
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong-persistent luminescence by host–guest Förster resonance energy transfer†
Hui-Li
Sun
a,
Qiang-Sheng
Zhang
b,
Zhong-Hao
Wang
a,
Yan-Ting
Huang
a and
Mei
Pan
 *a
*a
aMOE Laboratory of Bioinorganic and Synthetic Chemistry, Lehn Institute of Functional Materials, IGCME, GBRCE for Functional Molecular Engineering, School of Chemistry, Sun Yat-Sen University, Guangzhou 510006, China. E-mail: panm@mail.sysu.edu.cn
bHainan Provincial Key Laboratory of Fine Chem, School of Chemistry and Chemical Engineering, Hainan University, Haikou, P. R. China
First published on 26th September 2024
Abstract
In this study, Förster resonance energy transfer (FRET) is harnessed to construct a novel stimulus-responsive long-persistent luminescence (LPL) system. Two organic molecules, DPSD and DPOD, were initially found to have no afterglow under ambient conditions, but exhibited prolonged afterglow upon friction with paper, showing a significantly promoted transition of triplet excited states. Substituting paper with α-cellulose (the main composition of paper) reveals a novel host–guest long afterglow system and allows for a deeper investigation of the above paper-promoted LPL phenomenon. The activation of the LPL effect was achieved by matrixing these components through a grinding process, capitalizing on the efficient FRET from the host to the guest owing to the appropriate energy level match, and the robust intersystem crossing (ISC) capability of the guest. This model presents a new matrix strategy to achieve efficient LPL by a facile, low cost and easy-to-handle process. Furthermore, we successfully implemented anti-counterfeiting, encryption and decryption, decoration, and water/heat stimulus-responsive applications of the obtained materials. These advancements bring LPL materials one step closer to practical commercialization.
Introduction
Long persistent luminescence (LPL), or long afterglow, materials are a novel class of optical materials that can store the energy of incident light and continue emitting photons even after the illumination ceases, garnering widespread attention,1–7 especially in the domain of single-component systems.8 When an organic chromophore absorbs a photon, an electron is excited from the ground state (S0) to a higher singlet state (Sn) and then relaxes non-radiatively to the lowest singlet state (S1). Fluorescence results from the fast radiative decay from S1 to S0. The excited singlet state can also relax non-radiatively to a triplet state (T1) via intersystem crossing (ISC). Phosphorescence occurs when T1 returns to S0, but this process is slower due to forbidden transitions. In some organic crystals, triplet states can be stabilized, leading to LPL (100 ms–10 s).9 Despite the historical notion that transitioning to the triplet state is a challenging process for luminogens at room temperature (RT), recent breakthroughs have substantially improved intersystem crossing (ISC) efficiency. Notably, significant advancements have been achieved in extending LPL lifetimes from milliseconds to seconds or even minutes, rendering them easily discernible to the naked eye.10–12 This enhancement has been achieved by bolstering spin–orbit coupling (SOC) through the incorporation of heteroatoms,13–15 heavy atoms,16 and multimers.17 However, many of these advancements are contingent on special crystal structures,18 presenting challenges in terms of maintenance, cultivation, and practical applications, thereby limiting their widespread utility. Additionally, various multi-component phosphorescent systems have emerged, employing co-crystallization, rigid matrix encapsulation, hardening in polymer matrices, or interactions with molecules of similar or different types.10,19–22 While promising, the practical implementation of these multi-component materials is somewhat intricate, with specific conditions and flexibility constraints. Against this backdrop, LPL materials based on host–guest doping systems, independent of crystalline structures, offer a promising avenue to address these challenges. In comparison to conventional phosphorescent materials, LPL materials with stimulus-responsive properties, particularly those independent of specific crystal structures, offer distinct advantages and possess significant commercial value across various applications, including anticounterfeiting, sensing, and detection.23–27 Despite sporadic reports on such materials, comprehensive studies remain scarce,23,28 necessitating a more systematic exploration of their mechanisms and influencing factors in LPL generation.Paper, a remarkable invention originating from China and composed of cellulose, stands as a fundamental medium for communication, information storage, and cultural dissemination. Notably, recent advancements have led to the successful construction of LPL cellulose by various research groups.29–31 Despite the strides in LPL cellulose construction, there is still a notable gap in understanding how cellulose responds to stimuli in the context of LPL. Further research in this direction could provide valuable insights into the practical applications and potential improvements in the synthesis and processing of LPL cellulose. We report an unexpected finding of organic afterglow triggered by grinding on paper. Two organic molecules, DPSD and DPOD, showed no afterglow initially, but exhibited prolonged afterglow upon friction with paper, promoting the transition of triplet excited states. Substituting paper with α-cellulose allowed for a deeper investigation, revealing a novel host–guest long afterglow system. This LPL system, utilizing commercially available compounds, offers cost-effectiveness, the absence of halogen atoms, easy preparation, and high performance, promising widespread attention and further innovations.
Results and discussion
The synthesis of the D–A type organic molecules 4,4′-((2,5-dicyano-1,4-phenylene)bis(sulfanediyl))dibenzoic acid (DPSD) and 4,4′-((2,5-dicyano-1,4-phenylene)bis(oxy))dibenzoic acid (DPOD) is described in the ESI (Fig. S1–S5 and Table S1).† DPSD and DPOD have similar structures, but one contains two oxygen atoms (DPOD) and the other contains two sulfur atoms (DPSD). The electrostatic potential (ESP) surface reveals the charge distribution on its molecular surface (Fig. S6†).At RT, DPSD and DPOD do not exhibit long afterglow properties initially. However, when applied onto filter paper and ground, they show significantly long afterglow, with lifetimes of 7.77 ms and 62.25 ms, respectively (Fig. S7 and S8†). Compared to paper + DPSD and paper + DPOD, the paper's photoluminescence is negligible. Considering that paper's primary component is cellulose, we believe that the afterglow arises from energy transfer between cellulose (host) and organic molecules (guest) induced by grinding. To validate, we conducted research with α-cellulose (180–280 μm) instead of paper and observed long afterglow in α-cellulose + DPSD and α-cellulose + DPOD systems, with the afterglow colors of yellow and green, absolute quantum yields (QY) of 7.9% and 17.6%, and decay lifetimes of 11.47 and 535.21 ms, respectively (Fig. 1a and b, and Table S2†).
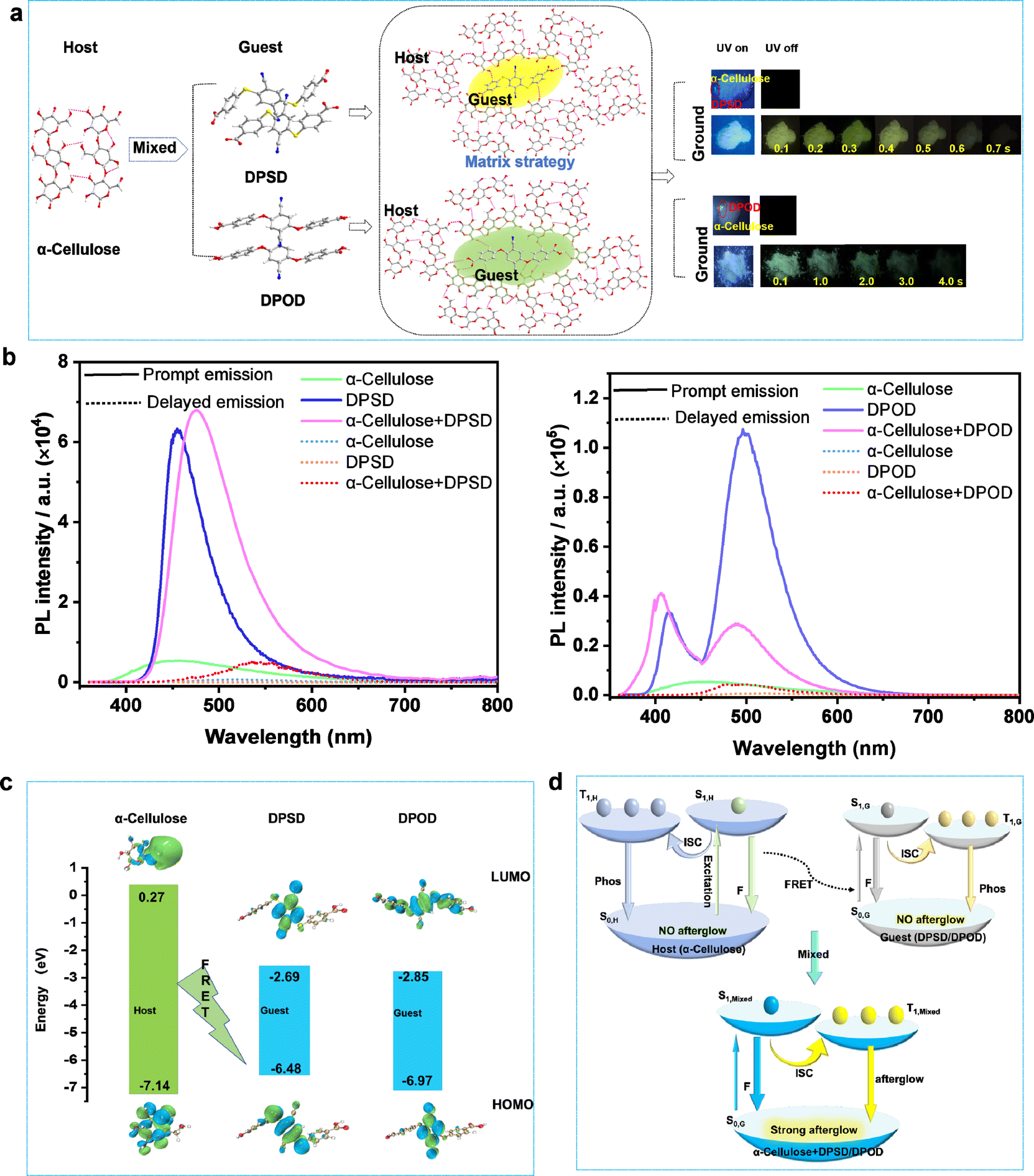 | ||
Fig. 1 (a) Schematic illustration of the preparation of α-cellulose + DPSD/DPOD LPL materials and LPL performance in the air; (b) prompt and delayed emission spectra of α-cellulose, DPSD, DPOD and α-cellulose + DPSD/DPOD (mass ratio = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under 340 nm excitation; (c) the calculated energy and molecular orbital (MO) diagrams of donors and acceptors; (d) proposed mechanism of photophysical processes based on ISC at RT (S, singlet; T, triplet; ISC, intersystem crossing; F, fluorescence; Phos, phosphorescence; FRET, Förster resonance energy transfer). :1) under 340 nm excitation; (c) the calculated energy and molecular orbital (MO) diagrams of donors and acceptors; (d) proposed mechanism of photophysical processes based on ISC at RT (S, singlet; T, triplet; ISC, intersystem crossing; F, fluorescence; Phos, phosphorescence; FRET, Förster resonance energy transfer). | ||
To achieve a stimulus-responsive LPL effect in a host–guest doping system based on FRET, three main criteria must be met: (1) compatible energy levels between the host and guest to facilitate the FRET process; (2) a rigid environment within the host molecules; and (3) efficient ISC from the singlet to the triplet state in the guest molecules to enable potential LPL emission.23,32 Based on these criteria, α-cellulose was chosen as the host due to its ability to form intermolecular hydrogen bonds. DPSD and DPOD were selected as the guests, as they have LPL emission in rigid environments.
We first investigated the energy levels between the host and guest, observing a spectral overlap between the host emission and guest absorption (Fig. S9†). The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the guests were entirely within the host (α-cellulose) (Fig. 1c). That indicates that the energy levels between the host and guest were matched. Next, the photoluminescence and corresponding excitation spectra of the α-cellulose host were recorded (Fig. S10†). As shown in Fig. S10,† its photoluminescence emission peak was located at 440 nm, while the corresponding best excitation wavelength was 425 nm. On this basis, its Stokes shift was calculated to be just 15 nm, indicating a rigid environment in host molecules.33–35 Third, the photoluminescence spectra of DPSD and DPOD were recorded at both room temperature and 77 K. At 77 K, while no LPL was observed at room temperature, significant LPL was detected with lifetimes of 49.13 ms for DPSD and 185.02 ms for DPOD, attributed to restricted molecular motion (Fig. S14–S19, Tables S6 and S12†). These findings confirm efficient ISC from singlet to triplet states in DPSD and DPOD, thereby validating the three criteria for the stimulus-responsive LPL effect. Based on these findings, we propose FRET as crucial in transferring energy from α-cellulose to DPSD and DPOD (Fig. 1d), thereby inducing interesting LPL properties in the matrixed system, which is further validated in the detailed photophysical studies below.
The photoluminescence spectra of α-cellulose are shown in Fig. S10–S12 and Tables S2–S4.† At RT, under 340 nm excitation, α-cellulose exhibited an emission band with a shorter lifetime (τ440nm = 3.38 ns), which can be referred to as its fluorescence emission (Fig. S10, S12a and Table S3†). Remarkably, the fluorescence emission peaks gradually red-shifted with increasing excitation wavelength, likely due to the polymorphism of excited states induced by different α-cellulose aggregates (Fig. S11a†). The emission band with a longer lifetime (τ470nm = 5.60 ms) is designated as its phosphorescence emission band (Fig. S11b–d, S12b and Table S4†). Since the photoluminescence intensity is too weak (QY = 6.3), we cannot observe its long afterglow properties at RT. Meanwhile, the fluorescence and phosphorescence lifetime of α-cellulose were enhanced at 77 K (Fig. S11a, S12 and Tables S3–S4†).
As illustrated in Fig. S13,† DPSD exhibits blue luminescence with a shorter lifetime (τ455nm = 1.52 ns), which is assigned to the singlet state fluorescence emission (Fig. S14a and Table S5†). In addition, at 77 K, time-gated spectra from 0–9.0 ms were recorded. We can observe a new emission peak with a longer lifetime (τ570nm = 49.13 ms) (Fig. S14b–d and Table S6†), manifesting phosphorescence properties. However, at RT, the phosphorescence is quenched by thermal deactivation, and we cannot observe its afterglow properties.
Through 35 s of grinding, we homogeneously mixed DPSD with α-cellulose, creating an α-cellulose + DPSD-matrixed system. Prompt and delayed emission spectra were recorded for different mass ratios of α-cellulose to DPSD (Fig. 2a–c). As the mass ratio varied from 0:1 to 200:1, the fluorescence intensity at 470 nm increased, while a new phosphorescence signal emerged around 554 nm. The phosphorescence intensity at 554 nm increased with α-cellulose concentration until reaching a mass ratio of 100:1 for α-cellulose + DPSD. Through grinding, the transition from the “afterglow OFF state” to the “afterglow ON state” of the host and guest has been achieved (Fig. 2c).
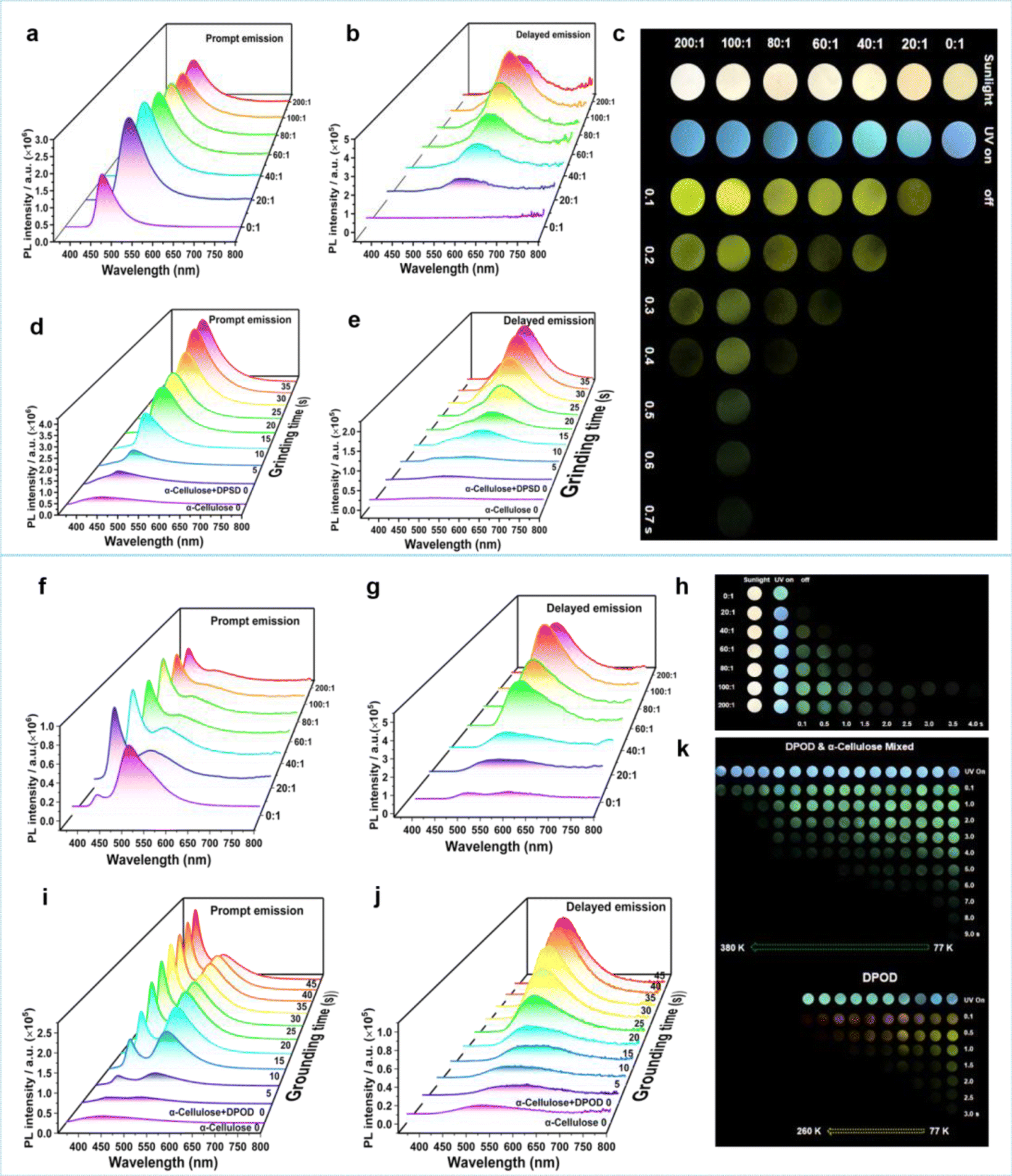 | ||
| Fig. 2 (a) Prompt and (b) delayed emission spectra of α-cellulose + DPSD (mass ratio = 0:1–200:1) under 340 nm excitation; (c) photos of α-cellulose + DPSD with different α-cellulose contents under 365 nm excitation; (d) prompt and (e) delayed emission spectra of α-cellulose and α-cellulose + DPSD after grinding for different times (mass ratio = 100:1); (f) prompt and (g) delayed emission spectra of α-cellulose + DPOD (mass ratio = 0:1–200:1) under 340 nm excitation; (h) photos of α-cellulose + DPOD with different α-cellulose contents under 365 nm excitation; (i) prompt and (j) delayed emission spectra of α-cellulose and α-cellulose + DPOD after grinding for different times; (k) photos of α-cellulose + DPOD (mass ratio = 100:1) and DPOD under 365 nm excitation at different temperatures. | ||
To investigate the potential force-responsive LPL effect, we ground α-cellulose + DPSD (100:1) (Fig. 2d and e). We observed a gradual decrease in the donor emission intensity at 440 nm, with little change after 35 s of grinding (Fig. 2d). Simultaneously, the corresponding fluorescence lifetime decreased from 3.93 ns to 1.85 ns (Table S7†). Additionally, we observed a gradual enhancement in the fluorescence of the acceptor at 470 nm, indicative of a typical FRET process (Fig. 2d).36,37 To quantify the FRET efficiency (EFRET), we estimated it based on the observed changes in the fluorescence lifetime of the donor (Table S7†). An increase in the grinding degree led to an enhanced resonance energy-transfer efficiency. After grinding for 5 s, the EFRET was 38.93%, which then increased to 47.58% at 15 s and reached a maximum of 52.93% at 35 s. At this point, the LPL emission was gradually increasing at 554 nm (Fig. 2e).
The observed LPL at 554 nm in α-cellulose + DPSD matches the phosphorescence of DPSD at 570 nm in the solid state at 77 K, confirming DPSD as the origin of the grind-induced LPL. The slight shifts in phosphorescence spectra compared to the DPSD solid suggest hydrogen bond interactions between DPSD and α-cellulose.38 Additionally, slight changes in the fluorescence spectra can be attributed to the grinding of DPSD (Fig. S15†). Temperature-dependent PL spectra and decay curves of α-cellulose + DPSD further support its phosphorescence properties at 554 nm (Fig. S16, S17, Tables S8 and S9†).
We then switched to another guest molecule DPOD. The photophysical characteristics of DPOD are first measured (Fig. S18–S20 and Tables S10–S12†). DPOD exhibited emission bands with a shorter lifetime (τ417nm = 0.61 ns) and a longer lifetime (τ500nm = 7.97 μs), attributed to its fluorescence and phosphorescence emissions, respectively (Fig. S18, S19a, b, Tables S10 and S11†). Furthermore, at 77 K, a time-gated spectral study within a range of 1–100 ms unveiled a new emission peak with a long lifetime (τ575nm = 185.02 ms) (Fig. S19c, S20 and Table S12†), accompanied by a yellow afterglow. However, this afterglow cannot be observed at RT. Consequently, we attribute the phosphorescence emission at 500 nm to the radiative decay of high-lying triplet excitons (T1). Upon cooling at 77 K, the afterglow at 575 nm stems from the lowest-lying triplet excited state 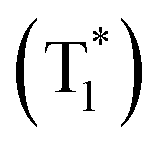 , as indicated by a red-shifted emission (Fig. S21†). From the single crystal structure perspective, DPOD has a tighter π⋯π stacking than DPSD, which may be one of the main factors inducing the
, as indicated by a red-shifted emission (Fig. S21†). From the single crystal structure perspective, DPOD has a tighter π⋯π stacking than DPSD, which may be one of the main factors inducing the 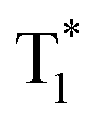 energy level of DPOD (Fig. S22†).
energy level of DPOD (Fig. S22†).
Then the emission spectra of α-cellulose + DPOD after grounding for 45 s were recorded. The phosphorescence emission intensity at 492 nm reached its highest peak at a mass ratio of 100:1 of α-cellulose + DPOD (Fig. 2f–h). Similarly, through grinding, we found that the fluorescence and phosphorescence intensity of DPOD increased with increasing grinding time, until grinding for 45 s (Fig. 2i–k). As the grinding time prolongs, the EFRET gradually increases. After grinding for 45 s, the EFRET reached 39.95% (Table S7†). It is crucial to highlight that the afterglow emission color of α-cellulose + DPOD differs from that of the DPOD solid at 77 K (Fig. 2k). The yellow afterglow at 575 nm is indicative of DPOD, and this may be attributed to the aggregated state of DPOD. Contrastingly, DPOD was present in a monomer state in α-cellulose + DPOD, so the green afterglow is observed. To validate this hypothesis, we investigated the solution state of DPOD in 2-MTHF and confirmed the absence of an emission peak at 575 nm, with only a long phosphorescence peak at 486 nm at 77 K (Fig. S23, S24 and Tables S13, S14†). Consequently, we posit that the grinding-induced LPL originates from the DPOD monomer state. The LPL of α-cellulose + DPOD was verified through temperature-dependent spectra and lifetime measurements (Fig. 2k, S25, S26, and Table S15, S16†). Interestingly, we can still observe its afterglow up to 380 K, indicating that the afterglow has high temperature tolerance (Fig. 2k).
Summarizing the above, by grinding, DPSD/DPOD is doped at a lower concentration into a non-conjugated pure organic host matrix (α-cellulose). This doping strategy utilizes the rigid matrix provided by the host to stabilize the triplet excitons of DPSD/DPOD, reduce the oscillator strength, and ultimately achieve efficient long afterglow luminescence. We successfully synthesized α-cellulose + DPSD and α-cellulose + DPOD materials, which exhibited yellow and green afterglow, respectively. Notably, α-cellulose + DPOD displayed a longer lifetime (535.21 ms) and higher QY (17.6%) compared to α-cellulose + DPSD (11.47 ms and 7.9%). Additionally, the afterglow of α-cellulose + DPOD showed high temperature tolerance, with its afterglow still observable at 380 K. By employing these two different guest molecules, we effectively tuned the long afterglow emission from yellow to green, enhancing both the afterglow lifetime and QY, as well as high temperature tolerance. These variations in the lifetime and emission color enable applications such as Morse code creation and the production of decorative items with diverse colors. Both compounds are inexpensive commercial reagents, making them advantageous for practical applications. To delve deeper into the correlation between host–guest interaction and the LPL effect, we conducted a comprehensive analysis using single crystals of α-cellulose, DPSD, and DPOD (Fig. 3 and Table S1†). Fig. 3a illustrates the crystal structure of α-cellulose, which provides a rigid environment. On one hand, the planar conformation of α-cellulose in the crystal significantly restricts molecular motion, including rotation and vibration.20,39,40 On the other hand, efficient intermolecular hydrogen bonding can be observed, forming a rigid network structure that hinders nonradiative decay processes. These factors make α-cellulose an excellent host for LPL.
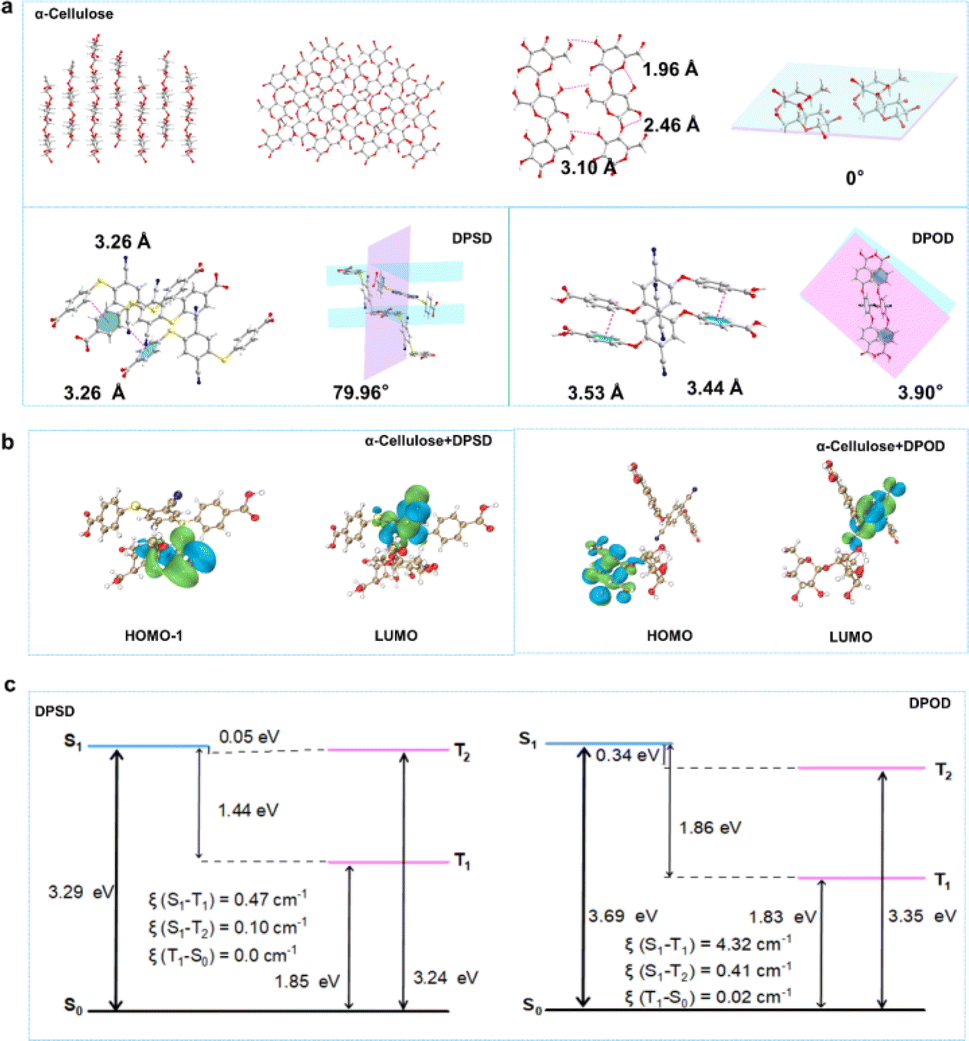 | ||
| Fig. 3 (a) Single-crystal structure of α-cellulose. α-Cellulose crystal showed a rigid environment with a planar conformation and efficient intermolecular hydrogen bonds, the dihedral angle was 0°; intermolecular interactions and packing modes in DPSD and DPOD crystals. In crystal, the molecules presented a twisted conformation, and the dihedral angles of the two benzene rings were 79.96° and 3.90°, respectively; (b) the calculated molecular orbital (MO) diagrams of α-cellulose + DPSD and α-cellulose + DPOD; (c) energy level diagram and SOCME (ξ) for DPSD and DPOD. | ||
In contrast, although intermolecular C–H⋯π exist, the twisted conformation with the torsion angle between two benzene ring units would result in free nonradiative motion.39,41,42 This twisted structure limits the observation of LPL in pure guests, despite achieving efficient ISC transitions from singlet to triplet states. As depicted in Fig. S27,† we can observe weak interactions between the host and guest through interaction region indicator (IRI) analysis. The oxygen atoms of the host and the hydrogen atoms of the guest form hydrogen bonding interactions, while van der Waals (vdW) forces also contribute to the host–guest interaction. The diverse weak interactions between the host and guest are crucial for maintaining the rigid conformation of the doped system and effectively limiting non radiative pathways by restricting the movement of the structure.42 Furthermore, grinding the materials enables effective intermolecular interactions between α-cellulose and the acceptor molecules, facilitating FRET processes between them.43 Therefore, applying distance sensitive FRET to host–guest doping systems can develop stimulus-responsive LPL materials. The host and guest molecules initially separate and gradually approach each other under force stimulation. As their molecular distance decreases, diverse weak interactions occur, while energy transfer between the host and guest occurs, resulting in excellent LPL performance.
Moreover, we observed a notable weakening of the PXRD of DPSD and DPOD crystals after grinding, suggesting that the guests are more susceptible to fragmentation under external stimuli, thereby facilitating their incorporation into the host. Additionally, α-cellulose, characterized by their relatively rigid structure, maintains strong PXRD even after grinding (Fig. S28†). Crucially, through the host–guest doping system, we successfully demonstrated the FRET effect, culminating in the LPL phenomenon.
To get an in-depth understanding of the photophysical properties, time-dependent density functional theory (TD-DFT) calculations were performed (Fig. 3b and c, and see the ESI† for details). The excitation energies, frontier molecular orbitals (FMOs) and spin-orbital coupling matrix elements (SOCMEs, ξ) were calculated to further elucidate the underlying transition characters.
We explored the energetic match of molecular pairs involving α-cellulose and DPSD/DPOD through TD-DFT calculations. In the case of the two-component self-assembled systems, α-cellulose + DPSD and α-cellulose + DPOD, the HOMO was observed to be situated on α-cellulose, while the LUMO was localized to DPSD/DPOD (Fig. 3b). This assignment of MO suggests the potential for energy transfer between the two components. For α-cellulose + DPSD, the energy of the S1 state is 2.13 eV, and the T1 state is at 1.80 eV, with a ΔE(S1, T1) of 0.33 eV. Similarly, for α-cellulose + DPOD, the energy of the S1 state is 2.28 eV, and the T1 state is at 1.97 eV, with a ΔE(S1, T1) of 0.31 eV (Fig. S29 and Tables S17 and S18†). At RT, notable ISC rates of 1.09 × 107 s−1 and 6.67 × 107 s−1 were observed for α-cellulose + DPSD and α-cellulose + DPOD, respectively. This indicates the efficient migration of excited electrons within the doping system, transitioning from singlet to triplet energy levels. These findings further elucidate why the most effective energy transfer occurs in the doping systems of α-cellulose + DPSD and α-cellulose + DPOD.
Following energy transfer, the triplet emission ability of the guest emerges as a crucial factor influencing the LPL in the matrixed systems. Notably, the HOMO and LUMO of all guests exhibit spatial separation (Fig. S30–S32†), implying the potential for intermolecular electron transitions upon light absorption. To delve deeper into these observations, the SOCMEs (ξ) between S1 and T1 for DPSD and DPOD were analyzed (Fig. 3c and Tables S19–S22†). A comparison reveals a larger coupling constant in DPOD (4.32 cm−1) compared to DPSD (0.47 cm−1). This disparity elucidates why α-cellulose + DPOD exhibits a prolonged lifetime and higher QY. Therefore, the ISC ability of the guest is deemed to exert a significant impact on phosphorescence efficiency and lifetime. Lastly, the favorable rigid environment provided by the host creates an optimal setting for the guest, effectively suppressing non-radiative transitions.
A novel Morse code-based encryption method was developed, utilizing differences in sample lifetimes. Short-lived α-cellulose + DPSD represented the Morse code's horizontal line, while the dot • was embodied by α-cellulose + DPOD with an extended lifetime. α-Cellulose denoted the interference symbol ×. This method effectively encrypted text and numbers, as demonstrated with the examples of “SYSU” and “1924” (Fig. 4a). The method's adaptability extends to the encryption of inverted information. For instance, the short lifetime conveyed point information, the long lifetime represented horizontal line information, and the original interference information described interruptions. Collectively, this information was utilized to encrypt the extensive statement “CUTE DOG” (Fig. 4b). Ornamental enhancements were achieved by encapsulating LPL materials in epoxy resin within a mold (Fig. 4c).
 | ||
| Fig. 4 (a) Analysis of the encryption and decryption process of the Morse code (b) and flipping the compilation process; (c) decorations were achieved by encapsulating the LPL materials α-cellulose + DPSD and α-cellulose + DPOD in epoxy resin in a mold; (d) photographs and water/heat stimulus-responsive repeated cycles for α-cellulose + DPOD (λex = 365 nm); (e) placing DPSD and DPOD on filter paper (mainly composed of cellulose), grinding the sample on the paper, and recording the photographs before and after grinding, respectively. | ||
Furthermore, the phosphorescence emissions of the matrixed system can change when exposed to water. We studied α-cellulose + DPOD tabletting under alternating water and heat stimulation. After exposure to water vapor, the tabletting showed reduced phosphorescence at 492 nm (Fig. S33†). However, heating at 80 °C for 10 minutes restored phosphorescence (Fig. S34†). The tabletting exhibited reversible water/heat responsiveness for at least five cycles without fatigue, making it promising for water detection and anti-counterfeiting (Fig. 4d). We replaced purchased α-cellulose with filter paper, observing the same luminescence phenomenon. Spreading DPSD and DPOD onto filter paper, grinding them together, and turning off the UV light source revealed enduring luminescence, similar to SYSU (Fig. 4e).
Conclusions
In summary, we designed a new matrix strategy to achieve efficient and stimulus-responsive LPL, by applying a paper (cellulose)-matrixed host–guest system attributed to FRET. The strategy starts with the discovery of the turn-on afterglow characteristics of DPSD and DPOD organic molecules upon friction with paper and deepens the investigation by replacing paper with its main chemical composition, α-cellulose. By grinding the guests DPSD and DPOD with the host α-cellulose gently, a new long afterglow system is established. The grinding process enables the efficient activation of the LPL effect by facilitating FRET from the host molecule to the guest molecule, which triggers the guest LPL at room temperature and sustains it up to 380 K. Further detailed experimental and theoretical research revealed that the efficient FRET from the host to the guest owing to the appropriate energy level match and the effective ISC ability of the guest due to the strengthening of the paper (cellulose) matrix are pivotal factors for achieving high phosphorescence efficiency. Furthermore, the newly designed LPL system is implemented for anti-counterfeiting, encryption/decryption, decoration, and water/heat stimulus-responsive applications. In general, this novel type of LPL-obtaining strategy, by utilizing commercially available matrices, offers cost-effectiveness, the absence of halogen atoms, easy preparation, and high performance, promising to attract widespread attention and drive further innovations.Data availability
All experimental, characterisation and calculation methods, as well as structural, photophysical and theoretical details, are available in the ESI.†Author contributions
H. S. and Q. Z. performed experimental and theoretical analysis. Z. W. and Y. H. performed spectral data analysis. M. P. supervised the study. H. S. and M. P. wrote the manuscript with contributions of all authors, and all authors have given approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NKRD Program of China (2021YFA1500401) and NSFC (22171291, 92261114, and 21821003). This paper is dedicated to Prof. Marcel Mayor on the occasion of his sixtieth birthday.Notes and references
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS.
- G. Xiao, Y. J. Ma, Z. Qi, X. Fang, T. Chen and D. Yan, Chem. Sci., 2024, 15, 3625–3632 RSC.
- B. Zhou and D. Yan, Matter, 2024, 7, 1950 CrossRef.
- C. Xing, B. Zhou, D. Yan and W. H. Fang, Adv. Sci., 2024, 11, 2310262 CrossRef CAS.
- C. Xing, Z. Qi, B. Zhou, D. Yan and W. H. Fang, Angew. Chem., Int. Ed., 2024, 63, e202402634 CrossRef CAS PubMed.
- C. Xing, B. Zhou, D. Yan and W.-H. Fang, CCS Chem., 2023, 5, 2866–2876 CrossRef.
- Q. Liao, Q. Gao, J. Wang, Y. Gong, Q. Peng, Y. Tian, Y. Fan, H. Guo, D. Ding, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2020, 59, 9946–9951 CrossRef CAS PubMed.
- A. Forni, E. Lucenti and C. Botta, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- S. Hirata and M. Vacha, J. Phys. Chem. Lett., 2016, 7, 1539–1545 CrossRef CAS PubMed.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef.
- S. Tian, H. Ma, X. Wang, A. Lv, H. Shi, Y. Geng, J. Li, F. Liang, Z. M. Su, Z. An and W. Huang, Angew. Chem., Int. Ed., 2019, 58, 6645–6649 CrossRef CAS.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- H. Zheng, P. Cao, Y. Wang, X. Lu and P. Wu, Angew. Chem., Int. Ed., 2021, 60, 9500–9506 CrossRef CAS.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed.
- L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS.
- Y. Tian, X. Yang, Y. Gong, Y. Wang, M. Fang, J. Yang, Z. Tang and Z. Li, Sci. China: Chem., 2021, 64, 445–451 CrossRef CAS.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- Y. Su, Y. Zhang, Z. Wang, W. Gao, P. Jia, D. Zhang, C. Yang, Y. Li and Y. Zhao, Angew. Chem., Int. Ed., 2019, 59, 9967–9971 CrossRef PubMed.
- Y. Wang, J. Yang, M. Fang, Y. Yu, B. Zou, L. Wang, Y. Tian, J. Cheng, B. Z. Tang and Z. Li, Matter, 2020, 3, 449–463 CrossRef.
- J. Yang, M. Fang and Z. Li, InfoMat, 2020, 2, 791–806 CrossRef CAS.
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, 1807887 CrossRef.
- Y. Zhou, W. Qin, C. Du, H. Gao, F. Zhu and G. Liang, Angew. Chem., Int. Ed., 2019, 58, 12102–12106 CrossRef CAS PubMed.
- J. Ren, Y. Wang, Y. Tian, Z. Liu, X. Xiao, J. Yang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2021, 60, 12335–12340 CrossRef CAS PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2020, 20, 175–180 CrossRef.
- M. Zeng, T. Li, Y. Liu, X. Lin, X. Zu, Y. Mu, L. Chen, Y. Huo and Y. Qin, Chem. Eng. J., 2022, 446, 136935 CrossRef CAS.
- Q. Gao, J. Rao, Z. Lv, M. Shi, M. Chen, G. Chen, X. Hao, B. Lü and F. Peng, Chem. Eng. J., 2023, 451, 138923 CrossRef CAS.
- X. Xu and B. Yan, Adv. Opt. Mater., 2022, 10, 2200451 CrossRef CAS.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- J. Wang, C. Wang, Y. Gong, Q. Liao, M. Han, T. Jiang, Q. Dang, Y. Li, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2018, 57, 16821–16826 CrossRef CAS PubMed.
- J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589–15593 CrossRef CAS.
- L. Gu, H. Shi, C. Miao, Q. Wu, Z. Cheng, S. Cai, M. Gu, C. Ma, W. Yao, Y. Gao, Z. An and W. Huang, J. Mater. Chem. C, 2018, 6, 226–233 RSC.
- P. T. Burks, A. D. Ostrowski, A. A. Mikhailovsky, E. M. Chan, P. S. Wagenknecht and P. C. Ford, J. Am. Chem. Soc., 2012, 134, 13266–13275 CrossRef CAS PubMed.
- C. A. M. Seidel, S. Kalinin, S. Sindbert, H. Vardanyan, A. Valeri, S. Felekyan and S. Müller, Biophys. J., 2011, 100, 1 CrossRef.
- D. Wang, H. Wu, J. Gong, Y. Xiong, Q. Wu, Z. Zhao, L. Wang, D. Wang and B. Z. Tang, Mater. Horiz., 2022, 9, 1081–1088 RSC.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef.
- L. Huang, L. Liu, X. Li, H. Hu, M. Chen, Q. Yang, Z. Ma and X. Jia, Angew. Chem., Int. Ed., 2019, 58, 16445–16450 CrossRef CAS.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589–15593 CrossRef CAS.
- B. W. van der Meer, D. M. van der Meer, and S. S. Vogel, in FRET-Förster Resonance Energy Transfer: From Theory to Applications, ed. I. L. Medintz and N. Hildebrandt, WileyVCH, 2012, vol. 492, pp. 234–238, DOI:10.1002/9783527656028.ch04.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2312419 and 2312421. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04746j |
| This journal is © The Royal Society of Chemistry 2024 |