
Effective enrichment of glycated proteome using ultrasmall gold nanoclusters functionalized with boronic acid†
Hongmae
Heo
 a,
Seonghyeon
Cho
bc,
Yuhyeon
Kim
a,
Soomin
Ahn
a,
Jeong-hun
Mok
d,
Hookeun
Lee
*b and
Dongil
Lee
*a
a,
Seonghyeon
Cho
bc,
Yuhyeon
Kim
a,
Soomin
Ahn
a,
Jeong-hun
Mok
d,
Hookeun
Lee
*b and
Dongil
Lee
*a
aDepartment of Chemistry, Yonsei University, Seoul 03722, Republic of Korea. E-mail: dongil@yonsei.ac.kr
bCollege of Pharmacy, Gachon University, Incheon 21936, Republic of Korea. E-mail: hklee@gachon.ac.kr
cBasil Biotech, 157-20 Sinsong-ro, Incheon 22002, Republic of Korea
dDepartment of Medical Device Management and Research, SAIHST, Sungkyunkwan University, Seoul 06355, Republic of Korea
First published on 7th October 2024
Abstract
Glycated proteins play a crucial role in various biological pathways and the pathogenesis of human diseases. A comprehensive analysis of glycated proteins is essential for understanding their biological significance. However, their low abundance and heterogeneity in complex biological samples necessitate an enrichment procedure prior to their detection. Current enrichment strategies primarily rely on the boronic acid (BA) affinity method combined with functional nanoparticles; however, the effectiveness of these approaches is often suboptimal. In this study, a novel nanocluster (NC)-based enrichment material was synthesized for the first time, characterized as Au22SG18 functionalized with 24 BA groups, in which SG is glutathione. The functionalized BA established a reversible covalent bond with the cis-dihydroxy group through pH adjustment, enabling selective enrichment of glycated peptides. After the optimization of the enrichment protocol, we demonstrated highly sensitive and selective enrichment of standard glycopeptides using the NC-based enrichment material, exhibiting excellent reusability. Efficient enrichment was also demonstrated for the glycated proteome from human serum. These results highlight the potential of the atomically well-defined ultrasmall Au NCs as a powerful tool for high-throughput analysis of glycated peptides.
1. Introduction
Glycation, also called non-enzymatic glycosylation, is one of the most common protein post-translational modifications resulting from the spontaneous covalent addition of reducing sugars to proteins through the Maillard reaction.1 The active aldehyde group of a reducing sugar reacts with the nucleophilic free amino group of a protein to form a reversible Schiff base, which is then rearranged to form stable Amadori compounds, defined as the early-stage glycation products. These compounds further convert to advanced glycation end products through several subsequent reactions.2 Indeed, Aberrant glycation is strongly associated with various human diseases, such as diabetes, Alzheimer's disease, cardiovascular disorders, renal disease, and cancer.3–7 An expanding body of research indicated that glycated proteins can serve as potential biomarkers for early disease diagnosis and prognosis. Thus, conducting a comprehensive analysis of the glycated proteome is crucial to provide a comprehensive understanding of the role of glycation in both normal physiological processes and pathological conditions, thereby discovering valuable new disease biomarkers.Mass spectrometry (MS)-based strategies and corresponding methods have substantially advanced glycoproteome studies.8,9 However, challenges such as low abundance, heterogeneity, broad dynamic range of protein glycation, and signal suppression in the presence of nonglycated peptides complicate their analysis.10,11 For comprehensive proteome research, the selective enrichment of glycated proteins from complex biological samples is an essential prerequisite to mass analysis. Among current glycated peptide enrichment techniques, boronic acid (BA) affinity chemistry demonstrated considerable potential in universally enriching glycated peptides owing to its selective and reversible covalent interaction with cis-diols.12–16 Although BA-functionalized materials have been widely applied in glycoproteome enrichment,17–19 their weak interactions limit their applications in complex samples. Several studies have focused on developing effective stationary phases with BA functional ligands to improve their enrichment techniques.20–23 Wu et al. developed an enrichment method using dendrimer beads conjugated with BA derivatives to enhance the glycopeptide enrichment, enabling large-scale analysis of glycoproteins.20 Cao et al. fabricated a BA-functionalized mesoporous graphene–silica composite for the efficient and selective enrichment of intact glycopeptides from complex samples.21 Teng et al. developed a strategy for preparing hollow magnetic nanospheres with large surface areas, realizing improved enrichment performance.22
Atomically precise Au nanoclusters (NCs) have emerged as a new class of materials owing to their unique physicochemical properties.24–28 The unique optical and electrochemical properties and well-defined atomic structures of ultrasmall Au NCs distinguish them from larger nanoparticle systems.29–35 In particular, water-soluble NCs have excellent biocompatibility, thereby attracting considerable research interest for biological applications.36–42 For example, water-soluble near-infrared (NIR)-emitting Au NCs that can effectively perform in vivo tumor-targeted imaging have been reported.36–39 In addition, we demonstrated the effective application of highly luminescent Au22 NCs functionalized with folic acids and a pH-responsive dye for bioimaging41 and intracellular pH sensing,42 respectively.
Inspired by these advantages, in this paper, we report the use of ultrasmall Au22 NCs as the stationary phase in BA-based enrichment technique for the first time. We synthesized densely BA-functionalized Au22 NCs and applied them to glycated peptide enrichment. The ultrasmall size of the NCs is expected to endow a significantly larger specific surface area than traditional larger nanoparticles, facilitating more effective interactions with target peptides. In addition, the high luminescent properties of Au22 NCs can visualize the isolation process, demonstrating a more effective enrichment. We developed a novel NC-based enrichment protocol and optimized the working conditions to maximize the interactions while minimizing nonspecific bindings. Finally, we demonstrated the effectiveness of this strategy in analyzing the glycated proteome of human serum.
2. Results and discussion
2.1. Synthesis of Au22 NCs functionalized with BA
Fig. 1 shows the synthesis scheme of BA-functionalized Au22 NCs (Au22-BA). The starting Au22SG18 NCs, where SG refers to glutathione, were synthesized following a previously reported procedure.43 Sequentially, the primary amine group of glutathione was protected with benzyl chloroformate (CBz-Cl) to prevent potential interparticle coupling.32 BA was then covalently conjugated to CBz-protected Au22SG18 (Au22-CBz) through a dicyclohexyl carbodiimide/N-hydroxysuccinimide (DCC/NHS) coupling reaction.44 Further details are provided in the Experimental section of ESI.† We synthesized Au22 NCs functionalized with two different BA derivatives, namely phenylboronic acid (PBA) and benzoboroxole (BX), with pKa values of 8.8 and 6.7, respectively (Fig. S1†). The enrichment performance (vide infra) of BX-functionalized Au22 NCs (Au22-BX) is better than that of the PBA-functionalized NCs (Au22-PBA); thus, we focused on Au22-BX NCs. | ||
| Fig. 1 Schematic of the BA-functionalization process of Au22 NCs. | ||
The chemical composition of the starting Au22SG18 NCs was confirmed by electrospray ionization (ESI) MS. As shown in Fig. 2a, the peaks at m/z 2460–2480 represent Au22SG18 ions containing various numbers of Na+ and H+ ions. The experimental isotope pattern of the most prominent peak at approximately m/z 2460 Da was superimposed with the simulated one of [Au22SG18-4H]4−, as depicted in the inset of Fig. 2a. After the coupling reaction, the purified Au22-BX was characterized by optical absorption spectrometry. As shown in Fig. 2b, Au22-BX exhibits a characteristic BX absorption peak, confirming the successful conjugation of BX to the Au22 NC. Moreover, the spectra of Au22-BX depicted a characteristic absorption of Au22SG18 NCs at 520 nm and its photoluminescent (PL) emission peak at approximately 680 nm (Fig. 2b, c and Fig. S2†), indicating the preserved integrity of Au22SG18 NCs during the coupling reaction. Remarkably, the synthesized Au22-BX exhibited intense NIR emission centered at 680 nm, with a quantum yield (QY) of approximately 38.5%, which is nearly five-fold brighter than the original Au22SG18 NCs (Fig. 2c and Fig. S3†). Such result is attributed to the rigidification effect after a series of surface modification processes.32,34,43
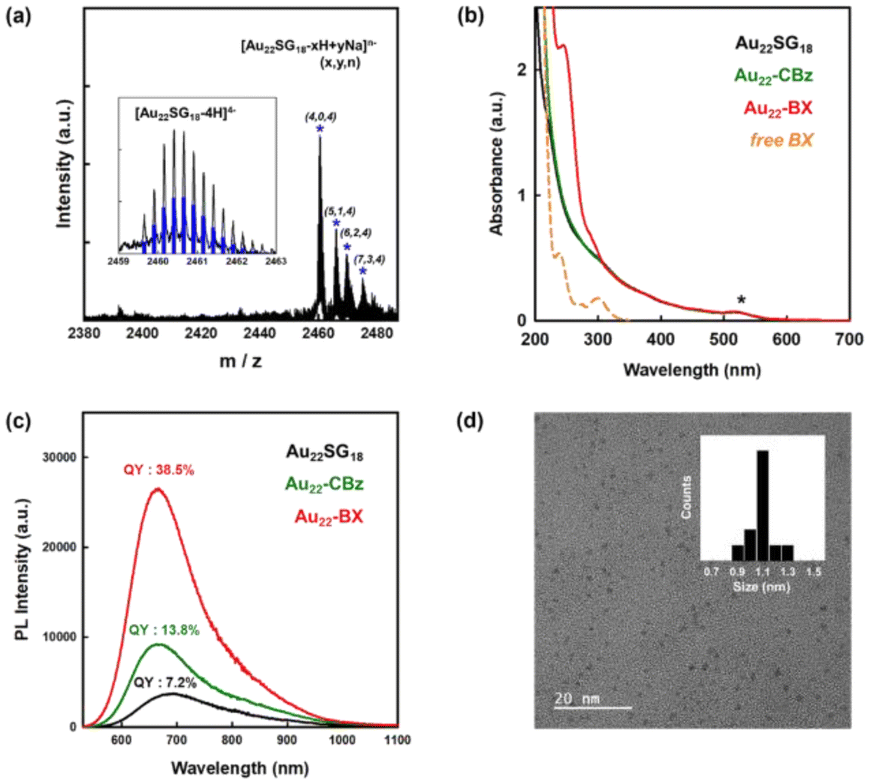 | ||
| Fig. 2 Characterization of Au22-BX. (a) Negative-ion mode ESI mass spectrum of synthesized Au22SG18. Inset: comparison between the experimental and calculated isotope pattern (blue bar). (b) Optical absorption spectra of Au22SG18, Au22-CBz, Au22-BX, and BX in water. (c) PL spectra (λEX = 520 nm) of Au22SG18, Au22-CBz, and Au22-BX in water. The QYs of the NCs were calibrated using indocyanine green as the standard. (d) TEM image of Au22-BX. The inset shows histograms of the NC core diameter. | ||
Although determining the chemical composition of as-synthesized Au22-BX using MS was challenging owing to its intricate fragmentation pattern, we quantified the number of BX groups conjugated to Au22SG18 using 1H nuclear magnetic resonance (NMR) spectroscopy. The quantity of conjugated CBz ligands was first determined by comparing the integration number of the CBz peak with the SG ligand peak. As shown in Fig. S4,† the peak at 7.2 ppm pertains to the conjugated CBz ligands, showing a 1.24-fold larger integration number than that of SG, which is centered at 2.2 ppm, indicating that all 18 primary amine groups are fully protected by CBz. Similarly, an average of 24 BX ligands were conjugated per Au22 NC. The polyacrylamide gel electrophoresis (PAGE) separation results in Fig. S5† confirm effective surface functionalization by demonstrating different mobilities of the synthesized NC products. Finally, the transmission electron microscopy (TEM) image depicted the spherical shape of Au22-BX, which is highly monodispersed with an average core size of 1.1 ± 0.1 nm, similar to that of Au22SG18 (Fig. 2d).44 Thus, Au22-BX was successfully synthesized. Furthermore, the synthesized Au22-BX NCs exhibit significantly improved stability compared to the initial Au22 NCs (Fig. S6†), ensuring their high potential for practical applications.
2.2. Development of glycated peptide enrichment method
BA and its derivatives can readily form reversible covalent bonds with cis-diols of sugars by controlling pH and has been widely employed for sugar detection.45–47 The peripheral BA of the as-prepared enrichment materials (Au22-BA) can selectively interact with the sugars of glycated peptides in an alkaline system, whereas the resulting covalent bonds can be reversibly broken to release glycated peptides under an acidic condition. The enrichment of glycated peptides by Au22-BA is illustrated in Scheme 1. The process consists of three essential procedures: incubation, washing, and elution (Experimental section in ESI† for details). During the elution process the bright PL of Au22-BA was utilized to achieve a more effective separation of peptides and NCs (Fig. S7; Experimental section in ESI†). As a proof of concept for this NC-based enrichment method, the capacity of the method was first investigated by employing teicoplanin, a widely recognized glycopeptide antibiotic with a single cis-2,3-diol site, as our standard model peptide (Fig. S8†). After completing the enrichment steps, the eluted sample was analyzed using matrix-assisted laser desorption ionization (MALDI) MS. As shown in Fig. S9,† the target peaks of teicoplanin are clearly visible in the mass spectra, demonstrating the feasibility of this method for enriching intact glycated peptides. Additionally, two standard glycopeptides, ramoplanin and bleomycin (Fig. S8†), were tested to further assess the sample applicability of our method. Like teicoplanin, ramoplanin with a cis-2,3-diol site was clearly detected in the mass spectra after enrichment (Fig. S10a†). The quantity of captured glycopeptides was also measured via optical absorption spectrometry (Fig. S11a†), and the effectiveness was determined by calculating the ratio of captured glycopeptides to its initial amount. As shown in Fig. S11b,† Ramoplanin displayed results comparable to teicoplanin, emphasizing the broad sample applicability of this enrichment method. In the case of bleomycin, which lacks a cis-diol site and is more challenging to capture, the results demonstrate that while the method remained effective (Fig. S10b†), the capture efficiency was significantly lower compared to the other two glycopeptides (Fig. S11b†), also reflecting sample specificity.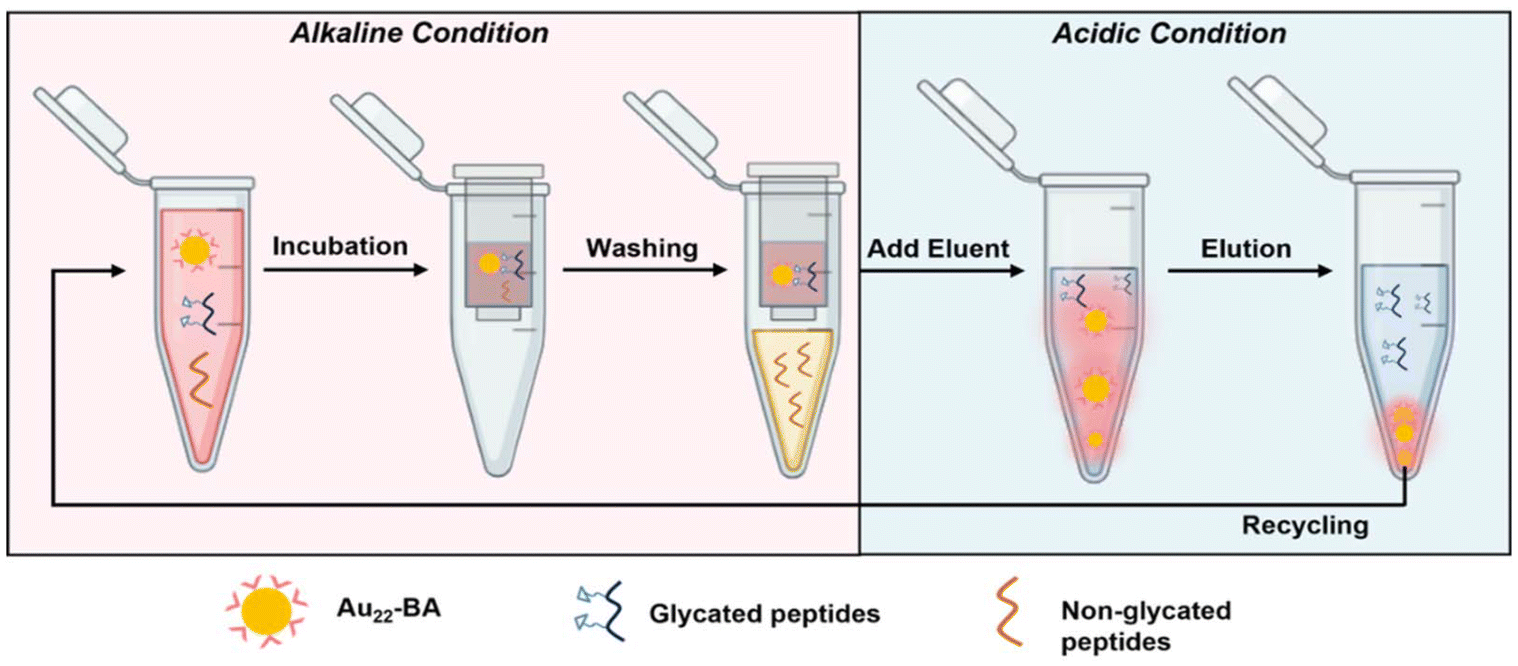 | ||
| Scheme 1 Schematic of the enrichment of glycated peptides based on Au22-BA. | ||
We further refined several crucial parameters in our glycated peptide enrichment protocols to achieve optimal enrichment. We implemented control experiments under various conditions using teicoplanin and evaluated the method's effectiveness using optical absorption spectrometry. As the affinity interaction of boronate is highly pH-sensitive, the pH value during incubation should be carefully considered. As such, we performed the incubation procedure under different pH values (7.4, 8.0, 9.0, and 10.0) to analyze the effect of pH. As shown in Fig. 3a, pH 9.0 yielded the most favorable result, aligning with the commonly accepted concept of more favorable covalent formation of boronate esters under high pH values.19,48 At pH 10.0, the performance notably dropped, which can be explained by the poor stability of the peptide samples under high pH conditions.49
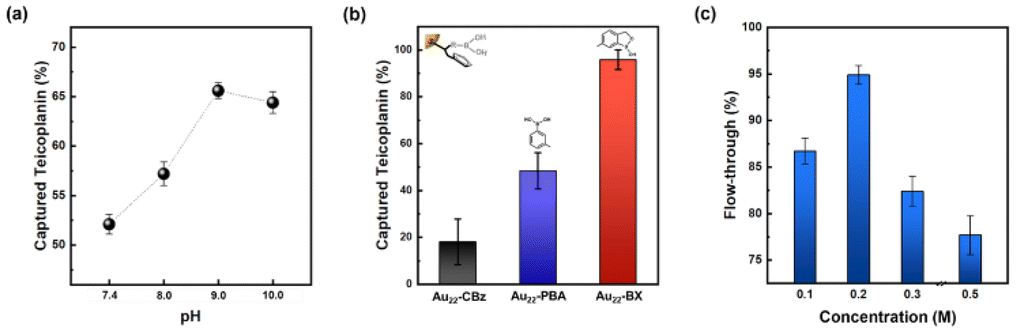 | ||
| Fig. 3 Optimization of enrichment method using standard peptides. (a) Effect of pH on the incubation process. (b) Structures of BA derivatives and their enrichment performance. (c) Effect of NaCl concentration on washing buffer. The results are monitored by the flow-through of vancomycin. The error bar represents the mean standard error, as calculated in triplicate experiments. | ||
With the pH condition fixed at 9.0, we optimized the functional ligands. As mentioned earlier, the effective interaction between the peripheral functional ligand and cis-diol of glycated peptide is vital for the overall effectiveness of the enrichment method. Although PBA has been previously used as a functional ligand, PBA-based methods often suffer from weak interactions.50 As such, we investigated BX, a potential BA derivative, as the functional ligand for the newly synthesized NCs. Both PBA- and BX-conjugated NCs were successfully prepared using similar methods (Fig. 1, 2 and Fig. S4, S12†). After conducting parallel experiments to assess the enrichment with standard peptides, Au22 NCs functionalized by BX achieved better enrichment performance compared to those functionalized by PBA (Fig. 3b). The general principle of boronate affinity involves the covalent formation of cyclic esters between a BA ligand and cis-diol containing species at pH conditions that are higher than the pKa of the BA ligand.19,48 As BX represents a unique class of BA ligands with a pKa that is significantly lower than that of general PBA ligands (Fig. S1†),51–53 Au22-BX exhibits better performance under the same pH conditions. Moreover, considering the stability of the biological samples, the BA ligand with a lower pKa value is more suitable for achieving a binding pH that is more suitable for physiological detection. Thus, in this study, we opted to use BX as the functional ligand to promote stronger interactions with target glycated peptides.
The Lewis base in the reaction environment facilitates esterification.54 As such, we incorporated a robust Lewis base F− ion into the incubation solution, as it is widely recognized as one of the most effective additives to promote the formation of boronic esters.55 As expected, the results confirmed the beneficial effect of F− ion addition with a notably higher binding efficiency (Fig. S13a†). In addition, we optimized other important parameters, such as incubation time and temperature (Fig. S13b and c; Experimental section in ESI†). An incubation time of 30 min and a temperature of 13 °C were deemed optimal for the incubation procedure.
Boronate affinity methods often encounter undesired nonspecific bindings, which can diminish their selectivity.56 The presence of negatively charged species on the NC surface can prompt an unexpected adsorption via electrostatic interactions. To alleviate this phenomenon, we incorporated NaCl into the washing buffer to enhance the ionic strength of the medium.57 In particular, we employed another glycopeptide antibiotic, namely vancomycin, for the control experiments. Vancomycin shares a similar structure with teicoplanin but without a cis-diol site, making it a suitable counterpart for the comparison (Fig. S8†). As shown in Fig. 3c, the approach is demonstrated to be effective, as indicated by the higher vancomycin content washed with a higher NaCl concentration. The observed drop at higher concentrations can be attributed to the decreased homogeneity owing to the salting-out effect, resulting in the optimal washing buffer concentration of 0.2 M NaCl. Additionally, the nonspecific bindings induced by charge transfer interactions can be further mitigated by NaF addition (10 mM). A more detailed description and results can be found in the Experimental section in ESI and Fig. S14,† respectively.
2.3. Evaluation of enrichment performance
After optimizing the enrichment protocol, the enrichment performance of our system was further investigated in terms of multiple aspects using standard model peptides. Firstly, considering the extremely low abundance of glycated peptides in biological samples, the evolution of the detection sensitivity is crucial for assessing the enrichment efficiency of materials. Teicoplanin at various concentrations was enriched using Au22-BX. Subsequently, the eluted sample was analyzed by MALDI MS to evaluate the sensitivity. Fig. 4 shows teicoplanin peaks with strong signal intensity, and signal peaks, which are detectable at a concentration of as low as 0.01 fmol μL−1 with a reasonable signal-to-noise ratio (S/N = 7.9). Such detection limit surpasses those reported for other composite materials in previous studies,21,22,58,59 including the best results obtained for glycated peptide enrichment using magnetic nanoparticles (1 fmol μL−1).58 This demonstrates the high sensitivity obtained, which adequately satisfies the requirements for enriching low-abundance glycated peptides. The enhanced sensitivity underscores the superior enrichment capability of our synthesized materials, which are mainly attributed to the ultrasmall size and densely functionalized surface of NCs, facilitating effective interactions with glycated peptides.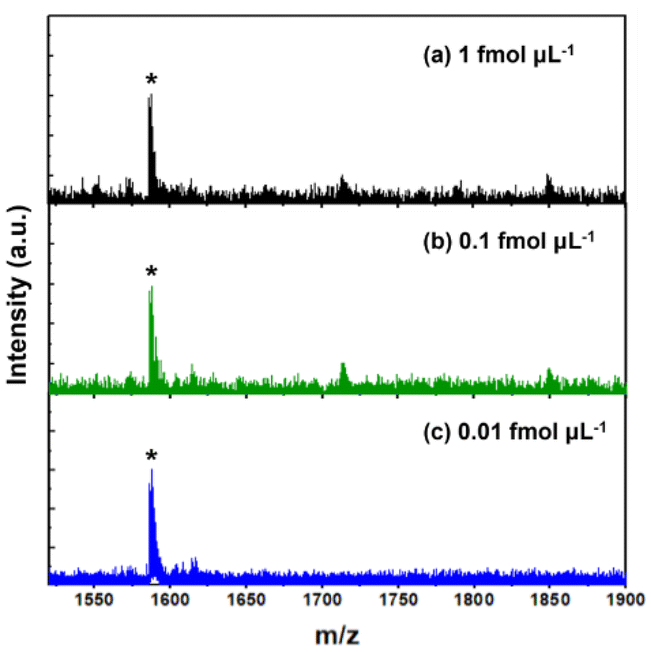 | ||
| Fig. 4 Sensitivity evaluation result. MALDI mass spectra of standard peptide (teicoplanin) at different concentrations: (a) 1 fmol μL−1, (b) 0.1 fmol μL−1, and (c) 0.01 fmol μL−1. * denotes a typical peak of teicoplanin. | ||
Enrichment techniques often suffer from undesired nonspecific binding owing to the complexity of biological samples, which can limit the detection of target species.56,60 As such, we focused on addressing this issue during the optimization of the washing processes, anticipating a high selectivity level. In detail, selectivity was assessed by employing the standard peptide vancomycin as the model interfering species. Teicoplanin was blended with vancomycin at different molar ratios and enriched using the optimized enrichment protocol. With teicoplanin and vancomycin molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, no teicoplanin signal is detected prior to enrichment owing to the substantial interference (Fig. S15a†). However, distinct desired peaks are noted in the mass spectra after enrichment, even with the extremely high molar ratio of teicoplanin and vancomycin to 1:1000 (Fig. S15c†). This selectivity rivals the best performance of previously reported enrichment materials,58,61 indicating the sufficiently high selectivity and robust anti-interference capability of our enrichment method.
:100, no teicoplanin signal is detected prior to enrichment owing to the substantial interference (Fig. S15a†). However, distinct desired peaks are noted in the mass spectra after enrichment, even with the extremely high molar ratio of teicoplanin and vancomycin to 1:1000 (Fig. S15c†). This selectivity rivals the best performance of previously reported enrichment materials,58,61 indicating the sufficiently high selectivity and robust anti-interference capability of our enrichment method.
Considering the significance of developing environmentally and economically sustainable materials across all industries, the reusability of enrichment materials is equally essential. Previous studies have assessed the reusability of their materials for performance evaluation.22,23,62 In this work, the reusability of as-prepared Au22-BX was assessed through repetitive Teicoplanin capturing experiments under identical conditions. Au22-BX that underwent the entire enrichment procedure were regenerated with an incubation buffer and purified several times by a 10 kDa desalting column. Subsequently, regenerated Au22-BX were reused to capture the Teicoplanin sample. The results revealed the maintained capture efficiency after five cycles (Fig. 5a and b). In particular, the absorption and PL spectra between the initial and regenerated Au22-BX NCs are maintained (Fig. 5c), confirming the structural stability of Au22-BX throughout the enrichment process. Moreover, the TEM image of regenerated Au22-BX exhibits a uniform distribution, similar to the initial sample, without any apparent aggregation after the enrichment process (Fig. 5d). The test implies the superior reusability of Au22-BX, highlighting their promising potential for practical applications. All these results indicate the outstanding performance of our enrichment method (Table S1†).
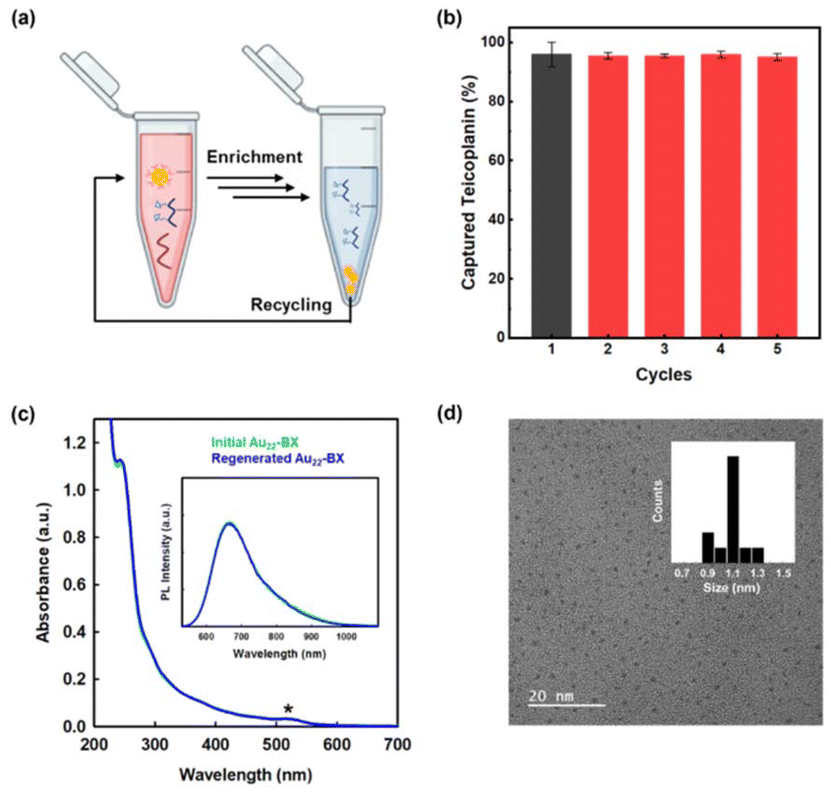 | ||
| Fig. 5 Evaluation of reusability. (a) Illustration of the recycling of enrichment. (b) Reusability of the materials tested by standard peptide (teicoplanin). The error bar represents the standard error of the mean, as calculated in triplicate. (c) Quality check of regenerated Au22-BX after enrichment experiment by optical absorption spectrometry. Inset: emission spectra (λEX = 520 nm) in water. (d) TEM images of the regenerated Au22-BX. The inset shows histograms of the NC core diameter. | ||
2.4. Selective enrichment of glycated peptide from human serum
The effective enrichment of the model glycopeptide has prompted us to explore the feasibility of the newly developed NC-based enrichment method in practical applications using human serum to promote the material's utilization in detecting biomarkers associated with disease diagnosis.11,60,63 Owing to the relatively low occurrence of glycation in vivo, certain glycation sites are difficult to identify, especially when working with limited sample quantities.64 Therefore, we incorporated human serum glycated in vitro to address this limitation. Digested glycated human serum sample was used in the analysis without pretreatment. After the enrichment by Au22-BX, the enriched glycated peptides were subjected to nanoscale liquid chromatography coupled to tandem MS (LC-MS/MS) analysis. The detected results of triplicate parallel experiments were compared with those of the initial samples (control) to assess the effectiveness of our NC-based enrichment method. The base peak chromatograms obtained from the LC-MS analysis of the peptides are shown in Fig. S16.† Compared with the control, the simplified profile of the enriched sample confirms the effectivity of the NC-based enrichment method for actual biological samples. The detailed results from each sample are summarized in Table 1. First, an average of approximately 82 unique glycated peptides were identified from triplicate experiments after enriching each human serum sample, which is a slightly smaller number than that identified from the control sample. However, the ratio of the glycated peptides to total peptides significantly increased by more than twofold for the enriched samples, confirming the effectiveness of our enrichment method. Furthermore, the number of identified glycated peptide-spectrum matches (PSMs), in which a peptide corresponds to a specific spectrum match, increases in the enriched samples compared to the control. Particularly, a ratio of 14.33% is achieved for glycated PSMs after enrichment, which is over four times greater than that of the control (3.37%). The increase in the number of PSMs indicates increased abundance, which indirectly correlates with the target protein quantity in terms of the precursor selection. Previous studies have evaluated the purity of species-specific peptides by examining the changes in the PSM ratios, particularly in the analysis of human-specific peptides.65 Therefore, the increase in PSM ratio signifies the increased detection effectivity of the glycated peptides and improved reliability of the peptide identification, indicating the effectiveness of the enrichment process for complex human serum samples.| Average counts | Control sample | Au22-BX enriched |
|---|---|---|
| Total peptides | 2433 | 898 |
| Number of glycated peptides | 106 | 82 |
| Ratio of glycated peptides | (4.31 ± 0.48)% | (8.89 ± 0.94)% |
| Total PSMs | 9824 | 3502 |
| Number of glycated PSMs | 322 | 495 |
| Ratio of glycated PSMs | (3.37 ± 0.62)% | (14.33 ± 0.93)% |
Table 1 shows some impressive enrichment results of Au22-BX. Moreover, critical aspects that need to be addressed in the future are revealed. First, the total PSM and peptide counts in the dataset after enrichment significantly decreased compared to those of the control group. This explains the slight increase in the abundance of glycated PSMs and the decrease in that of the glycated peptides after enrichment. These results can be ascribed to the presence of unseparated Au22-BX in the enriched PSM and peptide samples that can overestimate of quantities injected for the LC-MS/MS analysis. The enrichment protocol described in Scheme 1 is highly effective for standard peptides, but is less effective for the complex human serum even after several separation trials. Thus, a more effective separation method of Au22-BX should be devised.
Another important aspect of the enrichment result in Table 1 is the significant nonspecific bindings observed in the human serum sample. As discussed above, the Au22-BX enrichment method has excellent selectivity toward glycopeptides in the standard peptide system (Fig. S15†). However, the nonspecific bindings account for more than 91% and 85% of the peptides and PSMs, respectively, of the human serum sample. As those nonspecific bindings may mainly arise from the hydrophilic properties of the tripeptide-type ligand of Au22 NCs, ligand engineering30–32,66 of the NCs could be used to reduce the nonspecific bindings.
3. Conclusions
We developed a novel approach that combines BA affinity chemistry with ultrasmall Au NCs for the first time, achieving highly efficient enrichment of early-stage glycated peptides with exceptional selectivity and sensitivity. The new enrichment material (Au22-BX) with a pH response and large specific surface area was synthesized by functionalizing Au NCs with BX ligands using a facile post synthetic modification method. Au22-BX exhibited superior enrichment performance, including an ultralow detection limit (0.01 fmol μL−1), high selectivity (teicoplanin/vancomycin = 1/1000, w/w), and excellent reusability. Further experimental results substantiated the ongoing effectiveness of the strategy in enriching the glycated proteome, even when dealing with complex biological samples from human serum. Therefore, the novel enrichment strategy based on Au22-BX NCs provided a significant potential for global glycated proteome analysis. Further, their remarkable NIR emitting characteristics can be effectively used in visualizing the enrichment process. Notably, the advantages presented by these NCs could pave the way for their application in various proteomics fields, particularly in disease diagnostic and prognostic studies.Author contributions
H.H., H.L., and D.L. designed the project. H.H., Y.K., and S.A. conducted the NC synthesis and enrichment experiments. S.C. performed the LC-MS/MS analysis. J.M. assisted with the MS analysis. H.L. and D.L. supervised the project. H.H., H.L., and D.L. wrote and revised the manuscript. All authors discussed the results and provided comments on the manuscript at all stages.Data availability
Most data supporting this article have been included as part of the main text and the ESI.† Additional data that support the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korea Government (no. NRF-2022R1A2C3003610) and by Korea Evaluation Institute of Industrial Technology (KEIT) (no. 20018578).References
- P. Ulrich and A. Cerami, Recent Prog. Horm. Res., 2001, 56, 1–22 CrossRef CAS.
- A. Twarda-Clapa, A. Olczak, A. M. Białkowska and M. Koziołkiewicz, Cells, 2022, 11, 1312 CrossRef CAS.
- M. Wang, Y. Liang, K. Chen, M. Wang, X. Long, H. Liu, Y. Sun and B. He, Nanoscale, 2022, 14, 2119–2135 RSC.
- S. D. Yan, X. Chen, J. Fu, M. Chen, H. Zhu, A. Roher, T. Slattery, L. Zhao, M. Nagashima, J. Morser, A. Migheli, P. Nawroth, D. Stern and A. M. Schmidt, Nature, 1996, 382, 685–691 CrossRef CAS PubMed.
- M. A. Smith, S. Taneda, P. L. Richey, S. Miyata, S.-D. Yan, D. Stern, L. M. Sayre, V. M. Monnier and G. Perry, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5710–5714 CrossRef CAS.
- A. Simm, J. Proteomics, 2013, 92, 248–259 CrossRef CAS PubMed.
- M. N. Trujillo and J. J. Galligan, Nat. Chem. Biol., 2023, 19, 922–927 CrossRef CAS.
- T. Niwa, Mass Spectrom. Rev., 2006, 25, 713–723 CrossRef CAS.
- S. D'Aronco, S. Crotti, M. Agostini, P. Traldi, N. C. Chilelli and A. Lapolla, Mass Spectrom. Rev., 2019, 38, 112–146 CrossRef.
- R. Schmidt, D. Böhme, D. Singer and A. Frolov, J. Mass Spectrom., 2015, 50, 613–624 CrossRef CAS.
- S. Cho, V.-A. Duong, J.-H. Mok, M. Joo, J.-M. Park and H. Lee, Rev. Anal. Chem., 2022, 41, 83–97 CrossRef CAS.
- Q. Zhang, J. M. Ames, R. D. Smith, J. W. Baynes and T. O. Metz, J. Proteome Res., 2009, 8, 754–769 CrossRef CAS PubMed.
- L. Zhang, C.-W. Liu and Q. Zhang, Anal. Chem., 2018, 90, 1081–1086 CrossRef CAS.
- A. Frolov and R. Hoffmann, Ann. N. Y. Acad. Sci., 2008, 1126, 253–256 CrossRef CAS.
- Q.-H. Tong, T.-Y. Yan, T. Tao, L. Zhang, L.-Q. Xie and H.-J. Lu, Anal. Chem., 2018, 90, 3752–3758 CrossRef CAS PubMed.
- L. Wu, C. Fang, L. Zhang, W. Yuan, X. Yu and H. Lu, Anal. Chem., 2021, 93, 4398–4407 CrossRef CAS PubMed.
- L. Zhang, Y. Xu, H. Yao, L. Xie, J. Yao, H. Lu and P. Yang, Chem. – Eur. J., 2009, 15, 10158–10166 CrossRef CAS PubMed.
- W. Chen, J. M. Smeekens and R. Wu, Mol. Cell. Proteomics, 2014, 13, 1563–1572 CrossRef CAS.
- H. Li, H. He and Z. Liu, TrAC, Trends Anal. Chem., 2021, 140, 116271 CrossRef CAS.
- H. Xiao, W. Chen, J. M. Smeekens and R. Wu, Nat. Commun., 2018, 9, 1692 CrossRef PubMed.
- S. Kong, Q. Zhang, L. Yang, Y. Huang, M. Liu, G. Yan, H. Zhao, M. Wu, X. Zhang, P. Yang and W. Cao, Anal. Chem., 2021, 93, 6682–6691 CrossRef CAS.
- C. Zhang, X. Jin, L. Wang, C. Jin, X. Han, W. Ma, X. Li and G. Teng, ACS Appl. Mater. Interfaces, 2021, 13, 9714–9728 CrossRef CAS.
- L. Yang, Q. Zhang, Y. Huang, L. Lin, H. Schlüter, K. Wang, C. Zhang, P. Yang and H. Yu, Analyst, 2020, 145, 5252–5259 RSC.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- R. W. Murray, Chem. Rev., 2008, 108, 2688–2720 CrossRef CAS.
- P. Maity, S. Xie, M. Yamauchi and T. Tsukuda, Nanoscale, 2012, 4, 4027–4037 RSC.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- S. Li, W. Tian and Y. Liu, Nanoscale, 2021, 13, 16847–16859 RSC.
- S. M. Han, M. Park, J. Kim and D. Lee, Angew. Chem., Int. Ed., 2024, e202404387 CAS.
- K. Pyo, V. D. Thanthirige, S. Y. Yoon, G. Ramakrishna and D. Lee, Nanoscale, 2016, 8, 20008–20016 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- K. Pyo, H. Xu, S. M. Han, S. Saxena, S. Y. Yoon, G. Wiederrecht, G. Ramakrishna and D. Lee, Small, 2021, 17, 2004836 CrossRef CAS.
- Z. Liu, J. Chen, B. Li, D.-E. Jiang, L. Wang, Q. Yao and J. Xie, J. Am. Chem. Soc., 2024, 146, 11773–11781 CrossRef CAS.
- H. Liu, G. Hong, Z. Luo, J. Chen, J. Chang, M. Gong, H. He, J. Yang, X. Yuan, L. Li, X. Mu, J. Wang, W. Mi, J. Luo, J. Xie and X.-D. Zhang, Adv. Mater., 2019, 31, 1901015 CrossRef CAS.
- S. Li, J. Wei, Q. Yao, X. Song, J. Xie and H. Yang, Chem. Soc. Rev., 2023, 52, 1672–1696 RSC.
- J. Liu, M. Yu, C. Zhou, S. Yang, X. Ning and J. Zheng, J. Am. Chem. Soc., 2013, 135, 4978–4981 CrossRef CAS PubMed.
- M. Yu and J. Zheng, ACS Nano, 2015, 9, 6655–6674 CrossRef CAS PubMed.
- C. V. Conroy, J. Jiang, C. Zhang, T. Ahuja, Z. Tang, C. A. Prickett, J. J. Yang and G. Wang, Nanoscale, 2014, 6, 7416–7423 RSC.
- K. Pyo, N. H. Ly, S. Y. Yoon, Y. Shen, S. Y. Choi, S. Y. Lee, S. W. Joo and D. Lee, Adv. Healthcare Mater., 2017, 6, 1700203 CrossRef.
- K. Pyo, N. H. Ly, S. M. Han, M. B. Hatshan, A. Abuhagr, G. Wiederrecht, S.-W. Joo, G. Ramakrishna and D. Lee, J. Phys. Chem. Lett., 2018, 9, 5303–5310 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS.
- K. Pyo, S. M. Han, H. Xu, S. Saha, G. Ramakrishna and D. Lee, Sol. RRL, 2021, 5, 2000710 CrossRef CAS.
- J. Arnaud, A. Audfray and A. Imberty, Chem. Soc. Rev., 2013, 42, 4798–4813 RSC.
- S. Jin, Y. Cheng, S. Reid, M. Li and B. Wang, Med. Res. Rev., 2010, 30, 171–257 CrossRef CAS.
- D. Bruen, C. Delaney, D. Diamond and L. Florea, ACS Appl. Mater. Interfaces, 2018, 10, 38431–38437 CrossRef CAS PubMed.
- M. B. Espina-Benitez, J. Randon, C. Demesmay and V. Dugas, Sep. Purif. Rev., 2017, 47, 214–228 CrossRef.
- K. Arii, T. Kai and Y. Kokuba, Eur. J. Pharm. Sci., 1999, 7, 107–112 CrossRef CAS.
- M. Dowlut and D. G. Hall, J. Am. Chem. Soc., 2006, 128, 4226–4227 CrossRef CAS PubMed.
- X. Qin, Z. Zhang, H. Shao, R. Zhang, L. Chen and X. Yang, Analyst, 2020, 145, 7511–7527 RSC.
- W. L. A. Brooks, C. C. Deng and B. S. Sumerlin, ACS Omega, 2018, 3, 17863–17870 CrossRef CAS.
- T. Figueiredo, Y. Ogawa, J. Jing, V. Cosenza, I. Jeacomine, J. D. M. Olsson, T. Gerfaud, J.-G. Boiteau, C. Harris and R. Auzély-Velty, Polym. Chem., 2020, 11, 3800–3811 RSC.
- L. Ren, Z. Liu, M. Dong, M. Ye and H. Zou, J. Chromatogr. A, 2009, 1216, 4768–4774 CrossRef CAS PubMed.
- P. R. Westmark, L. S. Valencia and B. D. Smith, J. Chromatogr. A, 1994, 664, 123–128 CrossRef CAS.
- D. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- R. J. Carvalho, J. Woo, M. R. Aires-Barros, S. M. Cramer and A. M. Azevedo, Biotechnol. J., 2014, 9, 1250–1258 CrossRef CAS PubMed.
- L. Wu, W. Fei, Z. Liu, L. Zhang, C. Fang and H. Lu, Anal. Chem., 2022, 94, 5213–5220 CrossRef CAS PubMed.
- P. Su, M. Li, X. Li, X. Yuan, Z. Gong, L. Wu, J. Song and Y. Yang, J. Chromatogr. A, 2022, 1667, 462869 CrossRef CAS PubMed.
- Y. Zhang, J. Jiao, P. Yang and H. Lu, Clin. Proteomics, 2014, 11, 18 CrossRef PubMed.
- Z. Gao, R. Tang, S. Ma, S. Jia, S. Zhang, B. Gong and J. Ou, Anal. Methods, 2021, 13, 4515–4527 RSC.
- S. Saleem, M. S. Sajid, D. Hussain, F. Jabeen, M. Najam-ul-Haq and A. Saeed, Anal. Bioanal. Chem., 2020, 412, 1509–1520 CrossRef CAS.
- J. Jeon, J. Yang, J.-M. Park, N.-Y. Han, Y.-B. Lee and H. Lee, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1092, 88–94 CrossRef CAS PubMed.
- Q. Zhang, M. E. Monroe, A. A. Schepmoes, T. R. W. Clauss, M. A. Gritsenko, D. Meng, V. A. Petyuk, R. D. Smith and T. O. Metz, J. Proteome Res., 2011, 10, 3076–3088 CrossRef CAS.
- Z. Shi, B. Mao, X. Chen, P. Hao and S. Guo, Cancer Res. Commun., 2023, 3, 202–214 CrossRef CAS PubMed.
- B. Zhang, J. Chen, Y. Cao, O. J. H. Chai and J. Xie, Small, 2021, 17, 2004381 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr03283g |
| This journal is © The Royal Society of Chemistry 2024 |