
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed conformationally restricted alkylarylation enables divergent access to Csp3-rich N-heterocycles†
Kaixin
Chen‡
a,
Jie
Lin‡
a,
Jing
Jing
a,
Junda
Wang
a,
Jiayu
Hu
b,
Hong
Yi
 *b,
Aiwen
Lei
*b and
Jie
Li
*a
*b,
Aiwen
Lei
*b and
Jie
Li
*a
aKey Laboratory of Organic Synthesis of Jiangsu Province, MOE Key Laboratory of Geriatric Diseases and Immunology, Suzhou Key Laboratory of Pathogen Bioscience and Anti-infective Medicine, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: jjackli@suda.edu.cn
bCollege of Chemistry and Molecular Sciences, The Institute for Advanced Studies (IAS), Wuhan University, Wuhan, 430072, China. E-mail: hong.yi@whu.edu.cn; aiwenlei@whu.edu.cn
First published on 3rd September 2024
Abstract
Due to the intrinsic spatial orientation and structural novelty, Csp3-rich N-heterocycles have been recognized as increasingly sought-after scaffolds as compared to the aromatic ring-based moieties, which have generated considerable recent attention in drug discovery. Hence, we disclose a modular cobalt-catalyzed conformationally restricted alkylarylation strategy for the divergent access to Csp3-rich N-hetero(spiro)cycles. Herein, multiple effects, including radical rebound and conformational restriction, play critical roles in the stabilization of the stereospecific alkyl-cobalt-aryl intermediate. Under simple and mild reaction conditions, cobalt catalyst combines a range of polyfunctionalized cyclenyl bromides and organozinc pivalates to rapidly and reliably forge the architecturally complex Csp3-rich N-hetero(spiro)cycles (>70 examples, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), including but not limited to the [5,5]-, [5,6]-, [5,7]-, [5,12]-bicycles, tri- and tetracyclic N-heterocycles, as well as various novel N-heterospirocyclic scaffolds in one synthetic operation. Preliminary kinetic investigations suggested that the final reductive elimination might be the rate-determining step. Moreover, ample substrate scope, good functional group compatibility and facile derivatizations to the pharmaceutically active molecules show the potential applications of this technology to organic syntheses and drug discoveries in medicinal chemistry.
:1 dr), including but not limited to the [5,5]-, [5,6]-, [5,7]-, [5,12]-bicycles, tri- and tetracyclic N-heterocycles, as well as various novel N-heterospirocyclic scaffolds in one synthetic operation. Preliminary kinetic investigations suggested that the final reductive elimination might be the rate-determining step. Moreover, ample substrate scope, good functional group compatibility and facile derivatizations to the pharmaceutically active molecules show the potential applications of this technology to organic syntheses and drug discoveries in medicinal chemistry.
Introduction
Significant evidence proves that saturated and fused N-heterocyclic skeletons are important structural moieties due to their special three-dimensionality and rigidity, which can be commonly found in countless molecules of interest, including pharmaceuticals,1 lead candidates and biologically active entities (Scheme 1a),2 as well as organocatalysts (Scheme 1b).3 Although many efforts have been made during the last decades, crucial drawbacks, such as the lack of stereoselectivity and generality in this area, exist.4 Therefore, developing protocols that afford divergent polycyclic N-heterocycles in a stereo-controllable fashion is a longstanding goal in organic synthesis. Currently, Ru-NHC-catalyzed complete hydrogenation of indoles5 under 100 bar of H2 provides a platform to access octahydroindoles, while the relatively harsh conditions and the limited product type of [5,6]-fused N-heterocycles still represent drawbacks.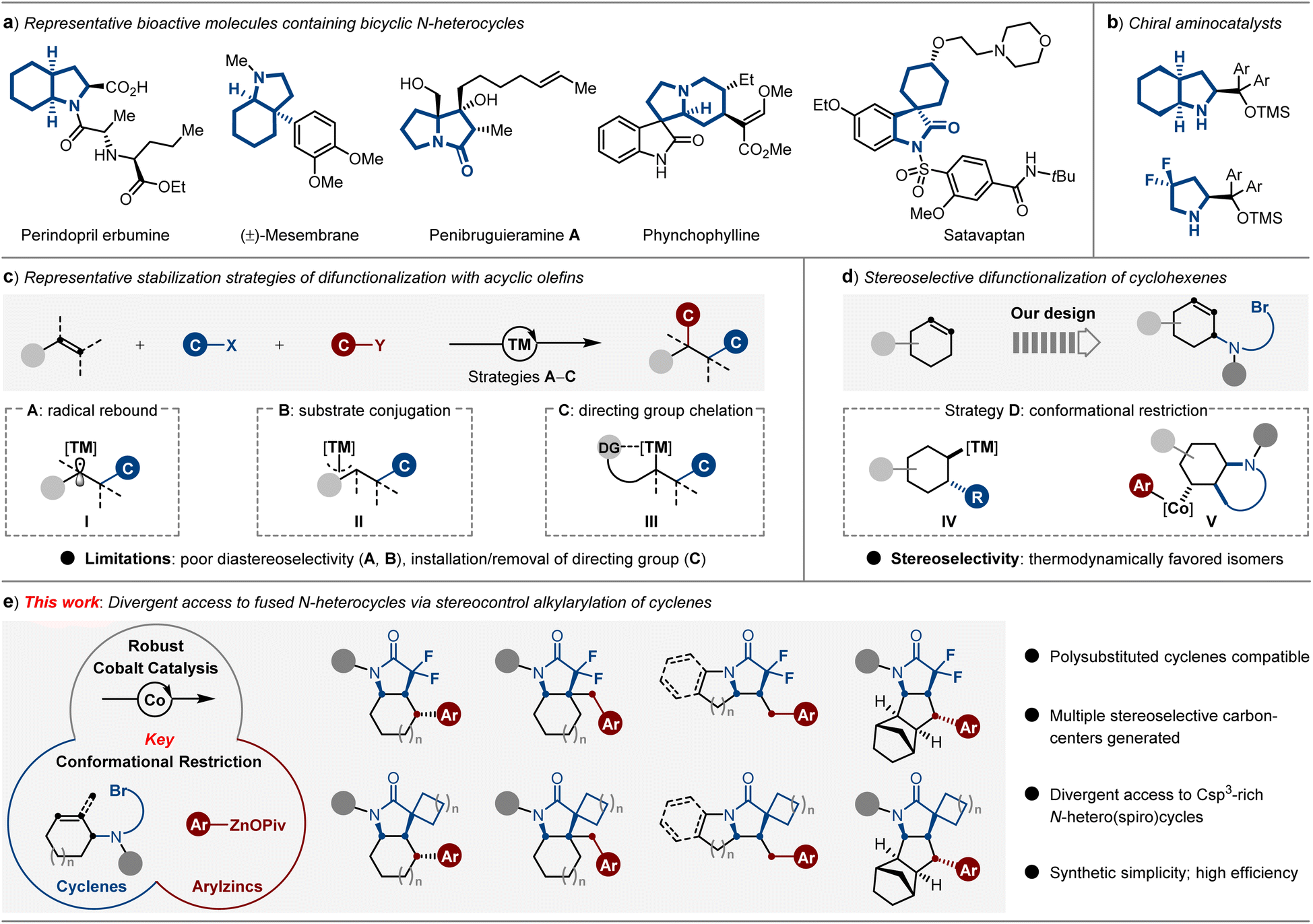 | ||
| Scheme 1 Modular access to fused N-heterocycles via metal-catalyzed alkene difunctionalization strategy. (a) Representative biologically active molecules containing fused bicyclic N-heterocycles and (b) chiral aminocatalysts. (c) Working modes of metal-catalyzed dicarbofunctionalization of acyclic olefins. (d) Dicarbofunctionalization of cyclohexenes via conformational restriction. (e) Cobalt-catalyzed conformationally restricted alkylarylation enables divergent access to Csp3-rich N-heterocycles. | ||
Considering that carbon–carbon bonds are the essential links in these Csp3-riched N-heterocycles, the construction of carbon–carbon bonds is the key to building such scaffolds. Notably, transition metal-catalyzed dicarbofunctionalization of olefins has been recognized as an increasingly powerful tool to establish complex molecular architectures from simple chemical feedstocks via sequentially forging twofold C–C bonds. Among these methods, it is a significant challenge to prevent β-hydride elimination of the key Csp3-[TM] intermediate. To this end, several strategies, such as radical rebound (I), substrate conjugation (II) and directing group chelation (III), as well as multiple effects operation in concert to enable the stabilization of Csp3-[TM] intermediates for achieving dicarbofunctionalization of acyclic olefins have been recently developed.6 However, representative drawbacks in controlling diastereoselectivity (strategies A and B) and pre-installation of directing groups (strategy C) still exist (Scheme 1c).
Recently, metal-catalyzed carboboration of endocyclic olefins has attracted considerable attention by offering considerable opportunities to the saturated (hetero)cyclic frameworks via conformational restriction strategy (IV),7 mostly affording the thermodynamically favored isomers as the products (Scheme 1d, left).8 Thus far, the substrate scope has largely been limited to the use of strained or activated endocyclic alkenes,9 while the stereoselective dicarbofunctionalization of unactivated cyclenes still remains challenging.10 Sporadic examples are shown to be highly dependent on the assistance of auxiliary directing groups.11 Notably, a directing group-free nickel-catalyzed reductive dialkylation of endocyclic olefins was only recently disclosed by the Huang group, while the products were obtained in rather modest diastereoselectivity.12
As a consequence, we believe that the dicarbofunctionalization of tethered cyclenes via the transition metal-catalyzed radical relay pathway involving the formation of a conformationally restricted intermediate, which will provide an attractive strategy for the rapidly divergent and stereoselective construction of fused Csp3-rich N-heterocycles (Scheme 1d, right). Indeed, intramolecular cyclizations of tethered cyclenes via atom transfer radical addition (ATRA) could occur in the absence of transition metals. However, the synthesis of Csp3-rich N-heterocycles bearing diverse carbogenic skeletons through sequence C–C/C–C bond formation still has challenges.13
As such, our approach of achieving this goal is to design active catalysis that operates the cascade dicarbofunctionalization of tethered cyclenes in a stereoselective fashion. Recently, the dramatic paradigms of carboxylate-coordination14 have been demonstrated in the preparation of solid organozinc pivalates,15 which possess relatively attenuated reducibility and show the distinct advantage of reactivity among conventional organozinc halides in the 3d transition-metal-catalyzed radical relay cross-coupling reactions.16 In this context, we detail the successful realization of divergent access to fused Csp3-rich N-hetero(spiro)cycles through the development of a conformationally restricted alkylarylation of cyclenes with readily available solid (hetero)arylzinc pivalates using industrially-friendly cobalt catalyst.17 Keys to our success of this strategy are the multiple effects of radical rebound and conformational restriction, which efficiently avoids the facile β-hydride elimination process, thus forming the stereospecific intermediate V. Under the optimal reaction conditions, the cobalt catalyst combines a range of polyfunctionalized cyclenyl bromides and organozinc pivalates to rapidly and reliably forge the architecturally complex Csp3-rich N-hetero(spiro)cycles with structural, functional, and physical features in medicinal chemistry (Scheme 1e).
Results and discussion
We began our studies by evaluating the cobalt-catalyzed diastereoselective alkylarylation of cyclohexene (1a) with salt-stabilized phenylzinc pivalate (2a). Preliminary experiments identified MeCN as the ideal reaction solvent and CoBr2 as the metal source of choice, which gave the arylated cis-octahydroindol-2-one 3a in 70% isolated yield with excellent diastereoselectivity (Fig. 1, top). Notably, to further examine the efficacy of cobalt catalysis, various chelating ligands such as bis(pyridine)s (L1–3), TMEDA (L4), tridentate 2,6-bis(N-pyrazolyl)pyridine (L5), dppbz (L6) and N-heterocyclic carbene (L7), could not lead to further increased yield; only neocuproine (L3) afforded the desired product 3a with equal efficiency to the former ligand-free conditions. With these results in hand, we wondered whether the high efficacy of this cobalt-catalyzed alkylarylation process was dominated by the phenylzinc pivalate. Hence, a series of different phenylzincs 2b–2f were prepared through transmetallation of Ph–MgCl (1.0 equiv) with corresponding ZnX2 (1.2 or 0.5 equiv; X = OPiv, Cl, Br, I). Thereafter, we tested their reactivity in the diastereoselective cobalt-catalyzed alkylarylation by parallel experiments. Notably, as compared to the solid Ph–ZnOPiv (2a),18 the other different phenylzinc halides 2b–2d, as well as the diphenylzincs 2e–2f, had a deleterious effect on the cobalt catalysis, thereby considerably restraining the desired reaction to occur. This observation is particularly noteworthy, as it suggests that the anion-coordination in organozinc pivalates promotes the catalytic activity of cobalt catalysts (Fig. 1). | ||
| Fig. 1 Optimization for diastereoselective cobalt-catalyzed alkylarylation of cyclohexene (1a). Reaction conditions: 1a (0.2 mmol, 1.0 equiv), 2a (3.0 equiv), CoBr2 (10 mol%), ligand (11 mol%), solvent (1.5 mL), 23 °C, 16 h. Yields are determined by GC analysis with dodecane as the internal standard. | ||
With the optimal reaction conditions established, we next turned our attention to examining the versatility of our stereoselective difluoroalkylarylation of cyclenes. As shown in Scheme 2, the scope of arylzinc pivalates (3–22) was largely insensitive to electronic and steric variations at the para-, meta-, and ortho-positions of appropriately aryl-substituted nucleophiles. Indeed, various functional groups such as SMe (5), TMS (6), OTBS (8), F (9, 12, 13, 15), Cl (10, 13, 14, 19), CF3 (11), dioxole (16–18) and ester (22) were well tolerated under the cobalt catalysis, furnishing the N-heterocycles in moderate to high yields with excellent stereoselectivity. Notably, our protocol could be extended to the construction of thiophene-, (di)benzothiophene- and (di)benzofuran-based backbones (23–26) when employing the corresponding solid heteroarylzinc pivalates as the nucleophilic partners; the structure of 25 was further validated by single-crystal X-ray diffraction. Furthermore, the current difluoroalkylarylation reaction can be conducted with a wide range of cyclenes. The ring size of the cyclic alkenes had no significant influence on the catalytic efficacy and five- and seven-membered olefins were well difluoroalkylarylated with arylzinc pivalates, leading to the bicyclic pyrrolidones 27–29 in 51%–65% yields. Gratifyingly, side chains of cyclohexene possessing phenyl (30), n-propyl (31), ester (32) and bismethyl (33) substituents posed no problem, thereby delivering the tetra-substituted cyclohexanes in a stereoselective manifold. Among them, the spatiotemporal selectivity of 30 was confirmed by single-crystal X-ray diffraction.
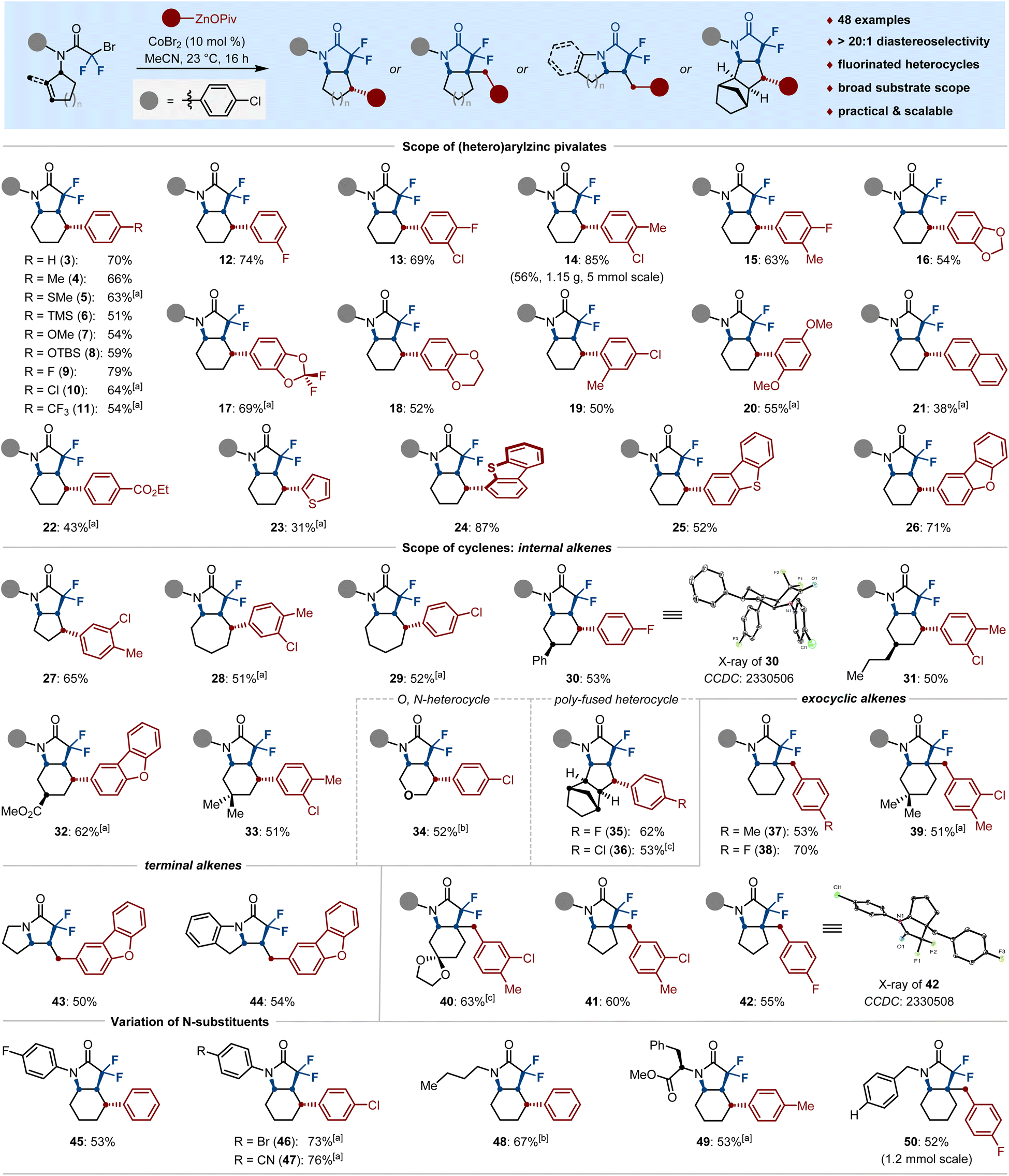 | ||
| Scheme 2 Divergent access to fluorinated Csp3-Rich N-heterocycles via cobalt-catalyzed difluoroalkylarylation. Reaction conditions: cyclenyl bromide (0.2 mmol, 1.0 equiv), (hetero)aryl–ZnOPiv (0.6 mmol, 3.0 equiv), CoBr2 (10 mol%), MeCN (1.5 mL), 23 °C, 16 h. [a]L3 (11 mol%) was used. [b] 0.1 mmol scale. [c] 0.15 mmol scale. High diastereoselectivity (dr > 20:1) for all the products was observed unless otherwise noted. | ||
Interestingly, the O-based cyclohexene, as well as the fused cyclopentene could be stereoselectively coupled to afford the novel N-heterocyclic scaffolds 34–36 in 52%–62% yields. As shown for 37–42, the reactions could be further extended to the more challenging exocyclic alkenes to generate the bicyclic N-heterocycles via building an all-carbon quaternary center and following Csp3–Csp2 bond formation with arylzinc pivalates. Remarkably, the pyrrolizines and their derivatives, which are important structural motifs of alkaloids with activities of relevance to biology or medicinal chemistry,2d,19 are easily within reach under identical reaction conditions; the desired fluorinated N-heterocycles 43–44 were obtained in 50–54% yields. Beyond that, a variation of the substitution pattern on the nitrogen atom was also investigated, such as aliphatic chain (48), removable benzyl group (50), as well as arenes bearing fluoro (45), sensitive bromo (46) and nitrile (47) functional groups, which proved to be suitable substrates. Even phenylalanine (49) could be aryl-bicyclized in satisfied yield with remarkably high diastereoselectivity.
Encouraged by the successful preparation of fluorinated bi-, tri-and tetracyclic N-heterocycles, we wondered whether it would be possible to enable a cyclized alkylarylation of the non-fluorinated cyclenyl bromides (Scheme 3). Substrates containing tertiary alkyl bromide resulted in stereoselective dicarbofunctionalization with various functionalized arylzinc pivalates, affording the [5,6]-, and [5,5]-bicyclic N-heterocycles 51–61 in 53%–97% yields with excellent diastereoselectivity (dr > 20:1). Interestingly, a 12-membered exocyclic alkene was proven to be a viable substrate as well, which smoothly underwent cyclized alkylarylation to afford the [5,12]-fused bicyclic heterocycle 62 bearing a quaternary center, albeit in a modest yield. Driven by the prevalence of N-heterospirocyclic motifs in a myriad of natural products and biologically relevant compounds,2a we were pleased to find that our protocol could be also effective for the divergent synthesis of spirocyclic pyrrolidinones 63–72 in one step via a facile spirocyclized alkylarylation, thus allowing us to dictate the site- and diastereoselective incorporation of the (hetero)aryl moiety at β-Csp3 sites within a spirocyclic N-heterocycle.
 | ||
| Scheme 3 Modular access to fused N-heterocycles and N-spirocycles via cobalt-catalyzed alkylarylation. Reaction conditions: cyclenyl bromide (0.2 mmol, 1.0 equiv), (hetero)aryl–ZnOPiv (0.6 mmol, 3.0 equiv), CoBr2 (10 mol%), MeCN (1.5 mL), 23 °C, 12 h. High diastereoselectivity (dr > 20:1) for all the products was observed unless otherwise noted. | ||
In addition, the lactam backbone of the resulting products could be easily subjected to the facile manganese-catalyzed deoxygenation using Ph2SiH2 as the reductant, the high-valued amines 73–75 and spirocyclic amine 76 were obtained in 54–92% yields. Notably, catalytic hydrogenation of indoles could not afford these scaffolds.5 Moreover, both MeLi and PhMgCl are suitable nucleophiles for the one-pot sequential reductive dimethylation (77) or arylation (78) processes (Scheme 4A). To further illustrate the synthetic versatility of our protocol in organic synthesis, large-scale reaction with 79 performed under the standard reaction conditions, and the resulting [5,6]-bicyclic lactam 81 successfully underwent stereoselective functionalization to afford the hydroxylated (82) and cyanated (83) products in a diastereoselective manifold, the stereoscopic conformation was validated by X-ray diffraction. Upon treatment of 83 with a stoichiometric amount of KOH, a monofluorinated product 84 was obtained in 75% yield. Moreover, we removed the protecting group of 85 to obtain a [5,6]-bicyclic amine 86, which is the analogue of the perindopril fragment (Scheme 4B). To access the mesembrane analogue 89 we devised a two-step reaction sequence consisting of the cobalt-catalyzed diastereoselective cyclic alkylarylation, along with manganese-catalyzed deoxygenation, which set the stage for the efficient preparation of the natural alkaloids, again with excellent regio- and diastereoselectivity (Scheme 4C).
 | ||
| Scheme 4 Synthetic applications. (A) Functionalization of amide-backbones, (B) late-stage synthesis of medicinally relevant molecules, (C) preparation of analogue of mesembrane. Reaction conditions: [a] Mn(CO)5Br (2.0 mol%), Ph2SiH2 (2.0 equiv), THF, 80 °C, 4 h. [b] MeLi (1.0 equiv), TiCl4 (1.0 equiv), and MeLi (2.0 equiv), Toluene. [c] (i) NaH (3.0 equiv), NaI (1.0 equiv), THF, 40 °C, 3 h; (ii) TMSCl (2.5 equiv), PhMgCl (2.0 equiv), 0 to 23 °C, 2 h. [d] (i) NaH (3.0 equiv), NaI (1.0 equiv), THF, 70 °C, 16 h; (ii) TMSCl (4.0 equiv), Bu4NCN (2.0 equiv), 23 °C, 5 h. [e] (i) DIBAL (1.3 equiv), THF, −35 °C, 2 h; (ii) TMSCN (5.0 equiv), −35 to 20 °C, 1 h. [f] (i) KOH (2.0 equiv) in H2O, 1,4-Dioxane, 115 °C, 3 h; (ii) then HCl. [g] TFA, anisole, 55 °C, 2 h. | ||
Following the evaluation of the scope and applications, we sought to disclose to unveil the underlying mechanism of the reaction. Our exploration began with a thorough kinetic analysis of diastereoselective cobalt-catalyzed cyclic alkylarylation coupling reactions performed in MeCN at 0 °C (Scheme 5a). Notably, when we varied the initial concentrations of cyclenyl bromide 90, the reaction rates remained nearly unchanged, indicating that the alkylarylation process does not depend on the concentration of cyclenyl bromide. These observations suggested the halogen atom abstraction via single-electron transfer from a cobalt catalyst is not the rate-limiting step. Similarly, altering the concentrations of arylzinc pivalate 91 did not considerably affect the reaction rates, reinforcing our understanding of the reaction dynamics (Scheme 5a-ii). However, a linear increase in the reaction rate with increasing concentrations of the [Co] catalyst was observed, pointing to a first-order dependence on the [Co] catalyst. Therefore, the above kinetic findings highlighted that reductive elimination might be the rate-determine step.20
 | ||
| Scheme 5 Mechanistic studies. (a) Kinetic profiles, (b) radical inhibition experiments, (c) EPR experiments and (d) Hammett correlation plot with different arylzinc pivalates. | ||
Further evidence supporting our mechanistic hypothesis came from control experiments using stoichiometric amounts of radical scavengers. These additives dramatically reduced the yield of the desired product 3 or completely halted the reaction (Scheme 5b), consistent with the results from EPR spin-trapping experiments. The detection of a carbon-centered radical intermediate 93 (g = 2.006, AN = 14.33 G, AH = 21.4 G), captured by 5,5-dimethyl-1-pyrroline N-oxide (DMPO), confirms the radical's role in the reaction pathway.21
To delve deeper insights into the influence of electronic effects on the transmetallation step, we employed Hammett correlation analysis (Scheme 5d). A negative ρ value (−1.30) revealed that the aryl-cobalt species behaves as a nucleophile, with electron-donating groups on arylzinc pivalates, enhancing the nucleophilicity of aryl-cobalt. This interaction facilitates the reaction with the in situ generated alkyl radical, shedding light on intricate electronic interactions governing this step.
On the basis of the above observations and previous insights,16a,c we proposed the catalytic cycle involves an initial intermolecular halogen atom transfer from alkyl bromide to the low-valent active species Co(I) A, thereby affording a Co(II) intermediate C and liberating an alkyl radical B. Subsequently, transmetallation reaction between C and arylzinc pivalate delivered the arylcobalt species E, while the alkyl radical B underwent intramolecular syn-addition to generate the intermediate D. At this stage, the dramatic effects of radical rebound and conformational restriction dominated the formation of intermediate F in a diastereoselective fashion. Finally, the reductive elimination process of F gave the desired Csp3-rich N-heterocycles as the products (Scheme 6).
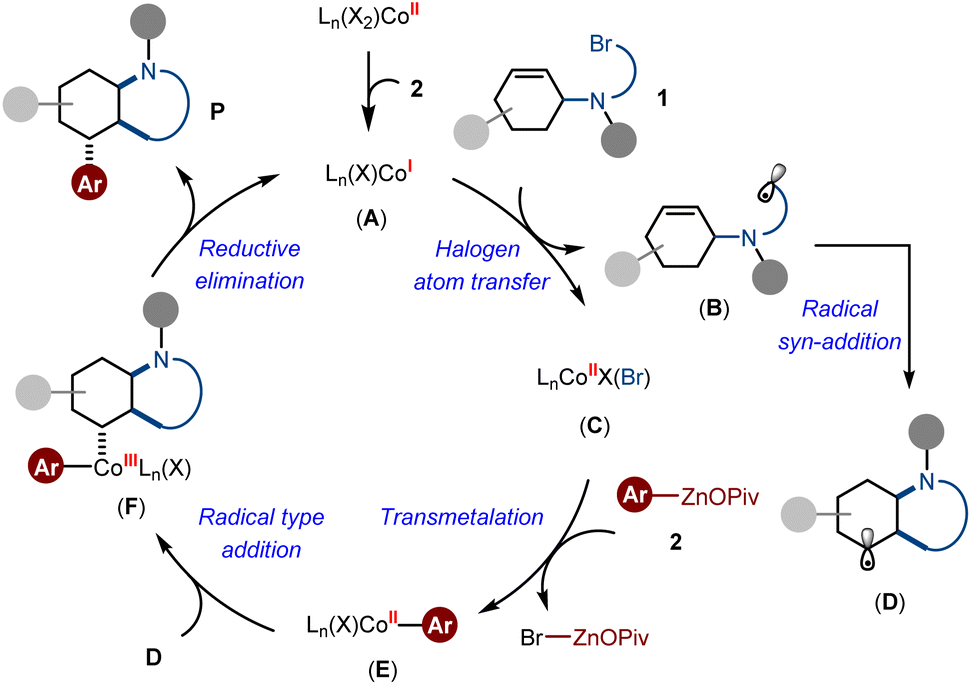 | ||
| Scheme 6 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we have documented a general, divergent cobalt-catalyzed domino cyclized alkylarylation of cyclenes, which enabled the construction of up to seven stereocenters. This method afforded a broad range of highly valuable Csp3-rich N-heterocycles and spirocyclic pyrrolidinones in good yields and diastereoselectivities. It is worth emphasizing that the newly formed cyclic alkyl radical features high stereospecific control, thereby undergoing conformational restriction strategy with the in situ formed aryl-cobalt nucleophile. Preliminary kinetic experiments suggested that the reductive elimination step might be the rate-limiting step. Moreover, the salient features of our protocol are the synthetic simplicity, wide scope and mild reaction conditions. The great potential of this protocol was illustrated by accessing the analogues of medicinally relevant molecules and aminocatalysts. We believe that this methodology will complement and expand the area of metal-catalyzed cascade radical relay cross-couplings and provide an efficient synthetic route to the preparation of Csp3-rich N-hetero(spiro)cycles, which are likely to be attractive in fragment-based lead identification projects.Data availability
The datasets supporting this article have been uploaded as parts of the ESI materials.† Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2330506 (30), 2330508 (42), 2330499 (82), and 2330494 (83). Copies of the data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/structures.Author contributions
J. L. conceived and directed the project and wrote the manuscript by all the other authors; K. C., J. Lin developed and performed the catalytic methods and the synthetic applications. H. Y., and A. L. designed and directed the mechanistic studies; K. C., and J. H. performed the mechanistic studies; K. C., J. J., and J. W. prepared the starting materials, all the authors were involved in the interpretation of the results presented in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22322108), Natural Science Foundation of Jiangsu Province (BK20231521, BK20221355) and Jiangsu Specially Appointed Professors Plan (SR10900122) for generous financial supports. We are grateful to Prof. Konstantin Karaghiosoff (Ludwig-Maximilians-Universität München) for his help in X-ray crystal analysis.Notes and references
- (a) M. Hurst and B. Jarvis, Drugs, 2001, 61, 867–896 CrossRef CAS; (b) D. N. Tran, A. Zhdanko, S. Barroso, P. Nieste, R. Rahmani, J. Holan, R. Hoefnagels, P. Reniers, F. Vermoortele, S. Duguid, L. Cazanave, M. Figlus, C. Muir, A. Elliott, P. Zhao, W. Paden, C. H. Diaz, S. J. Bell, A. Hashimoto, A. Phadke, J. A. Wiles, I. Vogels and V. Farina, Org. Process Res. Dev., 2022, 26, 832–848 CrossRef CAS; (c) C. Pascard, J. Guilhem, M. Vincent, G. Remond, B. Portevin and M. Laubie, J. Med. Chem., 1991, 34, 663–669 CrossRef CAS; (d) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances, Thieme, Stuttgart, 4th edn, 2001, pp. 1785–1787 Search PubMed.
- (a) K. Hiesinger, D. Dar’in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150–183 CrossRef CAS; (b) P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed; (c) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed; (d) S. Bindra, in The Alkaloids, ed. R. H. F. Manske, Academic Press, New York, 1973, pp. 84–121 Search PubMed.
- (a) M. Berger, D. Ma, Y. Baumgartner, T. H.-F. Wong and P. Melchiorre, Nat. Catal., 2023, 6, 332–338 CrossRef CAS; (b) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- (a) W. R. Bowman, M. O. Cloonan and S. L. Krintel, J. Chem. Soc., Perkin Trans. 1, 2000, 1–14 RSC; (b) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS; (c) V. B. Phapale, E. Buñuel, M. García-Iglesias and D. J. Cárdenas, Angew. Chem., Int. Ed., 2007, 46, 8790–8795 CrossRef CAS; (d) M. Kamlar, M. Urban and J. Veselý, Chem. Rec., 2023, 23, e202200284 CrossRef CAS.
- F. Zhang, H. S. Sasmal, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 15695–15701 CrossRef CAS PubMed.
- Selected reviews and examples: (a) Y.-C. Luo, C. Xu and X. Zhang, Chin. J. Chem., 2020, 38, 1371–1394 CrossRef CAS; (b) J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS PubMed; (c) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC; (d) R. Giri and S. Kc, J. Org. Chem., 2018, 83, 3013–3022 CrossRef CAS PubMed; (e) R. K. Dhungana, S. KC, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314–1340 CrossRef CAS; (f) J.-W. Gu, Q.-Q. Min, L.-C. Yu and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12270–12274 CrossRef CAS; (g) C. Xu, R. Cheng, Y.-C. Luo, M.-K. Wang and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18741–18747 CrossRef PubMed; (h) J. Derosa, V. A. van der Puyl, V. T. Tran, M. Liua and K. M. Engle, Chem. Sci., 2018, 9, 5278–5283 RSC; (i) J. Derosa, T. Kang, V. T. Tran, S. R. Wisniewski, M. K. Karunananda, T. C. Jankins, K. L. Xu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 58, 1202–1205 Search PubMed; (j) L. Liu, W. Lee, C. R. Youshaw, M. Yuan, M. B. Geherty, P. Y. Zavalij and O. Gutierrez, Chem. Sci., 2020, 11, 8301–8305 RSC; (k) A. García-Domínguez, R. Mondal and C. Nevado, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef PubMed; (l) S. Kc, R. K. Dhungana, N. Khanal and R. Giri, Angew. Chem., Int. Ed., 2020, 59, 8047–8051 CrossRef CAS; (m) J. Wang, D. Luo, Y. Hu, Z. Duan, J. Yao, K. Karaghiosoff and J. Li, Sci. China: Chem., 2024, 67, 360–367 CrossRef CAS; (n) D. Wang and L. Ackermann, Chem. Sci., 2022, 13, 7256–7263 RSC; (o) H.-Q. Ni, P. Cooper, S. Yang, F. Wang, N. Sach, P. G. Bedekar, J. S. Donaldson, M. Tran-Dubé, I. J. McAlpine and K. M. Engle, Angew. Chem., Int. Ed., 2022, 61, e202114346 CrossRef CAS.
- (a) K. M. Logan, S. R. Sardini, S. D. White and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 159–162 CrossRef CAS; (b) S. Joung, A. M. Bergmann and M. K. Brown, Chem. Sci., 2019, 10, 10944–10947 RSC; (c) C. Ding, Y. Ren, C. Sun, J. Long and G. Yin, J. Am. Chem. Soc., 2021, 143, 20027–20034 CrossRef CAS PubMed; (d) Y. Li, Y. Li, H. Shi, H. Wei, H. Li, I. Funes-Ardoiz and G. Yin, Science, 2022, 376, 749–753 CrossRef CAS; (e) W. Kong, Y. Bao, L. Lu, Z. Han, Y. Zhong, R. Zhang, Y. Li and G. Yin, Angew. Chem., Int. Ed., 2023, 62, e202308041 CrossRef CAS PubMed; (f) G. L. Trammel, P. B. Kannangara, D. Vasko, O. Datsenko, P. Mykhailiuk and M. K. Brown, Angew. Chem., Int. Ed., 2022, 61, e202212117 CrossRef CAS.
- (a) H. Ohmiya, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2006, 128, 1886–1889 CrossRef CAS PubMed; (b) T. Thaler, B. Haag, A. Gavryushin, K. Schober, E. Hartmann, R. M. Gschwind, H. Zipse, P. Mayer and P. Knochel, Nat. Chem., 2010, 2, 125–130 CrossRef CAS PubMed; (c) T. Thaler, L.-N. Guo, P. Mayer and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 2174–2177 CrossRef CAS; (d) J. Li, Q. Ren, X. Cheng, K. Karaghiosoff and P. Knochel, J. Am. Chem. Soc., 2019, 141, 18127–18135 CrossRef CAS PubMed.
- (a) L. Dian, D. S. Müller and I. Marek, Angew. Chem., Int. Ed., 2017, 56, 6783–6787 CrossRef CAS; (b) H. Li, M. Zhang, H. Mehfooz, D. Zhu, J. Zhao and Q. Zhang, Org. Chem. Front., 2019, 6, 3387–3391 RSC; (c) A. A. Kadam, T. L. Metz, Y. Qian and L. M. Stanley, ACS Catal., 2019, 9, 5651–5656 CrossRef CAS; (d) S. KC, R. K. Dhungana, N. Khanal and R. Giri, Angew. Chem., Int. Ed., 2020, 59, 8047–8051 CrossRef CAS PubMed; (e) Y. Ito, S. Nakatani, R. Shiraki, T. Kodama and M. Tobisu, J. Am. Chem. Soc., 2022, 144, 662–666 CrossRef CAS PubMed.
- S. Teng, Y. R. Chi and J. S. Zhou, Angew. Chem., Int. Ed., 2021, 60, 4491–4495 CrossRef CAS.
- (a) C. Xu, R. Cheng, Y.-C. Luo, M.-K. Wang and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18741–18747 CrossRef CAS; (b) V. Aryal, L. J. Chesley, D. Niroula, R. R. Sapkota, R. K. Dhungana and R. Giri, ACS Catal., 2022, 12, 7262–7268 CrossRef CAS; (c) M.-K. Wang, Y.-C. Luo, H.-Y. Zhao, Y. Zhang, D. Zhang and X. Zhang, ACS Catal., 2023, 13, 14090–14102 CrossRef CAS.
- C. Rao, T. Zhang, H. Liu and H. Huang, Nat. Catal., 2023, 6, 847–857 CrossRef CAS.
- (a) B. Giese, B. Kopping, T. Gobel, J. Dickhaut, G. Thoma, K. J. Kulicke and F. Trach, Org. React., 1996, 48, 301–856 CAS; (b) S. Z. Zard, Radical Reaction in Organic Synthesis, Oxford University Press, New York, 2003, selected examples CrossRef; (c) G. Storke and R. Mah, Heterocycles, 1989, 28, 723–727 CrossRef; (d) D. J. Babcock, A. J. Wolfram, J. L. Barney, S. M. Servagno, A. Sharma and E. D. Nacsa, Chem. Sci., 2024, 15, 4031–4040 RSC; (e) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS.
- A. Hernán-Gómez, E. Herd, E. Hevia, A. R. Kennedy, P. Knochel, K. Koszinowski, S. M. Manolikakes, R. E. Mulvey and C. Schnegelsberg, Angew. Chem., Int. Ed., 2014, 53, 2706–2710 CrossRef PubMed.
- (a) S. Bernhardt, G. Manolikakes, T. Kunz and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9205–9209 CrossRef CAS; (b) C. I. Stathakis, S. Bernhardt, V. Quint and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 9428–9432 CrossRef CAS PubMed; (c) S. M. Manolikakes, M. Ellwart, C. I. Stathakis and P. Knochel, Chem.–Eur. J., 2014, 20, 12289–12297 CrossRef CAS PubMed; (d) J. Wang, Z. Duan, X. Liu, S. Dong, K. Chen and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202202379 CrossRef CAS PubMed.
- (a) X. Cheng, X. Liu, S. Wang, Y. Hu, B. Hu, A. Lei and J. Li, Nat. Commun., 2021, 12, 4366 CrossRef CAS PubMed; (b) J. Lin, K. Chen, J. Wang, J. Guo, S. Dai, Y. Hu and J. Li, Chem. Sci., 2023, 14, 8672–8680 RSC; (c) X. Liu, H. Chen, D. Yang, B. Hu, Y. Hu, S. Wang, Y. Lan, A. Lei and J. Li, ACS Catal., 2023, 13, 9254–9263 CrossRef CAS.
- (a) A. Guérinot and J. Cossy, Acc. Chem. Res., 2020, 53, 1351–1363 CrossRef PubMed; (b) J. Li, Top. Organomet. Chem., 2023, 71, 113–144 CrossRef; (c) F. H. Lutter, S. Graßl, L. Grokenberger, M. S. Hofmayer, Y.-H. Chen and P. Knochel, ChemCatChem, 2019, 11, 5188–5197 CrossRef CAS; (d) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS; (e) C. Gosmini, J.-M. Bégouin and A. Moncomble, Chem. Commun., 2008, 3221–3233 RSC.
- Note: Ar–ZnOPiv•Mg(OPiv)X is abbreviated henceforth as Ar–ZnOPiv (X = Cl, Br, or I) for the sake of clarity.
- T. Aniszewski, Definition, Typology and Occurrence of Alkaloids, in Alkaloids-Secrets of Life, Elsevier, Amsterdam, 2007, pp. 1–59 Search PubMed.
- (a) L. Jin, H. Zhang, P. Li, J. R. Sowa Jr and A. Lei, J. Am. Chem. Soc., 2009, 131, 9892–9893 CrossRef CAS PubMed; (b) C. He, J. Ke, H. Xu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 1527–1530 CrossRef CAS.
- W.-M. Cheng, R. Shang and Y. Fu, Nat. Commun., 2018, 9, 5215 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2330506, 2330508, 2330499 and 2330494. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04056b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |