
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrostatic [FeFe]-hydrogenase–carbon nitride assemblies for efficient solar hydrogen production†
Yongpeng
Liu‡
 a,
Carolina
Pulignani‡
a,
Sophie
Webb
bc,
Samuel J.
Cobb§
a,
Santiago
Rodríguez-Jiménez
a,
Dongseok
Kim
a,
Ross D.
Milton
bc and
Erwin
Reisner
*a
a,
Carolina
Pulignani‡
a,
Sophie
Webb
bc,
Samuel J.
Cobb§
a,
Santiago
Rodríguez-Jiménez
a,
Dongseok
Kim
a,
Ross D.
Milton
bc and
Erwin
Reisner
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
bDepartment of Inorganic and Analytical Chemistry, University of Geneva, Geneva 41211, Switzerland
cNational Centre of Competence in Research (NCCR) Catalysis, University of Geneva, Geneva 41211, Switzerland
First published on 13th March 2024
Abstract
The assembly of semiconductors as light absorbers and enzymes as redox catalysts offers a promising approach for sustainable chemical synthesis driven by light. However, achieving the rational design of such semi-artificial systems requires a comprehensive understanding of the abiotic–biotic interface, which poses significant challenges. In this study, we demonstrate an electrostatic interaction strategy to interface negatively charged cyanamide modified graphitic carbon nitride (NCNCNX) with an [FeFe]-hydrogenase possessing a positive surface charge around the distal FeS cluster responsible for electron uptake into the enzyme. The strong electrostatic attraction enables efficient solar hydrogen (H2) production via direct interfacial electron transfer (DET), achieving a turnover frequency (TOF) of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 669 h−1 (4 h) and a turnover number (TON) of 198125 (24 h). Interfacial characterizations, including quartz crystal microbalance (QCM), photoelectrochemical impedance spectroscopy (PEIS), intensity-modulated photovoltage spectroscopy (IMVS), and transient photocurrent spectroscopy (TPC) have been conducted on the semi-artificial carbon nitride-enzyme system to provide a comprehensive understanding for the future development of photocatalytic hybrid assemblies.
669 h−1 (4 h) and a turnover number (TON) of 198125 (24 h). Interfacial characterizations, including quartz crystal microbalance (QCM), photoelectrochemical impedance spectroscopy (PEIS), intensity-modulated photovoltage spectroscopy (IMVS), and transient photocurrent spectroscopy (TPC) have been conducted on the semi-artificial carbon nitride-enzyme system to provide a comprehensive understanding for the future development of photocatalytic hybrid assemblies.
Introduction
Converting solar energy into clean chemical fuels, such as molecular hydrogen (H2), holds promise for advancing the concept of a circular economy.1 Among various photocatalysts, carbon nitride (CNX) has emerged as a particularly attractive candidate due to its unique advantages, including visible light absorption, cost-effective fabrication, scalability, and low toxicity.2 To further enhance the photocatalytic performance of CNX, significant efforts have been devoted to chemical modifications and the incorporation of co-catalysts.3 The introduction of ionic cyanamide functional groups into CNX (NCNCNX) has demonstrated substantial improvements in charge separation and photocatalytic activity, attributed to the prolonged lifetimes of photogenerated electrons.4,5 Moreover, the negatively charged cyanamide group provides a versatile platform for potential electrostatic interactions with co-catalysts.Nature has evolved enzymes as highly specific biological catalysts to facilitate essential processes in living organisms. Among these enzymes, hydrogenases (H2ases) stand out for their remarkable ability to catalyze the interconversion of protons and H2 with high efficiency at near-zero overpotential under mild conditions,6 surpassing the capabilities of synthetic catalysts.7 H2ases can be classified into three main types based on their metal cofactors: [NiFe]-H2ase, [FeFe]-H2ase, and [Fe]-H2ase, with [FeFe]-H2ase being generally the most active for the hydrogen evolution reaction (HER).6 The extensive investigation of H2ases as model biocatalysts8 has not only inspired the design of artificial systems such as synthetic Fe2S2(CO)6 catalysts that mimic the active site of the Fe2S2 subunit of the [FeFe]-H2ase (Fig. S1†),9,10 but also paves the way for developing biohybrid assemblies in semi-artificial photosynthesis systems.11,12
By interfacing CNX with H2ase, we combine the strengths of both artificial and biological approaches, resulting in unique properties that neither system can achieve individually.11,12 This integration opens up new avenues for exploring synergistic effects and unlocking unprecedented possibilities in solar energy conversion and catalysis. The activation of [FeFe]-H2ase by light can be considered as a model for the development of efficient bio-hybrid systems. Such a photocatalytic system has thus far been demonstrated using either toxic and expensive CdTe nanocrystals13 or carbon dots with a low turnover number (TON) of 20000 over 24 h.14 Direct electron transfer (DET) between graphitic carbon nitride (g-C3N4) and [NiFeSe]-H2ase has been established with non-specific interactions, resulting in a turnover frequency (TOF) of 4117 h−1 over 4 h.15 Subsequent improvements involved the incorporation of a non-diffusional electron mediator, TiO2, between g-C3N4 and [NiFeSe]-H2ase, leading to an enhanced TON (4 h) of 80000.16 In addition to its application in H2ase systems, CNX has predominantly been utilized for the regeneration of NADH in mediated electron transfer (MET) processes involving formate dehydrogenase17 or alcohol dehydrogenase.18
In this work, we present an approach for biological integration with CNX by demonstrating the electrostatic interaction with enzymes to form a functional biohybrid assembly. This method establishes a benchmark for solar H2 production, complemented by comprehensive interfacial characterizations utilizing a quartz crystal microbalance (QCM), photoelectrochemical impedance spectroscopy (PEIS), intensity-modulated photovoltage spectroscopy (IMVS), and transient photocurrent spectroscopy (TPC). Specifically, we coupled negatively charged NCNCNX with H2ases containing different surface charges for in vitro photocatalytic H2 production without an external electron relay. The adsorption process of H2ases on NCNCNX is quantified by QCM, whereas PEIS provides insights into the charge carrier dynamics at the biomaterial interface.
Results and discussion
The NCNCNX photocatalysts were synthesized using melamine and potassium thiocyanate following previously published methods.4,19 Detailed synthesis procedures and characterizations, including scanning electron microscopy (SEM), attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy, fluorescence spectroscopy, and ultraviolet-visible spectroscopy are provided in the ESI (Fig. S2–S5).† [FeFe]-H2ase from Clostridium pasteurianum (CpI, heterologously produced in Escherichia coli) and [NiFeSe]-H2ase from Desulfovibrio vulgaris Hildenborough (DvH) were expressed and purified under anaerobic conditions.20,21Fig. 1 illustrates the hypothesis that the negatively charged NCNCNX possesses the ability to activate enzymes with positively charged electron entry points, such as CpI [FeFe]-H2ase.22 Additionally, the unique property of NCNCNX in converting alcohols selectively into aldehydes offers an opportunity to monitor the clean oxidation reaction of 4-methylbenzyl alcohol (4-MBA) to 4-methylbenzaldehyde (p-tolualdehyde), allowing the quantification of stoichiometry from the products resulting from oxidation and reduction.5,23
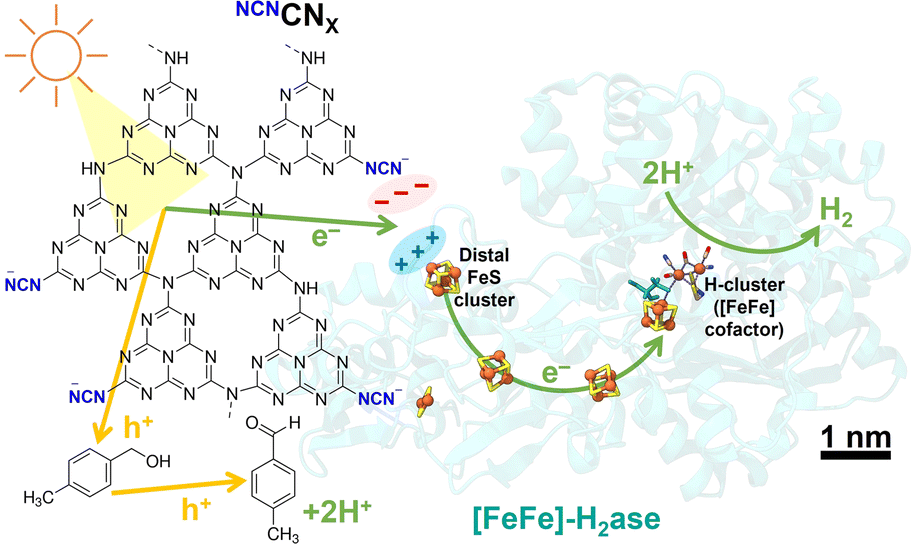 | ||
| Fig. 1 Schematic of photocatalytic H2 evolution coupled with alcohol oxidation to aldehyde using an electrostatic NCNCNX|[FeFe]-H2ase (CpI, PDB: 4XDC) assembly. Scale bar refers to enzyme and CNX is not shown to scale. | ||
To construct a photocatalytic system for solar H2 production coupled with selective alcohol oxidation, we interfaced [FeFe]-H2ase with NCNCNX in the presence of 50 mM 4-MBA in 1 mL aqueous MOPS buffer solution (0.1 M, pH 7). To determine the surface charge of NCNCNX, zeta potential measurements were performed. Notably, the presence of light-induced blue radicals (absorption band from 500–750 nm, Fig. S5†)5,24 did not impact the surface charge of NCNCNX (Fig. S6†), indicating that these radicals are long-lived and deeply trapped photoelectrons.23,25 The negatively charged surface is primarily attributed to the cyanamide group, which maintains a negative zeta potential even at a pH below 2 (Fig. S7†). CpI [FeFe]-H2ase was selected due to its distal FeS cluster ([4Fe-4S]) being surrounded by a positively charged region containing surface arginine and lysine residues. In vivo, this distal FeS cluster region is thought to interact with the negatively charged region of ferredoxin for electron transfer.22 By constructing electrostatic NCNCNX|[FeFe]-H2ase assemblies, we achieve efficient solar H2 production, with NCNCNX mimicking the role of ferredoxin to deliver electrons directly into [FeFe]-H2ase (Fig. 1).
Time-dependent photocatalytic H2 evolution using NCNCNX|[FeFe]-H2ase complexes is illustrated in Fig. 2a. The TON is determined by the ratio between the number of moles of product (H2) and the number of moles of catalyst (H2ase) and the TOF is calculated by the TON per hour. Notably, a nearly linear increase in H2 yield is observed during the initial 4 h, reaching 3.0 ± 0.3 μmol with a TOF of 18669 h−1. This TOF value is approximately 4.5 times higher than the previous benchmark (4117 h−1)15 and is even comparable to systems utilizing MET such as g-C3N4|TiO2 (TOF = 20000 h−1)16 (note that previous systems used Desulfomicrobium baculatum (Dmb) [NiFeSe]-H2ases). Continuous irradiation of the NCNCNX|[FeFe]-H2ase assemblies for 24 h yielded 7.9 ± 0.6 μmol of H2 (TON = 198125). The efficient DET between NCNCNX and [FeFe]-H2ase can be attributed to the specific electrostatic interaction at the interface, which will be further evaluated by QCM and PEIS. Furthermore, 4-MBA was selectively oxidized to p-tolualdehyde (Fig. S8–S10),† with a H2:p-tolualdehyde ratio of 0.77. The observed ratio indicates the deep trapping of some photogenerated electrons within the CNX polymeric structure in addition to some buffer (MOPS) oxidation (see below).
 | ||
| Fig. 2 (a) Time-dependent photocatalytic H2 evolution of NCNCNX with H2ases for direct electron transfer (DET) and mediated electron transfer (MET). (b) Schematic of electron transfer process. Conditions: 1 mL anaerobic MOPS buffer (0.1 M, pH 7) containing 50 mM 4-MBA, 2 mg NCNCNX, 40 pmol H2ase (either CpI [FeFe] (PDB: 4XDC) or DvH [NiFeSe] (PDB: 5JSH)), AM 1.5G irradiation, 600 rpm stirring, 25 °C. For MET experiments, 2 mM methyl viologen (MV) was used. Error bars represent the standard deviation for a sample size of 3. | ||
Recent transient spectroscopic and electron paramagnetic resonance (EPR) studies extensively characterized the deep traps stored in NCNCNX on the time scale from ps to s.23–26 EPR analysis showed that these long-lived and deeply trapped photoelectrons emerged as blue radicals, processing a symmetric Gaussian line near the free electron g value at X-band frequency of ∼9.6 GHz.24 These blue radicals can also be visualized by UV-vis spectroscopy (Fig. S5†) and qualitatively by the eye (Fig. S11†). The oxidation ability of NCNCNX has been further evaluated using glycerol (a waste product from the biodiesel industry) as the reductant on a model NCNCNX|Pt (2 wt%) system. The oxidation products are quantified as glyceraldehyde at 134.2 ± 5.7 μmol h−1 g−1 and dihydroxyacetone at 54.9 ± 6.8 μmol h−1 g−1 (Fig. S12–S15)† and the H2 yield is 94 ± 9 μmol h−1 g−1. Replacing 4-MBA with ethylenediaminetetraacetic acid (EDTA) led to a ∼16% enhancement of H2 yield (4 h) with NCNCNX|[FeFe]-H2ase assemblies (Table S1†). This indicates that, under DET conditions, the rate of 4-MBA oxidation is limiting compared to HER. Despite this observed higher activity, it is worth noting that EDTA is considered as a sacrificial electron donor, and its oxidation results in a range of products that cannot be easily characterized. As a result, the primary focus of this study is the conversion of 4-MBA to p-tolualdehyde, serving as a model reaction for the selective oxidation of alcohols to aldehydes.
Exclusion controls were conducted by removing individual components from the photocatalytic system (Table S1†). As depicted in Fig. 2a and S16,† the system exhibited significantly reduced efficiency in the absence of any component, with H2 yields below 0.3 μmol over 4 h. Minor H2 evolution activity was observed with NCNCNX|[FeFe]-H2ase assemblies even in the absence of 4-MBA, yielding 285 ± 24 nmol H2 in 4 h. This observation suggests that MOPS serves as a much less efficient electron donor in the photocatalytic reaction. NMR analysis (Fig. S17–S20†) provides evidence of MOPS oxidation during photocatalysis. However, it is noteworthy that alcohol oxidation on NCNCNX is so efficient and selective that the photocatalytic activity of MOPS oxidation is only ∼5% compared to 4-MBA oxidation in 24 h (Fig. 2a). Upon replacing MOPS buffer with pH 7 phosphate buffer, the reaction is drastically reduced in the absence of 4-MBA, accompanied by a ∼27% decrease in photocatalytic activity in the presence of 4-MBA (Fig. S21, Table S2†). This observation is in line with literature that MOPS as a standard Good's buffer can maintain high in vitro biochemical and biological activities.27 Notably, control experiments were also performed to validate the proposed electrostatic interactions using a DvH [NiFeSe]-H2ase, differing from the previously reported Dmb [NiFeSe]-H2ase.15,16 The distal FeS cluster of both [NiFeSe]-H2ases near its surface is surrounded by amino acids that lead to a local negative charge, serving as the electron entry point for interaction with the positively charged heme of cytochrome c3 during electron transfer in vivo.28 Interfacing DvH [NiFeSe]-H2ase with NCNCNX resulted in the production of 8.6 ± 0.33 nmol of H2 in 4 h, with a significantly lower TON of 215 (Fig. 2a). These results indicate that electrostatic repulsion prevents DET in this system. A detailed comparison among state-of-the-art photocatalytic systems combining carbonaceous photocatalysts and H2ase are listed in Table 1.
| Photocatalytic system (DET) | TOFa (h−1) | TONb | AQE | Ref. |
|---|---|---|---|---|
| a TOF is calculated from 4 h photocatalysis. b TON is calculated from 24 h photocatalysis. c TiO2 as a non-diffusional electron mediator. Illumination conditions. d AM 1.5G, 100 mW cm−2, Xe lamp. e 50 mW cm−2, LED lamp. | ||||
| NCNCNX|[FeFe] + MBAd | 18669 |
198125 |
0.35% | This work |
| g-C3N4|[NiFeSe] + EDTAd | 4117 | 36000 |
0.07% | 15 |
| CDs|[NiFeSe] + EDTAd | 3125 | 44000 |
0.36% | 29 |
| CDs|[FeFe] + TEOAe | 1500 | 19000 |
1.7% | 14 |
To determine the charge transfer efficiency of DET, methyl viologen (MV) as a soluble electron mediator is used to activate MET (Fig. 2a). Note that the presence of MV may suppress DET due to the kinetic and thermodynamic favorable one electron reduction of MV molecules to MV radicals.25,30 Upon the addition of MV (2 mM), the H2 yield reaches 24.0 ± 0.1 μmol for CpI [FeFe]-H2ase and 25.6 ± 1.3 μmol for DvH [NiFeSe]-H2ase after 24 h of irradiation. The comparable H2 yields, despite differences in specific activity, indicate that the rate limiting step during MET is not enzyme turnover but the photoactivity of NCNCNX, as evidenced by control experiments (Table S1†). Specifically, by doubling the H2ase loading from 40 pmol to 80 pmol, no significant changes in H2 yields were observed over both 4 hour and 24 hour periods under MET conditions. The efficiency of DET is qualitatively determined by the DET:MET ratio, defined by the ratio of H2 yield in the absence of MV (DET) and in the presence of MV (MET). In the case of [FeFe]-H2ase and [NiFeSe]-H2ase, DET:MET ratios of approximately 25% and 0.5% are observed, respectively (Fig. S22†). By comparing the results obtained from DET and MET, a schematic representation can be depicted in Fig. 2b. It highlights the establishment of efficient electron transfer directly between NCNCNX and [FeFe]-H2ase, with a DET:MET ratio of 25%. This finding emphasizes the effectiveness and benefits of electrostatic interactions in facilitating DET. Conversely, the electrostatic repulsion between [NiFeSe]-H2ase and NCNCNX prevents DET, resulting in a low DET:MET ratio of 0.5%. Notably, the apparent quantum efficiency (AQE) measured at 450 nm with CNCNX|[FeFe]-H2ase assemblies under DET and MET conditions are 0.35% and 1.4% (Table S3†), respectively. In contrast, a model NCNCNX|Pt (2 wt%) system yields 0.28 ± 0.05 mmol h−1 g−1 H2 and 0.51 ± 0.09 mmol h−1 g−1p-tolualdehyde with an AQE of 1.92% at 450 nm. In terms of the overall stability of the designed systems, the DET system exhibited a rather linear photocatalytic activity up to 12 h. While MET systems are fully inactive only after 20 h. Long-term experiments up to 36 h confirmed these trends, with no further H2 production in DET after 24 h and minimal H2 yield increases in MET (24.0 to 25.0 μmol for [FeFe] and 25.6 to 26.7 μmol for [NiFeSe]). These findings align with recent observations on carbon dot|[FeFe]-H2ase photocatalytic systems.14
To gain deep insights into the interaction between NCNCNX and H2ase, QCM analysis was conducted. As shown in the schematic in Fig. 3a, the Au-coated quartz chip was functionalized with a thin layer of NCNCNX by drop casting 0.5 mL of an ultrasonicated suspension (0.1 mg mL−1) of NCNCNX, to mimic the operando conditions during photocatalysis. By flowing a buffer solution containing enzymes on the chip, the adsorption process of H2ase at the surface of NCNCNX can be monitored as a function of time and quantified based on the Sauerbrey equation.31Fig. 3b shows the QCM analysis of CpI [FeFe]-H2ase and DvH [NiFeSe]-H2ase on the NCNCNX-modified chip. After establishing a stable baseline by circulating 0.1 M MOPS pH 7 buffer with 50 mM 4-MBA, the enzymes were introduced separately at the same concentration as in the photocatalysis experiments. The adsorption of both H2ases on NCNCNX exhibits in two distinct stages, a fast adsorption process before 1.5 h and slow adsorption after 1.5 h. Interestingly, a higher amount of [NiFeSe]-H2ase (39.5 pmol cm−2) is adsorbed onto NCNCNX compared to [FeFe]-H2ase (16.6 pmol cm−2) over 10 h. The observed adsorption profiles can be explained by the proposed electrostatic interactions in Fig. 3c. Based on the electrostatic potential maps (Fig. 3c), both H2ases exhibit distinct surface charge distribution. By indexing the specific protein structures, CpI [FeFe]-H2ase (PDB: 4XDC), and DvH [NiFeSe]-H2ase (PDB: 5JSH), within the protein dipole moment database,32 it is found that [NiFeSe]-H2ase possesses a larger dipole moment of 1972 D compared to [FeFe]-H2ase (1707 D), resulting in a stronger association (Fig. 3b). However, for [NiFeSe]-H2ase, DET can only be established via the negatively charged patch near the distal FeS cluster, which is unfavorable for the negatively charged NCNCNX and thus dramatically reduces DET to the enzyme active site for catalysis. Consequently, the strong association observed at the NCNCNX|[NiFeSe]-H2ase interface is non-specific and results mainly in inactive biohybrid assemblies. In contrast, absorbed [FeFe]-H2ase has positively charged distal FeS cluster that can specifically interact with negatively charged NCNCNX for DET.22,28 The presence of other positively charged regions in the [FeFe]-H2ase (Fig. 3c) might also attract NCNCNX. However, due to the rigidity of the heptazine-based NCNCNX (A–B′ stacking), proper orientation for DET near the distal FeS cluster could be hindered. This might explain the observed relatively low 25% DET/MET ratio. Therefore, QCM analysis provides valuable insights into the significance of specific interactions in facilitating DET.33
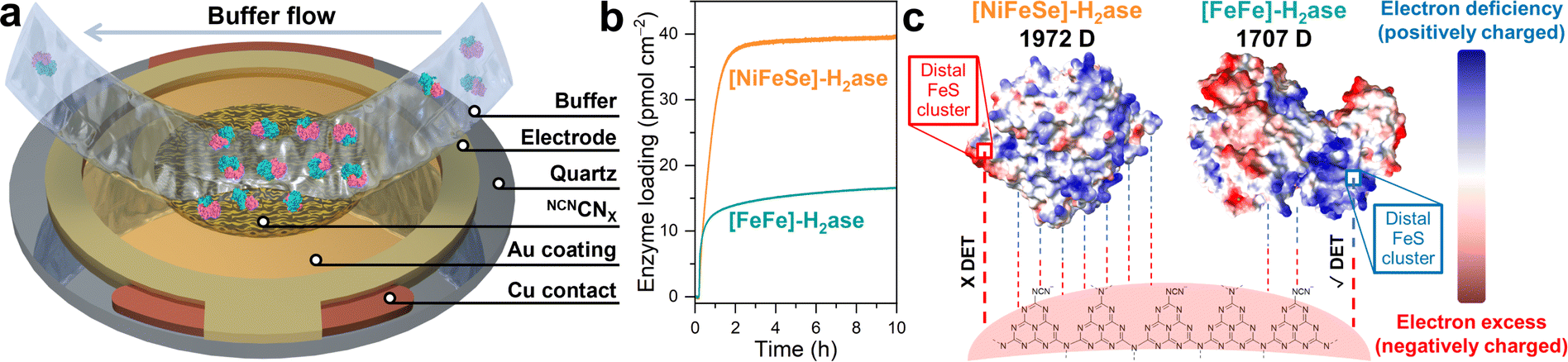 | ||
| Fig. 3 (a) Schematic illustration of a NCNCNX-coated quartz chip. Buffer containing [FeFe]-H2ase (PDB: 4XDC) is flowing towards the chip and [FeFe]-H2ase is adsorbed onto the surface. (b) QCM analysis of the adsorption process of H2ase on a NCNCNX-coated quartz chip. Conditions: 2 mL anaerobic MOPS buffer (0.1 M, pH 7) containing 50 mM 4-MBA, 80 pmol H2ase (either CpI [FeFe] or DvH [NiFeSe]), 25 °C. (c) Electrostatic potential maps of CpI [FeFe] (PDB: 4XDC) and DvH [NiFeSe] (PDB: 5JSH), and their interactions with NCNCNX. | ||
To investigate the charge carrier dynamics between H2ases and NCNCNX, PEIS was performed using a three-electrode configuration. By applying a sinusoidal potential modulation to the NCNCNX-modified working electrode, which was made by depositing a NCNCNX suspension (5 μL, 24 mg mL−1) on FTO-coated glass,23 the impedance was recorded as the ratio of the complex-valued potential and current.34 A Randles circuit consisting of a series resistance (RS) in parallel with a combination of bulk capacitance (Cbulk) and charge transfer resistance (Rct), was proposed to fit the impedance response (Fig. 4a).35 The Nyquist plot (Fig. 4a) of the impedance response measured at −0.1 V vs. the reversible hydrogen electrode (RHE), is dominated by a single semicircle with no indication of a Warburg diffusion element. The proposed equivalent circuit provided a good fit (r2 > 0.95) to the impedance response, enabling quantitative analysis of the charge transfer process.
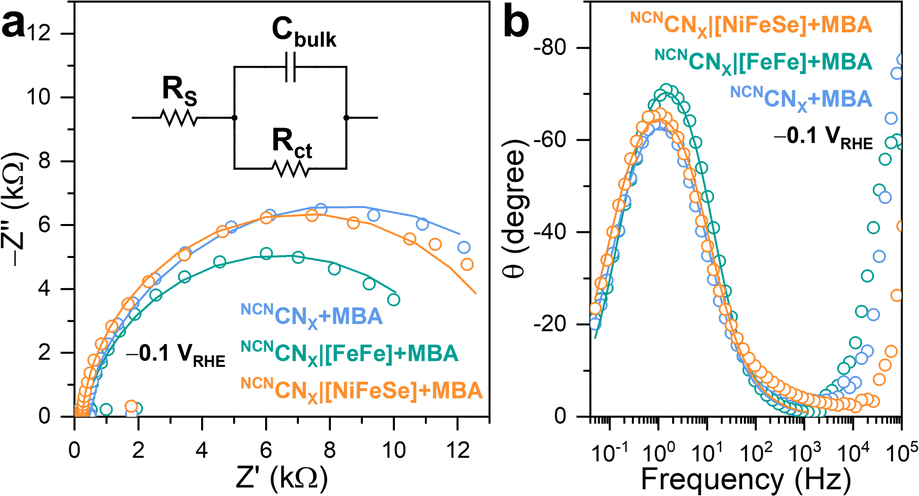 | ||
| Fig. 4 (a) Nyquist plots and (b) Bode phase plots of PEIS signal (open circles) with corresponding fitting curves (solid lines). Inset: proposed equivalent circuit to fit the impedance response. Conditions: 20 mL anaerobic MOPS buffer (0.1 M, pH 7) containing 50 mM 4-MBA, Ag/AgCl (3 M NaCl) reference electrode, Pt mesh counter electrode, AM 1.5G irradiation, 25 °C. | ||
Upon introduction of [FeFe]-H2ase on the working electrode, a decrease in the semicircle diameter is observed, corresponding to a decrease in Rct from 15975 Ω to 12317 Ω. This indicates that [FeFe]-H2ase, as a biocatalyst, facilitates the charge transfer from NCNCNX to the electrolyte for HER. Likewise, NCNCNX|[NiFeSe]-H2ase shows a Rct of 13950 Ω, similar to the bare NCNCNX. Such behaviors have been widely observed when incorporating synthetic co-catalysts onto semiconductors.36 The fitting results allowed determination of the pseudo first-order rate constant for charge transfer (kct), based on the phenomenological model developed for an illuminated photoelectrode.37,38 Specifically, the angular frequency at the maximum imaginary component of the semicircle in Nyquist plot (Fig. 4a) is equal to kct. The addition of [FeFe]-H2ase significantly enhances kct from 6.85 s−1 to 11.99 s−1, whereas [NiFeSe]-H2ase shows a negative impact on kct with a value of 5.02 s−1, further confirming the importance of specific interactions in facilitating the charge transfer process. The Bode phase plots (Fig. 4b) revealed that the charge transfer process occurred within the frequency range of 0.1 Hz to 1 kHz, consistent with the reported timeframe for photocatalytic HER using CNX.36 Within this range, the characteristic frequency at the maximum phase shift of NCNCNX|[FeFe]-H2ase are higher than NCNCNX|[NiFeSe]-H2ase and pristine NCNCNX, indicating [FeFe]-H2ase can initiate a faster charge transfer process for HER. Likewise, the characteristic frequencies of NCNCNX|[NiFeSe]-H2ase and pristine NCNCNX remain the same, meaning that DET cannot be established between NCNCNX and [NiFeSe]-H2ase. The voltage-dependent impedance response is illustrated in Fig. S23.† A more negative applied potential yields a diminished semicircular feature in the Nyquist plots, indicating reduced charge transfer resistance. This observation arises from the introduction of a larger band bending, resulting in improved separation of photogenerated charges.39,40 Consequently, a greater population of free charge carriers is localized within the semiconductor, increasing the conductivity of NCNCNX. Notably, the RC response of the conductive substrate forms a semicircle with a diameter of approximately 200 Ω in the Nyquist plots (Fig. S24a†) at high frequency region (10 kHz to 1 MHz, Fig. S24b†) in the Bode phase plots.23 This impedance study on CNX with H2ase demonstrates that a specific interaction enables efficient DET by decreasing in Rct and increasing in kct.
The charge carrier dynamics between H2ases and NCNCNX were further examined using IMVS and TPC techniques in a three-electrode setup. IMVS is a spectroelectrochemical method widely employed in assessing electron recombination processes in photovoltaics. It monitors the open circuit voltage response to the sinusoidally modulated incident light intensity. The characteristic frequency observed at the minimum point of the Nyquist plot (fmin) directly correlates to the time constant of electron recombination. This parameter can be calculated using the following equation, providing the first-order electron lifetime τn:41
| τn = (2πfmin)−1 |
This model has been recently expanded to photoanodes for solar water oxidation.42,43 Although IMVS is not operando due to the distinct differences between a photoelectrode and a photocatalyst, photoelectrochemical techniques have been widely employed to gain insights into charge carrier behaviors in photochemical systems. This equation can be applied to a NCNCNX-based photoelectrode due to it functions as a photoanode in the presence of 4-MBA and under open circuit conditions.23 As depicted in Fig. 5a, the IMVS response exhibits a distinct semicircle in quadrant IV of the Nyquist plot, suggesting rapid kinetics in 4-MBA oxidation, similar to the cases of sacrificial Na2SO3 and H2O2 oxidation on a hematite photoanode.42,43
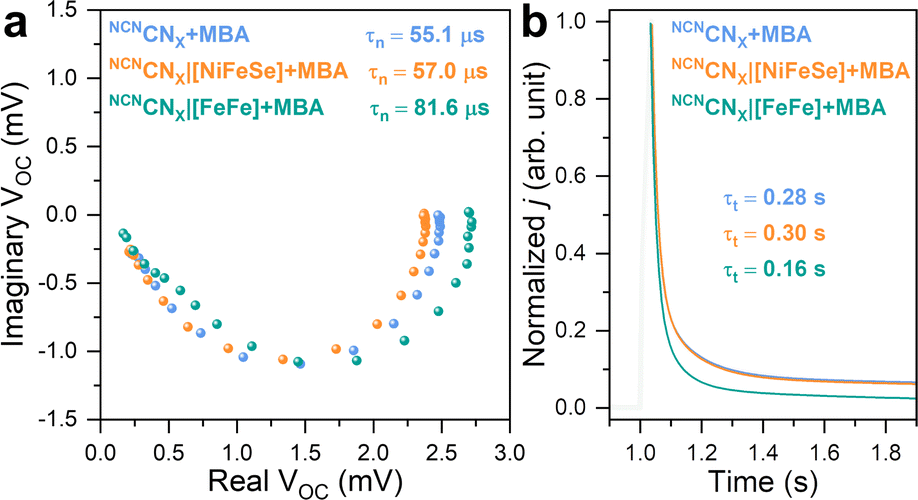 | ||
| Fig. 5 (a) Nyquist plots of IMVS response. (b) Normalized TPC response with corresponding exponential fitting curves. Conditions: 20 mL anaerobic MOPS buffer (0.1 M, pH 7) containing 50 mM 4-MBA, Ag/AgCl (3 M NaCl) reference electrode, Pt mesh counter electrode, AM 1.5G irradiation, 25 °C. | ||
The non-specific interaction between [NiFeSe]-H2ase and NCNCNX yields τn of 57.0 μs, whereas 55.1 μs is observed for pristine NCNCNX. This consistency indicates that the charge recombination process remains unaffected, matching with our previous observations of the absence of DET between [NiFeSe]-H2ase and NCNCNX. Upon the introduction of [FeFe]-H2ase onto NCNCNX, we observe a prolonged τn of 81.6 μs that can be explained as follows: Even under open circuit conditions, where no net electron exchange occurs at the NCNCNX|electrolyte interface, photogenerated electrons theoretically have the potential to react with protons via H2ases. Note that a TOF of 18669 h−1 (NCNCNX|[FeFe]-H2ase) corresponds to a frequency of 5.2 Hz. Therefore, all losses of photogenerated electrons within the measured frequency range (0.5 MHz to 0.5 Hz) are a combination of bulk recombination and catalytic reaction. The influence of a catalytic overlayer on IMVS response remains a topic of debate. Recent studies indicate that a co-catalyst overlayer can delocalize photogenerated charge carriers from the bulk photoelectrode, promoting charge separation and prolonging electron lifetime.42 Here, despite the absence of net exchange current, we hypothesize that photogenerated electrons can be stored in H2ase in the form of metal hydrides and reduced FeS clusters,44i.e., reversible intermediates for H2 evolution reaction. This storage mechanism reduces the probability of charge recombination with holes, resulting in an extended electron lifetime. IMVS observations are in line with the recent transient spectroscopic study on the impact of electron accumulation to charge recombination in NCNCNX.25 Thus, we demonstrate the use of IMVS on carbon nitride materials and on studying bio-hybrids.
Having gained insights into charge recombination, we conducted TPC measurements to assess the influence of H2ases on the electron extraction process of NCNCNX. The normalized TPC response, illustrated in Fig. 5b, reveals that the integration of [FeFe]-H2ase with NCNCNX leads to a significant reduction in electron transit time (τt) from 0.28 s to 0.16 s. This indicates that [FeFe]-H2ase, acting as a co-catalyst for NCNCNX, can effectively collect photoelectrons for HER, thereby facilitating electron transport within NCNCNX.45,46 Likewise, NCNCNX|[NiFeSe]-H2ase assemblies are non-specific, resulting in an unchanged τt of 0.30 s. The combined results from IMVS and TPC highlight the favorable effects of DET between [FeFe]-H2ase and NCNCNX on both charge recombination and transport. In contrast, the non-specifically interacted NCNCNX|[NiFeSe]-H2ase assemblies demonstrate minimal alterations in both τn and τt.
Conclusions
We present an electrostatic strategy for linking enzymes with carbon nitrides, demonstrating a benchmark for DET between NCNCNX and [FeFe]-H2ase for solar H2 production with a TON of 2 × 105 and a DET/MET ratio of 25% over 24 h. In contrast, the electrostatic repulsion between [NiFeSe]-H2ase and NCNCNX drastically reduced DET, leading to a DET/MET ratio of 0.5%. QCM analysis demonstrates that specific interactions play a pivotal role in enabling DET, irrespective of the observed differences in the adsorption profiles. Complementary spectroelectrochemical analysis using PEIS, IMVS, and TPC show that interfacing [FeFe]-H2ase with NCNCNX facilitates charge transfer and suppresses charge recombination, as evidenced by a 23% less resistive Rct, a 75% faster kct, a 48% longer τn, and a 43% shorter τt than bare NCNCNX. This study provides a promising and straightforward approach for achieving efficient electron transfer between carbon nitride and enzymes and serves as a reference for studying the charge carrier behavior of enzyme-photocatalyst assemblies using interfacial characterizations.Data availability
Data supporting the findings of this study are available from the Cambridge data repository: https://doi.org/10.17863/CAM.106936.Author contributions
Yongpeng Liu conceptualization, data curation, software, formal analysis, funding acquisition, investigation, visualization, methodology, writing – original draft, project administration, writing – review & editing; Carolina Pulignani conceptualization, resources, data curation, formal analysis, investigation, visualization, methodology, writing – original draft, writing – review & editing; Sophie Webb resources, investigation, methodology, writing – review & editing; Samuel J. Cobb data curation, investigation, methodology, writing – review & editing; Santiago Rodríguez-Jiménez formal analysis, investigation, methodology, writing – review & editing; Dongseok Kim investigation, methodology; Ross D. Milton conceptualization, resources, supervision, funding acquisition, validation, writing – original draft, project administration, writing – review & editing; Erwin Reisner conceptualization, resources, formal analysis, supervision, funding acquisition, validation, investigation, visualization, writing – original draft, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. L. gratefully acknowledges the Swiss National Science Foundation (SNSF) for the Postdoc. Mobility fellowship (grant number P500PN_202908) and the Isaac Newton Trust Early Career Fellowship (23.23(g)). We acknowledge support from the European Research Council (ERC) for a Consolidator Grant (MatEnSAP, 682833) and a UKRI/ERC Advanced Grant (EP/X030563/1). C. P. and E. R. acknowledge the European Union's Horizon 2020 project SOLAR2CHEM (Marie Skłodowska-Curie Actions with GAN 861151). S. W. and R. D. M. thank the NCCR Catalysis (grant number 180544) for support, a National Centre of Competence in Research funded by the SNSF. S. J. C. acknowledges The Leverhulme Trust for an Early Career Fellowship (ECF-2021-072) and Isaac Newton Trust (20.08(r)). S. R. J. gratefully acknowledges the European commission for a Horizon 2020 Marie Sklodowska-Curie individual Fellowship (GAN 891338). We thank Ana Margarida Coito and Prof. Inês A. C. Pereira for providing DvH [NiFeSe]-H2ase. We thank Dr Bidyut Bikash Sarma and Papa Kwakye Kwarteng for helpful discussions.References
- S. Nandy, S. A. Savant and S. Haussener, Chem. Sci., 2021, 12, 9866–9884 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- M. Kwak, J. Bok, B.-H. Lee, J. Kim, Y. Seo, S. Kim, H. Choi, W. Ko, W. H. Antink, C. W. Lee, G. H. Yim, H. Seung, C. Park, K.-S. Lee, D.-H. Kim, T. Hyeon and D. Yoo, Chem. Sci., 2022, 13, 8536–8542 RSC.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- H. Kasap, C. A. Caputo, B. C. M. Martindale, R. Godin, V. W. Lau, B. V. Lotsch, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2016, 138, 9183–9192 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331–4365 CrossRef CAS PubMed.
- G. Si, W.-G. Wang, H.-Y. Wang, C.-H. Tung and L.-Z. Wu, Inorg. Chem., 2008, 47, 8101–8111 CrossRef CAS PubMed.
- J.-X. Jian, C. Ye, X.-Z. Wang, M. Wen, Z.-J. Li, X.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2016, 9, 2083–2089 RSC.
- X. Fang, S. Kalathil and E. Reisner, Chem. Soc. Rev., 2020, 49, 4926–4952 RSC.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- K. A. Brown, S. Dayal, X. Ai, G. Rumbles and P. W. King, J. Am. Chem. Soc., 2010, 132, 9672–9680 CrossRef CAS PubMed.
- K. Holá, M. V. Pavliuk, B. Németh, P. Huang, L. Zdražil, H. Land, G. Berggren and H. Tian, ACS Catal., 2020, 10, 9943–9952 CrossRef.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. V. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 53, 11538–11542 CrossRef CAS PubMed.
- C. A. Caputo, L. Wang, R. Beranek and E. Reisner, Chem. Sci., 2015, 6, 5690–5694 RSC.
- Y. Zhang and J. Liu, Chem.–Eur. J., 2022, 28, e202201430 CrossRef CAS PubMed.
- S. Zhang, Y. Zhang, Y. Chen, D. Yang, S. Li, Y. Wu, Y. Sun, Y. Cheng, J. Shi and Z. Jiang, ACS Catal., 2021, 11, 476–483 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- Y. Liu, S. Webb, P. Moreno-García, A. Kulkarni, P. Maroni, P. Broekmann and R. D. Milton, JACS Au, 2023, 3, 124–130 CrossRef CAS PubMed.
- M. C. Marques, C. Tapia, O. Gutiérrez-Sanz, A. R. Ramos, K. L. Keller, J. D. Wall, A. L. De Lacey, P. M. Matias and I. A. C. Pereira, Nat. Chem. Biol., 2017, 13, 544–550 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- C. Pulignani, C. A. Mesa, S. A. J. Hillman, T. Uekert, S. Giménez, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2022, 61, e202211587 CrossRef CAS PubMed.
- V. W. Lau, D. Klose, H. Kasap, F. Podjaski, M.-C. Pignié, E. Reisner, G. Jeschke and B. V. Lotsch, Angew. Chem., Int. Ed., 2017, 56, 510–514 CrossRef CAS PubMed.
- W. Yang, R. Godin, H. Kasap, B. Moss, Y. Dong, S. A. J. Hillman, L. Steier, E. Reisner and J. R. Durrant, J. Am. Chem. Soc., 2019, 141, 11219–11229 CrossRef CAS PubMed.
- R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 5216–5224 CrossRef CAS PubMed.
- N. E. Good, G. D. Winget, W. Winter, T. N. Connolly, S. Izawa and R. M. M. Singh, Biochemistry, 1966, 5, 467–477 CrossRef CAS PubMed.
- P. M. Matias, C. M. Soares, L. M. Saraiva, R. Coelho, J. Morais, J. Le Gall and M. A. Carrondo, JBIC, J. Biol. Inorg. Chem., 2001, 6, 63–81 CrossRef CAS PubMed.
- G. A. M. Hutton, B. Reuillard, B. C. M. Martindale, C. A. Caputo, C. W. J. Lockwood, J. N. Butt and E. Reisner, J. Am. Chem. Soc., 2016, 138, 16722–16730 CrossRef CAS PubMed.
- M. Heyrovský, J. Chem. Soc. Chem. Commun., 1987, 1856–1857 RSC.
- G. Sauerbrey, Z. Med. Phys., 1959, 155, 206–222 CAS.
- C. E. Felder, J. Prilusky, I. Silman and J. L. Sussman, Nucleic Acids Res., 2007, 35, W512–W521 CrossRef PubMed.
- V. M. Badiani, C. Casadevall, M. Miller, S. J. Cobb, R. R. Manuel, I. A. C. Pereira and E. Reisner, J. Am. Chem. Soc., 2022, 144, 14207–14216 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Incorporated, 2nd edn, 2000 Search PubMed.
- J. E. B. Randles, Discuss. Faraday Soc., 1947, 1, 11–19 RSC.
- W. Jiang, Y. Zhao, X. Zong, H. Nie, L. Niu, L. An, D. Qu, X. Wang, Z. Kang and Z. Sun, Angew. Chem., Int. Ed., 2021, 60, 6124–6129 CrossRef CAS PubMed.
- K. G. U. Wijayantha, S. Saremi-Yarahmadi and L. M. Peter, Phys. Chem. Chem. Phys., 2011, 13, 5264–5270 RSC.
- P. Xu, C. L. Gray, L. Xiao and T. E. Mallouk, J. Am. Chem. Soc., 2018, 140, 11647–11654 CrossRef CAS PubMed.
- J. Bisquert, J. Phys. Chem. B, 2002, 106, 325–333 CrossRef CAS.
- Y. Liu, J. Quiñonero, L. Yao, X. D. C. Pereira, M. Mensi, R. Gómez, K. Sivula and N. Guijarro, J. Mater. Chem. A, 2021, 9, 2888–2898 RSC.
- J. Krüger, R. Plass, M. Grätzel, P. J. Cameron and L. M. Peter, J. Phys. Chem. B, 2003, 107, 7536–7539 CrossRef.
- Y. Liu, N. Guijarro and K. Sivula, Helv. Chim. Acta, 2020, 103, e2000064 CrossRef CAS.
- J. E. Thorne, J.-W. Jang, E. Y. Liu and D. Wang, Chem. Sci., 2016, 7, 3347–3354 RSC.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- F. Le Formal, K. Sivula and M. Grätzel, J. Phys. Chem. C, 2012, 116, 26707–26720 CrossRef CAS.
- Y. Liu, M. Xia, L. Yao, M. Mensi, D. Ren, M. Grätzel, K. Sivula and N. Guijarro, Adv. Funct. Mater., 2021, 31, 2010081 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00640b |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Chemistry, The University of Manchester, Manchester, M13 9PL, UK. |
| This journal is © The Royal Society of Chemistry 2024 |