
Sunlight-driven photocatalytic conversion of furfural and its derivatives
Qizhao
Zhang
,
Bang
Gu
 and
Wenhao
Fang
*
and
Wenhao
Fang
*
School of Chemical Science and Technology, Yunnan University, 2 North Cuihu Road, Kunming 650091, China. E-mail: wenhao.fang@ynu.edu.cn
First published on 17th April 2024
Abstract
Furfural (mainly furfural and 5-hydroxymethylfurfural) and its derivatives stand out as an important platform for C5 and C6 compounds from lignocellulosic biomass. Catalytic valorization of these renewable carbohydrates not only enables the manufacture of various high-valued chemicals but also enriches routes and theories for the direct use of crude biomass. Photocatalytic conversion of furfural and its derivatives using solar energy has recently received great attention. Such an environmentally friendly and energy-saving approach enables activation and transformation of the target chemical bonds under very mild reaction conditions using photoexcited charge carriers or photogenerated reactive species. However, highly selective photocatalysts are somewhat lacking and the reaction mechanisms usually remain obscure. Herein, this review article aims to systematically summarize the recent advances in sunlight-driven photocatalytic conversion of furfural and 5-hydroxymethylfurfural to sustainable building-block chemicals. In particular, the strategies of catalyst design and the unique roles of reactive species (i.e., photogenerated electrons and holes, hydroxyl and superoxide radicals, and singlet oxygen) are discussed. This allows insights into the catalytic mechanisms. In addition, the latest progress in the combination of such photooxidation with H2 evolution or CO2 reduction, as well as photo(electro)chemical conversion, is also reviewed. In addition to the common oxidation and reduction reactions, some nontraditional reaction paths (i.e., reductive coupling, oxidative decarboxylation and ring-opening) and promising products are introduced. Finally, challenges and future opportunities from the perspective of green chemistry are also analyzed. Therefore, a bright prospect can be foreseen in the near future for many more photocatalytic valorizations of furfural-like molecules.
1. Introduction
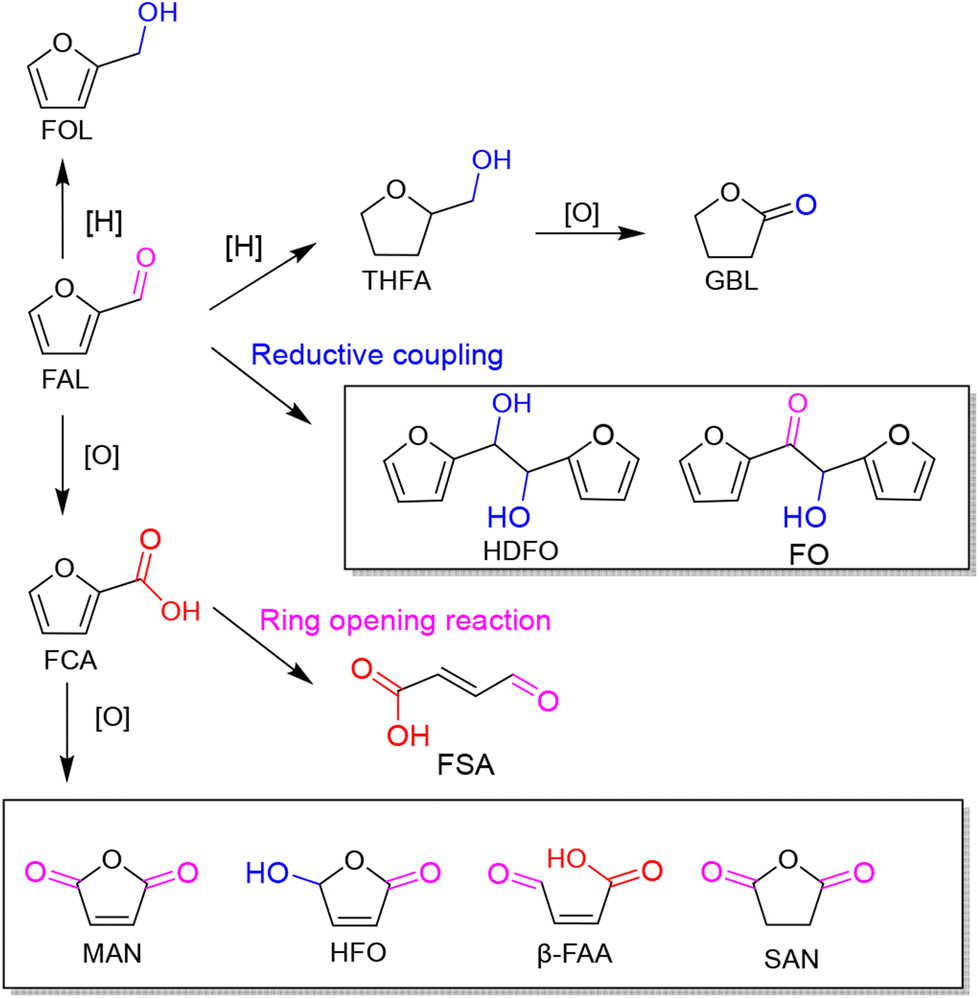
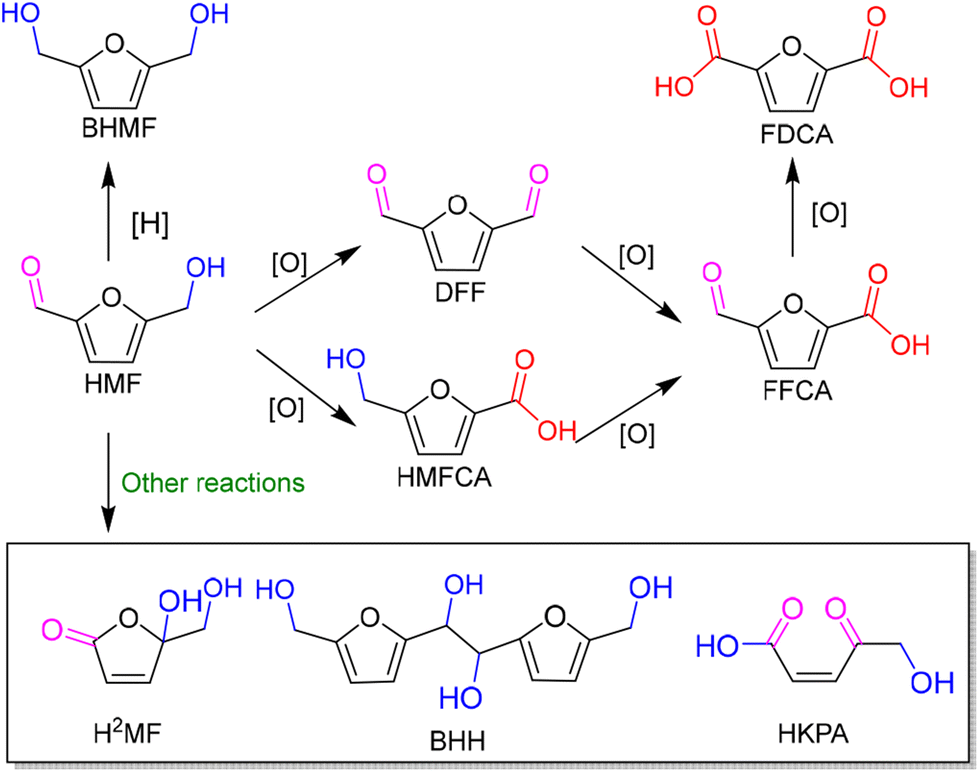
As an abundant and easily accessible resource, biomass provided the essential materials that have supported humanity for a long time until the rise of fossil resources.1 Currently, the world is increasingly dependent on non-renewable fossil fuels (i.e., coal, oil and natural gas) to meet the growing demand of development and the challenging requirements of modern industries.2 At the same time, the huge exploitation and burning of fossil resources leads to emission of large amounts of greenhouse gases that are already causing global climate change.3 Therefore, it is an urgent task and a major research challenge worldwide to find sustainable alternatives to the nonrenewable and petroleum-based energies and chemicals. In this context, the renaissance of biomass utilization in a much more efficient manner is expected.3 That is because carbon neutrality could be achieved in theory during biorefinery processes. The emitted greenhouse gases can be recycled and fed back to biomass resources through photosynthesis using solar energy. In light of this, innovation and complete use of abundant and renewable biomass resources are particularly important in the pursuit of a sustainable future.Plant materials consisting primarily of carbohydrate polymers (i.e., cellulose and hemicellulose) and aromatic polymers (i.e., lignin) are considered as lignocellulose and account for over 90% of all plant biomass.4 It is the most abundant source of renewable carbon and would not compete with food reserves.5 Thereby conversion of lignocellulosic biomass into value-added fuels and chemicals is a preferred option for harvesting sustainable energy and realizing carbon neutrality. In order to achieve this objective, bioplatform molecules, rather than crude biomass, are usually selected and studied in fundamental research due to the high complexity of biorefinary.6–9 Currently, furfural and its derivatives can be readily obtained from chemical and/or biological routes through hydrolysis and/or dehydrogenation of cellulose and hemicellulose. The biomass-derived furanic compounds mainly including furfural (FAL) and 5-hydroxymethylfurfural (HMF) are regarded as the key platform molecules in biorefining processes.10,11 They can be converted into a range of value-added building-block chemicals (Fig. 1 and 2), typically furfuryl alcohol (FOL),12 2,5-bis(hydroxymethyl)furan (BHMF),13 2,5-diformylfuran (DFF)14 and 2,5-furandicarboxylic acid (FDCA).15
 | ||
| Fig. 1 A schematic diagram of photocatalytic conversion of furfural (FAL) with major pathways and products. | ||
 | ||
| Fig. 2 A schematic diagram of photocatalytic conversion of 5-hydroxymethylfurfural (HMF) with major pathways and products. | ||
Among them, DFF, FDCA and BHMF with a symmetric structure have attracted much attention. That is because they are promising monomers for manufacturing furan-based polymers which could be alternatives to nonrenewable petroleum-based aromatic monomers. For instance, DFF can be widely used in the production of organic conductors and as intermediates for drugs and antifungal agents. FDCA can serve as ideal monomer instead of terephthalic acid to synthesize renewable bioplastics in the presence of ethylene glycol. Thus it has been listed within the top 12 value-added chemicals from biomass by the U.S. Department of Energy.16 FOL can be a building-block intermediate for manufacturing fibers, lubricants, and resins.12
Over the past two decades, many efforts have made to develop heterogeneous catalysts with the assistance of thermochemistry to achieve those transformations of furfural and its derivatives.17,18 However, the processes are usually and essentially operated by using high temperatures and/or pressures, resulting in considerable energy consumption and process cost. In contrast, the use of photocatalysis technology enables conversion of biomass to high-value organic chemicals under mild conditions and has attracted great interest due to its economic and environmental advantages. So far, preliminary studies on the photocatalytic conversion of FAL, HMF and their derivatives have been carried out.19–21 Notably, most of the photocatalysts are common ones already used in the classic photoreactions, e.g., water splitting. In some specific reactions, it still remains quite difficult to attain both good conversion and selectivity. For instance, titanium dioxide (TiO2), which was first applied to the photocatalytic oxidation of HMF, has an inherently large band gap.19 Hence TiO2 can only respond to ultraviolet (UV) irradiation. But the UV region only accounts for a very small part (ca. 3%) of the solar radiation spectrum. Unless visible light (ca. 44%) can participate in the photochemical reaction it is obviously a huge waste of solar energy.22 At the same time, inappropriate placement of the valence band of TiO2 can unfortunately promote the formation of excessive strong oxidants like hydroxyl radicals (˙OH). Such reactive species can usually lead to nonselective decomposition (e.g. mineralization) of organic compounds or side reactions. Besides, dedicated photoreactors are necessary for UV light-driven reactions, but these are expensive and environmentally unfriendly. Specifically, the high energy consumption for generating UV light and the size of customized reactors would severely restrict the application of large-scale photosynthesis. As a result, the focus in the field has gradually shifted to developing a range of visible-light-responsive photocatalysts with a suitable band location, in order to address the above issues.23–27
In addition, photogenic radical intermediates allow for novel and meaningful reaction paths (Fig. 1 and 2) that are unlikely to be achieved in conventional heterogeneous catalysis. For example, selective dimerization of FAL through photoreductive coupling enables molecules with increased carbon numbers to be further upgraded to gasoline-grade fuels.28,29 However, in conventional heterogeneous catalysis, the selective formation of vinyl radical intermediates is rather difficult because the reducing atmosphere usually results in hydrogenation at lower temperatures or hydrolysis at higher temperatures.30 Moreover, owing to the unique redox ability of photogenerated electrons (e−) and holes (h+) in the photocatalytic system, selective oxidation and reduction can simultaneously occur over the same catalyst. In such a context, H2 evolution31,32 or CO2 reduction33 can be simultaneously performed with photoconversion of FAL and HMF. Besides, the tandem photoreduction and oxidation of FAL can yield tetrahydrofurfuryl alcohol (THFA) and γ-butyrolactone (GBL).34 Photooxidative decarboxylation of FAL can be also realized by photocatalysis to form 5-hydroxy-2(5H)-furanone (HFO),35 maleic anhydride (MAN),36 succinic anhydride (SAN), maleic acid (MA),37 and other value-added products. Photocatalytic oxidative ring-opening can further obtain fumaric semialdehyde (FSA),38cis-β-formylacrylic acid (β-FAA), and (Z)-5-hydroxy-4-keto-2-pentenoic acid (HKPA)39 from furfural and its derivatives. The application values of these unconventional products are introduced in detail towards the end of this review.
In this review article, the recent advances in photocatalytic conversion of furfural and its derivatives into value-added products are summarized. To date, photooxidation of HMF to DFF has been extensively studied. But unsatisfactory selectivity of DFF is often observed due to an over-oxidation of the intermediates resulting from the formation of highly reactive substances. Hence the strategies and methods for improving the catalytic activity and selectivity are discussed. Moreover, the efforts on switching the oxidizing product from DFF to FDCA are summarized. In addition, bifunctional catalysts for simultaneous oxidation and H2 evolution of HMF and FAL or reduction of CO2 are also reviewed. This transformation has a very high atomic economy in line with the principles of green chemistry. Finally, some nontraditional reaction paths are introduced, such as reductive coupling, oxidative decarboxylation and ring-opening. Without exception these routes show higher selectivity and greater application value. Therefore, this large number of research topics indicates that the photocatalytic conversion of furfural and its derivatives has a broad prospect.
2. Photocatalytic oxidation of HMF to DFF
2.1. TiO2-based photocatalytic materials
TiO2 was first reported for the electrochemical photolysis of water among various semiconductor materials used as oxidation photocatalysts, starting a new era of photocatalysis.40 TiO2 has been regarded as the most reliable photocatalyst owing to its low cost, chemical inertness, high photoactivity and stability. Given the strong oxidizing power, TiO2 can easily lead to the direct degradation of organic compounds. Nevertheless, the wide band gap (ca. 3.2 eV) of TiO2 results in its intrinsic photoresponse being limited only to the UV light region. Therefore, many studies have been devoted to improving the product selectivity and visible-light response of TiO2-based photocatalysts.As summarized in Table 1, Yurdaka et al. successfully prepared amorphous TiO2 with different crystalline shapes and firstly explored their performances for the photocatalytic selective oxidation of HMF to DFF.19 The partial oxidizing ability of TiO2 photocatalyst was found to originate from its low crystallinity. The authors considered h+ or ˙OH radicals as the active species for this selective oxidation reaction. Ulyankina et al. prepared lowly crystallized TiO2 nanoparticles by electrochemical synthesis, which exhibited 33% selectivity of DFF in the presence of methanol.41 The value was found to be higher than previously prepared TiO2 catalysts by micro-emulsion and sol–gel methods. Interestingly, an increase in selectivity was observed during the recycling experiments due to the decreased TiO2 crystallinity. Hence this feature of TiO2 was further demonstrated to be beneficial for the enhancement of photocatalytic selectivity. Krivtsov et al. used non-metal doping with N into TiO2, which altered the surface chemistry of TiO2 and reduced the amount of OH groups on TiO2 surface.42 Unlike Yurdaka, Krivtsov believed that ˙OH radicals were nonselective. The N-doping can significantly diminish the surface OH groups, which enabled effective hindering of the generation of ˙OH species. Thus this promoted selective photooxidation of HMF to DFF. Allegri et al. also thought that h+ and ˙OH were responsible for the decomposition (i.e., complete oxidation) of HMF and products to CO2 and H2O, whereas the photogenerated e− and/or superoxide radicals (˙O2−) were the main species responsible for the selective conversion of HMF to HMFCA and DFF.43 However, surface hydroxylation of TiO2 is not the only way to generate ˙OH radicals. Khan et al. investigated the ligand–metal charge transfer-mediated selective oxidation of HMF to DFF over a TiO2 photocatalyst (SGH-TiO2) under visible light. The selectivity of DFF reached as high as 87% at 59% conversion of HMF.44 To demonstrate the important role played by OH groups in photocatalysis, SGH-TiO2 was chemically modified by surface fluorination followed by calcination to remove the surface OH groups, and the photocatalytic activity was completely blocked. Therefore, the surface hydroxyl groups of TiO2 played an important role in bonding the substrate and realizing the visible-light response. Similarly, Zhang et al. modified TiO2 with a visible-light-responsive p-type semiconductor (i.e., Cu2O).45 The obtained Cu2O∥TiO2 heterojunction photocatalyst achieved 52% conversion of HMF and over 44% selectivity of DFF in water. This may be the highest yield of DFF reported to date among TiO2-based catalysts in aqueous phase. In addition, the authors confirmed by free-radical scavenger experiments that in situ-generated ˙O2− and singlet oxygen (1O2) were the main active species for the selective photooxidation of HMF to DFF. As proposed in the reaction mechanism (Fig. 3), the formation of the p–n heterojunction can effectively coordinate the energy band positions and promote the separation of photogenerated e− and h+. As a result, the e− and h+ enrich in the conduction band of TiO2 and the valence band of Cu2O, respectively. As is known to all, the energy band position of a semiconductor is closely related to its redox capacity. Therefore, e− in the conduction band of TiO2 can effectively activate oxygen to generate ˙O2− which is then oxidized by h+ in the valence band of Cu2O to generate the final reactive 1O2 species, to realize the selective oxidation of HMF. Zhou et al. prepared a porous TiO2@UIO-67-Zr/Ti composite as photocatalyst by using microwave-assisted synthesis followed by metal exchange.46 The strong interactions between TiO2 and Zr/Ti-MOF material improved not only the transfer of photogenerated e− and h+ but also the light absorption range. 94% conversion of HMF and 73% selectivity of DFF were obtained in acetonitrile.
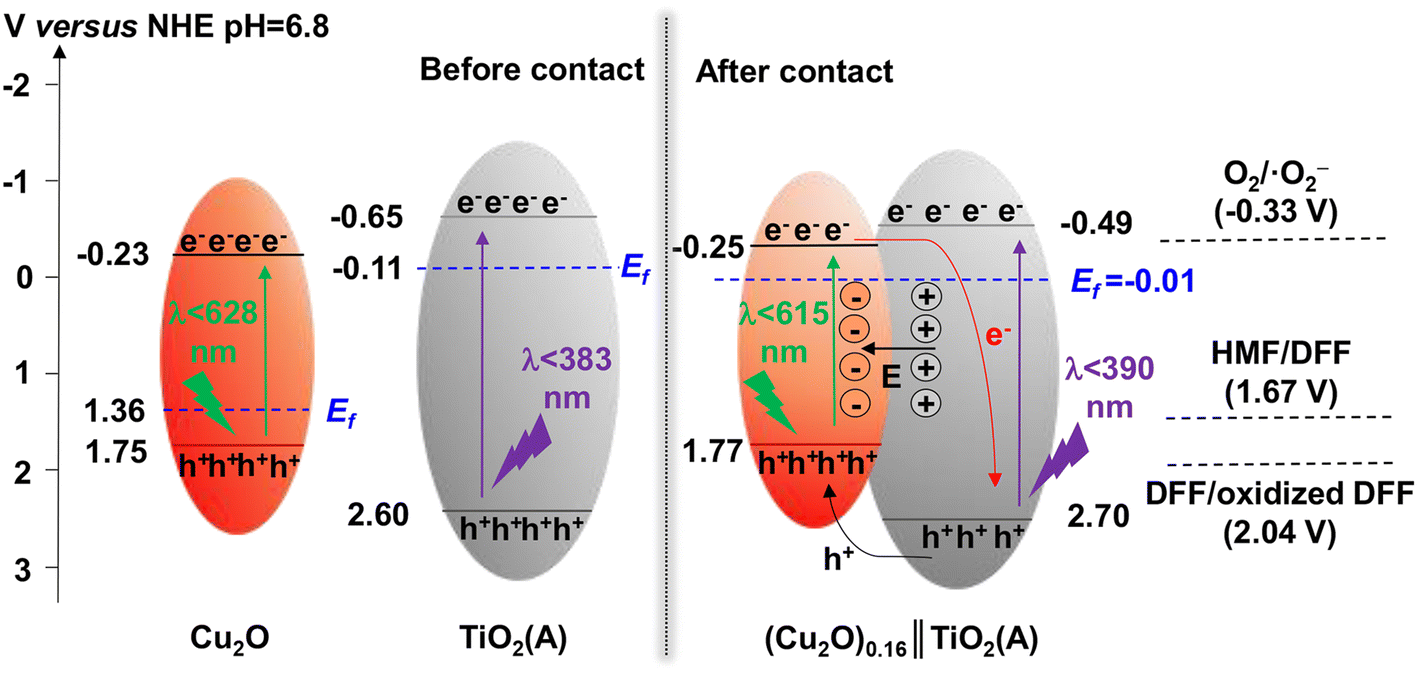 | ||
| Fig. 3 Schematic illustration of energy band and charge transfer process of the Z-scheme (Cu2O)0.16∥TiO2(A) photocatalyst. Reproduced from ref. 45, with permission of Elsevier, Copyright 2023. | ||
| Entry | Catalyst | Solvent | Reaction conditions | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | TiO2 | H2O | Near-UV radiation, O2 | 20 | 21–25 | 19 |
| 2 | Nano-TiO2 | H2O | Spotlight source (Hamamatsu, LC8) 365 nm (3.0 mW cm−2), O2 (3 mL min−1), 50 mM methanol | 15 | 33 | 41 |
| 3 | N-TiO2 treated at 400 °C | H2O | 500 W lamp with emission maximum at 365 nm, O2 | — | 30–40 | 42 |
| 4 | Au3Cu1/TiO2-m SFD | H2O | Simulated solar light, 1 atm O2, 1 h, 30 °C | 3 | 67 | 43 |
| 5 | SGH-TiO2 | CH3CN | visible light (λ = 515 nm), O2, 4 h | 59 | 87 | 44 |
| 6 | (Cu2O)0.16∥TiO2(A) | H2O | Xe lamp, 350–780 nm, 90 min, 35 °C | 52 | 44 | 45 |
| 7 | TiO2@MOF | CH3CN | 300 W Xe lamp, 4 °C, 5 h | 94 | 73 | 46 |
2.2. g-C3N4-based photocatalytic materials
Graphitized carbonitride (g-C3N4) is an organic-polymer semiconductor with rich surface properties, good thermal stability, chemical stability, and particularly narrow band-gap width compared with TiO2. Therefore, g-C3N4 has been viewed as a promising visible-light-responsive photocatalyst. As listed in Table 2, Krivtsov et al. firstly investigated different precursors to synthesize g-C3N4 as photocatalyst.47 These catalysts not only used sunlight more effectively but also displayed higher selectivity. Among them, MCN-540 (i.e., melamine as precursor calcined in air at 540 °C) showed the optimal photoactivity. The conversion of HMF reached 69% with 43% selectivity of DFF after 4 h of reaction.| Entry | Catalyst | Solvent | Reaction conditions | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | MCN-540 | H2O | UV lamp, O2, 4 h | 69 | 43 | 47 |
| 2 | PCN-H2O2 (TEO) | H2O | UV light/natural solar irradiation, O2, ∼4 h | 21/37 | 71/83 | 48 |
| 3 | US-CHNO3 | H2O | UV irradiation/natural solar light irradiation, O2, natural pH, 4 h | 27/80 | 32/22 | 49 |
| 4 | Metal-free g-C3N4 | CH3CN + PhCF3 | Xenon lamp (300 W, λ > 400 nm), 10 mL min−1 O2, 6 h | 31 | 86 | 50 |
| 5 | ZnPp-C3N4-TE | H2O | Natural solar light, natural pH, 4 h, 25 °C, 1 atm | 73 | 36 | 51 |
| 6 | Cu SAs/p-CNS | CH3CN | 20 W blue LED (λ = 455 ± 10 nm), O2 bubbling, 25 °C | 77 | 86 | 52 |
| 7 | CN-WO3@MnO2 | CH3CN | 420 nm LED light (10 W), 24 h | 78 | 80 | 53 |
| 8 | Ti3C2/g-C3N4 | PhCF3 | Visible light, O2, 10 h | 93 | 97 | 23 |
| 9 | WO3/g-C3N4 | CH3CN + PhCF3 | λ > 400 nm, 10 mL min−1 O2, 6 h, 30 °C | 28 | 87 | 54 |
| 10 | 12%Bi2WO6/mpg-C3N4 | CH3CN + PhCF3 | λ > 400 nm, 10 mL min−1 O2, 6 h | 59 | 84 | 55 |
| 11 | Nb2O5/g-C3N4 | CH3CN + PhCF3 | 300 W Xe lamp (AM 1.5G), 1 atm O2, 6 h | 53 | 99 | 56 |
| 12 | Ni/ACN | CH3CN | λ = 390 nm, 25 °C, 1 atm N2, 12 h | 85 | 97 | 57 |
| 13 | Polyimide M1P2 | CH3CN | 405 nm LED lamp (24 W), 1 atm air, 40 min | 61 | 91 | 58 |
Following that, additional treatment of g-C3N4 becomes a feasible way to improve its catalytic performance. Ilkaeva et al. prepared a PCN-H2O2 catalyst by treating g-C3N4 with H2O2.48 The bonding of H2O2 suppressed the formation of nonselective ˙OH radicals and further promoted the selectivity of DFF. Akhundi et al. modified g-C3N4 by two effective methods, i.e., HNO3 ultrasonication and thermal stripping.49 A higher yield was obtained for US-CHNO3 catalyst compared with the thermal stripping. Moreover, US-CHNO3 can expand the specific surface area of original g-C3N4 and increase the DFF yield. Wu et al. found water treatment of the calcined g-C3N4 can enlarge the specific surface area and pore volume, thus affecting the pristine photochemical property.50 Compared with the common g-C3N4, the modified catalyst presented lower e−–h+ recombination rate and better photocatalytic activity. Furthermore, García-López et al. loaded metalloporphyrins on g-C3N4 and obviously boosted absorption capacity of the photocatalyst upon visible light at higher wavelengths.51 Porphyrin molecules were found to benefit 1O2 generation and simultaneously inhibit ˙OH formation.
In order to promote the separation of photogenerated e− and h+, modification of g-C3N4 by introducing acceptors of e− and h+ are shown to be an effective strategy. Wang et al. synthesized a single-atom Cu/p-CNS catalyst by incorporating Cu as e− acceptor and S as h+ acceptor into g-C3N4. Cu-N4 and C–S–C dual atomic active sites were decorated on polymeric carbon nitride (Cu SAs/p-CNS). Cu and S diatomic sites cooperated to improve the separation of photogenerated carriers, thus enhancing photocatalytic activity.52 In the same way, Qian et al. prepared a CN-WO3@MnO2 photocatalyst.53 WO3 as electron transport channel and MnO2 as photoelectron acceptor can together strengthen the separation and transfer of photogenerated e− and h+. As illustrated in Fig. 4, such an effect was greatly boosted by Wang et al. on a Ti3C2/g-C3N4 photocatalyst by taking advantage of the strong interaction between Ti3C2 and g-C3N4. 97% selectivity of DFF at 93% conversion of HMF was attained in trichlorotoluene medium.23 This is the best result among g-C3N4-based photocatalysts in the literature to date.
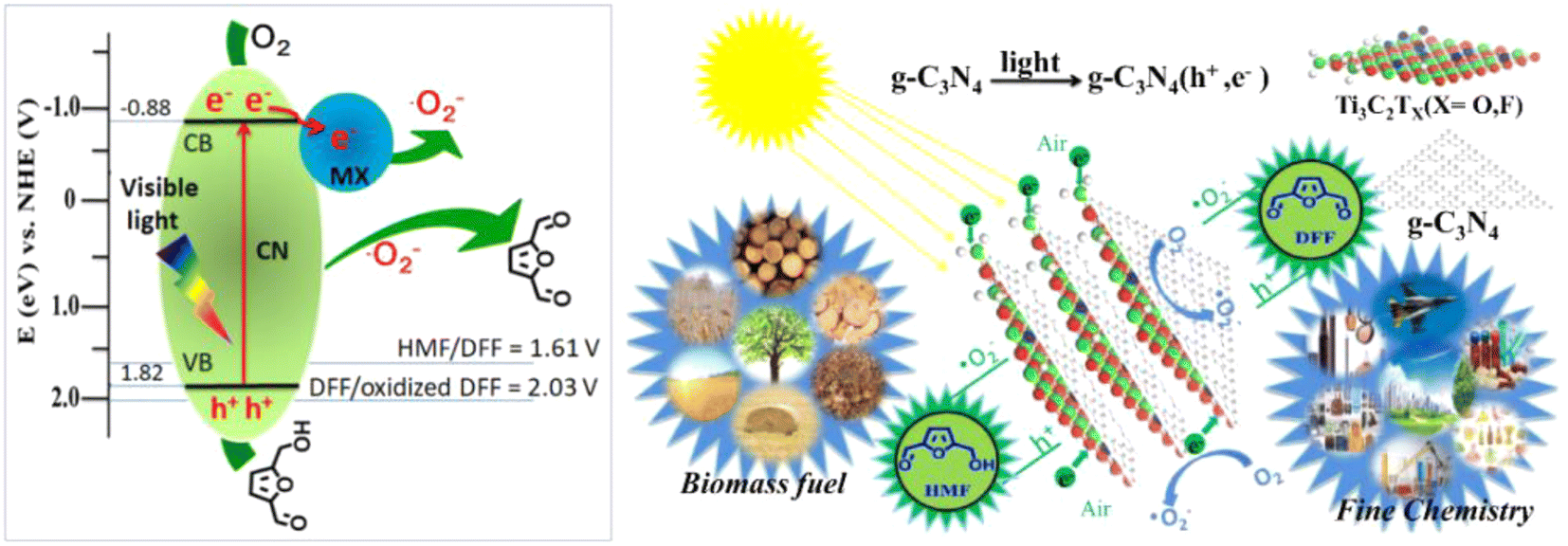 | ||
| Fig. 4 Illustration of the mechanism for selective photooxidation of HMF into DFF over the Ti3C2/g-C3N4 catalyst under visible light irradiation. Reproduced from ref. 23, with permission of Elsevier, Copyright 2020. | ||
Alternatively, constructing heterojuction structures on g-C3N4 can be very useful to facilitate charge separation. For instance, Zhang and Cheng, respectively, combined WO3 and Bi2WO6 with g-C3N4 to prepare Z-type WO3/g-C3N4 and Bi2WO6/g-C3N4 heterojunction.54,55 Li et al. integrated Nb2O5 to g-C3N4 to obtain II-type Nb2O5/g-C3N4 heterojunction.56 The experimental results showed an improved separation efficiency of photogenerated e− and h+ due to the existence of the built-in electric field in the heterojunction, thus boosting photocatalytic reaction performance. In addition, the energy band structure can be adjusted by the formation of the heterojunction. When the valence band position is lower than the redox potential of ˙OH radicals, the nonselective ˙OH cannot be produced. In light of this idea, Liang et al. adapted the band structure by coupling Ni with g-C3N4 to create a Schottky junction.57 Chu et al. modulated the energy band position of polyimide photocatalysts by polymerizing g-C3N4 with different molar ratios of anhydride.58 Both approaches successfully improved the selective photocatalytic conversion of HMF to DFF.
2.3. Metal sulfide-based photocatalytic materials
Metal sulfide semiconductors (mainly CdS and ZnIn2S4) have excellent properties such as narrow band gap (ca. 2.4 eV) and wide photoresponse range, and are very suitable catalysts for visible light-driven photocatalytic reactions. However, the high recombination rate of e−–h+ pairs and sensitivity to photocorrosion limit the further application and development of metal sulfides. Thereby various solutions have been proposed to overcome these issues (Table 3). Han et al. modified ultrathin CdS nanosheets with nickel as h+ acceptor for the first time and achieved photocatalytic oxidation of HMF to DFF under visible light irradiation.59 100% selectivity of DFF was obtained at 20% conversion of HMF. In order to further ameliorate the catalytic activity, Xia and Yuan combined Ru and chitosan-derived biochar as e− acceptors with CdS quantum dots to synthesize RuCdS and CdSQDs/Ch catalysts, respectively.60,61 Due to the effective separation of photogenerated e− and h+, both catalysts can reach over 90% selectivity of DFF in organic solvents. DiMeglio et al. directly introduced NO3− into a CdS catalytic system to act as a redox mediator in the solution.62 Nitrate-mediated alcohol oxidation proceeded through a mechanism involving the catalytic generation of ˙NO3− radicals, which enabled the photooxidation of HMF to DFF with >99% selectivity.| Entry | Catalyst | Solvent | Reaction conditions | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Ni/CdS | H2O | Blue LED (440–460 nm, 8 W), 10 mM HMF, N2, 22 h | 20 | 100 | 59 |
| 2 | Ru CdS | DMF | 300 W xenon lamp 100 mW cm−2, 16 h | 81 | 92 | 60 |
| 3 | CdS QDs/Ch | CH3CN | 100 mW cm−2, 25 °C, O2, 10 h | 79 | 97 | 61 |
| 4 | Nitrate-mediated CdS | CH3CN/Mn(NO3)2 | Blue LED light (447.5 nm, 170 mW cm−2), O2 | — | >99 | 62 |
| 5 | CdxInyS(x+1.5y) | H2O | λ > 420 nm, 20 °C, 6 h | 69 | 63 | 63 |
| 6 | MoS2/CdIn2S4 | H2O | 7 W COB LED lamp (>420 nm). 10 h | 62 | 81 | 64 |
| 7 | Ti3C2FxMXene/CdIn2S4 | H2O | λ > 400 nm, 20 °C, 12 h | 79 | 60 | 65 |
| 8 | (110)/(102)-ZnIn2S4 | CH3CN | Simulated solar light irradiation, O2, 1 h | 91 | 99 | 25 |
| 9 | ZnIn2S4 2D | CH3CN | Blue LED, 15 °C, air, 2 h | 95 | 70 | 66 |
| 10 | ZnIn2S4-TU | H2O | λ = 445 nm, 6 W, 9 h | 60 | 66 | 67 |
| 11 | CC/ZnIn2S4 | CH3CN | 400–780 nm, air, 4 h | 22 | 69 | 68 |
| 12 | SA-PDI/ZnIn2S4 | CH3CN | 300 W xenon lamp, O2, 0.5 h | 98 | 90 | 69 |
In addition to binary metal sulfides, ternary metal sulfides have been also studied extensively. Zhang et al. used a one-step hydrothermal method to controllably synthesize a CdxInyS(x+1.5y) catalyst.63 When the Cd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :In ratio exceeded 1:2, the catalyst displayed a 1D to 3D heterojunction structure composed of CdS nanorods and cubic CdIn2S4. This unique structure effectively promoted the separation of photoexcited e−–h+ pairs in CdxInyS(x+1.5y) and realized the cross-border transfer of photogenerated carriers. Likewise, Zhu and Zhang compounded MoS and Ti3C2Fx with CdIn2S4, respectively, to obtain the corresponding heterojunctions.64,65 Compared with the pristine CdIn2S4, the yields of DFF over MoS/CdIn2S4 and Ti3C2Fx/CdIn2S4 photocatalysts were increased by 20% and 30%, respectively.
:In ratio exceeded 1:2, the catalyst displayed a 1D to 3D heterojunction structure composed of CdS nanorods and cubic CdIn2S4. This unique structure effectively promoted the separation of photoexcited e−–h+ pairs in CdxInyS(x+1.5y) and realized the cross-border transfer of photogenerated carriers. Likewise, Zhu and Zhang compounded MoS and Ti3C2Fx with CdIn2S4, respectively, to obtain the corresponding heterojunctions.64,65 Compared with the pristine CdIn2S4, the yields of DFF over MoS/CdIn2S4 and Ti3C2Fx/CdIn2S4 photocatalysts were increased by 20% and 30%, respectively.
However, Cd is widely criticized in ecology because of its high toxicity to kidneys, lungs and bones. Because of the biological and environmental toxicity of Cd, Zhao et al. prepared Cd-free (110)/(102)-ZnIn2S4 nanosheets by crystal-face engineering strategy.25 As shown in Fig. 5 for the reaction mechanism, ESR characterization and scavengers experiment can verify the formation of ˙O2−, 1O2 and ˙OH species during the photoreaction and clarify the different roles they play in catalytic activity. 99% selectivity of DFF at 91% conversion of HMF is reported in O2 and acetonitrile, being the best result in the literature to date. By introducing Na2S, it can be found that the spontaneous replacement of O2− by S2− for photocatalytic regeneration can significantly improve the durability of ZnIn2S4. Following that, Ding et al. obtained 2D ZnIn2S4 nanosheets by adding an appropriate amount of sodium citrate during synthesis.66 This photocatalyst reached 95% conversion of HMF after only 2 h of blue LED irradiation. It was verified that the formation of ˙HMF radicals via oxidation at photogenerated h+ and the subsequent deprotonation of ˙HMF with 1O2 were the key to selectively oxidizing HMF to DFF. Pham et al. treated ZnIn2S4 nanoparticles by changing different sulfur sources such as thioacetamide or thiourea.67 It was disclosed that the latter can induce a greater number of Zn vacancies. This method effectively adjusted the band gap and promoted the separation of charge pairs, thereby improving the photocatalytic efficiency.
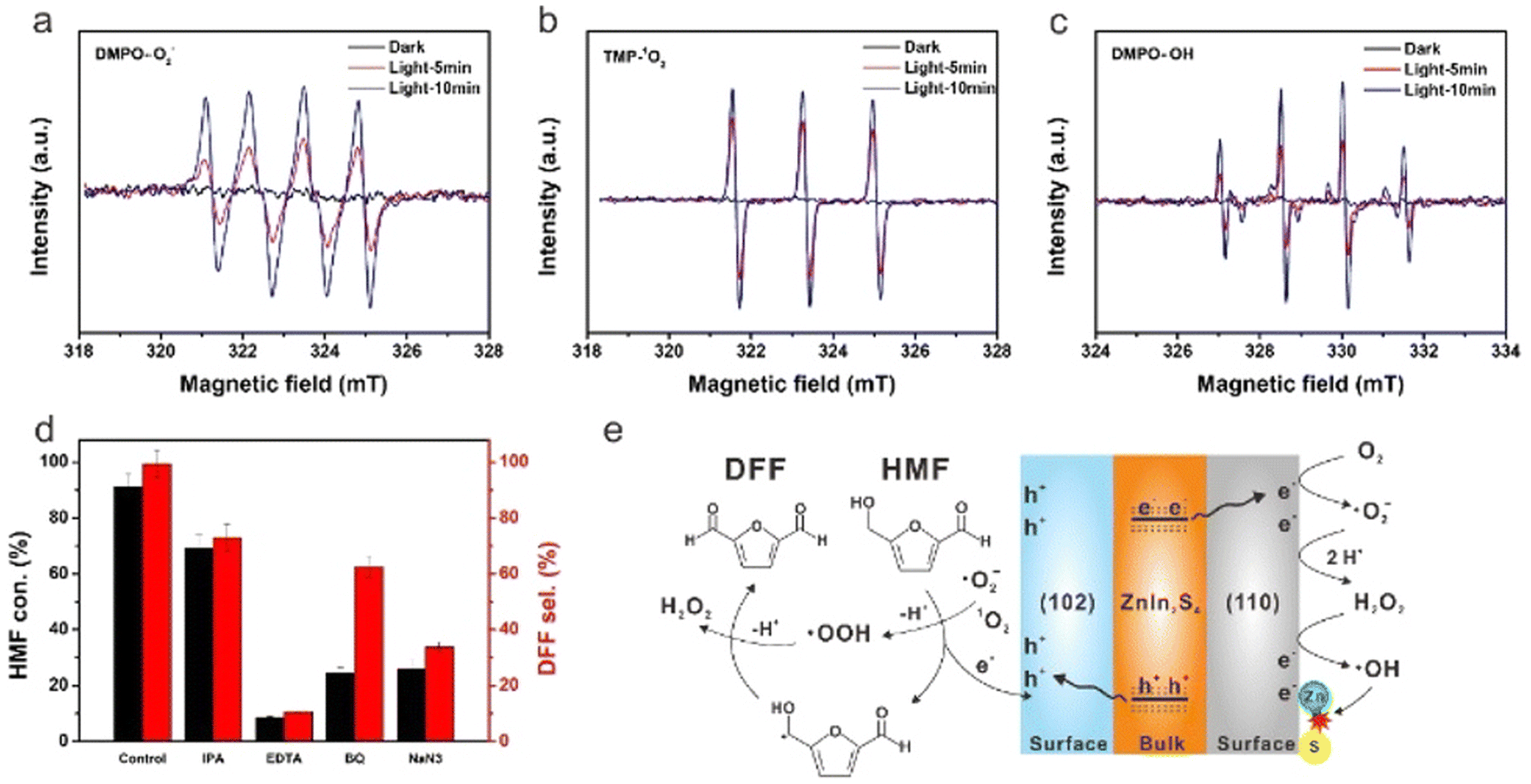 | ||
| Fig. 5 EPR signals of the reaction solution in the dark and simulated solar light irradiation with different time in the presence of DMPO and TMP as spin-trapping reagents for (a) ˙O2−, (b) 1O2 and (c) ˙OH. (d) Effect of different scavengers on HMF conversion and DFF selectivity. (e) Proposed photocatalytic mechanism for HMF conversion to DFF over the ZnIn2S4 catalyst. Reproduced from ref. 25, with permission of Royal Society of Chemistry, Copyright 2022. | ||
To further ameliorate the synthetic method, Qiu et al. combined cellulose-derived carbon dots with ZnIn2S4.68 It was disclosed that the added carbon dots can enhance the absorption of visible light, thus freely shuttling electrons to the carbon dots and improving e−–h+ pairs separation. The obtained CC/ZnIn2S4 photocatalyst showed an outstanding performance with a yield of DFF ca. 3.4 fold over the original ZnIn2S4. Similarly, Guo et al. integrated perylene imide supramolecular molecules as e− acceptors to ZnIn2S4.69 On the SA-PDI/ZnIn2S4 catalyst, the functionalized supramolecular served as active sites, which greatly accelerated the generation of ˙O2− radicals by absorbing O2 and concentrated the photogenerated e− on ZnIn2S4. Notably, the formation rate of DFF reached 1952 μmol g−1 h−1, which was nearly an order of magnitude higher than the previously reported photocatalytic technology. The yield of DFF after 0.5 h of illumination was 9.4 times higher than that of the bare ZnIn2S4.
2.4. Other photocatalytic materials
Some other semiconductor-based catalysts have also been reported for the photocatalytic selective oxidation of HMF to DFF (Table 4). Zhang et al. prepared a Nb2O5 by calcining at as high as 800 °C and applied it to the oxidation of HMF under visible light.70 It was found for the first time that the OH group of HMF could be adsorbed on Nb2O5 to form an alcoholized substance, lowering the band-gap energy of Nb2O5 to respond to visible light. This phenomenon can be explained by the ligand–metal charge transfer (LMCT) effect. Although 91% selectivity of DFF was obtained, the conversion of HMF was of only 19%. To improve the photoactivity, Zou et al. introduced Ce during catalyst preparation to enrich oxygen vacancy on the Nb2O5 surface, as illustrated in Fig. 6.71 This is believed to enhance the LMCT effect between HMF and the catalyst under visible light. However, the nonselective generation of H2O2 can easily occur in the aqueous phase, resulting in over-oxidation of the produced DFF. Thereby Pt nanoparticles are deposited on Ce-Nb2O5 to inhibit the accumulation of H2O2. Eventually the conversion of HMF can be increased to 36% with a superior selectivity (i.e., 93%). | ||
| Fig. 6 Proposed mechanism of photocatalytic selective oxidation of alcohols to aldehydes over the defective Nb2O5 with deposited Pt nanoparticles. Reproduced from ref. 71, with permission of Elsevier, Copyright 2021. | ||
| Entry | Catalyst | Solvent | Reaction conditions | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Nb2O5-800 | PhCF3 | Visible light (>400 nm), O2, 6 h | 19 | 91 | 70 |
| 2 | Pt-Ce-NbO-3 | H2O | λ > 420 nm 15 °C, pre-purged with air, 6 h | 36 | 93 | 71 |
| 3 | Rose-like Bi2WO6 | CH3CN | λ > 420 nm, 1 atm air, 10 h | 58 | 99 | 72 |
| 4 | Pt-Ov-BiOBr | CH3CN + TFME | 300 W xenon lamp with an AM1.5 filter, O2, 3 h | 90 | 78 | 73 |
| 5 | MAPbBr3 | CH3CN | Blue LED light (450 nm), air, 10 h, 15 °C | 100 | 90 | 74 |
| 6 | Cs2SnBr6/rGO | CH3CN | λ = 400 nm, 1 atm O2, 15 °C, 6 h | 100 | 88 | 75 |
| 7 | MnO2-NRs | CH3CN | UV light from LED system (365 nm), O2, 4 h, 39 °C | 99 | 100 | 76 |
| 8 | (Cr3+ or Co3+)-NiO | H2O | λ = 365 nm, 12 h | 60 | 95 | 77 |
In addition, Bi-based semiconductor materials have received much attention due to their stable crystal structures, excellent photovoltaic properties, and abundant reserves. Kumar et al. prepared and compared Bi2WO6, BiVO4 and Bi2MoO6 photocatalysts.72 Among those, the rose-like nanostructured Bi2WO6 presented higher photocurrent response, efficient charge-carrier separation, and suitable band-edge potential. These features enabled the optimal photocatalytic oxidation activity. Interestingly, BiOBr is composed of alternating [Bi2O2]2+ and Br− plates, of which oxygen atoms are exposed to the surface and easily produce oxygen vacancies (Ov). Gong et al. obtained a Pt-Ov-BiOBr catalyst by bonding Pt to the BiOBr surface.73 The Pt–O bond acted as electron transport channel to move the photogenerated e− from Ov-rich BiOBr to Pt, which efficiently caused generation of ˙O2− radicals. Meanwhile the photogenerated h+ would accumulate at the interface between Pt and Ov-BiOBr to oxidize HMF into ˙HMF radicals. The electron-rich Pt could better adsorb HMF, thus realizing a better contact of ˙O2− with reaction intermediates. Ultimately a higher conversion and selectivity were obtained.
In addition, organic–inorganic halide chalcogenide-type materials have been explored owing to their excellent photovoltaic properties. Zhang et al. studied lead halide chalcogenide (MaPbX3: Ma = CH3NH3+, X = Br−, I−) as photocatalyst.74 The conversion of HMF in acetonitrile reached 100% and the selectivity of DFF exceeded 90%. However, Pb-based chalcogenides would suffer from severe toxicity and long-term instability. It is well known that Pb can cause serious damage to the neurological, hematopoietic and digestive systems of humans and mammals. In light of that, Yin et al. synthesized a stable Pb-free chalcogenide Cs2SnBr6 modified with reduced graphene oxide (rGO).75 The Cs2SnBr6/rGO composite exhibited excellent photocatalytic activity. Almost full conversion of HMF and 88% selectivity of DFF were obtained in acetonitrile.
Interestingly, transition-metal oxides with various valence states have been also investigated. Giannakoudakis et al. prepared MnO2 nanorods (NRS) that allowed reaching almost 100% yield of DFF in acetonitrile under UV irradiation.76 The result is the best reported in the literature to date. As shown in Fig. 7, the charge transfer follows the Mn4+/Mn3+ redox cycle with O2 playing a key role. In the absence of O2, the conversion of HMF over MnO2-NRS within 6 h is only 21% and the selectivity of DFF is 67%. In this system, the mild heat and light leads to the adsorption of HMF via the C-OH moiety on the O-sites (deprotonated Brønsted sites) of MnO2-NRS and then it deprotonates twice to form DFF. The formation of water molecules sterically hinders/blocks the catalytic surface-centers by forming hydrogen bonds, which results in activity decrement with time on stream. On the other hand, aprotic and less polar organic solvents (e.g., acetonitrile) can facilitate the rate-determining step by promoting a higher O2 solubility and effective contact of O2 with the MnO2-NRS outer surface. Park et al. synthesized NiO nanoparticles bearing different concentrations of Ov by incorporating charge-mismatched (Cr3+ or Co3+) and charge-matched (Cr2+ or Co2+) dopants.77 Among these, Cr3+/Co3+-doped NiO had the highest amount of Ov and thus attained the best yield of DFF in aqueous phase under UV light.
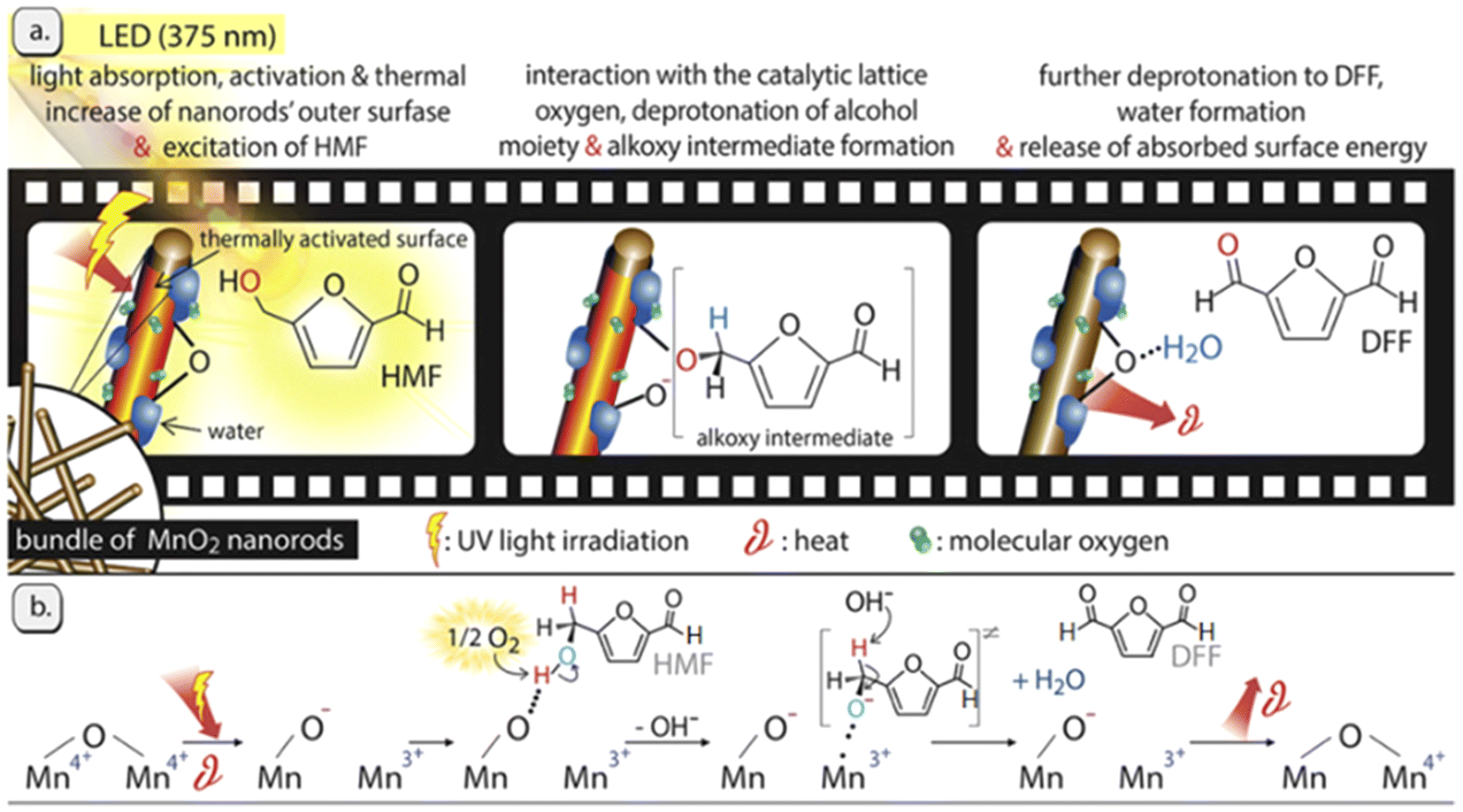 | ||
| Fig. 7 Illustrated representation of the light effect and the involved oxidation mechanism of HMF over the MnO2-NRs photocatalyst. Reproduced from ref. 76, with permission of Elsevier, Copyright 2019. | ||
Other than conventional semiconductors, some polymers are found to have semiconductor-like properties (Table 5). Heterogeneous photocatalytic materials based on conjugated microporous polymers (CMPs) have been reported for organic photoredox reactions. Ayed et al. reported a thiophene-containing covalent triazine backbone (CTF-Th) material for the partial oxidation of HMF to DFF in aqueous phase under visible light.26 Since CTF-Th can be highly dispersed in water during the photocatalytic process, a solid-phase polymerization of CTF-Th@SBA-15 was synthesized, and two possible pathways through ˙O2− or 1O2 were reported (Fig. 8). Under visible light irradiation, photogenerated e− reduce molecular oxygen to active form (˙O2−), and protons are extracted from HMF (1) to form 1a and highly active ˙OOH radicals. The alkoxy anion 1a formed by deprotonation of HMF can react with h+ through electron transfer to form the corresponding alkoxy radical 1b. Then it reacts with ˙OOH to release H2O2 and DFF (2). The 1O2 produced by intersystem crossover (ISC) can be inserted into the C–H bond of the alcohol functional group of HMF (1) to form (3). The intermediate can be formed by independent photolysis to form (4), and then further decomposed into the final product DFF (2) and H2O2 by-product. This catalyst showed a good performance under ambient conditions. >99% selectivity of DFF at 57% conversion of HMF was obtained after 30 h under visible light. Independently, Chen et al. synthesized three CMPs consecutively by C–H direct arylation polymerization using 2,4,6-(tris-2-thienyl)-1,3,5-triazine as a junction and different benzene derivatives as connectors.78 Therein the benzene-conjugated pTTT-Ben catalyst displayed a unique nanofibrous morphology, relatively wide band gap, and strong charge separation. Hence it showed the best photocatalytic performance in the selective oxidation reaction of HMF.
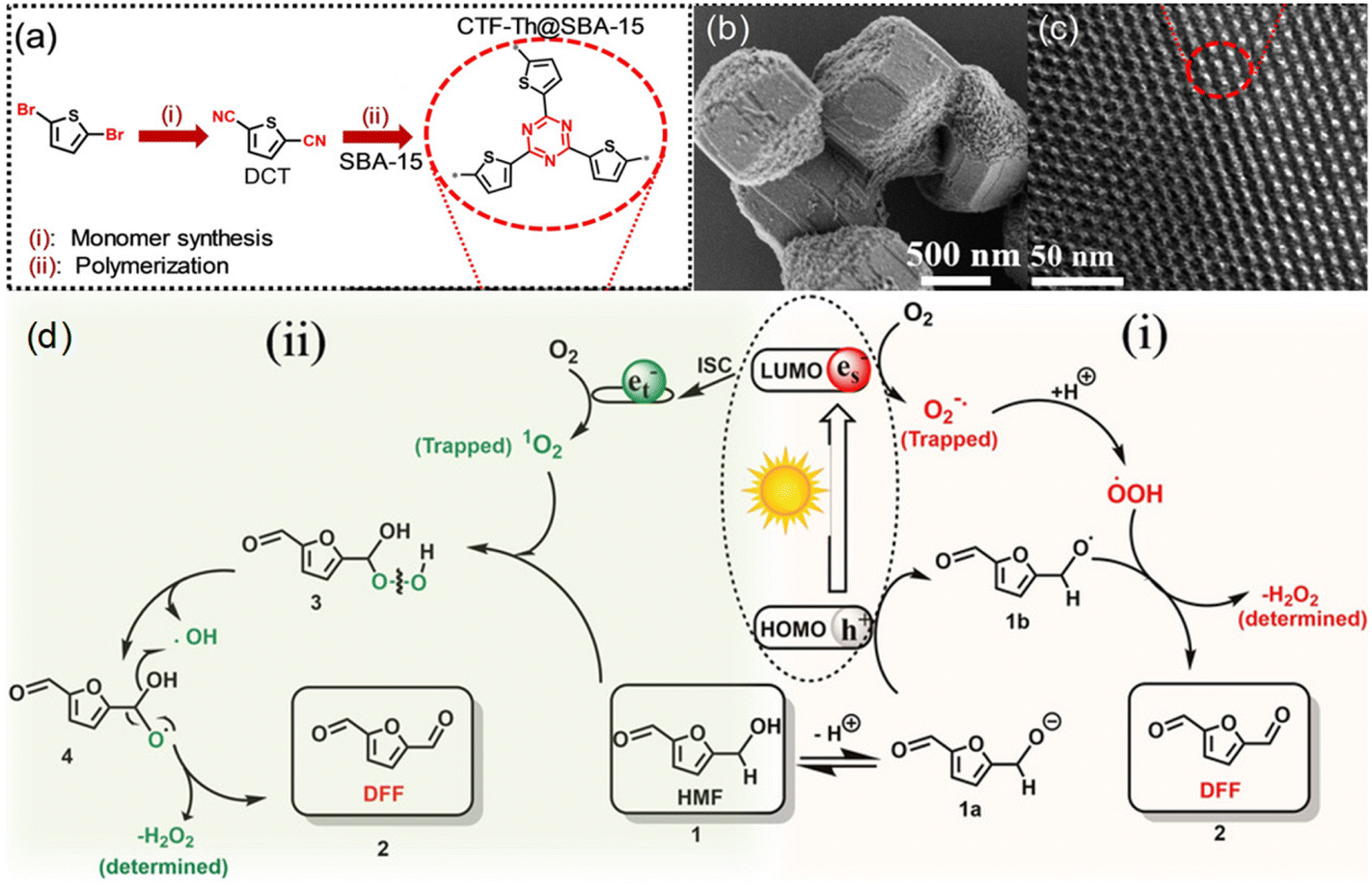 | ||
| Fig. 8 (a) Chemical structure and synthesis routes, (b) SEM and (c) TEM images of CTF-Th@SBA-15, (d) proposed reaction mechanism for the photocatalytic partial oxidation of HMF to DFF using CTF-Th@SBA-15 via two possible routes using either 1O2 or ˙O2−. ISC: Inter System Crossing. Reproduced from ref. 26, with permission of John Wiley & Sons, Inc, Copyright 2020. | ||
| Entry | Catalyst | Solvent | Reaction conditions | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | CTF-Th@ SBA-15 | H2O | Blue LED lamp (460 nm, 65 mW cm−2), 1 atm O2, 30 h, 25 °C | 57 | 99 | 26 |
| 2 | pTTT-Ben | H2O | LED lamp for 10 h, 30 °C | 25 | 100 | 78 |
| 3 | TMADT | CH3CN/6 M HBr | Visible light, 1 atm O2, 12 h, 25–28 °C | 83 | 81 | 79 |
| 4 | HBr | DMSO | Visible light, 1 atm O2, 8 h, 80 °C | 91 | 89 | 80 |
| 5 | Au–Ru/rGO | Toluene | Visible light, 5 bar O2, 8 h, 80 °C | 96 | 95 | 27 |
Different to CMPs, a simple and effective visible-light photocatalytic system using DT (deuterated tungstate)-HBr-based materials was reported by Yang et al. In the presence of acetonitrile and a concentrated HBr (6 mol L−1), conversion of HMF and selectivity of DFF could both reach ca. 82% after 12 h of photoexposure.79 It was demonstrated that HBr enabled effective utilization of water as an oxygen source for the oxidation reaction. Interestingly, a further study by Hu et al. confirmed that the cheap HBr acid could even catalyze this reaction under visible light in O2 and water.80 However, a higher temperature (80 °C) was required. 91% conversion of HMF and 89% selectivity of DFF were obtained in DMSO.
Ma et al. reported bimetallic AuRu nanoparticles on rGO as efficient photocatalyst for the partial oxidation of HMF to DFF in toluene.27 The synergistic interaction between the dual metal sites was demonstrated to promote photocatalytic activity upon visible light and even sunlight. Both conversion and selectivity could reach ca. 95%.
3. Combination of photocatalytic oxidation with H2 evolution or CO2 reduction
3.1. Photocatalytic oxidation of FOL combined with H2 evolution
The previous photocatalytic systems driven by solar energy basically use O2 as oxidant to partially oxidize HMF to DFF. However, over-oxidation byproducts such as FFCA and FDCA can be generated during the aerobic oxidation, resulting in poor selectivity and low atomic economy. In contrast, photocatalytic anaerobic oxidation of FOL or HMF combined with H2 evolution can be a desirable route. This strategy allows the use of photogenerated h+ to oxidize furfuryl compounds and to reduce protons by photogenerated e− for H2 formation. Particularly, FOL without C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group (vs. HMF) combined with H2 evolution can even bring higher efficiencies for H2 formation and selective oxidation. Thus a closed loop is established with high atomic economy. The absence of O2 basically excludes the formation of over-oxidation byproducts, which is beneficial for improving product selectivity.
O group (vs. HMF) combined with H2 evolution can even bring higher efficiencies for H2 formation and selective oxidation. Thus a closed loop is established with high atomic economy. The absence of O2 basically excludes the formation of over-oxidation byproducts, which is beneficial for improving product selectivity.
For instance (Table 6), the Ni/CdS catalyst can only convert 20% HMF to DFF under N2 and blue LED light, but it can completely convert FOL to FAL under exactly the same conditions.59 Combining semiconductor with cocatalyst, Yang et al. successfully prepared Ru-decorated Zn0.5Cd0.5S nanorods by a photodeposition method.81 The photocatalytic formation rate of H2 and FAL were 870 and 855 μmol h−1 g−1, respectively. The enhanced performance can be attributed to the efficient separation and transport of photogenerated e− and h+ promoted by the strong interaction between Ru cocatalyst and Zn0.5Cd0.5S semiconductor. In addition, a superior adsorption of Ru atoms toward the OH group in FOL accelerated the selective oxidation. After that, Liu et al. found that the amorphous Ru–RuOx hybrid structure had ultrafast h+ trapping ability compared with Ru atoms.82 Due to the abundant atomic interface, amorphous RuOx sites can inherently capture h+ within only 100 fs and promote the subsequent electron transfer (ca. 1.73 ps). As a result, this Ru–RuOx catalyst triggered a long-lived state of charge separation, eventually increasing the H2 evolution rate to 4240 μmol h−1 g−1. Apart from monometal modification, a bimetallic Ni–Au modified g-C3N4 photocatalyst was synthesized by a step-by-step photodeposition method.83 g-C3N4 was the main light-trapping material for generating e−–h+ pairs. The localized surface plasmon resonance effect can be generated when Au nanoparticles are in a specific size range, which broadens the optical absorption region to generate more charge carriers. Hence the plasma Au nanoparticles further improved the efficiency of light utilization over the catalyst. Ni was not only the e− trap but also the active site of H2 evolution. Benefiting from the synergistic effect among three components, H2 evolution rate over the Ni–Au/g-C3N4 photocatalyst reached 3 fold over Au/g-C3N4. In addition to using metal as co-catalyst, Yang et al. employed Mo2C as co-catalyst to accept photogenerated e− and the reaction sites of H2 evolution.84 The Mo2C@ZnIn2S4 catalyst showed a high formation rate of H2 (2800 μmol h−1 g−1) and FAL (11330 μmol h−1 g−1). The performance was much better than those over ZnIn2S4 and Pt/ZnIn2S4.
| Entry | Catalyst | Solvent | Reaction conditions | HER (μmol h−1 g−1) | FAL (μmol h−1 g−1) | Ref. |
|---|---|---|---|---|---|---|
| H2 evolution rate (HER in μmol h−1 g−1) and FAL formation rate (μmol h−1 g−1) are used to evaluate the catalytic performances.a Conv., 100%; select., 100%.b Conv., 3%; select., >99%. | ||||||
| 1 | Ni/CdSa | H2O | Blue LED (440–460 nm, 8 W), 10 mM FOL, N2, 22 h | — | — | 59 |
| 2 | 1 wt% Ru/Zn0.5Cd0.5S | H2O | 300 W Xe lamp with a UV cutoff filter | 870 | 855 | 81 |
| 3 | Ru-RuOx/C3N4 | H2O | 300 W Xe-lamp, Ar | 4240 | 4500 | 82 |
| 4 | Ni–Au/CNb | H2O | Xe lamp (200 mW cm−2) for 5 h, 1 atm Ar, 8 °C | 471 | — | 83 |
| 5 | MO2C@ZnIn2S4 | H2O | 300 W Xenon lamp (420 nm), 10 °C, 4 h | 2800 | 11330 |
84 |
| 6 | ReS2/ZnIn2S4-Sv | H2O | Xenon lamp (250 mW cm−2), 15 °C, remove air | 1080 | 710 | 85 |
| 7 | LaVO4/g-C3N4 | H2O | Xenon lamp (250 mW cm−2), remove air, 15 °C | 287 | 950 | 86 |
| 8 | ZnIn2S4/Tp-Tta COF | H2O | LED (420 nm, 80 W), 3 h, 1 atm N2 | 9730 | 12100 |
32 |
Besides, the preparation of heterojunctions is also a good way to develop bifunctional catalysts. Hu et al. designed a new type 3D/3D ReS2/ZnIn2S4-Sv heterojunction presenting a bifunctional cellular layered structure.85 ReS2 and S vacancies (Sv) increased the specific surface area, accelerated the charge separation and inhibited the recombination of carriers. Such features benefited the photocatalytic reaction. In another case, the 2D/2D LaVO4/g-C3N4 heterostructure prepared by Li et al. also showed a good photocatalytic activity.86 The production rate of H2 and FAL were 3 times higher than that of the original g-C3N4. In order to further improve the catalytic efficiency, a S-type Znln2S4/Tp-Tta COF (i.e., the organic 1,3,5-triformylphloroglucinol-4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline covalent organic framework) heterojunction was synthesized by Sun et al. using an in situ synthetic strategy.32 As shown in Fig. 9, the layered sandwich structure of the catalyst can expand the interfacial charge transfer and promote the multiple scattering of incident light. Besides, there is huge internal electric field and low charge transfer resistance in S-type heterojunctions. This can significantly facilitate the separation and transfer of photogenerated charges. Therefore, the overall redox performance of this photocatalytic system is greatly improved. The evolution rate of H2 and FAL reach 9730 and 12100 μmol h−1 g−1, respectively, the highest values reported in the literature to date.
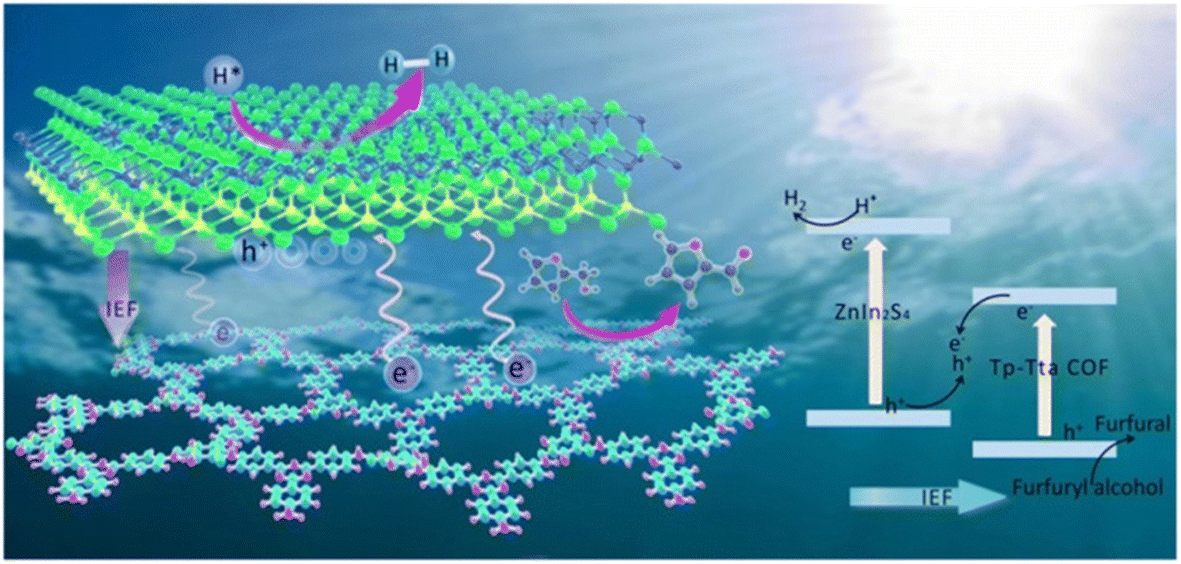 | ||
| Fig. 9 A possible mechanism of photocatalytic H2 release and FOL oxidation on the ZnIn2S4/Tp-Tta COF system. Reproduced from ref. 32, with permission of Royal Society of Chemistry, Copyright 2022. | ||
3.2. Photocatalytic oxidation of HMF combined with H2 evolution
As summarized in Table 7, Han et al. reported for the first time that Ni/CdS enabled selective oxidation of HMF to DFF under N2 atmosphere and visible light irradiation, while H2 formation was realized.59 This idea of combining semiconductor with cocatalyst aroused great interest for subsequent works. For instance, Pt/SGCN, Pt/CdS and Pt/Ti-MOF photocatalysts were successfully prepared by loading Pt as cocatalyst onto semiconductor carriers.29,87,88 As illustrated for Pt/CdS (Fig. 10), the formation of the Pt–CdS interface leads to e− transfer from semiconductor to Pt, due to the low work function of Pt nanoparticles. Therefore, HMF can be adsorbed on positively charged CdS through OH groups, while the furan ring of HMF can be adsorbed on the surface of Pt due to the strong interaction between Pt and π bonds in HMF. Under irradiation e− and h+ are generated on CdS semiconductor, and e− are moved to Pt. The adsorbed OH groups of HMF can be attacked by h+ in CdS, resulting in proton dissociation to form alkoxide radicals which further react with another h+ to produce the desired DFF. At the same time, protons released from HMF are transferred to nearby Pt nanoparticles, where these protons can be reduced by e− to generate H2. Therefore, the semiconductor-supported Pt achieves effective H2 evolution and selective formation of DFF (>99%, in select.) from HMF oxidation. Therein the Pt/CdS photocatalyst shows the highest conversion of 100% in the literature to date. Apart from semiconductors, Pt can be also loaded on donor–acceptor polymer, e.g., CMP-Tz (conjugated microporous polymers with triazine group as the central aryl unit).89 In the presence of ascorbic acid as a sacrificial agent, the Pt-CMP-Tz photocatalyst presented the highest H2 formation rate at 3207 μmol g−1 h−1. HMF, instead of ascorbic acid, can be also converted to DFF with a high selectivity. | ||
| Fig. 10 Proposed mechanism for photocatalytic dehydrogenation of HMF under visible light over the Pt/CdS nanocomposite. Reproduced from ref. 29, with permission of Elsevier, Copyright 2022. | ||
| Entry | Catalyst | Solvent | Reaction conditions | HER (μmol h−1 g−1) | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| H2 evolution rate (HER in μmol h−1 g−1), conversion of HMF and selectivity of DFF are used to evaluate the catalytic performances.a Presented in μmol h−1 m−2.b Obtained in CH3OH solvent.c The blank test without HMF. | |||||||
| 1 | Ni/CdS | H2O | Blue LED (440–460 nm, 8 W), 10 mM HMF/FOL, N2, 22 h | — | 20/100 | 100 | 59 |
| 2 | Pt/SGCN | H2O | LED (>400 nm, 100 mW cm−2), N2, 48 h | 36a | 38 | >99 | 87 |
| 3 | Pt/CdS | CH3CN + H2O | Blue LED, 14 h, N2 | 1393 | 100 | 99 | 29 |
| 4 | Pt/Ti-MOF | H2O | Visible light, 5 h | 727 | 56 | 99 | 88 |
| 5 | Pt-CMP-Tz | CH3CN + H2O | λ > 420 nm, 20 °C, ascorbic acid/20 mM HMF | 3207/1404 | 8 | 95 | 89 |
| 6 | NiS2/CdS | H2O | λ ≥ 420 nm, 25 °C10 h | 478 | 52 | 95 | 90 |
| 7 | NiS/Zn3In2S6 | H2O | Visible light (Xe lamp, 300 W), N2 | 120 | — | 94 | 91 |
| 8 | Cd0.7Zn0.3S/NiSe2 | CH3CN | 300 W Xe lamp (400–780 nm), 1 h, 1 atm Ar | 17 | 18 | 98 | 92 |
| 9 | P25-CuOx | CH3CN | UV-LED, 30 °C, Ar, 3h | ∼2b | 95 | 32 | 93 |
| 10 | Zn0.5Cd0.5S/1%MnO2 | H2O | 30 W white LEDs (λ ≥ 400 nm), 0.25 M Na2S/0.35 M Na2SO3, N2, 24 h, 25 °C | 25c | 0 | 0 | 95 |
| 11 | Zn0.5Cd0.5S/1%MnO2 | H2O | 30 W white LEDs (λ ≥ 400 nm), N2, 24 h, 25 °C | 1322 | 46 | 100 | 95 |
| 12 | 50% CdS/g-C3N4 | H2O | Visible light (Xe lamp, 300 W), 50 mL lactic acid solution | 263c | 0 | 0 | 94 |
| 13 | 50% CdS/g-C3N4 | PhCF3 | Visible light (Xe lamp, 300 W), O2, 2 h | — | 90 | 91 | 94 |
| 14 | ZnIN2S4/Nb2O5 | PhCF3 | 300 W xenon lamp, O2, 10 mL min−1, 3 h, 30 °C | — | 86 | 88 | 96 |
| 15 | ZnIN2S4/Nb2O5 | H2O | Simulated solar light, Pt as cocatalyst, N2, 3 h | 1286 | 22 | — | 96 |
| 16 | Zn0.5Cd0.5S–P | H2O | White LED light (30 × 3 W), Ar, 2 h | 419c | 0 | 0 | 97 |
| 17 | Zn0.5Cd0.5S–P | H2O | White LED light (30 × 3 W), Ar, 8 h | 786 | 40 | 65 | 97 |
| 18 | O-ZnInS-120 NSs | 1 M KOH | Visible light, 2.5 h | 1522 | 40 | 97 | 31 |
In addition to Pt as cocatalyst, inexpensive nickel compounds (i.e., NiS2, NiS and NiSe2) were coupled as cocatalysts with CdS, Zn3In2S6 and Cd0.7Zn0.3S semiconductors, respectively.90–92 NiS2, NiS and NiSe2 can form Schottky junctions with semiconductors because their metal properties are similar to those of precious metal Pt. Schottky junctions allow effective promotion of separation and transmission of photoexcited charges. The h+ in the semiconductor carriers can be captured by HMF to produce DFF and release protons, while the separated e−1 can react with protons on the surface of cocatalyst to produce H2. Besides, Giannakoudakis et al. confirmed that CuOx nanoclusters can serve as cocatalyst in a P25-CuOx (i.e., 2 nm CuOx dispersed on P25 surface) photocatalytic system.93 Therefore, it is reasonable to conclude that participation of photogenerated e− and h+ in the reaction is vital for the generation of DFF and H2. Hence an efficient separation of e− and h+ is the critical factor.
Instead of adding cocatalyst, the construction of heterojunctions is a more practical method. Wu et al. prepared a p–n heterojunction photocatalyst with Z-type mechanism by in situ growth of ultrasmall 2D CdS nanowires on the surface of 2D CN nanowires.94 This 2D/2D CdS/CN heterojunction exhibited superior photocatalytic behavior for oxidation and reduction. Both HMF conversion and DFF selectivity reached ca. 90% over the 50% CdS/CN composite after 2 h of visible light irradiation. In addition, Dhingra and Wang prepared Zn0.5Cd0.5S/MnO2 and ZnIn2S4/Nb2O5 p–n heterojunctions with Z-type mechanism.95,96 Both photocatalysts selectively oxidized HMF to DFF and exhibited H2 formation rate of ca. 1000 μmol g−1 h−1.
Alternatively, the separation of photogenerated e− and h+ can also be realized by surface modification of the catalyst. Ye et al. prepared a Zn0.5Cd0.5S–P photocatalyst and enriched S vacancies by doping P into ZnxCd1−-xS solid solution.97 Such surface vacancies were beneficial for electron accumulation and substrate adsorption, thus the H2 evolution rate was eight times higher than that of CdS-P. Zhu et al. treated ZnIn2S4 nanowires with oxygen plasma for 2 min and obtained a hierarchical porous O-ZIS-120 photocatalyst.31 It showed the highest H2 evolution rate of 1522 μmol h−1 g−1 in the current literature.
3.3. Photo(electro)catalytic oxidation of HMF to valuable chemicals combined with CO2 reduction
The use of fossil fuels has greatly promoted modern industrialization and economic development, but at the same time it has brought about serious environmental problems. It is worth noting that the concentration of CO2 in the atmosphere exceeds an astonishing value of 400 ppm (i.e., an increase of 120 ppm since the Industrial Revolution), resulting in global warming and a more extreme climate. In this context, the chemical reduction of CO2 with H2O to value-added compounds (i.e., CO, CH4, CH3OH and C2H6) by solar-driven semiconductors has been generally considered to be a promising technology to achieve carbon neutrality. However, the slow kinetics of semi-reaction by water oxidation leads to recombination of excited e− and unused h+, resulting in catalytic inefficiency. To solve this problem, a common strategy relies on using sacrificial e− donors as h+ cleaners. However, this method wastes the oxidation capacity of h+. Instead, using HMF as oxidation reactant and hydrogen donor can obtain value-added compounds and simultaneously improve the performance of CO2 reduction.As listed in Table 8, Fan et al. prepared a ZnNiFe-LDH (i.e., anionic compound of layered double hydroxides) catalyst by a coprecipitation method.33 As shown in Fig. 11, LDHs can store CO2 in the form of CO32− layers based on the strong affinity of LDHs to CO32−. In addition, the electron-deficient OH sites near the metal vacancies on the LDH layer contribute to adsorb and activate the OH group in HMF. The proximity of electron-rich Ni2+ to metal vacancies enables promotion of the activation of interlaminar CO32−. Therefore, HMF and interlayer CO32− can be composed for a redox reaction under light. After reaction, LDHs continually absorb CO2 in the air to supplement the CO32− consumed during reaction, and then use it in the photocatalytic redox reaction. The cumulative yields of CO and DFF reach 76 and 55 μmol g−1 after 4 h, respectively.
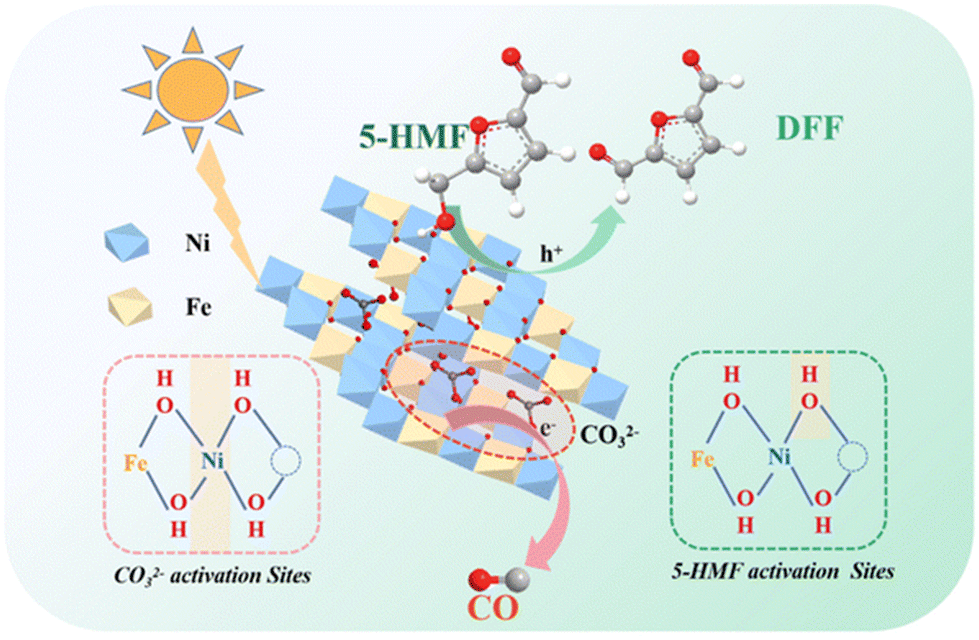 | ||
| Fig. 11 Proposed mechanism of the integration of CO2 photoreduction with oxidation of HMF. Reproduced from ref. 33, with permission of Royal Society of Chemistry, Copyright 2023. | ||
| Entry | Catalyst | Solvent | Reaction conditions | CO-ERa | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a CO evolution rate. b Faraday efficiency. | |||||||
| 1 | ZnNiFe-LDHs | CH3CN | 300 W Xe lamp, 30 °C, 0.2 MPa Ar, 4 h | 19 μmol h−1 g−1 | 25 | 71 (DFF) | 33 |
| 2 | TBP-CoPc/Py-TEMPO | 0.2 M Na2CO3 | CPE at 0.7 V vs. Ag/AgCl, 57.9 C | FEb: 97% | 100 | 91 (FDCA) | 98 |
Interestingly, Yang et al. used 4-(tert-butyl)-phenoxy-decorated cobalt phthalocyanine (TBP-CoPc) and pyrene-tethered 2,2,6,6-tetramethylpiperidin-1-oxy (Py-TEMPO) as reagents to construct a paired electrolysis system, which was non-covalently fixed on carbon nanotubes.98 As show in Fig. 12, a photovoltaic cell was used to catalyze the reduction of CO2 to CO and oxidize HMF to FDCA. In dual-electrode mode, the Faraday efficiencies (FE) of CO and FDCA were 97% and 91%, respectively, using a battery voltage at 2.5 V.
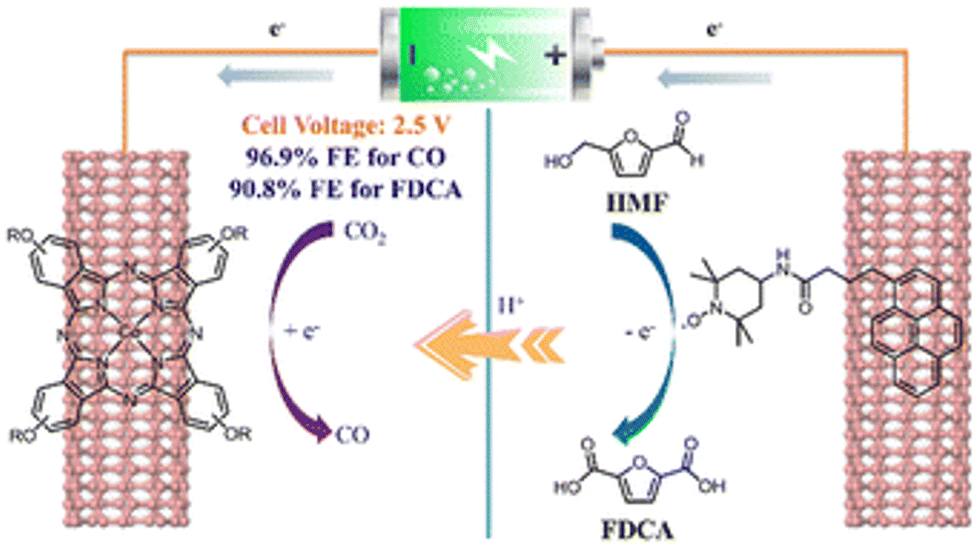 | ||
| Fig. 12 Schematic diagram of sunlight-driven paired electrolysis of the CO2RR and HMFOR by the TBP-CoPc/Py-TEMPOs photocatalyst. Reproduced from ref. 98, with permission of Royal Society of Chemistry, Copyright 2022. | ||
Obviously, the combination of photocatalytic HMF oxidation with CO2 reduction is of great significance to achieve carbon neutrality, but electrocatalytic CO2 reduction remains very challenging. Particularly, the thermodynamic stability and kinetic inertia of CO2 would lead to a slow reaction that highly competes with H2 evolution reaction. Therefore, there are very limited reports and further exploration is highly desired.
In short, the photocatalytic oxidation of furanic compounds (FOL and HMF) combined with H2 evolution or CO2 reduction shows a very high atom economy. It is greatly in line with green chemistry. However, the separation of photogenerated e− and h+ is essential for the catalyst to achieve the bi-objective. Thereby the catalyst design is often complicated. In addition, O2 molecules present in the system (an aerobic environment) can easily react with the removed protons from FOL/HMF to inhibit H2 evolution or CO2 reduction. Hence the reaction conditions also need to be O2-free. Therefore, further optimization of catalyst preparations and mechanistic studies are required.
4. Photocatalytic oxidation of HMF to FDCA, as well as HMFCA and FFCA
4.1. Photocatalytic oxidation of HMF to FDCA
Among various oxidation products from HMF, FDCA is undoubtedly the most valuable one. The symmetrical furan dicarboxylic acid shows very similar properties to petroleum-derived terephthalic acid, so it can be used as a substituted monomer for manufacturing renewable bioplastics. Obviously, photocatalytic oxidation of HMF to FDCA requires a stronger oxidation power (vs. DFF formation) of the catalytic system. But highly reactive oxidizing species can decompose HMF to undesired byproducts (i.e., CO2). Therefore, developing selective photocatalysts with a suitable energy-band structure is challenging yet important for the photooxidation of HMF to FDCA.As summarized in Table 9, Xu et al. combined metalloporphyrins with g-C3N4 to prepare CoPz/g-C3N4 as an efficient photocatalyst for oxidation of HMF in air under simulated sunlight.20 97% selectivity of FDCA at 99% conversion of HMF was achieved at 25 °C in alkaline aqueous solution (pH = 9.18). This may be the highest activity reported in the literature to date. As shown in Fig. 13, the strong interaction between CoPz and g-C3N4 not only improves the accessibility of the CoPz site and makes the catalyst recyclable, but also prevents the formation of ˙OH radicals by g-C3N4 and promotes the formation of reactive 1O2 at the CoPz site. These catalytic properties significantly boost the catalytic performance. Zhang et al. reported a visible light-driven Au–Ag/TiO2 catalytic system in Na2CO3 solution.99 Interestingly, the monometallic Ag nanocatalyst can only afford HMFCA, whereas Au and Au–Ag catalysts can produce FDCA under the same reaction conditions. This is because only Au nanoparticles can extract the H atom from the OH group in HMF.
 | ||
| Fig. 13 Possible mechanism for photocatalytic oxidation of HMF to FDCA over the CoPz/g-C3N4 catalyst. Reproduced from ref. 20, with permission of American Chemical Society, Copyright 2017. | ||
| Entry | Catalyst | Solvent | Reaction conditions | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | CoPz/g-C3N4 | H2O | Xe light (300–1000 nm, 0.5 W cm−2), Na2B4O7 buffer solution (40 mL), pH = 9.18, 1 bar air, 14 h, 25 °C | 99 | 97 | 20 |
| 2 | Ag1-Au2/TiO2 | H2O | λ > 420 nm, 0.3365 g Na2CO3, 5 h | 43 | 98 | 99 |
| 3 | Co3+@ZnO | H2O | λ ≥ 365 nm, air | 95 | 40 | 100 |
| 4 | (Cr3+or Co3+)-NiO | H2O | λ = 365 nm, 36 h | 95 | 50 | 77 |
| 5 | Fe@rGO | H2O | λ = 365 nm, 6 W, 36 h | 95 | 29 | 101 |
| 6 | Zn1−xCdxS | H2O | λ = 445 nm, 6 W, 24 h, air | 98 | 40 | 24 |
| 7 | ZnIN2S4-TU | H2O | λ = 445 nm, 6 W, 24 h | 92 | 43 | 67 |
| 8 | MCOF-Ti6BTT | CH3CN | Visible light (400–800 nm), 25 °C, 1 atm O2 | 100 | 95 | 102 |
Obviously, base additives are important for preventing the metal leaching with formation of carboxylate instead of free acid. But the extra acidification and purification of FDCA-salt would lead to a lower green index and a higher application cost. In order to promote the reaction without alkaline additives, Co3+@ZnO and (Cr3+or Co3+)-NiO catalysts were prepared by Pham and Park, respectively, by introducing charge-mismatched metal cations to semiconductors.77,100 Moderate selectivity of FDCA (40–50%) was obtained at a high conversion of HMF (95%) under base-free conditions. Pham et al. also reported a Cr-free and base-free Fe@rGO photocatalytic system.101 But this catalyst showed rather a low selectivity of FDCA (29%) at the same conversion of HMF. Pham et al. synthesized ZnxCd1−-xS nanoparticle photocatalysts with different concentrations of Zn2+ ions (x = 0.0, 0.3, 0.5 or 0.7).24 The approach was found to be effective for controlling the band gap and promoting e−–h+ separation, thereby improving the photocatalytic efficiency. In addition, the previous ZnIn2S4-TU catalyst with more Zn vacancies also enabled FDCA formation by extending the reaction time.67 However, the yield of FDCA (ca. 40%) was inadequate without alkaline additives. Instead, Chang et al. integrated Ti6-NH2 nano-sheet (i.e., a Ti cluster-based COF) with benzotrithiophene triformaldehyde (BTT) to obtain a MCOF-Ti6BTT photocatalyst.102 This integration provided strong visible-light absorption, effective e−–h+ separation efficiency, and appropriate photooxidation ability. Notably, the highest photocatalytic activity (i.e., >95% selectivity of FDCA at a full conversion of HMF) under base-free conditions has been reported to date in the literature.
4.2. Photocatalytic oxidation of HMF to HMFCA and FFCA
HMFCA is the paralleling intermediate to DFF during the first step of HMF oxidation. Thereby, it is difficult to selectively regulate the oxidation sequence of the CO or OH groups in HMF. Besides, the asymmetric structures of HMFCA and FFCA may hamper their direct use in downstream applications. Hence, there are only limited catalytic systems reported. As listed Table 10, Au/TiO2 and Ag/TiO2 catalysts enabled selective oxidation of HMF to HMFCA under visible light irradiation in Na2CO3 solution.103,104 Au/TiO2 reached 99% conversion and 95% selectivity, this being the best result in the literature to date. Notably, Xia et al. gained insight into the formation mechanism of HMFCA via selective oxidation of HMF over a Ru-CdS photocatalyst (i.e., Ru complex anchored onto CdS quantum dots) in N,N-dimethylformamide.60 As illustrated in Fig. 14, ˙OH or ˙O2− radicals can be generated over Ru-CdS under Ar or air atmosphere, respectively. Accordingly, such reactive species are found to be responsible for selectively oxidizing the OH or CO group in a HMF molecule. A high selectivity of HMFCA (93%) is obtained at 85% conversion of HMF under an air atmosphere.
 | ||
| Fig. 14 Schematic mechanism for selective photocatalytic oxidation of HMF to DFF or HMFCA by the Ru CdS catalyst. Reproduced from ref. 60, with permission of John Wiley & Sons, Inc, Copyright 2022. | ||
Table 11 displays the results for the photocatalytic oxidation of HMF to FFCA. A g-C3N4/NaNbO3 catalyst was prepared by calcining the mixture of NaNbO3 and melamine, showing 87% selectivity of FFCA at 36% conversion of HMF.105 As demonstrated in Fig. 15, ˙O2− radicals as the main active species can oxidize HMF to DFF with formation of adequate H2O2 which further oxidizes DFF to FFCA. The polarity of substrates greatly influences product selectivity. Since the polarity of DFF is weaker than those of HMF and FFCA, the oxidation rate of DFF is faster than that of HMF, promoting FFCA formation. Besides, Li et al. used polyoxometalate (i.e., a tetrabutylammonium salt of W10O324−) as photocatalyst to realize 88% conversion of HMF and 32% selectivity of FFCA in acetonitrile under UV irradiation.106
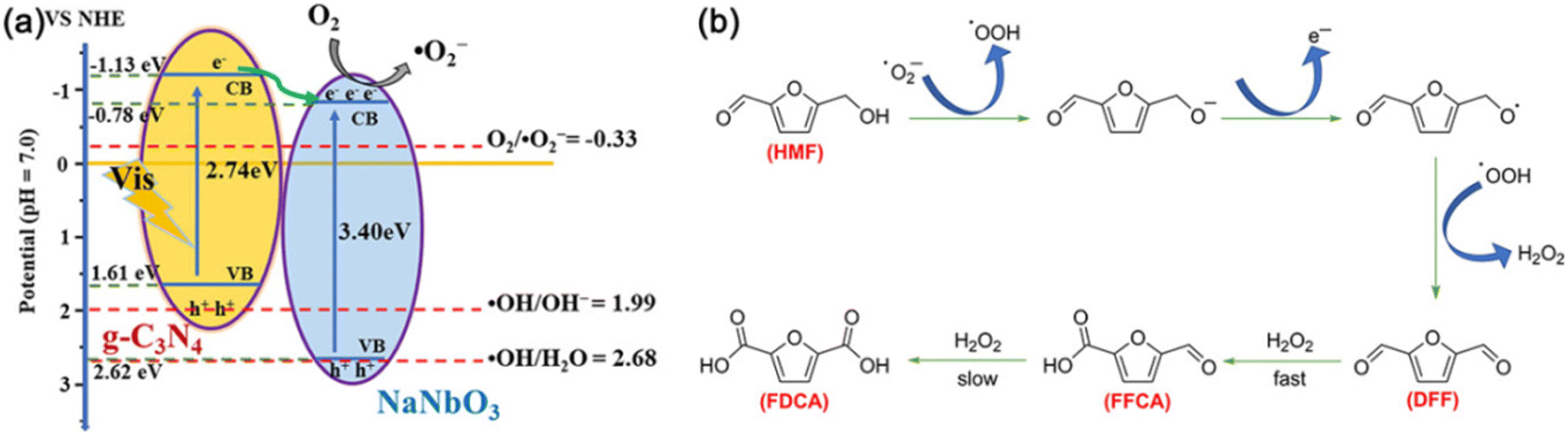 | ||
| Fig. 15 (a) Schematic diagram of e−–h+ pair separation over the g-C3N4/NaNbO3 heterojunction. (b) Proposed mechanism for the g-C3N4/NaNbO3-catalyzed oxidation of HMF under visible light. Reproduced from ref. 105, with permission of Elsevier, Copyright 2020. | ||
In conclusion, the addition of base additives and/or the use of organic solvents are often required to achieve better results in the photocatalytic selective oxidation of HMF to FDCA. Therefore, it is also necessary to further optimize the preparation of catalysts and modulate the appropriate energy-band positions in anticipation of higher FDCA yield in the base-free and aqueous phase. The studies on HMFCA and FFCA intermediates can help us to further understand the routes and mechanisms of the oxidation processes. It is of guiding significance to improve the construction of the theoretical system of photocatalysis.
5. Photo(electro)catalytic oxidation of HMF
Photo(electro)chemical cells can directly use photogenerated e−–h+ pairs in semiconductor electrodes for fuel production, just as nature does through photosynthesis. In a typical H2-producing photoelectrochemical cell, water reduction (H2 evolution) in the cathode is accompanied by anodic water oxidation (O2 evolution). However, water oxidation is not kinetically favorable and the produced O2 is of little value. Therefore, using photo(electro)chemical oxidation of HMF as an anodic reaction system would be a more efficient and practical scheme.As summarized in Table 12, Cha et al. constructed a photo(electro)chemical cell using photo(electro)chemical oxidation of HMF mediated by TEMPO as an anodic reaction.107 In this unit, the n-type nano-porous BiVO4 photoanode absorbs light and produces e−–h+ pairs (Fig. 16). The e− in the conduction band is transferred to the Pt counter electrode to reduce the water to H2. The h+ in the valence band is used for TEMPO-mediated HMF oxidation. Because the valence-band position of BiVO4 is 2.4 V (vs. RHE), the photogenerated h+ in the valence band of BiVO4 has sufficient overpotential to oxidize TEMPO before any external bias voltage is applied. Therefore, compared with electrochemical oxidation of HMF using metal electrodes, the use of photogenerated h+ in the valence band of BiVO4 can significantly lower the applied bias voltage to initiate TEMPO oxidation. The bias voltage applied is to enhance separation of e− and h+ so that the h+ on the semiconductor surface can be used in the oxidation reaction rather than changing the overpotential of the oxidation reaction. Surprisingly, this smart system has provided >99% conversion of HMF and selectivity of FDCA. The high FE (93%) of FDCA produced at 40 C (charge passage) indicated that TEMPO-mediated HMF oxidation is more beneficial for kinetics rather than water oxidation.
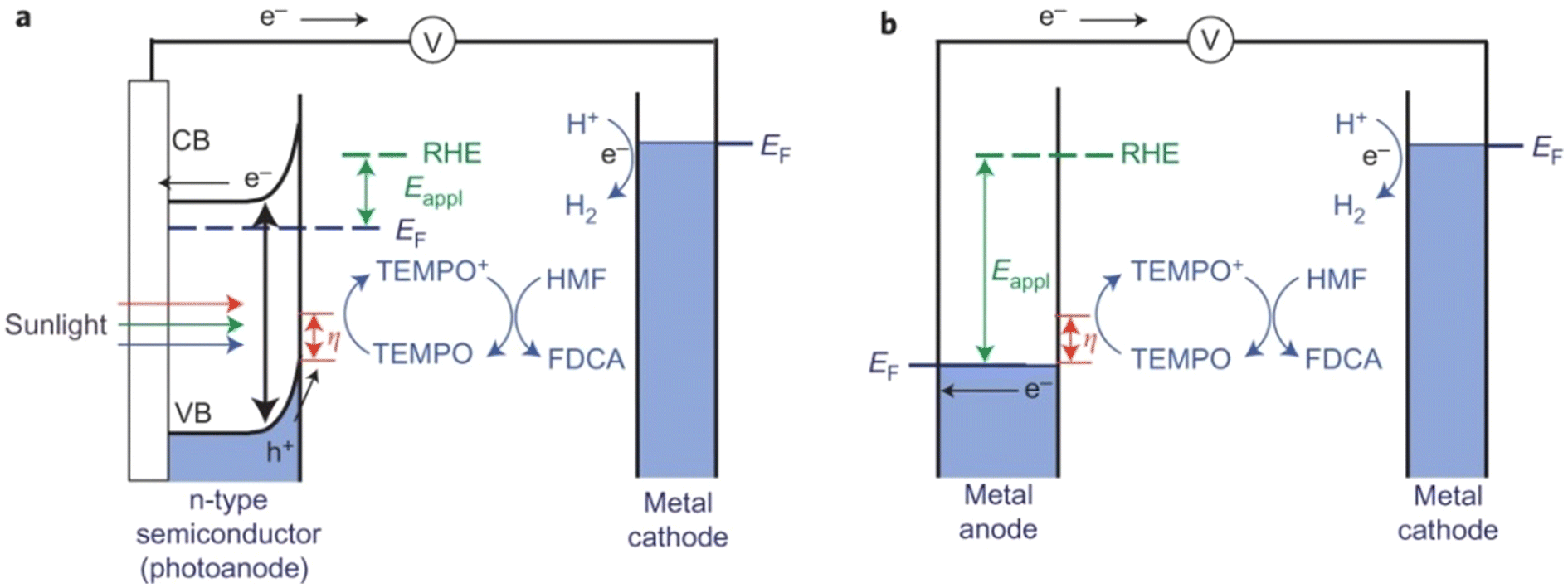 | ||
| Fig. 16 Schematic comparison of (a) photo(electro)chemical TEMPO-mediated HMF oxidation. (b) Electrochemical TEMPO-mediated HMF oxidation. CB, conduction band; EF, Fermi energy. Reproduced from ref. 110, with permission of Springer Nature, Copyright 2015. | ||
| Entry | Photo-anode | Reaction conditions | Main product | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | BiVO4 | AM 1.5 G illumination (100 mW cm−2), 1.04 V vs. RHE in a 0.5 M borate buffer solution (pH = 9.2) containing 7.5 mM TEMPO, 40 C | FDCA | >99 | >99 | 107 |
| 2 | BiVO4/CoPi-30 | Simulated solar illumination (AM 1.5 G, 100 mW cm−2), 0.64 V vs. RHE for 2.7 h, 5.0 mM TEMPO in sodium borate electrolytes (pH = 9.2), 12 C | FDCA | 99 | 89 | 108 |
| 3 | D-Lig-Fe/NiCoB | 5 mM HMF (1.6 V) 4 h, NiCoB catalyst-based electrocatalysis | FDCA | 99 | 80 | 109 |
| 4 | Ti/TiO2NT-1h-500-Pt | Fluorescent lamps (Philips, 8 W) emitting mainly at 365 nm, 150 mL 50 mM Na2SO4, 0.50 V bias and at pH = 5, 3 h, 25 °C | DFF | 59 | 40 | 110 |
On this basis, Chadderdon et al. used BiVO4 modified by CoPi as photoanode to drive TEMPO-mediated oxidation of HMF to FDCA.108 Compared with BiVO4, BiVO4/CoPi-30 lowered the potential required for TEMPO oxidation by ca. 0.5 V. To reducing the cost that originated from the TEMPO additive, Zhao et al. effectively converted lignin into photothermal materials (D-Lig-Fe) through demethylation of lignin and coordination with Fe3+, with a photothermal efficiency of 36%.109 Solar-thermal-electric conversion was realized by coupling D-Lig-Fe with a thermoelectric generator. Then, the generated voltage was applied to convert HMF to FDCA with a high conversion (99%) and selectivity (80%). Different from FDCA as the final product, Özcan et al. used Ti/TiO2NT as a photoanode for the first time to synthesize DFF.110 This photo(electro)chemical cell showed 59% conversion of HMF with 40% selectivity of DFF at a bias voltage of 0.5 V.
To sum up, electrochemical oxidation can provide the advantages of controlling oxidation potential and monitoring reaction rate by current, which may provide additional insights into the mechanism. However, there have been very few reports so far on photo(electro)catalytic oxidation of HMF to value-added chemicals. Because water oxidation is the main competitive reaction, it could limit the FE of HMF oxidation. Therefore, the developed photoanode needs to be able to selectively adsorb and activate HMF so as to effectively reduce the oxidation overpotential of HMF and eliminate the interference of water oxidation.
6. Photo(electro)catalytic reduction of HMF and FAL
Reports on the photo(electro)catalytic reduction of HMF and FAL are relatively scarce compared with their oxidation reactions (Table 13). There is only one article to date on the photo(electro)chemical reduction of HMF. Roylance et al. constructed a photo(electro)chemical cell by using BiVO4 semiconductor as anode and AgGD as cathode.111 As shown in Fig. 17, the h+ on BiVO4 surface is used to oxidize water to O2 and the e− in BiVO4 conduction band is transferred to the AgGD cathode to reduce HMF to BHMF. The electrode can diminish energy input by directly using solar energy. In electrochemical batteries, it is necessary to provide sufficient overpotential for the cathodic reaction (i.e., HMF reduction) and anodic reaction (i.e., water oxidation) through external bias voltage. The FE and selectivity of BHMF both reach ca. 95% when the bias voltage is 1.0 V.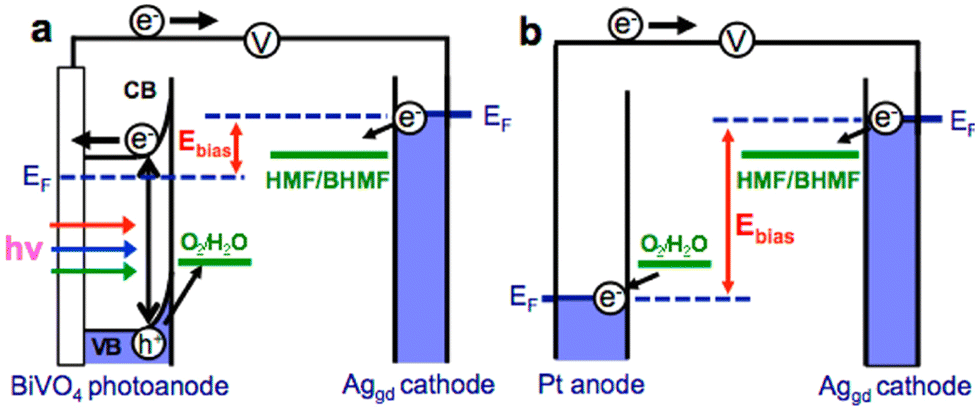 | ||
| Fig. 17 Schematic diagrams comparing the external bias necessary to achieve HMF reduction for (a) a photo(electro)chemical cell and (b) an electrochemical cell. Reproduced from ref. 111, with permission of American Chemical Society, Copyright 2016. | ||
| Entry | Catalyst | Solvent | Reaction conditions | Main product | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Presented in yield (%). b Presented in productivity (mmol g−1 h−1). | |||||||
| 1 | BiVO4 | H2O | AM 1.5 G illumination (100 mW cm−2), 1.0 V, 0.5 M borate buffffer solution (pH = 9.2) electrolytes, 5 C | BHMF | — | 95 | 111 |
| 2 | Pt/g-C3N4 | H2O + TEOA | Visible light (210 W xenon lamp), TEOA (0.5 mL), 4 h, 80 °C | BHMF | 7a | 112 | |
| 3 | NiTiO3/ZnIn2S4 | H2O + TEOA | λ > 420 nm, 10 v/v% TEOA, ultrasonicated for 15 min and saturated with N2 for 30 min | BHMF | >99 | 100 | 113 |
| 4 | PtNi/TiO2 | H2O + CH3OH | 300 W Xe lamp (λ = 320–780 nm), 1 atm Ar, 30 °C, CH3OH (25 mL) | BHMF | 100 | 100 | 114 |
| 5 | WO3/PdOx@C | H2O/CH3OH (3:1) |
Visible light (≥420 nm, 250 W, tungsten lamp, with a UV cutoff filter), KOH to adjust the pH = 7.5–8.2 | BHMF | 95 | 87 | 115 |
| 6 | A-TiO2 | EtOH | 25 W UV light for 1 h, 1 atm N2, 25 °C | FOL/BHMF | 99/>99 | 99/99 | 116 |
| 7 | Au/SiC | IPA | Xe lamp (400–800 nm, 1.0 W cm−2), Ar, KOH (20 mg), 4 h, 20 °C | FOL/BHMF | 100/90 | 100/93 | 21 |
| 8 | Pd/MIL-101(Fe)-NH2 | CH3CN | Xe lamp, TEA (0.6 mL), HCOOH (0.4 mL), 6 h, 25 °C | FOL/BHMF | 47/35 | 62/77 | 117 |
| 9 | Pt/SnNb2O6 | CH3OH | Xe lamp irradiation (λ ≥ 400 nm), 2 h, 1 atm H2 | FOL | 100 | 100 | 119 |
| 10 | Cu@C-600 | IPA | Visible light (0.5 W cm−2), 1 atm H2, 24 h, 100 °C | FOL | 99a | 118 | |
| 11 | TiO2 | 2-Pentanol | UV light, hydrogen-free conditions, 25 °C, 30 min | FOL | >99 | >99 | 120 |
| 12 | Cu/Cu2O-MC | IPA | Visible light, N2, K2CO3 (1.44 equiv.), 14 h | FOL | 94 | 96 | 121 |
| 13 | CTP-Th | H2O | Xe lamp (300 W, λ > 420 nm, 100 mW cm−2), Ar, 5 mmol ascorbic acid, pH = ∼1.6 | FOL | 0.5b | 122 | |
| 14 | ZnIn2S4-ADH | PBS solution | Visible light irradiation, pH = 8.0, 0.5 mM of [Cp*Rh(bpy)H2O]2+, 100 mM of L-ascorbic acid, and 1.0 mM of NAD+ | FOL | 100 | 79 | 123 |
Guo and Dhingra successfully photoreduced HMF to BHMF over a Pt/g-C3N4 and a NiTiO3/ZnIn2S4 catalyst, respectively.112,113 The reactions were driven by visible light in water with triethylamine as sacrificial e− donor. The NiTiO3/ZnIn2S4 catalyst presented 100% selectivity of BHMF at almost full conversion of HMF. That was the highest yield reported in the literature to date using water as hydrogen source. As illustrated in Fig. 18, the high photocatalysis of NiTiO3/ZnIn2S4 benefits from the Z-type heterojunction between NiTiO3 and ZnIn2S4, which promotes an easy separation and migration of interfacial charge carriers.
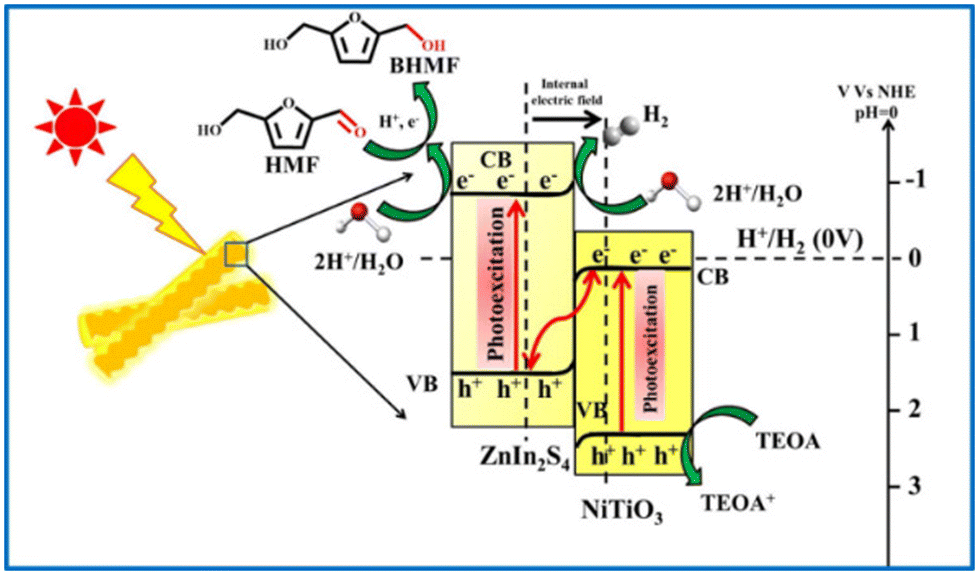 | ||
| Fig. 18 Pictorial representation of Z-scheme mechanism for photocatalytic H2 generation and subsequent HMF reduction by NiTiO3/ZnIn2S4 hierarchical nanostructures. Reproduced from ref. 113, with permission of Elsevier, Copyright 2022. | ||
However, using water as hydrogen donor would easily generate reactive oxygen species to interfere with the hydrogenation of HMF. In this context, Cui and Kumar, respectively, obtained BHMF over the PtNi/TiO2 and WO3/PdOx@C photocatalysts using methanol as hydrogen donor.114,115 The strong electron coupling between PtNi alloy and TiO2 not only ameliorated the charge transfer, but also promoted the evolution of surface chemical bonds during photoreduction of HMF. As a result, a highest yield of BHMF (100%) can be obtained on PtNi/TiO2, and the reaction conditions (i.e., base-free, H2-free) were among the mildest in the reported literature.
Interestingly, a few catalysts have been shown to enable photoreduction of both HMF and FAL into BHMF and FOL. Qiao et al. obtained BHMF and FOL on the amorphous TiO2 (A-TiO2) photocatalyst using ethanol as hydrogen source.116 The yields of desired products achieved around 99%. As displayed in Fig. 19, the surface of A-TiO2 stores e− under UV irradiation while the h+ can deprotonate ethanol into acetaldehyde. When the system is switched to a “dark” state, the CO group in HMF is attracted by the hydrogen bond interaction with the surface OH group. At the same time, the stored h+–e− pairs are subjected to proton-coupled electron transfer to achieve highly selective hydrogenation of HMF. Besides, Hao et al. used tert-butanol as hydrogen donor for Au/SiC catalyzed photoreduction of HMF and FAL.21 Therein 100% yield of FOL was attained. Similarly, Dong et al. realized BHMF and FOL formation on a Pd/MIL-101(Fe)-NH2 photocatalyst using formic acid as hydrogen source.117
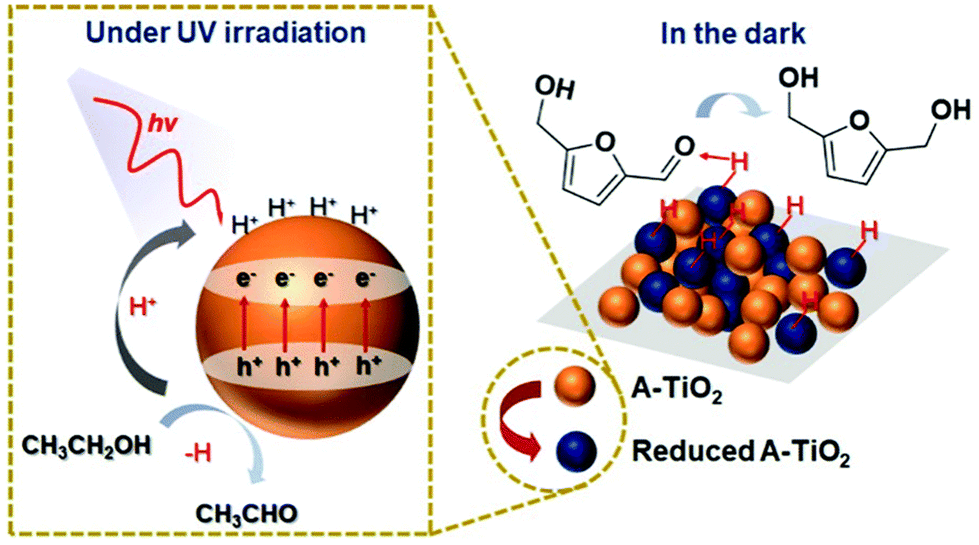 | ||
| Fig. 19 Hypothetical mechanism of HMF hydrogenation to BHMF over A-TiO2. Reproduced from ref. 116, with permission of Royal Society of Chemistry, Copyright 2019. | ||
FAL in comparison with HMF has no interference from the OH group attached to furan ring, thereby photohydrogenation of FAL may be easier. Firstly, molecular H2 can serve as reductant and hydrogen donor. Wang et al. prepared a Cu@C-600 catalyst that achieved 99% yield of FOL under 1 atm of H2 and visible light irradiation.118 However, a high temperature of 100 °C was required. Also under a hydrogen atmosphere, ultrathin SnNb2O6 nanosheets with functional Pt nanoparticles allowed the use of H2 to selectively phororeduce FAL to FOL. This composite photocatalyst showed the highest performance (i.e., 100% yield of FOL) without any additives.119
However, the use of H2 would bring certain safety risks and cost problems. In light of this, the catalytic transfer hydrogenation using organic solvents as hydrogen donor could be alternative route. Nakanishi et al. investigated the selective hydrogenation of FAL to FOL on a TiO2 catalyst at room temperature in 2-pentanol.120 FAL was almost completely converted to FOL under ultraviolet irradiation for 30 min. It was also found that 2-pentanol was oxidized to 2-pentanone by photogenerated h+. Zhang et al. prepared a Cu/Cu2O-MC (mesoporous carbon) photocatalyst which can directly convert FAL (94%) into FOL (96%) in iso-propanol at room temperature.121
Other than the metal photocatalysts, Hu et al. reported a covalent triazine polymer containing thiophene (CTP-Th) as visible-light responsive catalyst for photohydrogenation of FAL to FOL.122 The thiophene and triazine units can be expressed as electron donor (D) and receptor (A), respectively. The D–A structure promoted the delocalization of π electrons and enhanced charge separation. Under visible light irradiation (λ > 420 nm), CTP-Th can hydrogenate FAL to FOL at a rate of ca. 0.5 mmol g−1 h−1 using ascorbic acid as a h+ sacrificial agent. In addition, Zhao et al. constructed a sustainable photoenzyme coupling system.123 Instead of the conventional active cells, a ZnIn2S4 photocatalyst enabled driving of the regeneration of biocoenzyme nicotinamide adenine dinucleotide (NADH) with a rate of 90%. NADH served as the reductant to realize the selectively catalytic reduction of FAL. Full conversion of FAL with 79% selectivity of FOL was obtained in water. This result showed a high compatibility between photobased and bio-based catalysts.
In short, most of the reported catalysts could photoreduce HMF and FAL with superior conversion. However, expensive precious metals are needed, the preparation cost is high, and various additives are required. Therefore, novel catalysts with better selectivity and efficiency under lower costs and milder conditions are still highly desired.
7. Photocatalytic reductive coupling of FAL and HMF
FAL and HMF can be upgraded by chain-extension reaction to high-energy density diesel or jet fuels. It is particularly difficult to selectively produce biofuel precursors through homo-coupling or cross-coupling of furfuryl platform molecules, and despite the significance of this type of reaction, only limited success has been achieved.124 Therefore, the photocatalytic reductive coupling of FAL and HMF still remains challenging for biomass valorization. As listed in Table 14, Wu et al. reported for the first time that the crystal plane of TiO2 can regulate the product selectivity during photocatalytic reductive coupling of HMF.28 As demonstrated in Fig. 20, the surface oxygen vacancy plays a decisive role. The catalyst without surface oxygen vacancy (i.e., the (110) facet) is beneficial for the reductive coupling of FAL to hydrofuroin (HDFO) or furoin (FO), i.e., high-quality fuel precursor. The selectivity of coupling products can reach as high as 100%. On the other hand, the catalyst with abundant oxygen vacancies (i.e., the (001) facet) gives priority to the hydrogenation of FAL to FOL.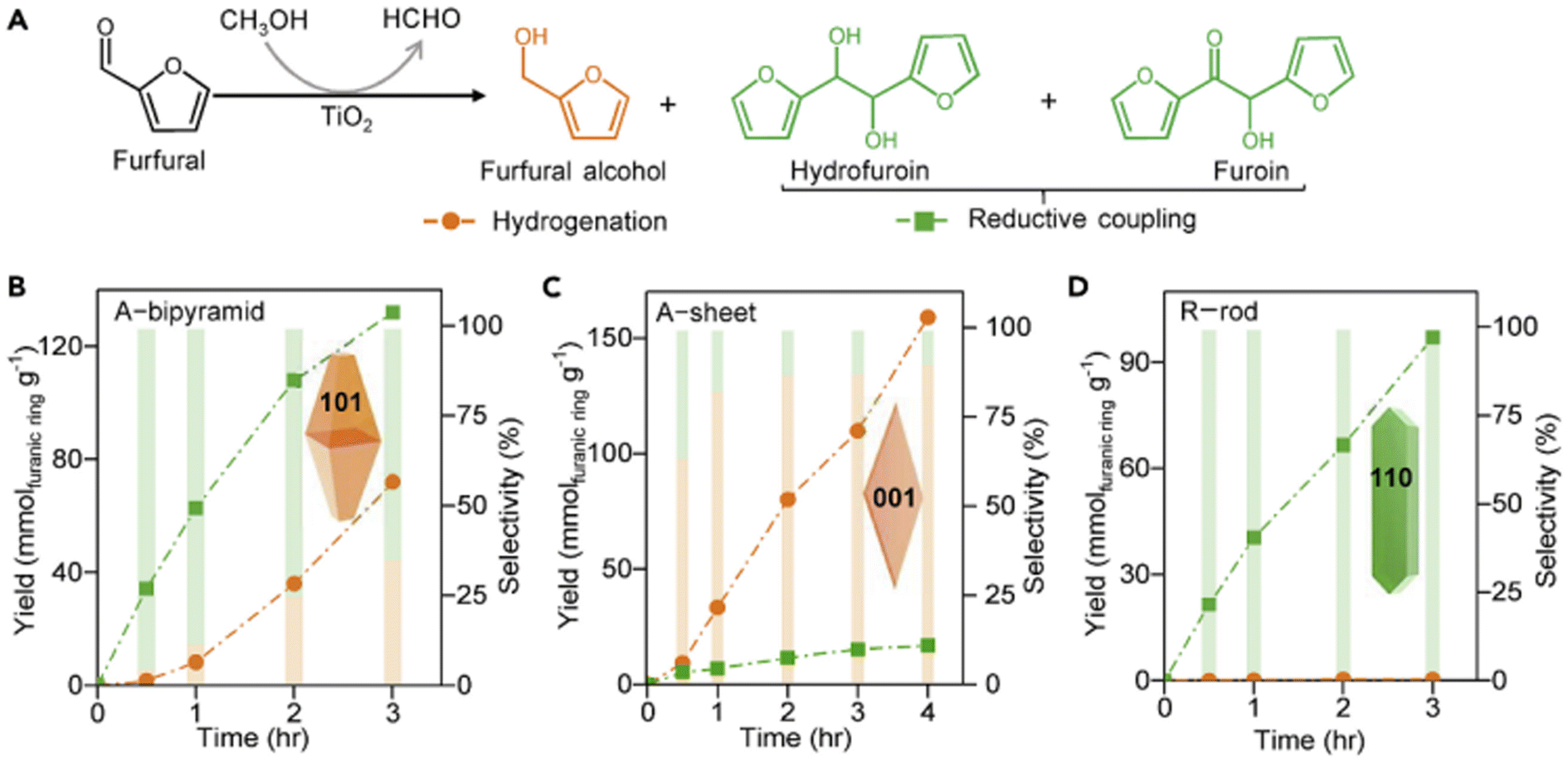 | ||
| Fig. 20 Photocatalytic conversion of FAL over TiO2 nanocrystals. (A) Structures of products from hydrogenation of CO group and reductive coupling of FAL. Variations of product yield and selectivity with time over (B) A-bipyramid, (C) A-sheet, and (D) R-rod. Reproduced from ref. 28, with permission of Elsevier, Copyright 2020. | ||
| Entry | Catalyst | Solvent | Reaction conditions | Main product | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | TiO (110) | CH3OH | Xe lamp (300 W, 320–780 nm, 600 mW cm), N2, 4 h, 40 °C | HDFO + FO | — | 100 | 28 |
| 2 | 1% Cu/P25 | CH3OH | λ = 365 ± 5 nm, CH3OH (1 mL), 1 atm Ar, 6 h | HDFO | 100 | 50 | 125 |
| 3 | Ag1.0In1.5Zn0.3S3.3 | D2O | 10 W LED (528 nm), 25 °C, p-toluenethiol (7.5 mmol), 6 h | HDFO | 95 | 100 | 126 |
| 4 | 1% MoS2/CdS | CH3CN/H2O | Blue LED, 12 h, N2 | BHH | 35 | 9 | 29 |
Following that, Lv et al. prepared a series of metal-supported P25 catalysts (Cu, Ni, Pt and Pd).125 Among them, 1% Cu/P25 could completely convert FAL in methanol to HDFO (50%, in select.) via C–C coupling. Notably, when 50 μL of H2O was added to the reaction mixture, the selectivity of HDFO significantly dropped. Interestingly, this phenomenon was observed over all prepared catalysts. Further characterizations revealed that a trace of H2O increased the charge transfer resistance, which led to a slow interfacial charge transfer and inhibited reductive coupling reaction. Therefore, facilitating the interfacial charge transfer is the key to improving the selectivity of coupling products. Kowalik et al. prepared non-stoichiometric Ag1.0In1.5Zn0.3S3.3 nanocrystals for photoreduction of FAL using p-toluenethiol as reductant and D2O as solvent.126 It was disclosed that base additives (i.e., AcOCs, Et3N, and tert-BuOK) were essential for the selective formation of FOL. But the absence of alkali can totally switch the product to HDFO through C–C coupling, at a high conversion of FAL (95%).
Regarding the photoreductive coupling of HMF, only a MoS2/CdS nanocomposite catalyst has been reported in the literature.29 The OH group in HMF was adsorbed on CdS and attacked by the proton-coupled electron transfer steps to form keto free radicals. The coupling of two keto free radicals gave rise to the production of 5,5′-bis(hydroxymethyl)hydrofuroin (BHH), in addition to the main product of DFF. However, the selectivity of BHH would be rather low (9%).
In summary, it can be concluded from these limited reports that the adsorption state of carbon center free radicals on the surface of a photocatalyst is the key factor in determining the selectivity of the reductive coupling reaction. Therefore, modifying the surface structure of the catalyst and regulating the adsorption energy are effective approaches.
8. Other unconventional and promising photocatalytic reactions of FAL and HMF
Last but not least, some unconventional and promising photocatalytic reactions of FAL and HMF, such as oxidative decarboxylation, ring-opening and so forth, have emerged and received attention very recently. As summarized in Table 15, Esser et al. first reported that FAL could be converted to 5-hydroxy-2 (5H)-furanone (HFO) in ethanol through oxidative decarboxylation using organic dyes as photocatalysts.35 Furanone is a ubiquitous component found in a variety of natural products and can be used as synthetic intermediate for many bioactive chemicals, such as pesticides, prostaglandins and alkaloids.127 In light of this pioneering study, Hermens et al. used methylene blue as a photocatalyst to obtain 100% conversion of FAL and 85% selectivity of HFO.128 In the same way, Huang et al. reported the photocatalytic conversion of FAL over organic dyes (i.e., rose bengal (RB), eosin Y (EY) and methylene blue (MB)) in water.38 Unfortunately, a low yield of HFO (<35%) was obtained in organic solvents with the self-made photoreactor. Compared with FAL, 2-furoic acid (FCA) was the preferred substrate for the photocatalytic synthesis of HFO. Moreover, the solvent effect was significant. The yields of HFO obtained from FCA over the series photocatalysts in water medium still remained very low (22–27%). However, the yield can be fairly good (>90%) in organic solvents. Interestingly, using a phosphate buffer together with acetone enabled a further increase in the yield of HFO up to 94%.| Entry | Catalyst | Solvent | Reaction conditions | Main product | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Presented in yield (%). | |||||||
| 1 | Organic dyes | EtOH | Lights, in alcohols, <40 °C | HFO | >99 | >95 | 35 |
| 2 | Methylene blue | MeOH | 10 W LED, 1 L flask | HFO | 100 | 85 | 128 |
| 3 | EY/RB | H2O | 30 W green LEDs (530–540 nm), 4 mL of phosphate buffer (0.2 M, pH = 7), 25 °C, 9 h | HFO | 97 | 99 | 38 |
| 4 | EY@DVB | H2O | 30 W green LEDs (530–540 nm), 25 °C, 200 rpm, 48 h | FSA | 94 | 99 | 38 |
| 5 | SGCN | CH3CN | Solar simulator (AM 1.5 G, 100 mW cm−2), 30 h, 25 °C | MAN/HFO/SAN/β-FAA | >95 | 42/33/8/11 | 36 |
| 6 | P(Pd)-PDMA-PPEGMA | MeOH | λ = 350–650 nm, purged with O2 for 30 min | HFO | 90 | 100 | 133 |
| 7 | 1%M·CN | Solvent free | Sunlight (3 days), 36 ± 3 °C, 82.4–94.5 mW cm−2, H2O2 (1.2 mmol)/O2/O2/O2 | MA/FU/MA/HFO | 98/94/94/94 | 100/37/40/23 | 37 |
| 8 | 1%Pd@E-g-C3N4 | IPA | Light source (150 W LED), IPA (5 mL), 2 bar H2, 1 h | FOL | 65 | 90 | 34 |
| 9 | 3%Pd@E-g-C3N4 | IPA | Light source (150 W LED), IPA (5 mL), 2 bar H2, 5.5 h | THFA | 100 | 100 | 34 |
| 10 | 3%Pd@E-g-C3N4 | CH3CN | Light source (150 W LED), THFA (0.1 mmol), CH3CN (3 mL), 8 h, 1 bar O2 | GBL | 75 | 100 | 34 |
| 11 | RuphenPy | H2O | 525 nm LED, 25 °C, 9 h, air | HMFO | 85 | 94 | 39 |
| 12 | RuphenPy | H2O | 525 nm LED, ice bath, 9 h, air, NaOH (45 mM) | HKPA | 62a | 39 | |
In order to realize a more efficient conversion of FAL to HFO in the aqueous phase, a whole-cell biocatalyst (i.e., E. coli co-expression of vanillin dehydrogenase (VDH2) and NADH oxidase (NOX), E. coli_CtVDH2_NOX) was developed to oxidize FAL to FCA. Therefore, E. coli_CtVDH2_NOX cells were combined with RB to synthesize HFO from FAL. Notably, this unique mode attained an excellent result under green LED irradiation, i.e., 97% conversion of FAL and 99% selectivity of HFO. In addition, the combination of E. coli_CtVDH2_NOX cells and EY@DVB (i.e., weakly basic anionic resin based on styrene-divinylbenzene (DVB) copolymer) can be used for synthesis of fumaric semialdehyde (FSA). FSA is a versatile intermediate usually used in the synthesis of value-added chemicals because it can be easily oxidized and reduced.38 As shown in Fig. 21, FAL is completely oxidized to FCA by E. coli_CtVDH2_NOX cells within 2 h, and FCA is subsequently ring-opened to FSA with an outstanding yield (93%) by 1O2 photooxygenation on EY@DVB. This is the very first report on FSA formation from FAL ring-opening by photocatalytic bio-oxidation.
 | ||
| Fig. 21 Schematic illustration of photocatalytic FAL to HFO over organic dye. Reproduced from ref. 35, with permission of John Wiley & Sons, Inc., Copyright 1994. Organic dye combined with biological and chemical photocatalysts for FAL to HFO or FSA. Reproduced from ref. 38, with permission of Royal Society of Chemistry, Copyright 2021. | ||
Based on the above work, HFO with one less carbon has been obtained from FAL by photooxidative decarboxylation. However, the specific reaction mechanism remains unclear. Chauhan et al. reported a mesoporous graphite carbon nitride (SGCN) as photocatalyst.36 It showed a superior FAL conversion (>95%) with selectivity of 42% and 33% for maleic anhydride (MAN) and HFO, respectively. The selectivity of MAN can be greatly increased up to 66% even under natural sunlight, which became a groundbreaking approach for the sustainable production of MAN. MAN is an important raw material for the synthesis of unsaturated polyester resins.129 In addition, the plausible mechanism of photooxidation of FAL was investigated by scavenger experiments and electron spin resonance spectroscopy. As demonstrated in Fig. 22, FAL is initially converted to FCA via photooxidation by 1O2 and then decarboxylated to furan. Further oxidation gives rise to the formation of 2(5H)-furanone which is then oxidized to HFO. Finally, MAN is produced by further oxidation of HFO. Besides, HFO shows partial tautomerism. Hence, succinic anhydride (SAN) and cis-β-formylacrylic acid (β-FAA) are formed by in situ rearrangement during the reaction. SAN is used as an intermediate in the chemical, pharmaceutical and food industries, while β-FAA is an intermediate for synthesizing furanones.130,131 MAN can be continually transformed to maleic acid (MA) by photooxidation by 1O2 species.
 | ||
| Fig. 22 (a) Generation of 1O2 on the surface of SGCN via h+, (b) 1O2 generation via triplet state energy transfer, and (c) plausible mechanistic pathway of FAL photooxidation. Reproduced from ref. 36, with permission of Royal Society of Chemistry, Copyright 2022. | ||
MA as an unsaturated dicarboxylic acid is a critical raw material in the manufacture of unsaturated polyester resins, surface coatings, plasticizers, pharmaceuticals, and fine chemicals.132 In order to further improve the yield of MA, Ebrahimian et al. combined expired metformin drugs with urea to prepare a g-C3N4 photocatalyst with a high N/C ratio.37 Benefiting from the high specific surface area and the efficient separation of photogenerated e−-h+ pairs, this photocatalyst achieved 98% conversion of FAL and 100% selectivity of MA using H2O2 as oxidant.
In order to further promote the yield of HFO, Seo et al. synthesized a Pd-containing porphyrin core (P(Pd)) amphiphilic block copolymer (P(Pd)-PDMA-PPEGMA) by Ru-catalyzed atom transfer radical polymerization.133 This copolymer had a hydrophilic outer layer composed of poly(dodecyl methacrylate) (PDMA) and polyethylene glycol methacrylate (PEGMA). As illustrated in Fig. 23, the relatively hydrophobic FAL molecule diffuses into hydrophobic blocks and is adsorbed through the hydrophobic–hydrophobic interaction. After that, the adsorbed FAL reacts with the 1O2 species formed by the porphyrin core, leading to hydrophilic products (i.e., HFO and methyl formate). As a result, the hydrophilic product is desorbed, which accelerates the catalytic reaction. This catalytic system showed 90% conversion of FAL and 100% selectivity of HFO within 10 h.
 | ||
| Fig. 23 (a) Scheme for oxidation of FAL, (b) detailed reaction mechanism of FAL oxidation utilizing amphiphilic block copolymer, reaction kinetics for FAL oxidation, (c) using porphyrin-based copolymers, and (d) using P(Pd)-PDMA-PPEGMA with varying block fraction. Reproduced from ref. 133, with permission of Elsevier, Copyright 2022. | ||
In addition to photoreduction of the CO group in FAL, Ghalt et al. firstly reported photoreduction of the CC bond in furan ring, that is, the selective photosynthesis of tetrahydrofurfuryl alcohol (THFA) from FAL.34 THFA is not only used as an environmentally sound solvent but also can be converted into useful linear polyols by hydrogenolysis.134 Pd nanoparticles decorated on E-g-C3N4 (i.e., a high-surface-area g-C3N4 obtained by ultrasonic peeling) showed superior photo(electro)chemical performance due to effective separation of charge carriers. FAL was selectively hydrogenated to value-added FOL and THFA. Specifically, 0.5% Pd@E-g-C3N4 selectively photoreduced FAL to FOL, whereas 3% Pd@E-g-C3N4 directly photoreduced FAL to THFA under 2 bar of H2. As shown in Fig. 24, photogenerated e− are transmitted and accumulated on Pd nanoparticles. These e− can promote desorption of the desorbed-H atom from the Pd site, thus reducing the FAL adsorbed on the E-g-C3N4 surface. The higher Pd content provides additional sites for the adsorption of FOL through furan ring, which promotes the over hydrogenation of FOL to THFA. Hence, the one-step conversion of FAL to THFA is realized. Furthermore, THFA can be selectively converted to γ-butyrolactone (GBL) over the same catalyst under O2 atmosphere under 150 W LED light. GBL shows a wide range of biological characteristics and can be used as a lead structure for new drugs.135 It is also used to synthesize degradable polymers.136 In addition, it can be used as an auxiliary electrolyte to supplement insufficient characteristics.137
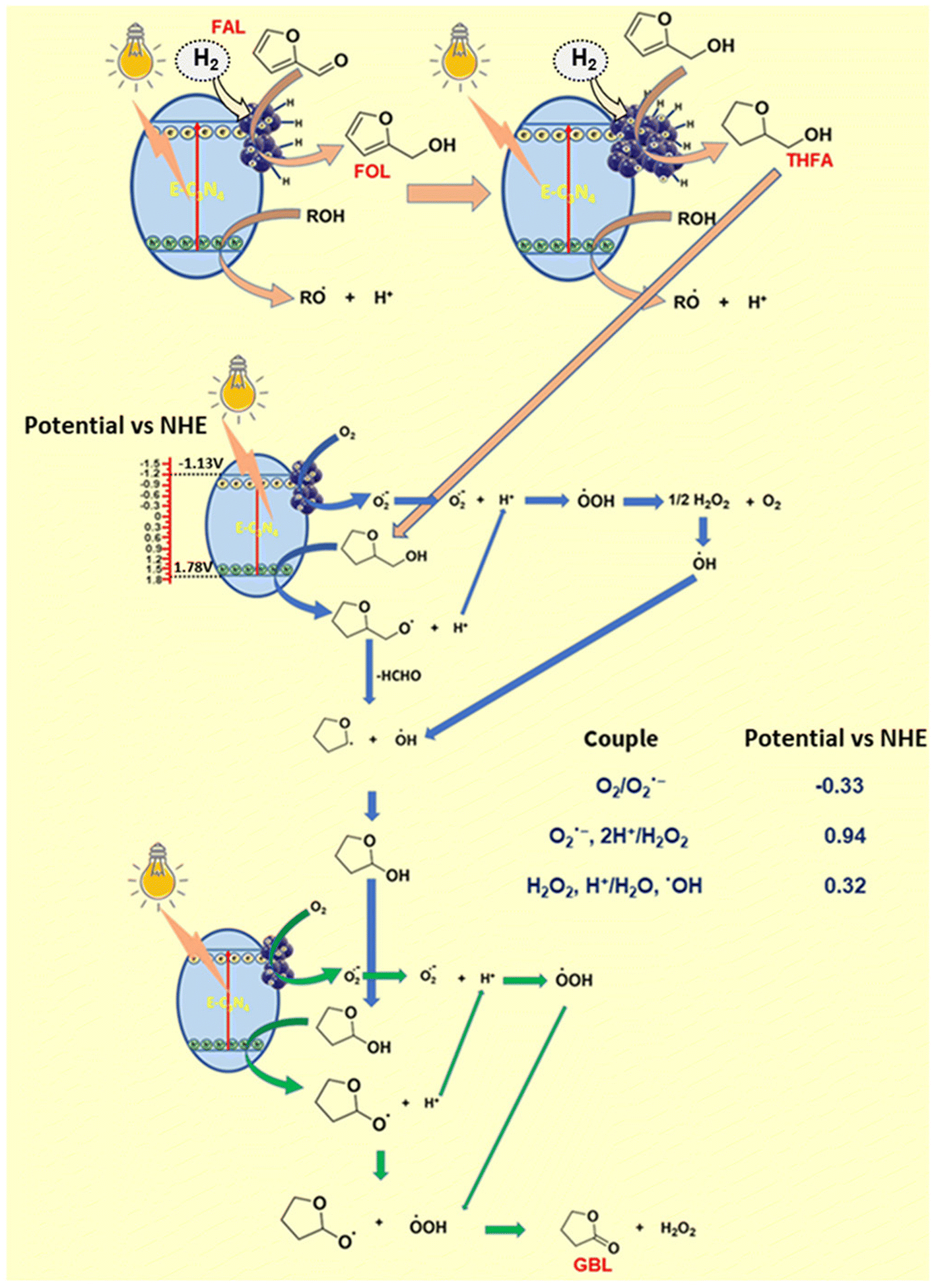 | ||
| Fig. 24 Proposed mechanism and reaction steps associated with photocatalytic conversion of FAL to THFA and THFA to GBL. Reproduced from ref. 34, with permission of Royal Society of Chemistry, Copyright 2023. | ||
In addition to FAL, other photocatalytic reactions of HMF have been also reported. Sell et al. synthesized two structure-related water-soluble Ru complexes (RubpyPy and RuphenPy).39 The couple groups were decorated with covalently attached pyrene chromophores. The triplet energy of the latter one was slightly lower than that of the metal complexes, thus prolonging the lifetime of the so-called triplet reservoir and excited state by up to two orders of magnitude. Taking advantage of the excellent sensitization of 1O2 of RuphenPy, this dyad was used as a photocatalyst for laboratory-scale reactions. As presented in Fig. 25, when HMF (30 mM) aqueous solution is irradiated with green light at 25 °C in a continuous air flow, two main stoichiometric products (i.e., formic acid and unsaturated lactone 5-hydroxy-5-(hydroxymethyl)furan-2(5H)-one (H2MF)) are observed. 85% conversion of HMF and 94% selectivity of HMFO can be obtained after 9 h. Furthermore, the photooxidation of HMF with NaOH additive was investigated in ice bath. It is interesting to find that the main product switches to a ring-opening product, i.e., (Z)-5-hydroxy-4-keto-2-pentenoic acid (HKPA). The yield can reach 62%. H2MF and HKPA can be used to produce polyHKPA, a novel biopolymer.138
 | ||
| Fig. 25 1H NMR spectra of photooxidation of HMF (30 mM) in air-saturated water with RuphenPy as sensitizer (0.2 mol%) before and after 9 h of irradiation with color-coded peaks of key species. Reproduced from ref. 39, with permission of Royal Society of Chemistry, Copyright 2022. | ||
In a word, this section strongly implies great potential for photocatalytic conversion of FAL and HMF. More unexpected high-value products could probably be obtained by photocatalysis. Therefore, it is particularly important to develop more general and accurate analytical methods (apart from the conventional NMR and GC-MS) for rapid separation and detection of those complicated products.
9. Conclusion and outlook
The conversion of biomass-derived furfuryl compounds into value-added chemicals through photocatalysis has recently gained significant attention due to its potential for achieving high efficiency, low pollution, and low-cost performance under mild reaction conditions. Since the first report in 2013 on the photocatalytic selective oxidation of HMF to DFF using TiO2, an increasing number of photocatalysts have been developed for the photocatalytic oxidation of HMF. At present, there are already over sixty photocatalytic systems reported in the literature. Interestingly, about one-fourth of them show that H2 evolution or CO2 reduction can be carried out at the same time as oxidizing HMF to DFF. This strategy makes full use of photogenerated e− and h+ so it has higher economic value and better efficiency. For instance, an O-ZIS-120 photocatalyst exhibits the best performance, i.e., a DFF yield of 1624 μmol h−1 g−1 with a selectivity >97%, along with a H2 evolution rate of 1522 μmol h−1 g−1. In addition to the typical photocatalytic oxidation of HMF and FOL, some unconventional reactions such as reduction, coupling, decarboxylation, ring-opening have been also studied. Many unexpected but promising high-value products can be obtained in these ways. This shows an enormous potential in the photocatalytic transformation of furfural and its derivatives.Unraveling the reaction mechanism is undoubtedly a central task in addition to developing novel photocatalysts. Some important conclusions can be drawn by summarizing the state of the art. Firstly, photogenerated e− have reducing ability, and the magnitude of this competence is determined by the position of the conduction band (LUMO orbitals). Accordingly, photogenerated h+ have oxidizing ability, the magnitude of which is decided by the position of the valence band (HOMO orbitals). Therefore, developing a catalyst with an appropriate energy band position is crucial for achieving photocatalytic selective oxidation and/or reduction of furfuryl compounds. Specifically, the energy band position can be rationally adjusted by optimizing preparation methods, doping elements, and constructing heterojunctions. Meanwhile, the plasma resonance effect and the ligand metal electron transfer effect (LMCT) can achieve visible light response of a catalyst. In addition, the influence of solvents and reaction atmospheres should not be ignored. Notably, using water as solvent often has a significantly negative impact on the reduction reactions. Moreover, the roles played by various active oxygen species in the oxidation systems must be seriously distinguished. For example, hydroxyl radicals (˙OH) with quite a strong oxidizing ability can usually over-oxidize reactants or products. Therefore, suppressing the generation of ˙OH can probably improve product selectivity in the oxidation reactions. Instead, superoxide anion radicals (˙O2−) and singlet oxygen (1O2) are widely accepted as the key active species in achieving highly selective photocatalytic oxidations.
Although some breakthroughs have been reported, there are still many issues and significant challenges that need to be well addressed. Owing to the numerous advantages of photocatalysis, future research should focus on the following aspects particularly in line with the principles of green chemistry:
(1) Atom economy. Develop photocatalytic systems that can simultaneously and fully utilize photogenerated e− and h+, such as selective oxidation of HMF combined with H2 evolution or CO2 reduction. Establish highly atom-economical closed-loop processes.
(2) Design for energy efficiency. Develop photocatalytic systems that can respond to visible light at room temperature and under atmospheric pressure. It is preferable to use catalysts that are simple and readily available, and optimize the preparation methods to reduce costs and maximize the utilization of solar energy.
(3) Low or non-toxic metal elements. Catalytic systems containing toxic elements such as Cr, Cd and Pb should preferably be avoided. This will allow us to boost the green footprint of the photocatalytic transformations.
(4) Safe solvents and additives. Develop photocatalysts that can work in environmentally friendly solvents, especially water, without the assistance of any additives. This can reduce environmental pollution and operational maintenance costs.
(5) Use of renewable feedstocks. Platform chemicals such as FAL and HMF are derived from acid-catalyzed dehydration of lignocellulosic biomass. Therefore, it would be significant to develop photocatalytic systems that can directly obtain high-value chemicals from the crude starting materials, promoting the industrialization of photocatalysis.
In addition to fulfilling the above principles of green chemistry, there should be further understanding of the reaction mechanisms and expansion of the types of reactions. Currently, the photooxidation of HMF is intensively studied and has already obtained some advances. But investigation of some unconventional reactions (such as reduction, coupling, ring-opening, decarboxylation, etc.) is still in the very early stages. Meanwhile, the development of more universal and accurate analytical methods for detecting complicated reaction products is crucial and essential. Furthermore, a unified unit of measurement is vital for convenient and rigorous comparison studies on different photocatalytic systems. Therefore, the apparent quantum efficiency (AQE) should be used to measure catalyst performances, yet this is often overlooked in most works.
Last but not least, the isolation and purification of the target products need to be considered. This is particularly important for industrial applications. The concentration of substrates in photocatalysis is usually much lower compared with traditional catalysis. As expected, the concentration of products can be also low. Therefore, developing low-cost enrichment, separation, and purification strategies is essential.
In summary, the concept of carbon neutrality has received much attention in recent years, and the prospect of biomass-based photocatalytic transformations is very promising. It is necessary to continuously develop photocatalysts with better selectivity and efficiency for upgrading furfural and its derivatives under mild reaction conditions. However, many challenges still remain. We hope this review will provide better perspectives, from materials design to reaction mechanism and to practical application. Finally, further studies can be stimulated on sunlight-driven catalytic valorization of furfural molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22272149 and 22062025), the Yunnan Fundamental Research Projects (202101AT070171), the Yunnan University's Research Innovation Fund for Graduate Students (KC-23234000), the Workstation of Academician Chen Jing of Yunnan Province (202105AF150012), and the Free Exploration Fund for Academician (202205AA160007). The authors thank the Advanced Analysis and Measurement Center of Yunnan University for the sample testing service.References
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- A. V. Puga, Coord. Chem. Rev., 2016, 315, 1–66 CrossRef CAS.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- M. Wang and F. Wang, Adv. Mater., 2019, 31, 1901866 CrossRef PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5835 RSC.
- Y. Wang, Y. Lu, Q. Cao and W. Fang, Chem. Commun., 2020, 56, 3765–3768 RSC.
- Y. Wang, Y. Wang, Y. Lu, Q. Cao and W. Fang, Chem. Commun., 2021, 57, 1742–1745 RSC.
- R. Zhang, Y. Wang, W. Zhang, Y. Lu, Q. Tang, Q. Cao, B. Gu and W. Fang, Chem. Commun., 2023, 59, 9134–9137 RSC.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- P. Pal and S. Saravanamurugan, ChemSusChem, 2019, 12, 145–163 CrossRef CAS PubMed.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- X. Kong, Y. Zhu, Z. Fang, J. A. Kozinski, I. S. Butler, L. Xu, H. Song and X. Wei, Green Chem., 2018, 20, 3657–3682 RSC.
- Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS.
- S. Yurdakal, B. S. Tek, O. Alagöz, V. Augugliaro, V. Loddo, G. Palmisano and L. Palmisano, ACS Sustainable Chem. Eng., 2013, 1, 456–461 CrossRef CAS.
- S. Xu, P. Zhou, Z. Zhang, C. Yang, B. Zhang, K. Deng, S. Bottle and H. Zhu, J. Am. Chem. Soc., 2017, 139, 14775–14782 CrossRef CAS PubMed.
- C.-H. Hao, X.-N. Guo, Y.-T. Pan, S. Chen, Z.-F. Jiao, H. Yang and X.-Y. Guo, J. Am. Chem. Soc., 2016, 138, 9361–9364 CrossRef CAS PubMed.
- G. Lu, F. Chu, X. Huang, Y. Li, K. Liang and G. Wang, Coord. Chem. Rev., 2022, 450, 214240 CrossRef CAS.
- X.-X. Wang, S. Meng, S. Zhang, X. Zheng and S. Chen, Catal. Commun., 2020, 147, 106152 CrossRef CAS.
- V. N. Pham, S. Lee, H. Lee and H. S. Kim, Inorg. Chem., 2023, 62, 3703–3711 CrossRef CAS PubMed.
- H. Zhao, D. Trivedi, M. Roostaeinia, X. Yong, J. Chen, P. Kumar, J. Liu, B.-L. Su, S. Larter, M. G. Kibria and J. Hu, Green Chem., 2023, 25, 692–699 RSC.
- C. Ayed, W. Huang, G. Kizilsavas, K. Landfester and K. A. I. Zhang, ChemPhotoChem, 2020, 4, 571–576 CrossRef CAS.
- B. Ma, Y. Wang, X. Guo, X. Tong, C. Liu, Y. Wang and X. Guo, Appl. Catal., A, 2018, 552, 70–76 CrossRef CAS.
- X. Wu, J. Li, S. Xie, P. Duan, H. Zhang, J. Feng, Q. Zhang, J. Cheng and Y. Wang, Chem, 2020, 6, 3038–3053 CAS.
- J. Wang, Y. Yuan, K. Ren, B. Wang and Z. Li, J. Catal., 2023, 417, 178–184 CrossRef CAS.
- Y.-B. Huang, Z. Yang, J.-J. Dai, Q.-X. Guo and Y. Fu, RSC Adv., 2012, 2, 11211–11214 RSC.
- Y. Zhu, W. Deng, Y. Tan, J. Shi, J. Wu, W. Lu, J. Jia, S. Wang and Y. Zou, Adv. Funct. Mater., 2023, 2304985 CrossRef CAS.
- L. Sun, W. Wang, T. Kong, H. Jiang, H. Tang and Q. Liu, J. Mater. Chem. A, 2022, 10, 22531–22539 RSC.
- J. Fan, Y. Zhao, Q. Wang, M. Gao, X. Li, D. Li and J. Feng, Dalton Trans., 2023, 52, 1950–1961 RSC.
- R. Ghalta and R. Srivastava, Sustainable Energy Fuels, 2023, 7, 1707–1723 RSC.
- P. Esser, B. Pohlmann and H.-D. Scharf, Angew. Chem., Int. Ed. Engl., 1994, 33, 2009–2023 CrossRef.
- D. K. Chauhan, V. R. Battula, A. Giri, A. Patra and K. Kailasam, Catal. Sci. Technol., 2022, 12, 144–153 RSC.
- M. R. Ebrahimian, M. Tavakolian and M. Hosseini-Sarvari, J. Environ. Chem. Eng., 2023, 11, 109347 CrossRef CAS.
- Y.-M. Huang, G.-H. Lu, M.-H. Zong, W.-J. Cui and N. Li, Green Chem., 2021, 23, 8604–8610 RSC.
- A. C. Sell, J. C. Wetzel, M. Schmitz, A. W. Maijenburg, G. Woltersdorf, R. Naumann and C. Kerzig, Dalton Trans., 2022, 51, 10799–10808 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Ulyankina, S. Mitchenko and N. Smirnova, Processes, 2020, 8, 647 CrossRef CAS.
- I. Krivtsov, M. Ilkaeva, E. Salas-Colera, Z. Amghouz, J. R. García, E. Díaz, S. Ordóñez and S. Villar-Rodil, J. Phys. Chem. C, 2017, 121, 6770–6780 CrossRef CAS.
- A. Allegri, V. Maslova, M. Blosi, A. L. Costa, S. Ortelli, F. Basile and S. Albonetti, Molecules, 2020, 25, 5225 CrossRef CAS PubMed.
- A. Khan, M. Goepel, A. Kubas, D. Łomot, W. Lisowski, D. Lisovytskiy, A. Nowicka, J. C. Colmenares and R. Gläser, ChemSusChem, 2021, 14, 1351–1362 CrossRef CAS PubMed.
- Q. Zhang, H. Zhang, B. Gu, Q. Tang, Q. Cao and W. Fang, Appl. Catal., B, 2023, 320, 122006 CrossRef CAS.
- Y. Zhou, J. Liu and J. Long, J. Solid State Chem., 2021, 303, 122510 CrossRef CAS.
- I. Krivtsov, E. I. García-López, G. Marcì, L. Palmisano, Z. Amghouz, J. R. García, S. Ordóñez and E. Díaz, Appl. Catal., B, 2017, 204, 430–439 CrossRef CAS.
- M. Ilkaeva, I. Krivtsov, E. I. García-López, G. Marcì, O. Khainakova, J. R. García, L. Palmisano, E. Díaz and S. Ordóñez, J. Catal., 2018, 359, 212–222 CrossRef CAS.
- A. Akhundi, E. I. García-López, G. Marcì, A. Habibi-Yangjeh and L. Palmisano, Res. Chem. Intermed., 2017, 43, 5153–5168 CrossRef CAS.
- Q. Wu, Y. He, H. Zhang, Z. Feng, Y. Wu and T. Wu, Mol. Catal., 2017, 436, 10–18 CrossRef CAS . The water treatment was to add 5 mL of water to g-C3N4 obtained from the primary calcination. After the secondary calcination, the modified g-C3N4 catalyst showed a large specific surface area and pore volume.
- E. I. García-López, F. R. Pomilla, E. Bloise, X.-f. Lü, G. Mele, L. Palmisano and G. Marcì, Top. Catal., 2021, 64, 758–771 CrossRef.
- G. Wang, R. Huang, J. Zhang, J. Mao, D. Wang and Y. Li, Adv. Mater., 2021, 33, 2105904 CrossRef CAS PubMed.
- H. Qian, Q. Hou, W. Zhang, Y. Nie, R. Lai, H. Ren, G. Yu, X. Bai, H. Wang and M. Ju, Appl. Catal., B, 2022, 319, 121907 CrossRef CAS.
- H. Zhang, Z. Feng, Y. Zhu, Y. Wu and T. Wu, J. Photochem. Photobiol., A, 2019, 371, 1–9 CrossRef CAS.
- L. Cheng, D. Huang, Y. Zhang and Y. Wu, Appl. Organomet. Chem., 2021, 35, e6404 CrossRef CAS.
- H. Li, H. Zhao, Y. Dong, Y. Zhu and J. Li, Adv. Energy Sustainability Res., 2022, 3, 2200116 CrossRef CAS.
- W. Liang, R. Zhu, X. Li, J. Deng and Y. Fu, Green Chem., 2021, 23, 6604–6613 RSC.
- S. Chu, J. Shao, H. Qu, X. Wang, R. Xiao and H. Zhang, ChemSusChem, 2023, e202300886 CrossRef CAS PubMed.
- G. Han, Y.-H. Jin, R. A. Burgess, N. E. Dickenson, X.-M. Cao and Y. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CrossRef CAS PubMed.
- T. Xia, W. Gong, Y. Chen, M. Duan, J. Ma, X. Cui, Y. Dai, C. Gao and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202204225 CrossRef CAS PubMed.
- C. Yuan, Q. Liu, M. Wei, S. Zhao, X. Yang, B. Cao, S. Wang, A. E.-F. Abomohra, X. Liu and Y. Hu, Fuel, 2022, 320, 123994 CrossRef CAS.
- J. L. DiMeglio, A. G. Breuhaus-Alvarez, S. Li and B. M. Bartlett, ACS Catal., 2019, 9, 5732–5741 CrossRef CAS.
- M. Zhang, Z. Yu, J. Xiong, R. Zhang, X. Liu and X. Lu, Appl. Catal., B, 2022, 300, 120738 CrossRef CAS.
- Q. Zhu, Y. Zhuang, H. Zhao, P. Zhan, C. Ren, C. Su, W. Ren, J. Zhang, D. Cai and P. Qin, Chin. J. Chem. Eng., 2023, 54, 180–191 CrossRef CAS.
- M. Zhang, Y. Zhang, L. Ye, Z. Yu, R. Liu, Y. Qiao, L. Sun, J. Cui and X. Lu, Appl. Catal., B, 2023, 330, 122635 CrossRef CAS.
- S. Ding, J. B. G. Filho, T. Peppel, S. Haida, J. Rabeah, N. Steinfeldt and J. Strunk, Sustainable Energy Fuels, 2023, 7, 4396–4400 RSC.
- V. N. Pham, S. Lee, D. T. Hoang, J. Baik, H. S. Kim and H. Lee, Inorg. Chem., 2023, 62, 12913–12919 CrossRef CAS PubMed.
- J. Qiu, L. Zhang, D. Dai, G. Xia and J. Yao, ChemSusChem, 2022, 15, e202200399 CrossRef CAS PubMed.
- Y. Guo, B. Liu, J. Zhang, G. Wang, C. Pan, H. Zhao, C. Wang, F. Yu, Y. Dong and Y. Zhu, Appl. Catal., B, 2024, 123217 CrossRef CAS.
- H. Zhang, Q. Wu, C. Guo, Y. Wu and T. Wu, ACS Sustainable Chem. Eng., 2017, 5, 3517–3523 CrossRef CAS.
- Y. Zou, Y. Hu, A. Uhrich, Z. Shen, B. Peng, Z. Ji, M. Muhler, G. Zhao, X. Wang and X. Xu, Appl. Catal., B, 2021, 298, 120584 CrossRef CAS.
- A. Kumar and R. Srivastava, ACS Appl. Nano Mater., 2021, 4, 9080–9093 CrossRef CAS.
- M. Gong, H. Zhao, C. Pan, Y. Dong, Y. Guo, H. Li, J. Zhang, G. Wang and Y. Zhu, New J. Chem., 2023, 47, 7118–7126 RSC.
- M. Zhang, Z. Li, X. Xin, J. Zhang, Y. Feng and H. Lv, ACS Catal., 2020, 10, 14793–14800 CrossRef CAS.
- L. Yin, Y. Wu, X. Bao, X. Liu, D. Dai, M. Zhang, Z. Wang, Z. Zheng, Y. Liu, H. Cheng, Y. Dai, B. Huang and P. Wang, Chem. – Eur. J., 2023, 29, e202300999 CrossRef CAS PubMed.
- D. A. Giannakoudakis, V. Nair, A. Khan, E. A. Deliyanni, J. C. Colmenares and K. S. Triantafyllidis, Appl. Catal., B, 2019, 256, 117803 CrossRef CAS.
- Y. Park, V. N. Pham, K. Lee and H. Lee, Inorg. Chem., 2023, 62, 13428 CrossRef CAS PubMed.
- B. Chen, L. Chen, Z. Yan, J. Kang, S. Chen, Y. Jin, L. Ma, H. Yan and C. Xia, Green Chem., 2021, 23, 3607–3611 RSC.
- B. Yang, W. Hu, F. Wan, C. Zhang, Z. Fu, A. Su, M. Chen and Y. Liu, Chem. Eng. J., 2020, 396, 125345 CrossRef CAS.
- W. Hu, J. She, Z. Fu, B. Yang, H. Zhang and D. Jiang, RSC Adv., 2021, 11, 23365–23373 RSC.
- F. Yang, S. Liu, T. Tang, S. Yao and C. An, Catal. Sci. Technol., 2023, 13, 2469–2474 RSC.
- D. Liu, T. Ding, L. Wang, H. Zhang, L. Xu, B. Pang, X. Liu, H. Wang, J. Wang, K. Wu and T. Yao, Nat. Commun., 2023, 14, 1720 CrossRef CAS PubMed.
- Q. Yang, T. Wang, F. Han, Z. Zheng, B. Xing and B. Li, J. Alloys Compd., 2022, 897, 163177 CrossRef CAS.
- J. Yang, X. Zhang, Z. Zeng, C. Han and Y. Liang, Inorg. Chem. Front., 2023, 10, 6308–6319 RSC.
- J. Hu, X. Li, J. Qu, X. Yang, Y. Cai, T. Yang, F. Yang and C. M. Li, Chem. Eng. J., 2023, 453, 139957 CrossRef CAS.
- X. Li, J. Hu, T. Yang, X. Yang, J. Qu and C. M. Li, Nano Energy, 2022, 92, 106714 CrossRef CAS.
- V. R. Battula, A. Jaryal and K. Kailasam, J. Mater. Chem. A, 2019, 7, 5643–5649 RSC.
- P. Qiu, Y. Yao, S. Lu, L. Chen, Y. Chen and X. Liao, Fuel, 2023, 351, 129043 CrossRef CAS.
- Y. Yan, X. Yu, C. Shao, Y. Hu, W. Huang and Y. Li, Adv. Funct. Mater., 2023, 2304604 CrossRef CAS.
- S. Liu, B. Zhang, Z. Yang, Z. Xue and T. Mu, Green Chem., 2023, 25, 2620–2628 RSC.
- S. Meng, H. Wu, Y. Cui, X. Zheng, H. Wang, S. Chen, Y. Wang and X. Fu, Appl. Catal., B, 2020, 266, 118617 CrossRef CAS.
- T. Shan, L. Luo, T. Chen, L. Deng, M. Li, X. Yang, L. Shen and M.-Q. Yang, Green Chem., 2023, 25, 2745–2756 RSC.
- D. A. Giannakoudakis, A. Qayyum, V. Nair, A. Khan, S. R. Pradhan, J. Prekodravac, K. Rekos, A. P. LaGrow, O. Bondarchuk, D. Łomot, K. S. Triantafyllidis and J. C. Colmenares, Mol. Catal., 2021, 514, 111664 CrossRef CAS.
- H. Wu, S. Meng, J. Zhang, X. Zheng, Y. Wang, S. Chen, G. Qi and X. Fu, Appl. Surf. Sci., 2020, 505, 144638 CrossRef CAS.
- S. Dhingra, T. Chhabra, V. Krishnan and C. M. Nagaraja, ACS Appl. Energy Mater., 2020, 3, 7138–7148 CrossRef CAS.
- Y. Wang, X. Kong, M. Jiang, F. Zhang and X. Lei, Inorg. Chem. Front., 2020, 7, 437–446 RSC.
- H.-F. Ye, R. Shi, X. Yang, W.-F. Fu and Y. Chen, Appl. Catal., B, 2018, 233, 70–79 CrossRef CAS.
- Z.-W. Yang, J.-M. Chen, L.-Q. Qiu, W.-J. Xie and L.-N. He, Catal. Sci. Technol., 2022, 12, 5495–5500 RSC.
- W. Zhang, Q. Li and H. Xia, Appl. Surf. Sci., 2023, 613, 156036 CrossRef CAS.
- V. N. Pham, H. Jeon, S. Hong and H. Lee, Inorg. Chem., 2022, 61, 16887–16894 CrossRef CAS PubMed.
- V. N. Pham, H. Jeon, S. Han, S. Hong and H. Lee, Adv. Sustainable Syst., 2022, 6, 2200355 CrossRef CAS.
- J.-N. Chang, Q. Li, Y. Yan, J.-W. Shi, J. Zhou, M. Lu, M. Zhang, H.-M. Ding, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202209289 CrossRef CAS PubMed.
- B. Zhou, J. Song, Z. Zhang, Z. Jiang, P. Zhang and B. Han, Green Chem., 2017, 19, 1075–1081 RSC.
- W. Zhang, X. Li, S. Liu, J. Qiu, J. An, J. Yao, S. Zuo, B. Zhang, H. Xia and C. Li, ChemSusChem, 2022, 15, e202102158 CrossRef CAS PubMed.
- Y. Zhu, Y. Zhang, L. Cheng, M. Ismael, Z. Feng and Y. Wu, Adv. Powder Technol., 2020, 31, 1148–1159 CrossRef CAS.
- Z. Li, M. Zhang, X. Xin and H. Lv, ChemCatChem, 2021, 13, 1389–1395 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- D. J. Chadderdon, L.-P. Wu, Z. A. McGraw, M. Panthani and W. Li, ChemElectroChem, 2019, 6, 3387–3392 CrossRef CAS.
- X. Zhao, L. Shi, B. Tian, S. Li, S. Liu, J. Li, S. Liu, T. D. James and Z. Chen, J. Mater. Chem. A, 2023, 11, 12308–12314 RSC.
- L. Özcan, P. Yalçın, O. Alagöz and S. Yurdakal, Catal. Today, 2017, 281, 205–213 CrossRef.
- J. J. Roylance, T. W. Kim and K.-S. Choi, ACS Catal., 2016, 6, 1840–1847 CrossRef CAS.
- Y. Guo and J. Chen, RSC Adv., 2016, 6, 101968–101973 RSC.
- S. Dhingra, M. Sharma, V. Krishnan and C. M. Nagaraja, J. Colloid Interface Sci., 2022, 615, 346–356 CrossRef CAS PubMed.
- E. Cui, Q. Li, X. Wang, N. Xu, F. Zhang, G. Hou, M. Xie, Z. Wang, X. Yang and Y. Zhang, Appl. Catal., B, 2023, 329, 122560 CrossRef CAS.
- A. Kumar and R. Ananthakrishnan, ACS Appl. Energy Mater., 2022, 5, 2706–2719 CrossRef CAS.
- S. Qiao, Y. Zhou, H. Hao, X. Liu, L. Zhang and W. Wang, Green Chem., 2019, 21, 6585–6589 RSC.
- S. Dong, Z. Liu, R. Liu, L. Chen, J. Chen and Y. Xu, ACS Appl. Nano Mater., 2018, 1, 4247–4257 CrossRef CAS.
- R. Wang, H. Liu, X. Wang, X. Li, X. Gu and Z. Zheng, Catal. Sci. Technol., 2020, 10, 6483–6494 RSC.
- Y. Shi, H. Wang, Z. Wang, C. Liu, M. Shen, T. Wu and L. Wu, J. Energy Chem., 2022, 66, 566–575 CrossRef CAS.
- K. Nakanishi, A. Tanaka, K. Hashimoto and H. Kominami, Chem. Lett., 2017, 47, 254–256 CrossRef.
- M. Zhang and Z. Li, ACS Sustainable Chem. Eng., 2019, 7, 11485–11492 CrossRef CAS.
- Y. Hu, W. Huang, H. Wang, Q. He, Y. Zhou, P. Yang, Y. Li and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 14378–14382 CrossRef CAS PubMed.
- H. Zhao, Q. Zhu, Y. Zhuang, P. Zhan, Y. Qi, W. Ren, Z. Si, D. Cai, S. Yu and P. Qin, GreenChE, 2022, 3, 385–394 Search PubMed.
- A. Gumidyala, B. Wang and S. Crossley, Sci. Adv., 2016, 2, e1601072 CrossRef PubMed.
- S. Lv, H. Liu, J. Zhang, Q. Wu and F. Wang, J. Energy Chem., 2022, 73, 259–267 CrossRef CAS.
- P. Kowalik, P. Bujak, M. Penkala, W. Tomaszewski, W. Lisowski, J. W. Sobczak, D. Siepietowska, A. M. Maroń, J. Polak, M. Bartoszek and A. Pron, Chem. Mater., 2023, 35, 6447 CrossRef CAS.
- B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502–10566 CrossRef CAS PubMed.
- J. G. H. Hermens, A. Jensma and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202112618 CrossRef CAS PubMed.
- X. Li, B. Ho and Y. Zhang, Green Chem., 2016, 18, 2976–2980 RSC.
- L. Pu, L. Ye and Y. Yuanqi, J. Mol. Catal. A: Chem., 1999, 138, 129–133 CrossRef CAS.
- P. Rapado, L. Faba and S. Ordóñez, J. Environ. Chem. Eng., 2023, 11, 111466 CrossRef CAS.
- R. Wojcieszak, F. Santarelli, S. Paul, F. Dumeignil, F. Cavani and R. V. Gonçalves, Sustainable Chem. Processes, 2015, 3, 9 CrossRef.
- J. Y. Seo, J. E. Kim, Y. J. Kwon, S. H. Kim, S. Cho, D. H. Choi, K. Y. Cho and K.-Y. Baek, Mater. Lett., 2022, 311, 131577 CrossRef CAS.
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS.
- M. Seitz and O. Reiser, Curr. Opin. Chem. Biol., 2005, 9, 285–292 CrossRef CAS PubMed.
- T. Moore, R. Adhikari and P. Gunatillake, Biomaterials, 2005, 26, 3771–3782 CrossRef CAS PubMed.
- H.-Y. Oh, S.-J. Park and S.-J. In, J. Mol. Liq., 2020, 314, 113627 CrossRef CAS.
- T. S. A. Heugebaert, C. V. Stevens and C. O. Kappe, ChemSusChem, 2015, 8, 1648–1651 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |