
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free diboration and silaboration of alkenes and alkynes using bis(9-heterofluorenyl)s†
Jannik
Gilmer
ab,
Timo
Trageser
a,
Luis
Čaić
a,
Alexander
Virovets
 a,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a,
Felipe
Fantuzzi
*b and
Matthias
Wagner
*a
a,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a,
Felipe
Fantuzzi
*b and
Matthias
Wagner
*a
aInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, D-60438 Frankfurt am Main, Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
bSchool of Chemistry and Forensic Science, University of Kent, Park Wood Rd, Canterbury CT2 7NH, UK. E-mail: f.fantuzzi@kent.ac.uk
First published on 31st March 2023
Abstract
Diboration and silaboration reactions are prominent tools to introduce valuable functional groups into organic substrates. To date, most diboranes(4) and silylboranes used for this purpose are electronically and/or kinetically stabilized and require activation by a catalyst. We show here that the tetraaryl (μ-hydrido)diborane(4) anion [3]− and the silyl (hydrido)borate ([4]−)/Me3SiBr system react spontaneously with the archetypal olefin ethylene in the absence of a catalyst. The actual active species in both cases are the valence isoelectronic intermediates [FluB–B(H)Flu]− ([1]−) and FluB–Si(H)Flu (2), which consist of two 9-heterofluorenyl halves that get attached to the 1 and 2 positions of ethylene. At room temperature, [1]− is present in a dynamic equilibrium with its isolable isomer [3]−, while 2 has to be released in situ at low temperatures by H− abstraction from [4]−. Quantum-chemical calculations show qualitatively identical reaction mechanisms for [1]− and 2. Since the reactions start with π coordination of the ethylene molecule to a vacant B(pz) orbital, the high Lewis acidity and low steric hindrance of the 9-borafluorenyl fragments are the keys to success. As the reaction proceeds, back-donation from the B–E bond into the ethylene π* orbital becomes increasingly important (E = B, Si). The scope of the reactions has been extended to tBu(H)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and tBuC
CH2 and tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH on the one hand and FluB–Si(Cl)Flu as well as FluB–Si(Cl)Ph2 on the other.
CH on the one hand and FluB–Si(Cl)Flu as well as FluB–Si(Cl)Ph2 on the other.
Introduction
Hydroborations are addition reactions of an H–BR2 bond to alkenes or alkynes.1 The resulting organoboranes are versatile precursors for further functionalization or C–C coupling. The ease with which hydroboration reactions proceed depends critically on the Lewis acidity of the hydroborane and, in turn, on the nature of its R substituents: while the dialkyl borane 9-borabicyclo[3.3.1]nonane (9-BBN) smoothly adds to unsaturated organic substrates already at RT,2 significantly elevated temperatures are required for hydroborations with the less Lewis acidic catecholborane (HBcat) or pinacolborane (HBpin).1c As a consequence, considerable research efforts have been devoted to the development of efficient hydroboration catalysts, such as [ClRh(PPh3)3] (Wilkinson's complex).1,3Switching from hydroboration to diboration doubles the number of boryl groups that can be introduced in a single step. However, this advantage comes at a price because strongly Lewis acidic diboranes(4) such as B2Cl4, which spontaneously adds to ethylene or acetylene,4 are difficult to prepare and impractical to handle.5 Thus, the most commonly used diboranes(4) today are the commercially available compounds B2cat2 and B2pin2, which routinely require activation by catalysts (Scheme 1).6 These can be as diverse as noble metal complexes7 and plain alkoxide ions.8 The latter act through tetracoordination of one B center to form B(sp2)–B(sp3) diboranes with strongly polarized B–B bonds.9
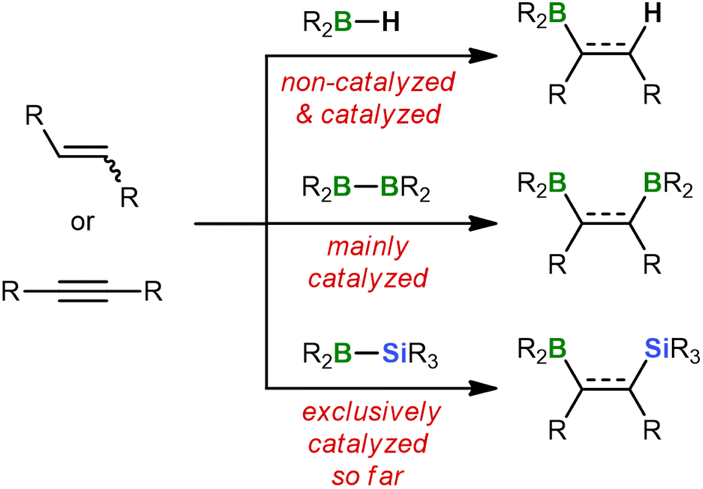 | ||
| Scheme 1 General schemes for hydroboration, diboration, and silaboration reactions on alkenes and alkynes. | ||
Diborations typically provide products containing two identical functionalizable groups that are afterwards difficult to address individually. To remedy this deficiency, silaboration reactions have been developed during which two chemically distinct substituents of largely orthogonal reactivity are introduced – a boryl and a silyl group.10 Similar to the B–B bond in B2pin2, the B–Si bonds of most silylboranes are rather inert and consequently need to be activated by transition-metal complexes or nucleophiles.6c,11,12 This is because the vast majority of available silylboranes carry electronically tamed or kinetically protected boryl moieties like Bpin,10a Bcat,13 or BMes2.14 As a result, boron tetracoordination by pristine alkenes or alkynes hardly takes place, which explains why these substrates are unable to activate the B–Si bonds on their own.
Can an unprecedented catalyst-free silaboration be achieved by purposefully increasing the Lewis acidity of the boryl residue while decreasing its steric demand? For the following reasons, we relied on our recently reported borafluorenyl derivative 2 to answer this question (Fig. 1a):15 (i) the B center of 2 is part of a formally antiaromatic borole ring and therefore a particularly good electron acceptor, and (ii) it is exposed to its environment due to the planarized 2,2′-biphenylylene ligand. Moreover, the silafluorenyl part of 2 renders it a structural analog of corresponding bis(9-borafluorenyl) anions [1]− (Fig. 1a), which we have already investigated in different contexts in the recent past.16 The [1]−/2 pair now offers the unique opportunity to compare diborations and silaborations on the basis of two compounds that could not be any more alike. The results of our combined experimental and theoretical study are reported herein – among them the first spontaneous 1,2-addition of a silylborane to a substrate as simple as ethylene.
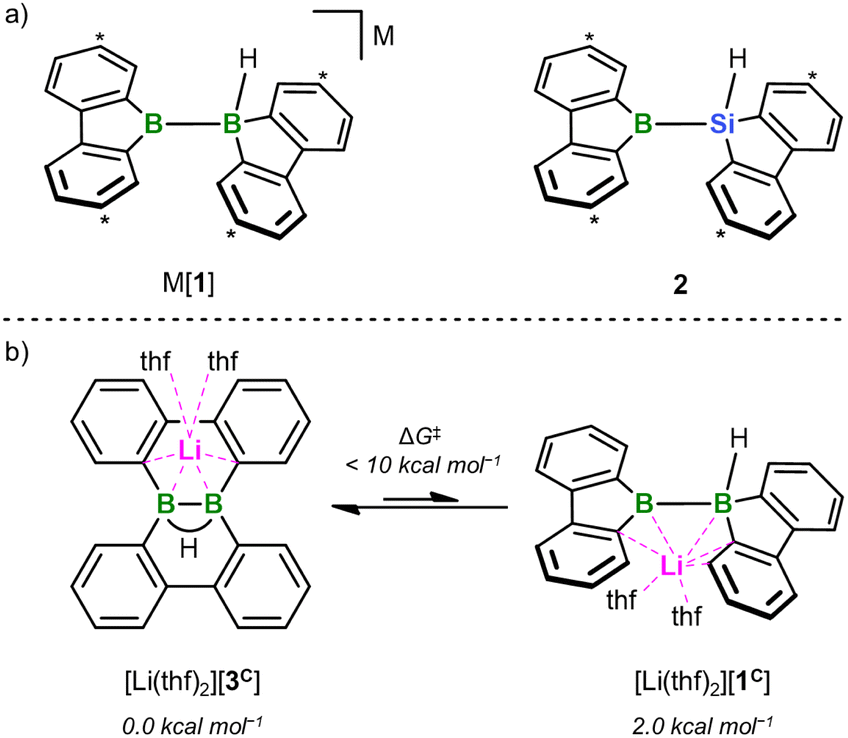 | ||
| Fig. 1 (a) The two valence isoelectronic key intermediates M[1] and 2 of the catalyst-free diboration and silaboration reactions, respectively (M = Li, K). M[1] is in a dynamic equilibrium with its isolable isomer M[3]; 2 forms in situ through thf-ligand dissociation from the NMR-detectable adduct 2(thf) (ΔGdiss = 10.5 kcal mol−1). Carbon atoms marked with “*” bear tBu groups. (b) DFT calculations (SMD(THF)/ωB97XD/def2-QZVPP//SMD(THF)/ωB97XD/6-31+G**) predict that [Li(thf)2][1C] is energetically less favorable by 2.0 kcal mol−1 than [Li(thf)2][3C] (ΔG‡ < 10 kcal mol−1). Compound numbers with a superscript “C” indicate calculated structures for which the Ar-bonded tBu groups have been replaced by H atoms. For clarity, schematic representations of the calculated structures are shown; full graphical representations are included in the ESI.† | ||
Results and discussion
All reactions and NMR measurements were carried out in THF or THF-d8; all DFT calculations were performed at the SMD(THF)/ωB97XD/def2-QZVPP//SMD(THF)/ωB97XD/6-31+G** level of theory. Throughout this work, calculated structures XC differ from the corresponding experimental structures X in that all aryl-bonded tBu groups are replaced by H atoms, in order to reduce CPU time and the number of potential conformers.Since the immediate, valence isoelectronic starting materials [1]− and 2 are not (yet) available, we first required suitable synthesis equivalents that are either in equilibrium with [1]−/2 or capable of releasing the actual reactive species in situ. An appropriate synthesis equivalent for M[1] should be its isolable isomer M[3] (M = Li, K; Scheme 2), because M[3] behaved as if it were M[1] already in previous reactivity studies.16d,f Moreover, quantum-chemical calculations indicate that (i) [Li(thf)2][3C] is only 2.0 kcal mol−1 more stable than [Li(thf)2][1C] and (ii) the rearrangement [3C]− → [1C]− is associated with barriers <10 kcal mol−1 (Fig. 1b; more details in Scheme S2†).17 On the BSi side, the thf adduct 2(thf) of the elusive 2 can straightforwardly be generated from Na[4] through H−-abstraction with Me3SiBr at low temperatures (Scheme 2; 2C(thf) → 2C + THF; ΔGdiss = 10.5 kcal mol−1).15,17
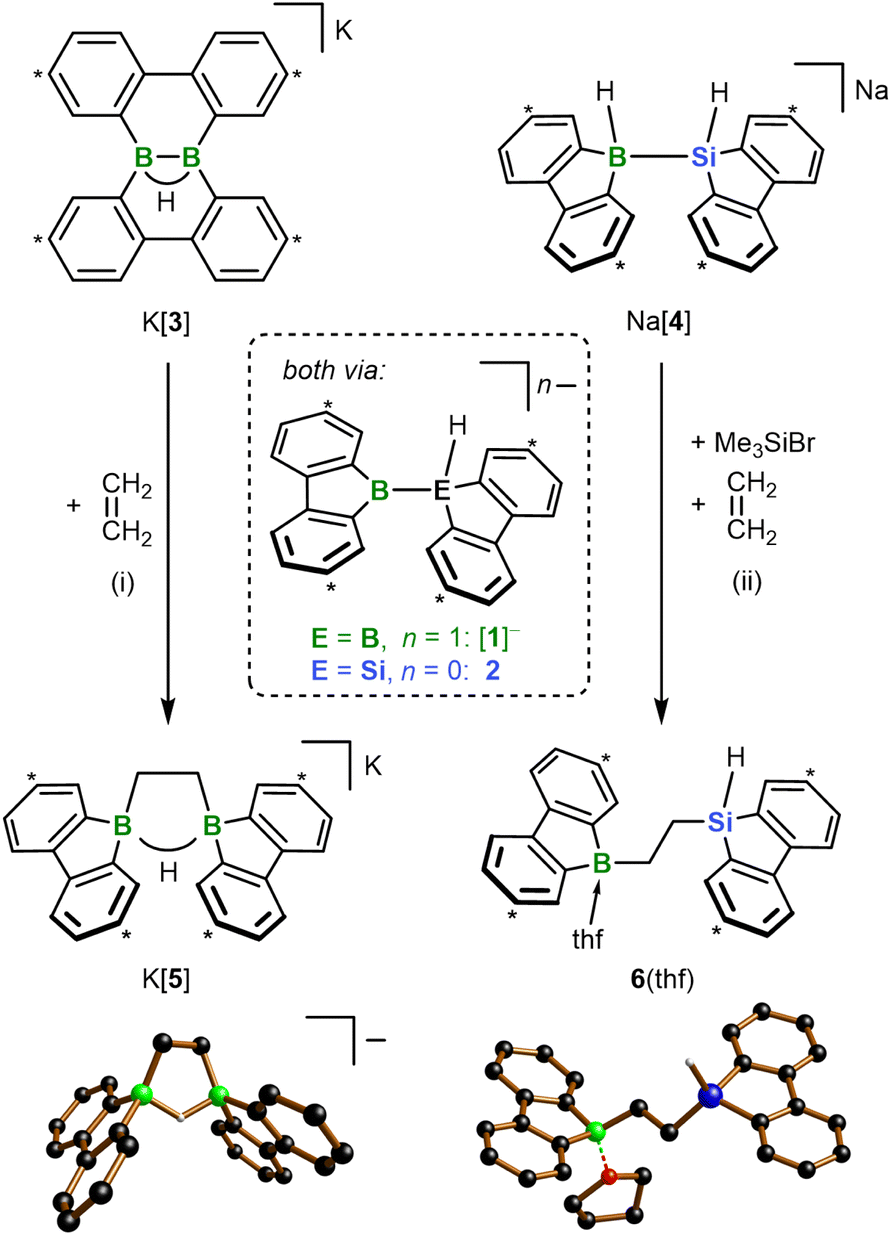 | ||
| Scheme 2 Diboration and silaboration of ethylene, starting from K[3] or Na[4]/Me3SiBr and furnishing K[5] and 6(thf), respectively. The reactions proceed via the valence isoelectronic intermediates [1]− and 2. (i) THF-d8, −196 °C to RT, 99% yield; (ii) THF, −78 °C to RT, 56% yield. Carbon atoms marked with “*” bear tBu groups. In the crystal-structure plots, the [K(thf)2]+ counter cation of [5]−, C-bonded H atoms, and Ar-bonded tBu groups are omitted for clarity; H: gray, B: green, C: black, O: red, Si: blue. | ||
According to our experience, the reactivity of anionic aryl boranes is often strongly influenced by the nature of their counter cations.15,18 We have therefore conducted all transformations involving the [1]− anion with both Li[3] and K[3]. Since in the present context the counter-cation dependence turned out to be marginal, we will henceforth refer only to the K+ salts, as these gave the qualitatively best crystallographic analyses (all results obtained with the Li+ salts are provided in the ESI†).
In a first experiment, we assessed the behavior of K[3] and the Na[4]/Me3SiBr system toward ethylene, the archetypal olefin. In both cases, the reaction was complete within minutes at T ≤ RT and furnished the 1,2-bis(9-borafluorenyl)ethane salt K[5] and the 1-(9-silafluorenyl)-2-(9-borafluorenyl)ethane 6(thf), respectively (Scheme 2). While the formation of K[5] was quantitative (99% yield), a somewhat lower product selectivity of 90% was determined by NMR in the case of 6(thf) (56% yield), which we attribute to issues associated with the in situ release of 2. The 1H NMR spectrum of K[5] shows a signal at 1.47 ppm (4H) assignable to the chemically equivalent protons of the 1,2-ethanediyl bridge. A severely broadened 13C resonance at 15.2 ppm is characteristic of B-bonded alkyl-C atoms.19 Only one tBuC6H3 signal set is observed in both the 1H and the 13C{1H} NMR spectrum, testifying to a high (average) symmetry of [5]− in solution. The computed 13C chemical shifts of [5]− deviate on average by <2 ppm from the experimentally determined values (a significantly larger mean deviation >4 ppm was determined for the computed 13C shifts of a hypothetical diboration product with a dibenzo[g,p]chrysene-type framework; cf. [3]−).17 The 1,2-bis(9-borafluorenyl)ethane structure of [K(thf)2][5] was ultimately proven by single-crystal X-ray diffraction (SCXD; Scheme 2). As sole discrepancy, the B⋯B-bridging H− ion of the twisted cyclic HB2C2 core, which was located in the difference Fourier map and freely refined, was not detectable even in the 1H{11B} NMR spectrum. Also, no doublet splitting was observable in the 11B NMR spectrum, although the resonance clearly appeared in the spectral region of tetracoordinated 11B nuclei (9.3 ppm).19 To confirm that these spectral peculiarities do not contradict our structural proposal, but are inherent to K[5], we first compared its 1H NMR data with those of the known FluB–CH2CH2–BFlu (BFlu = 2,7-di-tBu-9-borafluorenyl), a neutral analog of [5]− devoid of the B⋯B bridging H− ion.20 We found differences in the chemical shift values of corresponding protons of up to 0.5 ppm, but after addition of 1 equiv. Li[HBEt3] the spectra of the reaction mixture and of K[5] became essentially superimposable. Unlike K[5], the less symmetric 6(thf) leads to two different CH2 and tBuC6H3 signal sets in the 1H as well as the 13C{1H} NMR spectrum. Only one of the two 1,2-ethanediyl 13C resonances is broadened. The signal of the Si-bonded H atom appears as a triplet at 4.68 ppm (1H, 3JH,H = 3.5 Hz) and shows 29Si satellites (1JH,Si = 192 Hz). Its well-resolved shape points against a bonding interaction with the adjacent B atom in solution, even though B(μ-H)Si structural motifs are known.21 The pronounced shielding of the 11B nucleus in 6(thf) (13.6 ppm) is therefore likely due to thf coordination. In line with this, 1H NMR spectra (THF-d8) recorded on a sample of 6(thf) isolated from THF-h8 revealed the presence of 1 equiv. of THF-h8. The solid-state structure of 6(thf) features a terminal Si–H atom, a thf ligand attached to the B atom, and an approximate s-trans configuration about the 1,2-ethanediyl bridge (B–C–C–Si = 167°).
According to quantum-chemical calculations, the reaction mechanisms underlying the formation of K[5] and 6(thf) starting from K[1] and 2 are strikingly similar (Scheme 3): first, ethylene interacts with [1C]− or 2Cvia mutually analogous transition states [TS1C]− (+11.4 kcal mol−1) and TS2C (+6.2 kcal mol−1), respectively. The fact that [TS1C]− is 5.2 kcal mol−1 more uphill agrees well with the comparatively lower Lewis acidity of [1C]− as judged from calculated F− affinities: [1C]− = 45 kcal mol−1vs.2C = 70 kcal mol−1.17 The subsequent insertion of the C2 entity into the B–B or B–Si bond results in similar energy gains of −35.6 ([8C]−) and −36.5 kcal mol−1 (gauche-6C). In the case of gauche-6C formation, a connecting shallow minimum 7C can be located (−5.5 kcal mol−1), while this was not possible in the case of [8C]−, albeit similar structures are encountered in the course of intrinsic reaction coordinate (IRC) calculations. Mere rotation about a B–C/C–C bond leads from [8C]−/gauche-6C to [5C]−/anti-6Cvia [TS3C]−/TS4C (ΔG‡ = 7.8/5.2 kcal mol−1; Scheme 3b). All in all, the formation of the final products [5C]− and 6C(thf) is thermodynamically downhill by −25.4 and −30.4 kcal mol−1, respectively.22
 | ||
| Scheme 3 (a) Computed reaction mechanism for the diboration and silaboration of ethylene, starting from [1C]− and 2C and leading to the primary intermediates [8C]− and gauche-6C, respectively. (b) Conformational changes and subsequent adduct formations furnishing the final products [5C]− and 6C(thf). Compound numbers with a superscript “C” indicate calculated structures for which the Ar-bonded tBu groups have been replaced by H atoms. All energies in kcal mol−1 at the SMD(THF)/ωB97XD/def2-QZVPP//SMD(THF)/ωB97XD/6-31+G** level of theory. Prox, dist: proximal, distal with respect to the B⋯E core. | ||
For each of the two transition states [TS1C]−/TS2C underlying the actual ethylene activation, the distances between the two olefinic C atoms and the respective B atom differ by <0.1 Å. Moreover, no Cδ+Cδ− charge separation is observed in natural bond orbital (NBO) calculations, which supports the overall picture of symmetric side-on coordination.23 At the same time, the B–E bond polarization is inverted between the starting materials [1C]−/2C (ΔqNBO = q[B(sp2)] − q[E(sp3)] = 1.15/−0.59), indicating that the observed ethylene activations are not necessarily triggered by a specific B–E bond polarization.17 Thus, to further characterize the ethylene activation cascade, we calculated several intrinsic bond orbitals (IBOs) based on different structures obtained from IRCs along the reaction paths [TS1C]− → [8C]− and TS2C → gauche-6C.24 These IBOs (selected ones are depicted in Fig. 2, the full set is shown in the ESI†) can be interpreted for both mechanisms in the same fashion as follows: initially, the main interaction takes place between the filled ethylene π orbital and the vacant B(pz) orbital. As the olefin progresses closer, the filled B–E σ orbital interacts increasingly more strongly with the π* orbital of the ethylene. This back-bonding is symmetry-allowed because the nodal plane of the π* orbital finds its counterpart in the nodal plane of the B–E σ orbital that passes through the B atom (yellow and red lobes in Fig. 2). Such concerted type of activation is also found for singlet carbenes25 and fits with our previously reported view on [1]− as 9-borafluorene-stabilized boryl anion.16d,f,20 At even later stages, the orbital structure underlying the back-bonding changes to a geometry reminiscent of a closed 2-electron-3-center (2e3c) bond between the proximal olefinic C atom and both heteroatoms (cf. structure 7C).26 Simultaneously, the former π contributions of the C2 fragment gradually merge into a newly formed B(sp2)–C σ orbital at the distal olefinic C atom. Finally, the 2e3c-bond orbital transforms to a pure E(sp3)–C σ-bond orbital.27
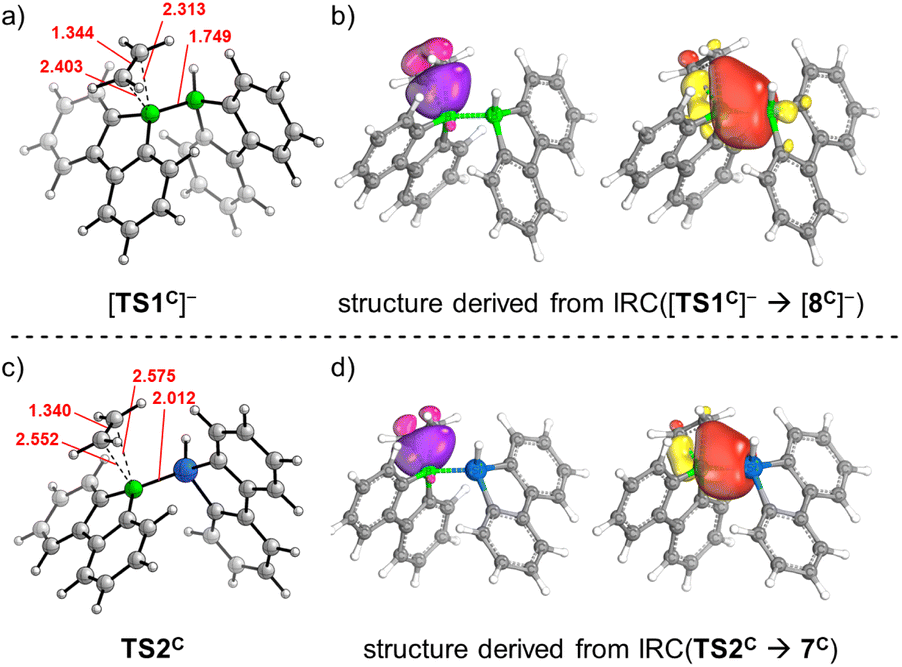 | ||
| Fig. 2 (a) Computed geometry of [TS1C]−. (b) Two IBOs observed on a structure at 2.582 bohr amu1/2 along the IRC([TS1C]− → [8C]−) path; IBO contributions: left: C(dist) = 0.77, C(prox) = 0.76, prev. B(sp2) = 0.43, others = 0.04; right: C(dist) = 0.08, C(prox) = 0.11, prev. B(sp2) = 0.90, B(sp3) = 0.74, others = 0.17. (c) Computed geometry of TS2C. (d) Two IBOs observed on a structure at 15.076 bohr amu1/2 along the IRC(TS2C → 7C) path; IBO contributions: left: C(dist) = 0.76, C(prox) = 0.65, prev. B(sp2) = 0.55, others = 0.04; right: C(dist) = 0.10, C(prox) = 0.17, prev. B(sp2) = 0.92, Si(sp3) = 0.72, others = 0.08. Highlighted in red are selected bond lengths and atom⋯atom distances (in Å). Prox, dist: proximal, distal with respect to the B⋯E core; prev. = previously. | ||
With the aim of replacing the in situ generated 2 with an isolable analog of precisely controllable composition and purity, we switched from the Na[4]/Me3SiBr system to the chlorosilane 9(thf) (Scheme 4).15 As further asset, nucleophilic Cl-substitution reactions on the chlorosilyl-containing products could be included, in addition to hydrosilylation reactions, in the pool of possible late-stage derivatizations. Similar to 2, treatment of 9(thf) with ethylene under ambient conditions resulted in 1,2-silaboration within minutes to afford 10(thf).28 As anticipated, the product selectivity was enhanced so that 10(thf) could be harvested in 96% yield. Given this background, we used 9(thf) as preferred starting material for subsequent silaboration reactions.
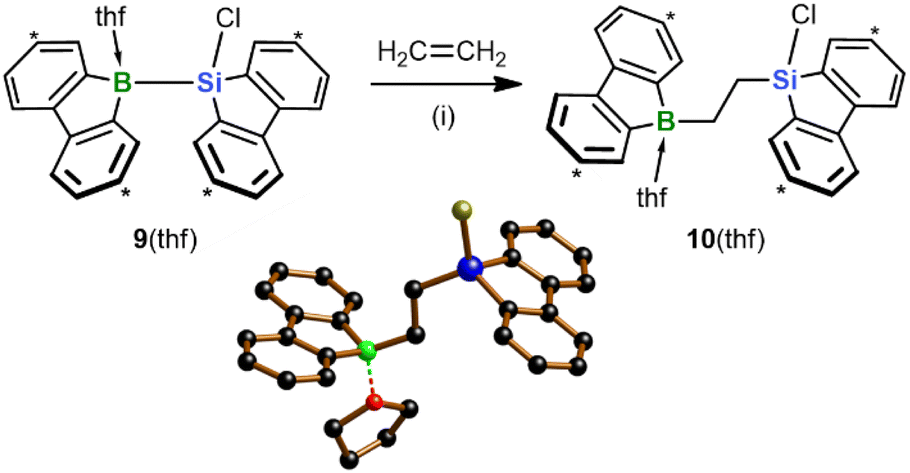 | ||
| Scheme 4 Silaboration of ethylene, starting from the chlorosilane 9(thf) and furnishing 10(thf). Carbon atoms marked with “*” bear tBu groups. In the crystal-structure plot, C-bonded H atoms and Ar-bonded tBu groups are omitted for clarity; B: green, C: black, O: red, Si: blue, Cl: yellow. (i) THF-d8, −196 °C to RT, 96% yield. | ||
In previous works, we had prepared bis(9-borafluorenyl)methane anions of the form [12]− (Scheme 5a) through twofold hydroboration of tBuCCH, followed by the addition of the H− donor [HBEt3]−.20,29 Since these species can also be regarded as formal 1,1-diboration products of 3,3-dimethyl-but-1-ene (tBu(H)CCH2), we next addressed the question of whether the 1,2-diboration/1,2-silaboration reaction of ethylene with K[3]/9(thf) can be shifted toward a 1,1-addition scenario by mere introduction of a bulky tBu group into the olefin (Scheme 5a). For the diboration reaction we found that the diborylethane K[11] is formed together with its diborylmethane isomer K[12] in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (RT, quantitative conversion). Silaboration of tBu(H)CCH2 gave exclusively the 1,2-addition product 13, with the tBu group adjacent to the boryl substituent. Upon heating to 50 °C for 5 d, K[11] underwent quantitative rearrangement to K[12];30 consistent with this, [12C]− is predicted to be thermodynamically more stable than [11C]− by 6.0 kcal mol−1. 13 remained inert at 50 °C, although its 1-boryl-1-silyl-isomer 14 (Scheme 5a) should also be thermodynamically favored by 5.5 kcal mol−1 (cf. 13Cvs.14C). Since rearrangement cannot be enforced by applying higher temperatures (prolonged heating of 13 to 100 °C results in decomposition), we alternatively resorted to the successive hydrosilylation/hydroboration of tBuCCH to prove that a stable 14 exists in principle (Scheme 5c).31 Given the established access routes to [12]− and 14via hydroboration of 9-vinyl-9-heterofluorenes, it is reasonable to propose that the [11]− → [12]− isomerization can proceed along a dehydroboration/rehydroboration sequence. Assuming an analogous scenario for a putative 13 → 14 rearrangement, either the transition state associated with the dehydroboration step is too high to be overcome at T < 100 °C or additional high-temperature reaction channels open after dehydroboration. As the most obvious distinguishing feature, [11]− generates four sets of tBuC6H3 signals and [12]− only two, in both the 1H and 13C{1H} NMR spectrum. Due to the stereogenic center in [11]−, the protons of the 1,2-ethylenediyl group represent an AMX spin system, whereas the tBuH2C–CH linker of [12]− has an A2X spin system. In [11]−, both BC−CB 13C resonances are severely broadened; in [12]−, this applies solely to the CB2 signal. At RT, all NMR signals of the 9-borafluorenyl group of 13 are broadened (almost) beyond detection, which we attribute to a coalescence phenomenon induced by rotation of the substituent about the B–C bond. Indeed, cooling of the sample to T = −30 °C results in the expected four tBuC6H3 signal sets. The chemical shift values of the two 13C sets corresponding to the 9-borafluorenyl moiety show characteristic deviations from those of 6(thf) and similarities to those of tricoordinate 9-bromo-9-borafluorene,32 indicating the absence of a B-coordinating thf ligand or of a B⋯Si-bridging Cl atom. The coalescence temperature of 9-borafluorenyl rotation decreases from RT to approximately −30 °C upon going from 13 to 14, pointing to the tBu group as major origin of rotational hindrance. Thus, the room-temperature 1H NMR spectrum of 14 shows three tBuC6H3 signal sets, one of them with double intensity. Gas-phase diffusion of n-hexane into a THF solution of K[11]/K[12], that had never been exposed to temperatures above RT, resulted in the growth of co-crystals of composition [K(thf)2]{[11]0.73[12]0.27}. Almost all atoms of both species coincide in the crystal lattice except for their {B2(μ-H)(μ-CH2CHtBu)} and {B2(μ-H)(μ-CHCH2tBu)} fragments, which overlap partly (shown in blue and red, respectively, in Scheme 5b). From this superposition it becomes obvious that [12]− is the sterically less encumbered isomer. The solid-state structure of 13 confirms the 1,2-addition mode of the silaboration reaction as well as its proposed regioselectivity. Furthermore, the SCXD structure of free 13 bears striking resemblance with the computed intermediate gauche-6C (Scheme 3), in line with the proposition that the initially formed gauche-6C is favored over anti-6C as long as no thf coordination occurs.
:1 ratio (RT, quantitative conversion). Silaboration of tBu(H)CCH2 gave exclusively the 1,2-addition product 13, with the tBu group adjacent to the boryl substituent. Upon heating to 50 °C for 5 d, K[11] underwent quantitative rearrangement to K[12];30 consistent with this, [12C]− is predicted to be thermodynamically more stable than [11C]− by 6.0 kcal mol−1. 13 remained inert at 50 °C, although its 1-boryl-1-silyl-isomer 14 (Scheme 5a) should also be thermodynamically favored by 5.5 kcal mol−1 (cf. 13Cvs.14C). Since rearrangement cannot be enforced by applying higher temperatures (prolonged heating of 13 to 100 °C results in decomposition), we alternatively resorted to the successive hydrosilylation/hydroboration of tBuCCH to prove that a stable 14 exists in principle (Scheme 5c).31 Given the established access routes to [12]− and 14via hydroboration of 9-vinyl-9-heterofluorenes, it is reasonable to propose that the [11]− → [12]− isomerization can proceed along a dehydroboration/rehydroboration sequence. Assuming an analogous scenario for a putative 13 → 14 rearrangement, either the transition state associated with the dehydroboration step is too high to be overcome at T < 100 °C or additional high-temperature reaction channels open after dehydroboration. As the most obvious distinguishing feature, [11]− generates four sets of tBuC6H3 signals and [12]− only two, in both the 1H and 13C{1H} NMR spectrum. Due to the stereogenic center in [11]−, the protons of the 1,2-ethylenediyl group represent an AMX spin system, whereas the tBuH2C–CH linker of [12]− has an A2X spin system. In [11]−, both BC−CB 13C resonances are severely broadened; in [12]−, this applies solely to the CB2 signal. At RT, all NMR signals of the 9-borafluorenyl group of 13 are broadened (almost) beyond detection, which we attribute to a coalescence phenomenon induced by rotation of the substituent about the B–C bond. Indeed, cooling of the sample to T = −30 °C results in the expected four tBuC6H3 signal sets. The chemical shift values of the two 13C sets corresponding to the 9-borafluorenyl moiety show characteristic deviations from those of 6(thf) and similarities to those of tricoordinate 9-bromo-9-borafluorene,32 indicating the absence of a B-coordinating thf ligand or of a B⋯Si-bridging Cl atom. The coalescence temperature of 9-borafluorenyl rotation decreases from RT to approximately −30 °C upon going from 13 to 14, pointing to the tBu group as major origin of rotational hindrance. Thus, the room-temperature 1H NMR spectrum of 14 shows three tBuC6H3 signal sets, one of them with double intensity. Gas-phase diffusion of n-hexane into a THF solution of K[11]/K[12], that had never been exposed to temperatures above RT, resulted in the growth of co-crystals of composition [K(thf)2]{[11]0.73[12]0.27}. Almost all atoms of both species coincide in the crystal lattice except for their {B2(μ-H)(μ-CH2CHtBu)} and {B2(μ-H)(μ-CHCH2tBu)} fragments, which overlap partly (shown in blue and red, respectively, in Scheme 5b). From this superposition it becomes obvious that [12]− is the sterically less encumbered isomer. The solid-state structure of 13 confirms the 1,2-addition mode of the silaboration reaction as well as its proposed regioselectivity. Furthermore, the SCXD structure of free 13 bears striking resemblance with the computed intermediate gauche-6C (Scheme 3), in line with the proposition that the initially formed gauche-6C is favored over anti-6C as long as no thf coordination occurs.
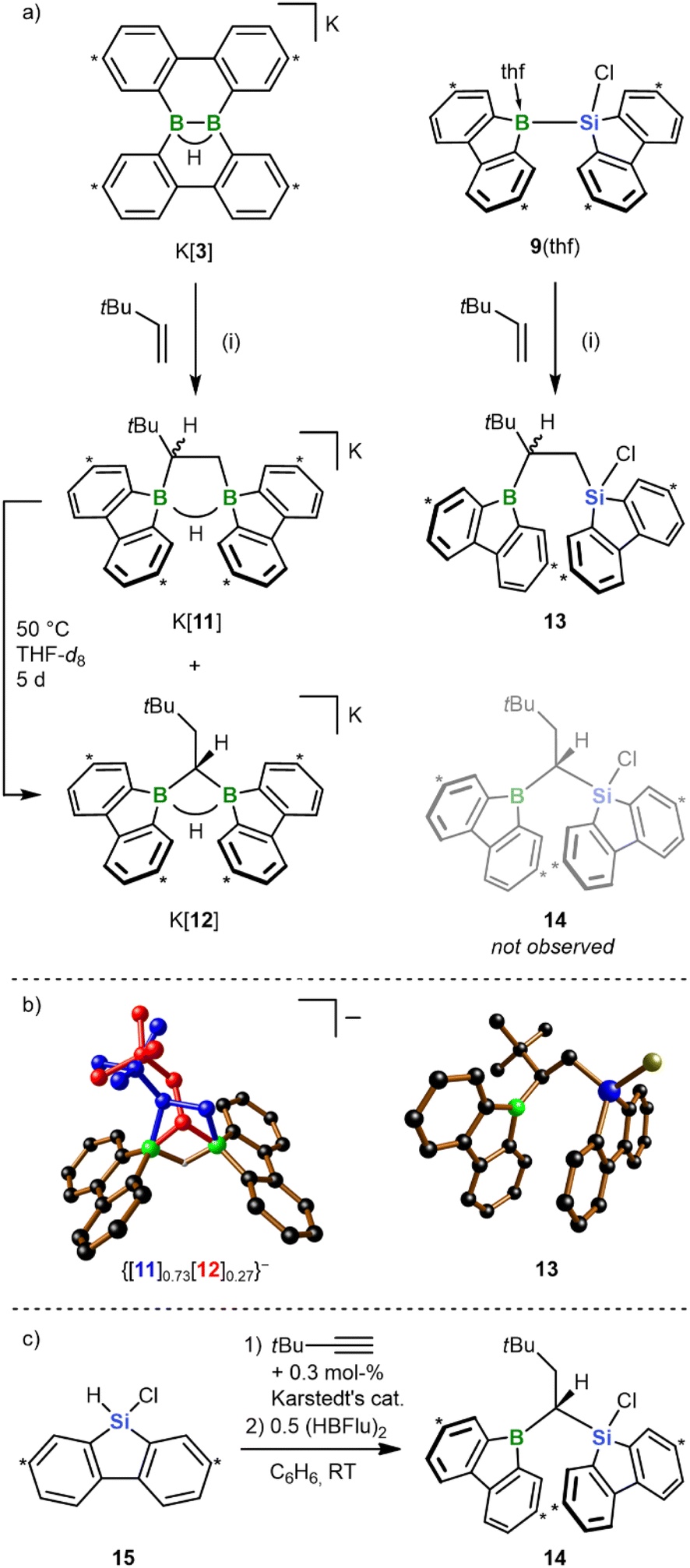 | ||
| Scheme 5 (a) Diboration and silaboration of tBu(H)CCH2, starting from K[3] or the chlorosilane 9(thf) and furnishing K[11]/K[12] (ratio 10:1) and 13, respectively. (i) THF-d8, RT, quant. conv. by NMR. Upon heating, the mixture K[11]/K[12] is quantitatively converted to the 1,1-diboration product K[12]. The putative 1,1-silaboration product 14 was not observed, but obtained via a different route (cf. (c)). (b) Solid-state structures of co-crystallized {[11]0.73[12]0.27}− and of 13. The [K(thf)2]+ counter cation of {[11]0.73[12]0.27}−, C-bonded H atoms, and Ar-bonded tBu groups are omitted for clarity; H: gray, B: green, C: black, Si: blue, Cl: yellow. (c) Synthesis of 14 from 15, tBuCCH, and (HBFlu)2via a hydrosilylation/hydroboration sequence [(HBFlu)2 = 2,7-di-tert-butyl-9H-9-borafluorene dimer]. Carbon atoms marked with “*” bear tBu groups. | ||
In view of the fact that the diborations/silaborations with K[3] and 9(thf) proceed uncatalyzed and with remarkable ease, the next question was: can the scope of the two reactions be extended even to alkynes? As a first trial, we treated both starting materials with tBuCCH and observed clean formation of the 1,2-cis-addition products K[16] and 17, respectively, within minutes at RT (Scheme 6). As in 13, the tBu group of 17 ends up next to the 9-borafluorenyl unit. These structural assignments are based on SCXD and (2D) NMR experiments. The configuration of 17 would be considered “abnormal” in most transition metal-catalyzed silaboration reactions of alkynes RCCH, where the silyl group is usually introduced adjacent to the R substituent.33 There, inverting this “normal” regioselectivity is hard to achieve and requires elaborate catalyst design.34 A notable NMR feature of ditopic organoborane K[16] is that both the B(μ-H)B and the CCH signals are split into well-resolved doublets in the 1H{11B} NMR spectrum due to pronounced 3JH,H coupling of 8.1 Hz.
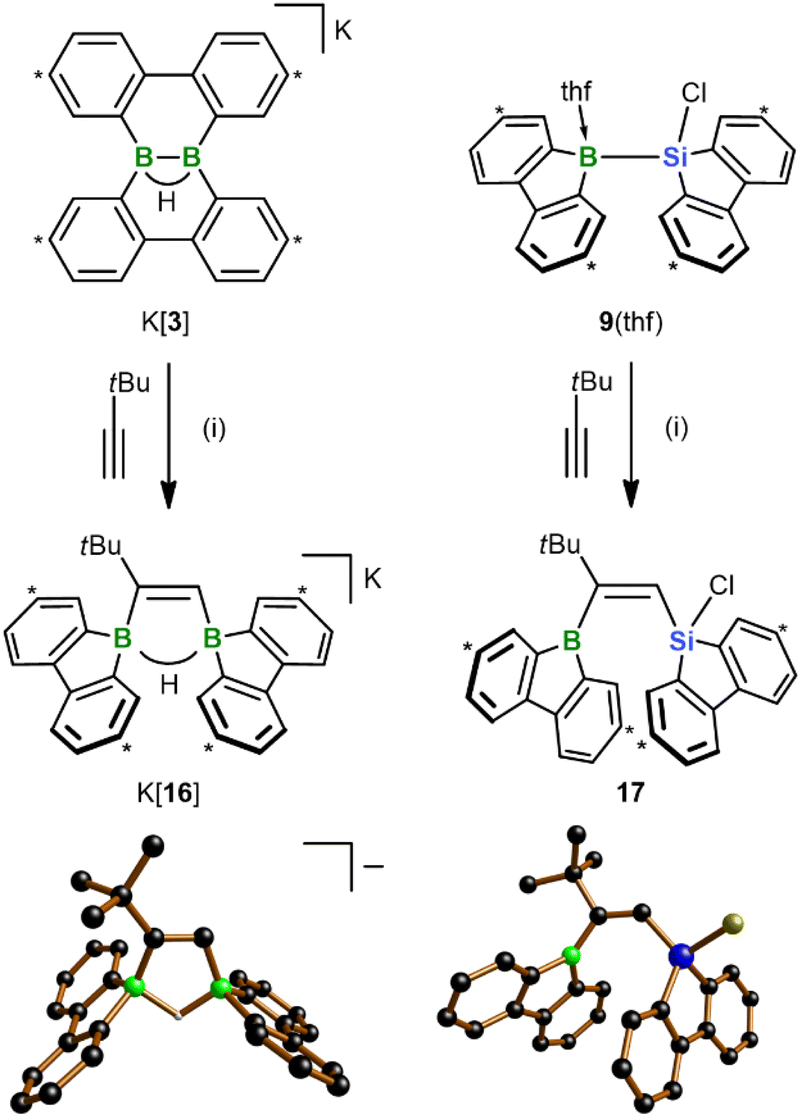 | ||
| Scheme 6 Diboration and silaboration of tBuCCH, starting from K[3] or the chlorosilane 9(thf) and furnishing K[16] and 17, respectively. Carbon atoms marked with “*” bear tBu groups. In the crystal-structure plots, the [K(thf)2]+ counter cation of [16]−, C-bonded H atoms, and Ar-bonded tBu groups are omitted for clarity; H: gray, B: green, C: black, Si: blue, Cl: yellow. (i) THF-d8, RT, quant. conv. by NMR. | ||
Finally, we investigated the extent to which our silaboration reaction depends on (i) the sterically undemanding planar framework of the 9-silafluorenyl fragment and (ii) an enhanced Lewis acidity of the Si center due to the compressed endocyclic C–Si–C angle.35 To this end, we formally cut the biphenyl backbone of the 9-silafluorenyl and switched to an Si(Cl)Ph2 substituent (cf. 18(thf); its synthesis is analogous to that of 9(thf) and described in the ESI;†Scheme 7). We found that the reaction of 18(thf) with ethylene and tBuCCH proceeds with similar ease and selectivity as in the case of 9(thf) to give the final products 19(thf) and 20 (see the ESI for the SCXD results†). We conclude that neither of the factors (i) and (ii) is critical for the reaction to proceed. In addition to this fundamental insight, an advantage of 18(thf) in terms of applications is the easier availability of the starting material Cl(H)SiPh2 compared to Cl(H)SiFlu (15).15
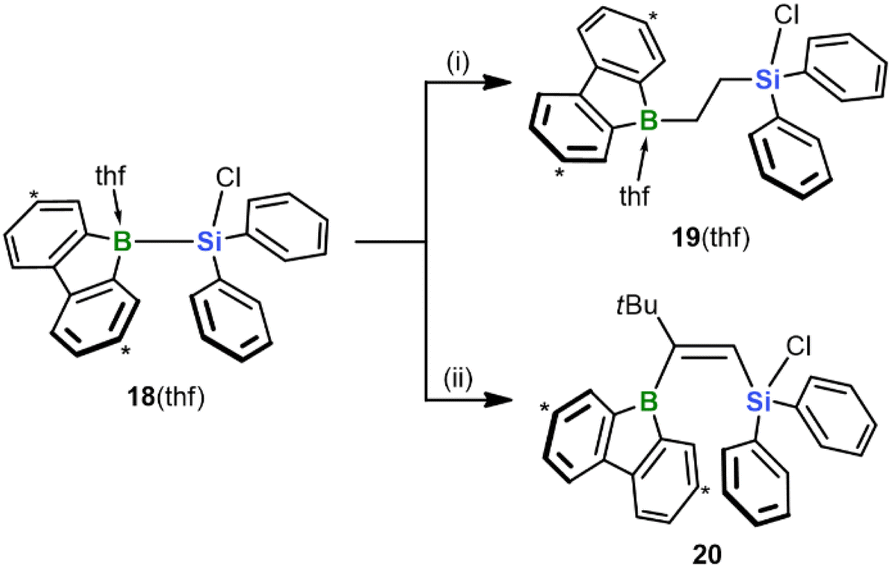 | ||
| Scheme 7 Silaboration of ethylene or tBuCCH, starting from diphenylsilane 18(thf) and furnishing 19(thf) or 20, respectively. Carbon atoms marked with “*” bear tBu groups. (i) exc. ethylene, THF-d8, −196 °C to RT, quant. conv. by NMR; (ii) 1 equiv. tBuCCH, THF-d8, RT, quant. conv. by NMR. | ||
Conclusions
We have successfully employed a pair of otherwise isostructural B–B- and B–Si-bonded compounds ([1]− and 2) to compare diboration and silaboration reactions under essentially identical steric conditions. Irrespective of their different charge, both molecules undergo spontaneous 1,2-addition to CC double and CC triple bonds without the need for a catalyst. Such reactivity is rare for electron-precise diboranes and unprecedented for silylboranes. We attribute it to the high Lewis acidity and the low steric shielding of the installed planar borafluorenyl units. In the case of the substrate tBu(H)CCH2, also the 1,1-diboration product [12]− was observed at RT, albeit to a small degree of less than 10%. Upon heating to 50 °C, the corresponding 1,2-diboration product [11]− quantitatively rearranges to [12]−. We never encountered an analogous 1,1-silaboration product – not even at elevated temperatures. Using quantum-chemical calculations, we developed a mechanistic proposal that is consistent with all experimental facts. The crucial activation step occurs via simultaneous (i) coordination of a filled substrate π orbital to the empty B(pz) orbital and (ii) back-bonding from the filled B–E σ orbital to the substrate π* orbital (E = B, Si). Apart from the insights into catalyst-free diboration and silaboration reactions gained from the studies on our diboranes and silylboranes, the products obtained are promising precursors for future syntheses of B,Si-doped PAHs.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
J. G. performed the experimental studies, characterized all new compounds, and performed the quantum-chemical calculations. T. T. optimized the synthesis of M[5] and performed the syntheses of M[11] as well as M[12]. L. C. assisted with the synthesis of 14. M. B. performed the X-ray crystal structure analyses of the compounds 9(thf), [K(thf)2]{[11]0.73[12]0.27}, [Li(thf)4][12], 14, 20. A. V. performed the X-ray crystal structure analyses of the compounds [K(thf)2][5], 6(thf), 13, [K(thf)2][16], and 17, and further enhanced the structures [K(thf)2]{[11]0.73[12]0.27}, [Li(thf)4][12], and 20. H. W. L., F. F., and M. W. supervised the project. The manuscript was written by J. G. and M. W. and edited by all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. G. thanks the German Academic Exchange Service for a research grant. We thank the center for scientific computing (CSC) Frankfurt and the University of Kent for providing HPC resources that contributed to the computational investigation of this work.Notes and references
- (a) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS; (b) I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS; (c) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS; (d) S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877–8922 RSC.
- H. C. Brown and J. C. Chen, J. Org. Chem., 1981, 46, 3978–3988 CrossRef CAS.
- D. Männig and H. Nöth, Angew. Chem., Int. Ed., 1985, 24, 878–879 CrossRef.
- (a) G. Urry, J. Kerrigan, T. D. Parsons and H. I. Schlesinger, J. Am. Chem. Soc., 1954, 76, 5299–5301 CrossRef CAS; (b) P. Ceron, A. Finch, F. Frey, J. Kerrigan, T. Parsons, G. Urry and H. I. Schlesinger, J. Am. Chem. Soc., 1959, 81, 6368–6371 CrossRef CAS; (c) C. Pubill-Ulldemolins, E. Fernánez, C. Bo and J. M. Brown, Org. Biomol. Chem., 2015, 13, 9619–9628 RSC.
- Further selected examples of catalyst-free diboration reactions: (a) H. Klusik, C. Pues and A. Berndt, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1984, 39, 1042–1045 CrossRef; (b) S. Luckert, E. Eversheim, U. Englert, T. Wagner and P. Paetzold, Z. Anorg. Allg. Chem., 2001, 627, 1815–1823 CrossRef CAS; (c) C. Kojima, K.-H. Lee, Z. Lin and M. Yamashita, J. Am. Chem. Soc., 2016, 138, 6662–6669 CrossRef CAS PubMed; (d) J. Böhnke, H. Braunschweig, A. Deißenberger, T. Dellermann, R. D. Dewhurst, J. O. C. Jiménez-Halla, S. Kachel, H. Kelch and D. Prieschl, Chem. Commun., 2017, 53, 12132–12135 RSC.
- (a) T. B. Marder and N. C. Norman, Top. Catal., 1998, 5, 63–73 CrossRef CAS; (b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (c) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415–430 RSC; (d) S. Ding, L. Xu and Z. Miao, Molecules, 2019, 24, 1325 CrossRef PubMed; (e) L. Tendera, F. Fantuzzi, T. B. Marder and U. Radius, Chem. Sci., 2023, 14, 2215–2228 RSC.
- (a) T. Ishiyama, N. Matsuda, N. Miyaura and A. Suzuki, J. Am. Chem. Soc., 1993, 115, 11018–11019 CrossRef CAS; (b) R. T. Baker, P. Nguyen, T. B. Marder and S. A. Westcott, Angew. Chem., Int. Ed., 1995, 34, 1336–1338 CrossRef CAS.
- (a) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 CrossRef CAS PubMed; (b) N. Miralles, J. Cid, A. B. Cuenca, J. J. Carbó and E. Fernández, Chem. Commun., 2015, 51, 1693–1696 RSC.
- R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC.
- (a) M. Suginome, H. Nakamura and Y. Ito, Chem. Commun., 1996, 2777–2778 RSC; (b) M. Suginome, H. Nakamura and Y. Ito, Angew. Chem., Int. Ed., 1997, 36, 2516–2518 CrossRef CAS; (c) S. Onozawa, Y. Hatanaka and M. Tanaka, Chem. Commun., 1997, 1229–1230 RSC; (d) J. Carlos Araujo Da Silva, M. Birot, J.-P. Pillot and M. Pétraud, J. Organomet. Chem., 2002, 646, 179–190 CrossRef; (e) H. Ito, Y. Horita and E. Yamamoto, Chem. Commun., 2012, 48, 8006–8008 RSC; (f) E. Yamamoto, R. Shishido, T. Seki and H. Ito, Organometallics, 2017, 36, 3019–3022 CrossRef CAS; (g) Y. Morimasa, K. Kabasawa, T. Ohmura and M. Suginome, Asian J. Org. Chem., 2019, 8, 1092–1096 CrossRef CAS; (h) Y. Gu, Y. Duan, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 2061–2065 CrossRef CAS PubMed.
- (a) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef CAS PubMed; (b) C. Moberg, Synthesis, 2020, 52, 3129–3139 CrossRef CAS; (c) J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010–2073 RSC.
- Rare exceptions requiring particular substrates are reported in: (a) M. Suginome, T. Fukuda, H. Nakamura and Y. Ito, Organometallics, 2000, 19, 719–721 CrossRef CAS; (b) K. Oshima, T. Ohmura and M. Suginome, Chem. Commun., 2012, 48, 8571–8573 RSC; (c) M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 1970–1974 CrossRef CAS PubMed.
- M. Suginome, T. Matsuda, H. Nakamura and Y. Ito, Tetrahedron, 1999, 55, 8787–8800 CrossRef CAS.
- E. Bonnefon, M. Birot, J. Dunogues, J.-P. Pillot, C. Courseille and F. Taulelle, Main Group Met. Chem., 1996, 19, 761–767 CAS.
- J. Gilmer, M. Bolte, A. Virovets, H.-W. Lerner, F. Fantuzzi and M. Wagner, Chem.–Eur. J., 2023, 29, e202203119 CrossRef CAS PubMed.
- (a) T. Kaese, A. Hübner, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2016, 138, 6224–6233 CrossRef CAS PubMed; (b) T. Kaese, H. Budy, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2017, 56, 7546–7550 CrossRef CAS PubMed; (c) J. Gilmer, H. Budy, T. Kaese, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2020, 59, 5621–5625 CrossRef CAS PubMed; (d) T. Trageser, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2020, 59, 7726–7731 CrossRef CAS PubMed; (e) H. Budy, J. Gilmer, T. Trageser and M. Wagner, Eur. J. Inorg. Chem., 2020, 2020, 4148–4162 CrossRef CAS; (f) T. Trageser, D. Bebej, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2021, 60, 13500–13506 CrossRef CAS PubMed.
- See the ESI for more details†.
- (a) E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2019, 141, 6082–6091 CrossRef CAS PubMed; (b) H. Budy, T. Kaese, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2021, 60, 19397–19405 CrossRef CAS PubMed.
- H. Nöth and B. Wrackmeyer, Nuclear Magnetic Resonance Spectroscopy of Boron Compounds, in NMR Basic Principles and Progress, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer Verlag, Berlin, Heidelberg, New York, 1978 Search PubMed.
- T. Kaese, T. Trageser, H. Budy, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Sci., 2018, 9, 3881–3891 RSC.
- (a) B. Wrackmeyer, W. Milius and O. L. Tok, Chem.–Eur. J., 2003, 9, 4732–4738 CrossRef CAS PubMed; (b) H. Gao, R. Müller, E. Irran, H. F. T. Klare, M. Kaupp and M. Oestreich, Chem.–Eur. J., 2022, 28, e202104464 CAS.
- In addition to the diboration mechanism outlined in Scheme 3, we also considered alternative mechanisms starting from isomers of [3C]− other than [1C]−. All of these have considerably higher overall barriers and should therefore contribute less to the overall reaction scenario (see the ESI for further details†).
- For steric reasons, this is different to the proposed mechanism for the addition of propylene to the bulkier [pinB–B(OMe)pin]−, where initially only one of the two olefinic C atoms coordinates to the anionic diborane (ref. 8a).
- G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed.
- N. G. Rondan, K. N. Houk and R. A. Moss, J. Am. Chem. Soc., 1980, 102, 1770–1776 CrossRef CAS.
- S. P. Gnanasekar and E. Arunan, Aust. J. Chem., 2020, 73, 767–774 CrossRef CAS.
- The NBO calculations show a similar picture: The transition states [TS1C]−/TS2C are mainly stabilized by the interaction of the ethylene π orbital with the pz orbital of the respective sp2-hybridized borafluorenyl moiety (60.8/43.8 kcal mol−1). An interaction between the B–E σ orbital and the π* orbital of ethylene is small at these stages (8.2/1.9 kcal mol−1). However, the latter becomes increasingly pronounced as the reaction course progresses.
- Ohmura, Suginome, and co-workers reported a transition metal-catalyzed silaboration that also tolerates an SiCl function (ref. 34a).
- A. Hübner, Z.-W. Qu, U. Englert, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2011, 133, 4596–4609 CrossRef PubMed.
- Reaction of Li[3] with 3,3-dimethyl-but-1-ene at RT in C6D6 gives Li[12] with 70% selectivity, which precipitates from an n-hexane/C6D6 solution at −30 °C. Following this procedure, Li[12] can be isolated in higher purity compared to the thermal rearrangement protocol starting from M[11] (M = Li, K).
- We have confirmed that Karstedt’s catalyst does not promote the rearrangement 13 → 14.
- A. Hübner, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2012, 51, 12514–12518 CrossRef PubMed.
- Assuming that the alkyne insertion into the B–Si bond of 9(thf) proceeds via a transition state analogous to TS2C (cf.Scheme 3), we anticipate that the observed regioselectivity is due to steric effects: to avoid unfavorable interactions with the “three-dimensional” silyl group, the sterically demanding tBu substituent of the incoming alkyne must be oriented above the planar borafluorene moiety, which ultimately promotes the observed “abnormal” regioselectivity.
- (a) T. Ohmura, K. Oshima, H. Taniguchi and M. Suginome, J. Am. Chem. Soc., 2010, 132, 12194–12196 CrossRef CAS PubMed; (b) T. Ohmura, K. Oshima and M. Suginome, Angew. Chem., Int. Ed., 2011, 50, 12501–12504 CrossRef CAS PubMed; (c) K. Kojima, Y. Nagashima, C. Wang and M. Uchiyama, ChemPlusChem, 2019, 84, 277–280 CrossRef CAS PubMed; (d) M. Zhao, C.-C. Shan, Z.-L. Wang, C. Yang, Y. Fu and Y.-H. Xu, Org. Lett., 2019, 21, 6016–6020 CrossRef CAS PubMed.
- (a) A. H. J. F. de Keijzer, F. J. J. de Kanter, M. Schakel, R. F. Schmitz and G. W. Klumpp, Angew. Chem., Int. Ed., 1996, 35, 1127–1128 CrossRef CAS; (b) S. Deerenberg, M. Schakel, A. H. J. F. de Keijzer, M. Kranenburg, M. Lutz, A. L. Spek and K. Lammertsma, Chem. Commun., 2002, 4, 348–349 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2247211–2247221. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01395b |
| This journal is © The Royal Society of Chemistry 2023 |