, Eur. J. Inorg. Chem., 2021, 2643–2653 CrossRef CAS.
- K. Thum, A. Friedrich, J. Pahl, H. Elsen, J. Langer and S. Harder, Chem.–Eur. J., 2021, 27, 2513–2522 CrossRef CAS PubMed.
- L. M. Engelhardt, S. Harvey, C. L. Raston and A. H. White, J. Organomet. Chem., 1988, 341, 39–51 CrossRef CAS.
- C. Jones, L. McDyre, D. M. Murphy and A. Stasch, Chem. Commun., 2010, 46, 1511–1513 RSC.
- J. J. Brooks, W. Rhine and G. D. Stucky, J. Am. Chem. Soc., 1972, 94, 7346–7351 CrossRef CAS.
- T. X. Gentner, B. Rösch, K. Thum, J. Langer, G. Ballmann, J. Pahl, W. A. Donaubauer, F. Hampel and S. Harder, Organometallics, 2019, 38, 2485–2493 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Doug Stephan on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic details including ORTEP plots, 1H and 13C NMR spectra, details for the DFT calculations including XYZ-files. CCDC 2240705–2240709. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00909b |
| This journal is © The Royal Society of Chemistry 2023 |