
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular purification of aromatic polyester monomers from chemical depolymerization†
Gavan W.
Lienhart
,
Thomas
Palisin
,
William
Gross
,
Amelia
Moll
and
James M.
Eagan
 *
*
School of Polymer Science and Polymer Engineering, The University of Akron, Akron, Ohio 44325-3909, USA. E-mail: eagan@uakron.edu
First published on 11th October 2024
Abstract
The separation of poly(ethylene terephthalate) (PET) comonomers produced from the chemical recycling and depolymerization of PET is a critical step in realizing polyester material circularity. Various grades of PET (e.g., fiber, bottle, tire-cord) each require precise degrees of crystallinity which is controlled by the introduction of isomeric phthalates, comonomers which then contaminate secondary monomer streams. The use of supramolecular interactions as a method of extracting and separating these comonomers is herein described. The binding constants (Kb) between various-sized cyclodextrins with dimethyl terephthalate/dimethyl isophthalate and bis-2-hydroxyethyl terephthalate/bis-2-hydroxyethyl isophthalate are measured by fluorescence quenching and titration measurements. It is shown that beta-cyclodextrins most favorably bind the monomers (Kb up to 830 M−1) and that methyl ester comonomers bind more strongly than the glycolysis products. Competitive binding measurements between isomeric aromatic esters indicate alpha cyclodextrin binds dimethyl terephthalate (Kb = 328 M−1) 6-times more strongly than the dimethyl isophthalate isomer (Kb = 55 M−1). Crosslinking of the cyclodextrins with diisocyanate or perfluorinated bis-cyanobenzene moieties resulted in particles that could effectively remove the monomers from depolymerization solutions (up to 0.6 mmol of monomer per gram of particle). The results suggest supramolecular extraction and separation may be a scalable means of improving the purity and atom recovery of secondary recycled polyester feedstocks.
Sustainability spotlightEnsuring plastics are responsibly produced and disposed of requires the development of chemical recycling methods that enable the recovery of pure monomer feedstocks. Polyethylene(terephthalate) (PET) is the most abundant polyester plastic and is used in bottles, fibers, and in engineering applications such as tire-cords. Whereas the typical PET beverage bottle contains an isophthalate comonomer to reduce crystallinity and impart clarity, tire-cord PET requires high crystallinity polymers that are free of the isophthalate linkage. This work investigates supramolecular interactions as a means of extracting and separating polyester comonomers without high-energy distillation or chromatography. Our work emphasizes the importance of sustainable consumption and production patterns (SDG 12), industry innovation and infrastructure (SDG 9), and climate action (SDG 13). |
Introduction
Polyesters are high-strength chemically and mechanically recyclable materials that are widely used in fiber and blow molding applications. They are pervasive in society in the form of beverage bottles, textiles, and reinforcing cords. However, the molecular composition of each of these materials differ in regards both their comonomer content and degree of polymerization. For example, tire-cord poly(ethylene terephthalate) (PET) is a high molecular weight homopolymer with high degrees of crystallinity; whereas PET bottles contain approximately 2 wt% of an isophthalate comonomer which serves to disrupt crystallinity and impart clarity to the plastic products.1 Material flow between these applications in post-consumer recycling processes makes it difficult to produce high quality material from recycled polyester. As a result, mechanical recycling is often limited to either bottle or low-value textile applications, and is not straightforward for high-modulus low-shrink (HMLS) tire cords.2–4 Alternatively, chemical depolymerization enables purification by recrystallization of depolymerized materials, but can suffer from low yields and sequential procedures that add to cost and difficulty.5 Separation of isomeric polyesters remains a noteworthy barrier to circular recycling of PET.One desirable feature of polyesters is the scalable depolymerization through alcoholysis to afford the monomer constituents.6–8 Both methanolysis to dimethyl terephthalate (DMT, 1) and dimethyl isophthalate (DMI, 2) as well as glycolysis to the bis-(2-hydroxyethyl)terephthalate (BHET, 3) and bis-(2-hydroxyethyl)isophthalate) (BHEI, 4) are industrially practiced.9,10 While the catalysis of depolymerization has made significant advances in recent years,11 the purification of minor components with similar physical properties also requires advances for improving the circularity of polyester materials.12–15
We hypothesized that host–guest binding interactions might be suitable for selectively interacting with isomeric monomers and thus expand the methods available for purification. In this work, the binding interactions of cyclodextrin hosts with polyester monomers have been studied using fluorescence spectroscopy along with the efficacy of extracting monomers from mixtures with crosslinked cyclodextrin adsorbents.
Cyclodextrins are a family of cyclic oligosaccharides with varying numbers of glucose units possessing a bowl shape with a hydrophobic interior and hydrophilic exterior. This allows them to serve as host molecules, forming stable complexes with smaller molecules of the appropriate shape and size.16 Three different sizes of naturally occurring cyclodextrin were studied in this work: α, β, and γ with 6, 7, and 8 glucose units respectively. Cyclodextrins have found numerous uses in polymer synthesis, pharmaceuticals, personal care products, food formulations, and water purification.17–26 They were selected for this analysis because they are readily available at low cost and because they have previously shown large differences in binding affinity for different sets of regioisomers.27,28
Cyclodextrin adsorbents have been extensively developed for extracting pollutants from water and the environment. Akashi and coworkers have studied urethanes and ester crosslinked cyclodextrins for removing polychlorobiphenyl contaminates in oil with excellent efficiency and recovery.23,29 Crini et al. demonstrated the extraction of similar aromatic pollutants from aqueous solutions with epichlorohydrin crosslinked β-cyclodextrin.30 Wilson also studied polyester cyclodextrin materials for adsorbing nitrophenol, and discovered the existence of dual sorption mechanisms into both the cyclodextrin cavities and the polymer network.31 Dichtel and coworkers showed the surface area of cyclodextrin sorbents could be significantly increased with rigid aryl crosslinkers;19,20 and subsequently the importance of the crosslinker chemistry on the sorption capacity of polyfluoroalkyl substances.24
Most relevant to our extraction investigations, Murai et al. studied the binding of phthalate ester plasticizers with β-cyclodextrin using NMR and fluorescence spectroscopy.14 When crosslinked with epichlorohydrin, the cyclodextrin particles removed dimethyl and dipropyl phthalates from methanolic solutions. Similarly, Chen et al. demonstrated chitosan bound α-cyclodextrin could extract long-chain alkyl plasticizer phthalates with a sorption capacity of 3.21 mg g−1 (mg phthalate per g of sorbent).12
However, none of these studies evaluated the competitive selectivity in separating isomeric phthalates as a method of purification. Mendicuti quantified the thermodynamics of complexation of dimethyl isomeric esters (Fig. 1) in α and β-cyclodextrins using fluorescence spectroscopy.27 They showed that at 25 °C β-cyclodextrin and dimethyl terephthalate (1: DMT) favorably bind (Kb = 923 M−1) compared to dimethyl isophthalate (2: DMI) with β-cyclodextrin (Kb = 343 M−1). They further quantified that both esters had higher affinity for β-cyclodextrin relative to α-cyclodextrin. The only supramolecular separation of isomeric esters we are aware of was reported by McNair and Khaled.28 Their approach utilized a capillary zone electrophoresis setup and a buffer solution of borate and various cyclodextrins to resolve the isophthalate and terephthalate isomeric carboxylic acids.
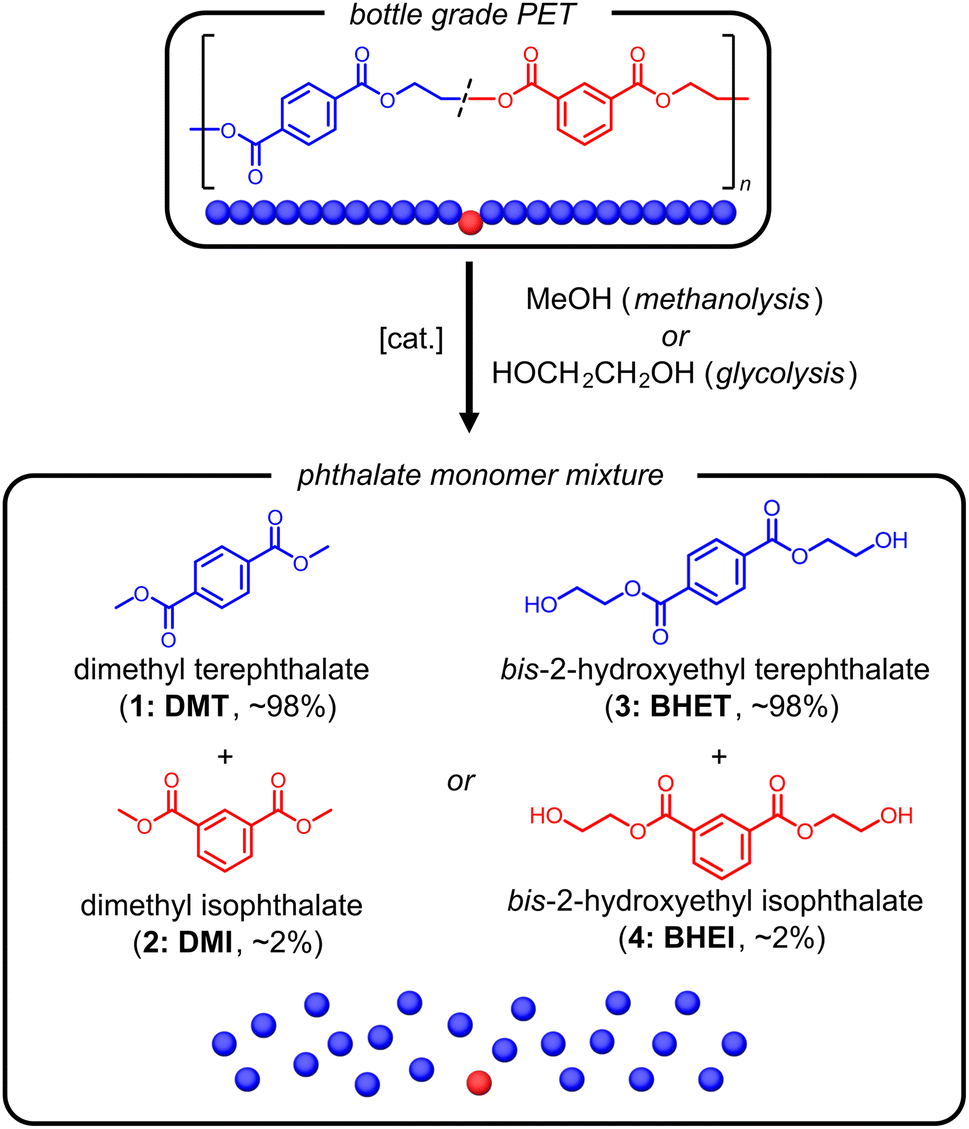 | ||
| Fig. 1 Depolymerization of PET bottles to monomer by methanolysis or glycolysis to produce mixtures of terephthalate and isophthalate monomers for purification. | ||
Herein we expand the understanding of binding constants between cyclodextrins and polyester glycolysis products (3: BHET and 4: BHEI), and the extraction of the polyester monomers by crosslinked cyclodextrin adsorbents. Our analysis of selective binding purification was carried out with both homogenous cyclodextrin binding constant measurements and heterogeneous crosslinked cyclodextrins. Fluorescence spectroscopy was used to determine binding constants of methyl and glycol terephthalate and isophthalate esters (DMT, DMI, BHET, BHEI; 1–4) with various diameter cyclodextrins (α-CD, β-CD, γ-CD). Crosslinked cyclodextrin adsorbents were synthesized using both diisocyanate (MDI, HMDI) and perfluorinated bis-cyanobenzene (TFTN, TFIN) crosslinkers. Typical selective extraction procedures took place with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratios of isomeric monomers, with additional experiments conducted at various 3:1 and 1:3 compositions to evaluate effects of monomer ratio. Removal of isomeric impurities was carried out through filtering depolymerized solution through the adsorbent and collecting either the filtrate or the bound material with solvent.
:1 ratios of isomeric monomers, with additional experiments conducted at various 3:1 and 1:3 compositions to evaluate effects of monomer ratio. Removal of isomeric impurities was carried out through filtering depolymerized solution through the adsorbent and collecting either the filtrate or the bound material with solvent.
Results and discussion
Complexation, or binding, is an equilibrium process and the degree to which it occurs is measured with a binding constant Kb as defined by: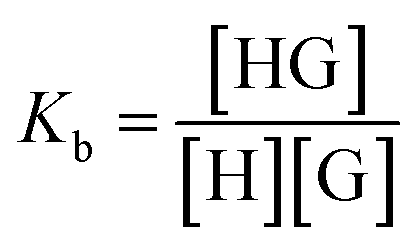 | (1) |
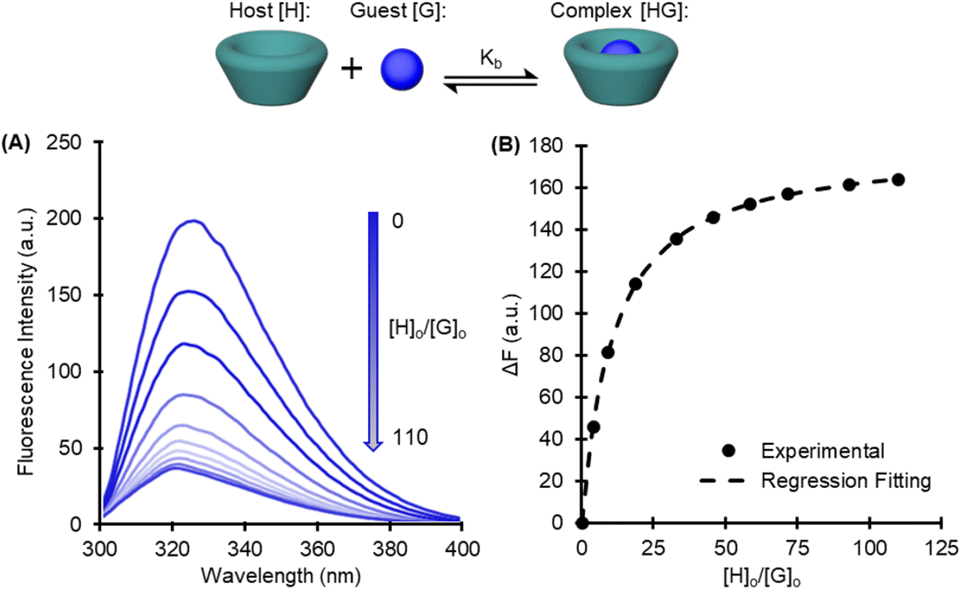 | ||
| Fig. 2 Example determination of binding constant for DMT and β-CD by chemical titration and fluorescence spectroscopy. (A) Decreases in fluorescence intensity observed for solutions with increasing equivalents of the host. (B) Nonlinear regression of fluorescence intensity changes ΔF to determine a binding constant. | ||
To assess the viability of separating isophthalate and terephthalate monomers by host–guest binding chemistry, the binding constants were measured for each pairing. The goal was to find a set of monomers (glycolysis or methanolysis products) that show large discrepancies in Kb between the isophthalate and terephthalate species.
Solution-phase binding affinities
To measure the Kb, a chemical titration method was adapted from Mendicuti,27 using fluorescence spectroscopy to correlate complexation induced changes in fluorescence intensity to the binding constant by a nonlinear regression. Binding constants were measured in triplicate for each combination of cyclodextrin and monomer, and the results are displayed in Table 1 (Fig. S8 and S19†).| Methanolysis | Glycolysis | ||||
|---|---|---|---|---|---|
| Host | Guest | K b (M−1) | Host | Guest | K b (M−1) |
| a BHEI/α-CD showed low binding constant values with a standard deviation exceeding the measured absolute value. | |||||
| α-CD | DMT | 328 ± 5 | α-CD | BHET | 15 ± 2 |
| DMI | 55 ± 3 | BHEI | <1a | ||
| β-CD | DMT | 830 ± 38 | β-CD | BHET | 339 ± 10 |
| DMI | 206 ± 6 | BHEI | 98 ± 2 | ||
| γ-CD | DMT | 84 ± 4 | γ-CD | BHET | 45 ± 2 |
| DMI | 55 ± 2 | BHEI | 14 ± 4 | ||
The most apparent finding from the Kb studies is that, in all instances, terephthalate monomers DMT and BHET show greater binding over corresponding isophthalates DMI and BHEI. This suggests a more favorable inclusion complex wherein the ester moieties effectively thread the cyclodextrin cavity, whereas the 1,3-aromatic diester moiety prevents the nonpolar moiety from maximizing the non-covalent interactions regardless of the cyclodextrin diameter. Additionally, all monomers displayed the greatest binding affinity toward the β-cyclodextrin. The estimated width of the phthalate aromatic ring is approximately 4.3 Å, which accommodates the β-CD cavity diameter (ca. 6.2 Å) while also maximizing hydrophobic interactions. Meanwhile the γ-CD (ca. 8.0 Å) is too large, and the hydrophobic interactions are lost. Interestingly, the larger cavity did not more favourably accommodate the BHEI width, relative to BHET, possibly due to the ester polarity competing with the hydrophobic binding mechanism. We speculate that α-CD (ca. 4.7 Å) may accommodate the aromatic ring but sacrifices too much entropic energy in this limited number of host/guest conformations. We also found that methanolysis produced monomers DMT and DMI with greater levels of binding relative to the glycolysis products BHET and BHEI. We propose this is a consequence of the increased hydrophilicity of the glycol monomer relative to the methyl ester which detracts from the host/guest affinity for BHET and BHEI.
Regarding the largest discrepancy in binding constant, we found that α-CD was the most selective host, preferentially binding DMT with nearly a 6-fold higher Kb than DMI. Although α-CD also exhibited high selectivity between BHET and BHEI, the Kb values were low confidence due to the low absolute values and sensitivity of the measurements. This result raises the issue of balancing selectivity simultaneously with the overall binding constant. In this case, the ratio of β-CD binding constants with terephthalate and isophthalate isomers were 4.0 times and 3.5 times higher for methanol and glycol esters, respectively.
Recycled monomer extraction with crosslinked cyclodextrins
With selective binding established for the terephthalate monomers, a selective extraction method was developed to separate the comonomers. Crosslinked adsorbents were synthesized spanning different cyclodextrin types, crosslinker chemistries, and crosslink densities – described in Fig. 3 and Table 2. Synthetic procedures were adapted from previous reports.19,23 All crosslinked cyclodextrins were purified by sequential Soxhlet extractions and mechanically pulverized into powders to ensure accurate UV-vis measurements (Fig. S6†). Structures were confirmed with infrared spectroscopy and the degree of cyclodextrin incorporation in mmol of cyclodextrin per g of particle was determined by elemental analysis.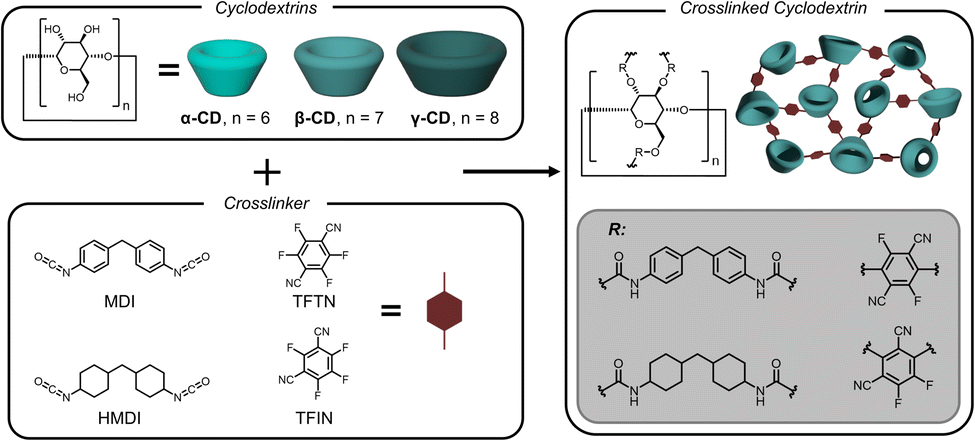 | ||
| Fig. 3 Crosslinking with diisocyanates and perfluorinated bis-cyanobenzenes. MDI: methylene diphenyl diisocyanate; HMDI: 4,4′-methylene dicyclohexyl diisocyanate; TFTN: tetrafluoroterephthalonitrile; TFIN: tetrafluoroisophthalonitrile. | ||
| Entry (#) | Cyclodextrin (α, β, or γ) | Crosslinker (NCO/ArF4) | Degree of crosslinkinga | Cyclodextrinb (mmol g−1) |
|---|---|---|---|---|
| a Determined from functional group ratio of [OH] to electrophilic moieties of crosslinkers, [NCO] or [F]. b Incorporated cyclodextrin determined from elemental analysis of the crosslinked particles. | ||||
| 1 | β | HMDI | 1.05 | 0.071 |
| 2 | β | HMDI | 1.98 | 0.344 |
| 3 | β | HMDI | 3.46 | 0.440 |
| 4 | α | HMDI | 3.53 | 0.301 |
| 5 | γ | HMDI | 3.51 | 0.434 |
| 6 | α | MDI | 3.70 | 0.407 |
| 7 | β | MDI | 3.65 | 0.415 |
| 8 | γ | MDI | 3.49 | 0.344 |
| 9 | β | TFTN | 1.93 | 0.601 |
| 10 | β | TFIN | 1.95 | 0.527 |
The adsorbents were used in a series of experiments to investigate the selective extraction of terephthalate monomers. Briefly, an aqueous solution of equimolar DMT and DMI (0.05 mM each) was treated with 100 mg of the absorbent. The solution was then filtered, and UV-vis was used to determine the change in concentration of each species after the extraction. Fig. 4 presents the selectivity results as the ratio of extracted terephthalate to isophthalate removed (Δ[DMT]/Δ[DMI]).
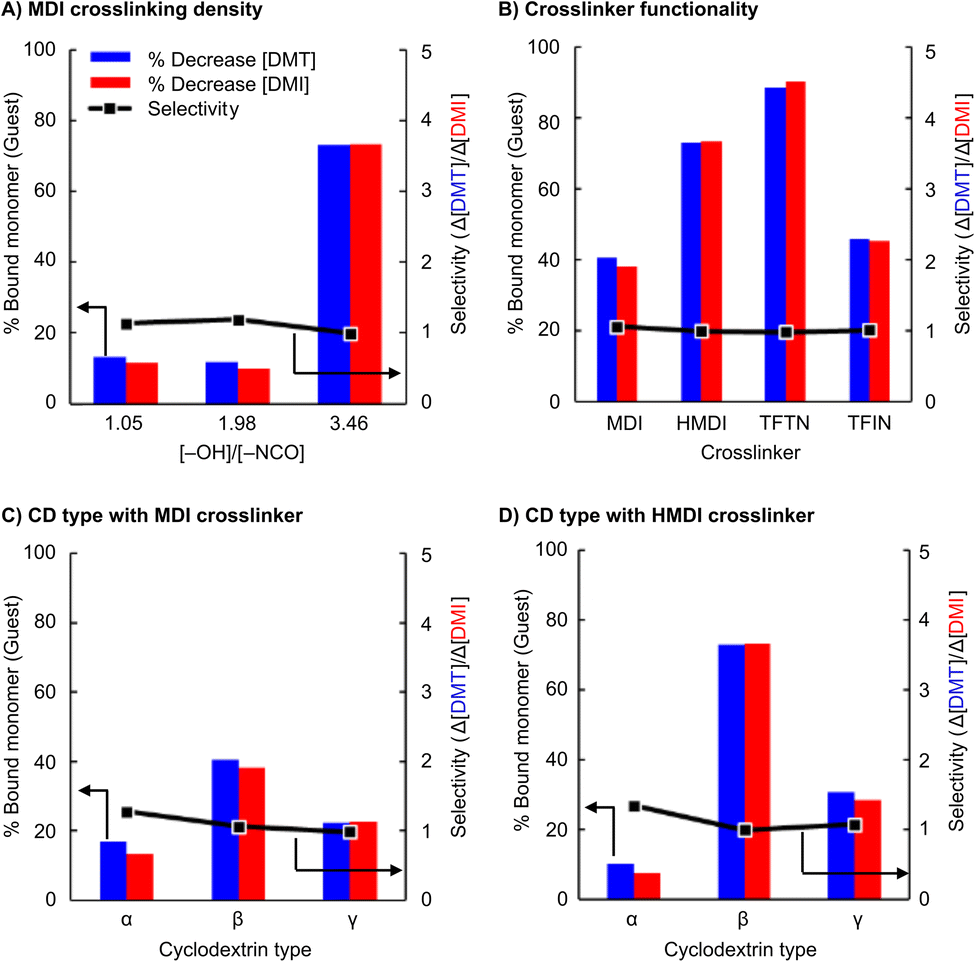 | ||
| Fig. 4 All data for DMT and DMI monomer mixtures (A) effect of crosslink density on selectivity with β-CD crosslinked with MDI. Functional group feed ratio used in place of crosslink density. (B) β-CD with various crosslinkers. (C) Different cyclodextrins with crosslinker MDI at [–OH]/[–NCO] = 3.5. (D) Different cyclodextrins with crosslinker HMDI. | ||
The initial experiment, performed with HMDI crosslinked β-CD, unexpectedly showed a near complete lack of selectivity. Despite DMT having a 4-fold greater Kb than DMI, nearly the same amount of each monomer was extracted. Our initial hypothesis was that the crosslinking reaction created the loss of expected selectivity. Because this sample used a nearly 1:1 functional group ratio (Table 2, entry 1), it was plausible that too many of the cyclodextrin hydroxyl groups were converted into urethanes which compromises the native structure of the cyclodextrin and renders the measured Kb values irrelevant in the urethane networks. It is also expected that the identity of the crosslinker plays a role in determining selectivity, as has been reported previously, although the origin of mechanism is not fully elucidated.24 For the present application the cyclodextrin was envisioned to provide the selectivity, but the amorphous, porous, and high-surface-area nature of the resins also provides secondary non-covalent binding interactions outside of the CD cavities. With this consideration, urethane and perfluorinated bis-cyanobenzene crosslinks were evaluated for monomer extraction and selectivity.
The effects of crosslink density are presented in Fig. 4A, and suggests little correlation to selectivity. The extraction ratio remained near 1.0 for all samples. However, the extraction volume grew significantly when less crosslinker (HMDI) was used. Nearly 75% of all monomers were extracted when using an adsorbent synthesized with a [–OH]/[–NCO] feed ratio of ∼3.5, a notable increase from the 15% measured in the more densely crosslinked systems. All subsequent urethane systems studied were synthesized with this functional group feed ratio (i.e., 3.5 = [–OH]/[–NCO]).
The evaluation of different cyclodextrin types produced more promising selectivity results. Fig. 4B and C show extractions with all three cyclodextrins crosslinked with isocyanates HMDI and MDI, respectively. Overall, the differentiation between DMT and DMI was still low, and all crosslinked particles exhibit selectivities <1.5. The highest selectivity observed was with α-CD particles, but this selectivity came at the cost of lower extraction efficiency, possibly owing to the lower cyclodextrin content (Table 2, entries 4 & 6).
Similar experiments were performed using adsorbents synthesized with perfluorinated bis-cyanobenzene crosslinkers. Although these materials did not overcome the selectivity challenge, they did show a significant increase in extraction efficiency. TFTN crosslinked β-CD, in particular, removed 90% of all monomers from solution with a single exposure.
Additional experiments were performed to evaluate the effects of the initial monomer feed ratio. The above selective extraction experiments were performed with a DMT mole fraction (χDMT) of 0.50, though a χDMT of 0.98 is more representative for depolymerized PET bottles. Initial χDMT of 0.25, 0.50 and 0.75 were tested to determine if selectivity behavior is altered with one component in excess (Fig. 5A). The resulting χDMT of the extracted material nearly matched the feed χDMT, indicating no additional selectivity.
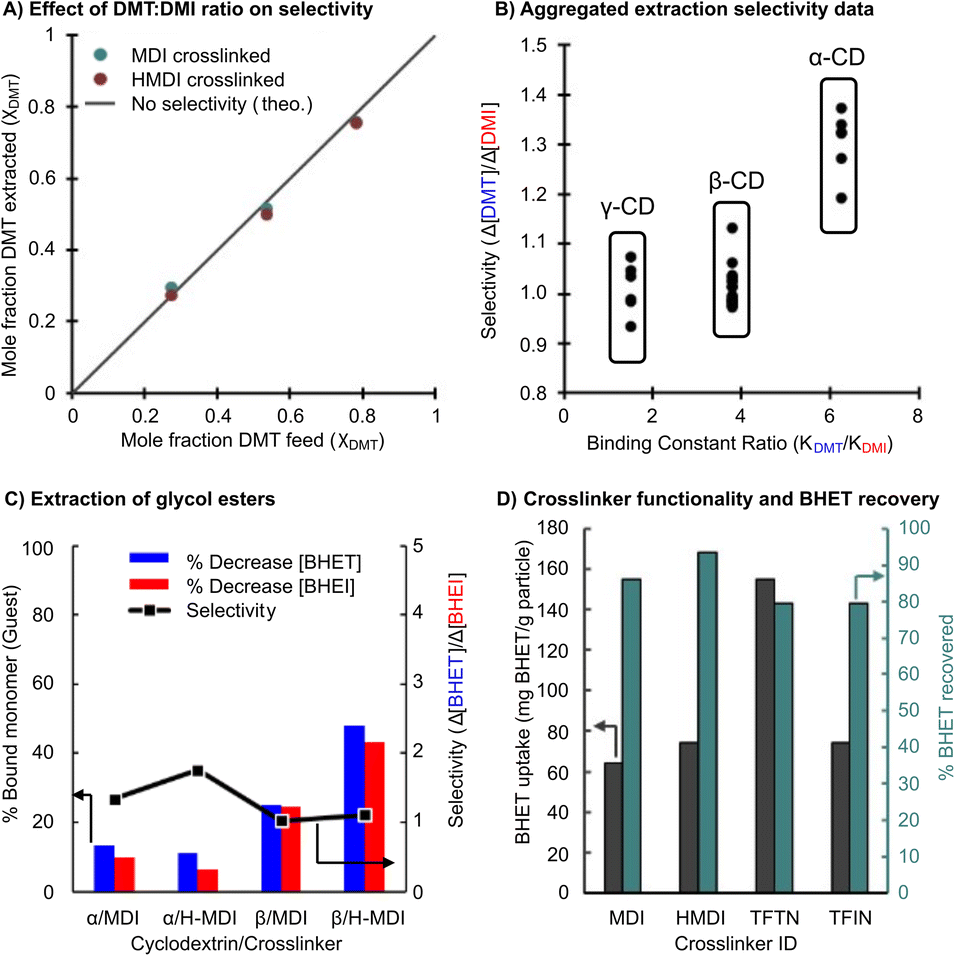 | ||
| Fig. 5 (A) Feed vs. extracted mole fractions of DMT used in place of selectivity. (B) Aggregate selectivity data for all trials, including those at 40 °C. (C) Glycolysis monomers selectivity. (D) Bulk BHET extraction using β-CD with various crosslinkers and the monomer recovery by methanol rinse. | ||
We next evaluated the selectivity in extracting BHET from a mixture of the glycolysis products (Fig. 5B). This experiment gave the greatest selectivity of any system at 1.7 (70 mol% more BHET than BHEI extracted) with an α-CD-based adsorbent.
The selectivity data presented across all trials clearly indicates that the cyclodextrin-based adsorbents underperform the selectivity expectations set by the differences in Kb values. However, the expected competitive binding is reflected in the data when presented in aggregate (Fig. 5C). In terms of an industrial separation though, it is unlikely that these methods will surpass recrystallization as the solitary means of monomer purification. However, it is common for recrystallization to generate low yields of 60–70%, especially for more soluble glycol esters, and requires sequential procedures to obtain desirable results. Given the extraction efficiencies obtained from cyclodextrin based adsorbents it may be pragmatic to employ them as a recovery agent for the leftover monomer from a recrystallization procedure. Tests for this are shown in Fig. 5D where TFTN crosslinked β-CD extracts 160% of the adsorbents mass in BHET. Monomer recovery is also respectably efficient, with 79–94% recovered from a single methanol wash.
In order to demonstrate the proposed supramolecular recovery process under more representative conditions, a glycolysis depolymerization was conducted on a shredded post-consumer PET beverage bottles with 1 wt% Zn(OAc)2 as a catalyst in ethylene glycol (10 wt equiv.). The crude products were precipitated in water to afford BHET in 61% yield as a mixture of BHET and BHEI. The aqueous glycol solution which did not crystallize was filtered over 4 wt% (relative to original PET) of the heterogeneous β-CD/TFTN particles. The solids were then rinsed with methanol and the solution was evaporated to afford additional BHET/BHEI solids. The process was repeated an additional three times with the same β-CD/TFTN particles, and the wt% of monomer recovered relative to the extraction mass recorded (Fig. 6). We observe that the total BHET recovered could be increased from the 61 mol% of aqueous crystallization to 72 mol% with supramolecular extraction. Notably the higher phthalate concentration (ca., 0.25 M) and excess ethylene glycol in this recycling recovery process did produce different extraction efficacies than under the binding conditions (50 mM, Fig. 5D). Instead of extracting 160 wt% BHET relative to the particle mass, 95 wt% was observed in the first extraction, and then 75–80 wt% for the next three extractions. No drying or particle regeneration steps were applied between cycles which may explain the decrease from 95 to 80%. The larger discrepancy between the dilute binding study and recycling experiment can be explained due to the excess of ethylene glycol. While this does affect the binding solvent properties, it also occupies binding sites in the heterogeneous particles, both within the CD and within the porous framework. This is evidenced by ethylene glycol observed in the methanol extracts, which was removed under heated vacuum prior to recording the BHET yield.
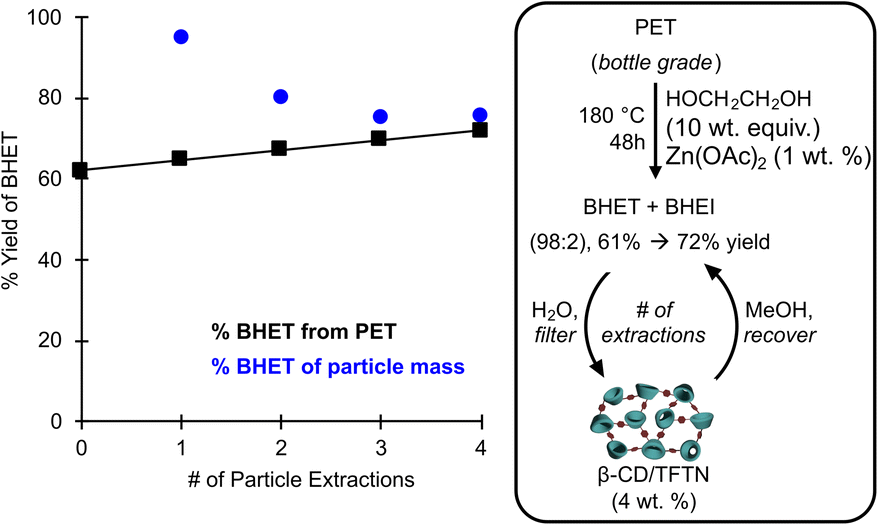 | ||
| Fig. 6 Demonstration of enhanced monomer yield from glycolysis of post-consumer PET bottles by solid-phase extraction with β-CD/TFTN particles in water and MeOH rinses. | ||
Collectively this work indicates that selective removal of isophthalates with supramolecular adsorbents is possible, but the prepared particles lacked the selectivity desired for an industrial scale regioisomeric separation. Alternatively, the efficacy at recovering soluble monomers from aqueous environments or depolymerization solutions were favourable and may find applications in increasing the yield of chemical recycling processes.
Experimental
General considerations
All spectroscopy experiments were carried out using ultra-pure water from a Millipore Synergy purification system. 1.0 × 1.0 cm quartz cuvettes were used for all experiments. Between each sample the cuvettes were triple rinsed with the subsequent sample, wiped down with a Kimwipe, and visually inspected for any debris. Fluorescence spectroscopy measurements were made on an Agilent Cary Eclipse fluorimeter. Experiments were performed in emission mode with an excitation wavelength of 289 nm, emission range of 300–600 nm, excitation and emission slit widths of 5 nm, 120 nm per minute scan speed, 1.0 nm data interval, and Savitzky–Golay 5 factor data smoothing. UV-vis spectra were taken on an Agilent Cary 60 spectrometer. Absorbance measurements were made between 200 and 800 nm with a scan speed of 120 nm per minute and data interval of 1.0 nm. Infrared spectra were collected on a PerkinElmer Frontier spectrometer equipped with an attenuated total reflection (ATR) system. Elemental composition of crosslinked cyclodextrins was determined using a LECO TruSpec CHNS Micro elemental analyzer. Samples were run in triplicate using approximately 1–2 mg of material each.Materials
4,4′-Methylene diphenyl diisocyanate (98%), 4,4′-methylene dicyclohexyl diisocyanate (90% mixture of isomers), p-toluenesulfonic acid monohydrate (p-TSA, >98.5%) and tetrafluoroterephthalonitrile (99%) were purchased from Millipore Sigma and used as received. Tetrafluoroisophthalonitrile (98%) was obtained from AmBeed and used as received. Anhydrous, amine-free dimethyl formamide (DMF, >99.9%) was purchased from Beantown Chemicals and used as received. Isophthalic acid (99%) was from Alfa Aeser and used as received. Bis-(2-hydroxyethyl)terephthalate (>85%) was purchased from TCI America and recrystallized from ethyl acetate and hexanes prior to use. Dimethyl terephthalate (99%) was purchased from Acros and recrystallized from ethanol and water. Dimethyl isophthalate (>99%) from TCI America was sublimed at atmospheric pressure before use. α-Cyclodextrin (97%) and γ-cyclodextrin (98%) were purchased from AmBeed and dried in a vacuum oven at 60 °C for two days. β-Cyclodextrin (98%) was obtained from Acros and dried in a vacuum oven at 60 °C for two days. Potassium carbonate (K2CO3, 99%) from Millipore Sigma was dried in a vacuum oven at 60 °C for two days. Ethylene glycol (Certified grade) was purchased from Fisher and stored over 4 Å molecular sieves. Tetrahydrofuran (THF, >99.9% anhydrous and inhibitor free) was purchased from VWR and dried in a VigorTech USA solvent purification system.Synthetic procedures
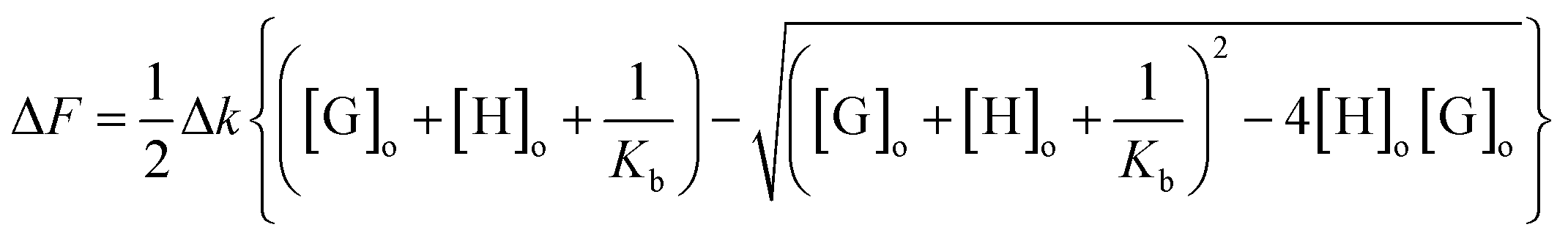 | (2) |
Non-linear regression of fluorescence data was performed using eqn (2) above, where ΔF is the pure guest solution ([H] = 0), Δk is the change in fluorescence constant upon complexation, [G]o is the initial guest concentration (constant), and [H]o is the initial host concentration. Regression fitting using Microsoft Excel's solver function to best fit Δk and Kb.
Selective extraction experiments
A stock solution was prepared with equimolar amounts of isophthalate and terephthalate monomers, each at 5 × 10−5 M. The materials were dissolved in ultra-pure water using gentle heat and stirring, then cooled to room temperature. A graduated cylinder was used to measure out 25 mL of the solution into Erlenmeyer flasks containing a known mass of crosslinked cyclodextrin (∼100 mg). The solutions were gently swirled to expose all the adsorbent to the solution then were allowed to sit for 5 minutes. Crosslinked cyclodextrin was then removed by gravity filtration using Cytiva Whatman #2 filter paper that was prerinsed with copious amounts of ultra-pure water. Each experiment also included sample of the pure stock solution, and another where (∼100 mg) of the crosslinked cyclodextrin was washed with 25 mL of ultra-pure water, each similarly filtered. These solutions enable us to determine changes in concentration before and after the extraction as well as confirm that the adsorbent is not contributing significantly to the solution background.All solutions from the experiment were subsequently diluted by a factor of 5 and analyzed by UV-vis to determine the changes is concentration. Isophthalate monomers were determined using their peak at 211 nm. Terephthalate monomers were analyzed at a wavelength of 256 nm. These wavelengths were selected to minimize the effect of one monomer on the others measures concentration. For every monomer system and wavelength, the desired component showed a molar absorptivity ∼16-fold greater than the other component. Because all solutions measured had approximately the same concentration of isophthalates and terephthalates, even after extractions, correcting for the concentration of the other was unnecessary in standard experiments.
Concentration changes of each monomer Δ[G] were calculated as a percentage difference from the stock solution used for each experiment. Rather than calculate individual concentrations with a calibration curve, the % change is simply the difference in absorbance between stock and experimental solutions at the desired wavelength.
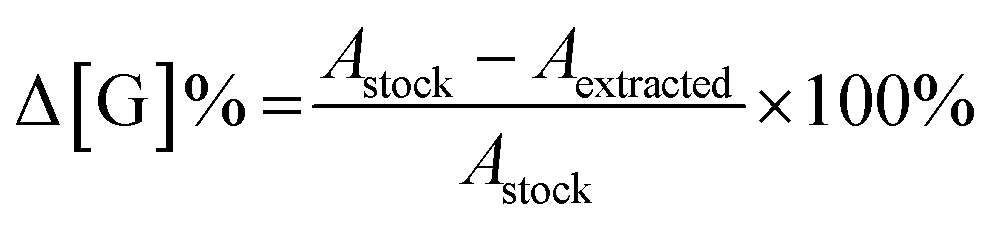 | (3) |
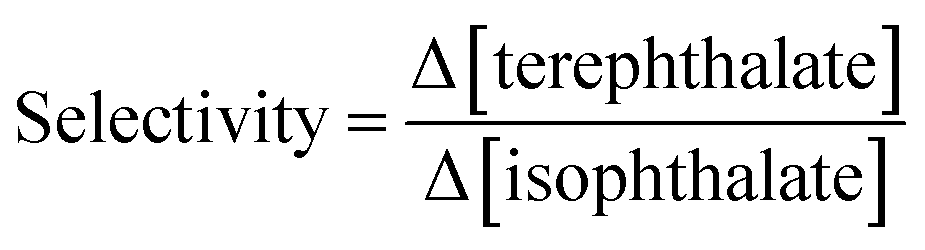 | (4) |
Selective extraction at different initial monomer concentrations
Similar extraction experiments were performed on stock solutions with different starting ratios of DMT and DMI. The total concentration of each was kept at 10−4 M and monomer ratios of 1:3, 1:1 and 3:1 were used. Because the selectivity term defined above is only valid for the extraction of equimolar mixtures, it was decided to express differences in affinity for crosslinked cyclodextrin by comparing the initial mol fraction of DMT, χDMT (feed), to that removed by the absorbent, χDMT (extracted).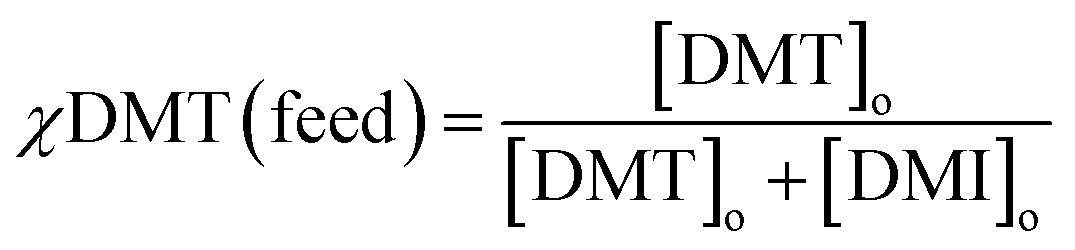 | (5) |
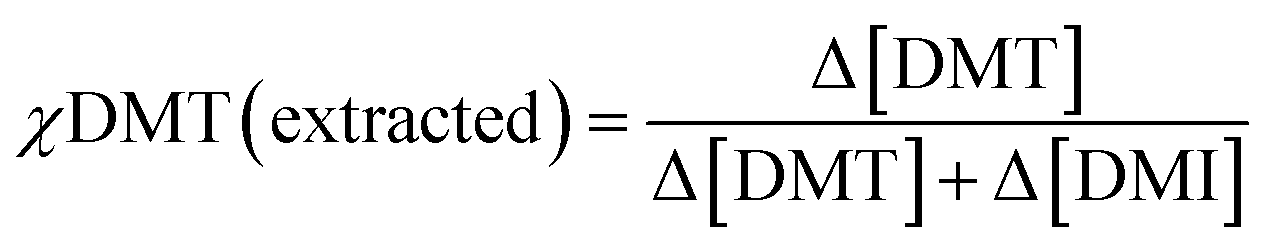 | (6) |
In addition to modifying the expression of selectivity, the concentrations measured by UV-vis were corrected for the residual absorbance of the comonomer in the mixture. An equation was derived for accurate concentrations. Absorbance values at 256 and 211 nm can be expressed as a linear combination of the absorbances from both DMT and DMI at these respective wavelengths as in eqn (7) and (8). Solving these as a system of linear equations yields expressions for the concentration of DMT and DMI, eqn (9) and (10).
| A256 = εDMT256[DMT] + εDMI256[DMI] | (7) |
| A211 = εDMT211[DMT] + εDMI211[DMI] | (8) |
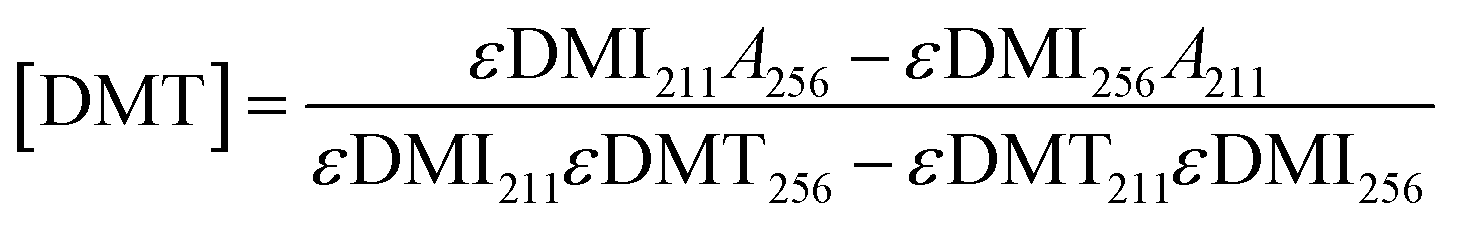 | (9) |
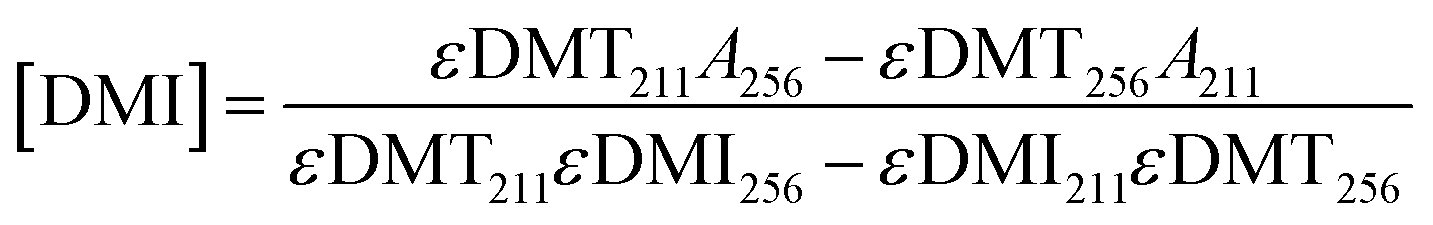 | (10) |
| Term (guest, wavelength) | Molar absorptivity (M−1 cm−1) |
|---|---|
| εDMT256 | 9660 |
| εDMT211 | 2295 |
| εDMI256 | 563 |
| εDMI211 | 36141 |
Bulk extraction efficiency experiment
A 7.47 × 10−3 M solution of BHET was prepared in ultra-pure water using heat and gentle stirring. This concentration is near the saturation point, and BHET was observed to slowly crystalize if allowed to sit for more than 24 hours. To avoid this problem, the solution was cooled quickly by submerging it in a room temperature water bath and performing experiments shortly after.As with earlier experiments, a known mass (165 mg) of crosslinked cyclodextrin was transferred to an Erlenmeyer flask and subsequently treated with 25.0 mL of the concentrated solution. A control flask without any adsorbent was also prepared. Gentle swirling was used to expose all absorbent to the solution. The flasks were then sealed using parafilm and aluminum foil to prevent evaporation and were left for 15 hours without stirring or shaking. Each sample was then gravity filtered using filter paper that had been prerinsed with substantial amount of ultra-pure water. For UV-vis, 120 μL of each filtrate was diluted to 50 mL total volume. Absorbance measurements were taken at the 244 nm peak instead of 256 nm because there was no concern about signal overlap with comonomers in a pure guest solution.
The mass of BHET extracted per gram of absorbent was calculated as below. Changes in absorbance between stock and extracted solutions were again used in place of constructing a calibration curve.
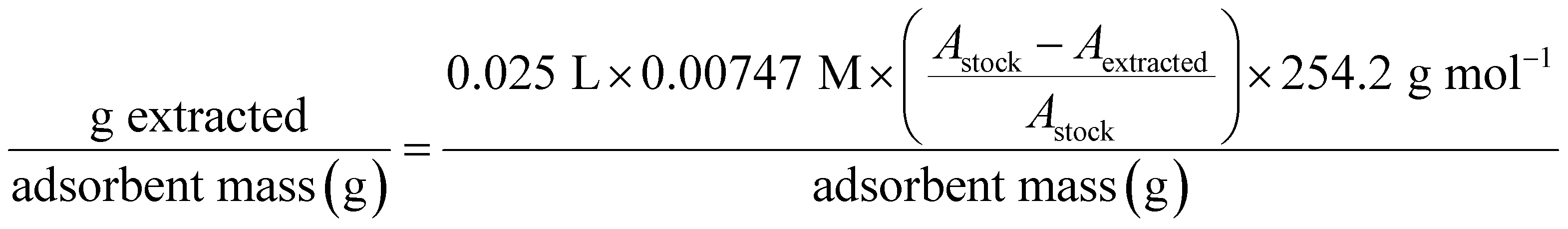 | (11) |
Monomers were recovered from the cyclodextrin adsorbents by transferring a known mass of them to an Erlenmeyer flask and shaking them overnight in 15 mL of methanol. 333 μL aliquots were taken, evaporated, then redissolved in 50.0 mL of water. Concentrations were determined via spectroscopy and the percentage recovery was calculated based on the total amount removed from solution in the initial extraction.
Depolymerization and monomer recovery experiment
A post-consumer generic brand PET beverage bottle was shredded to particle sizes of <1 × 1 cm using a mechanical shredder, dried in a vacuum oven at 60 °C, and 5.0 g of the plastic was charged into a 100 mL round-bottom flask. Ethylene glycol (50 g) and Zn(II) acetate (50 mg) were sequentially added, the vessel was fixed with a reflux condenser, and heated to 180 °C for 48 hours under an ambient atmosphere with stirring. Once the reaction was homogeneous, the vessel was cooled and poured into 50 mL of deionized water to precipitate the phthalate glycol esters. The suspension was filtered to afford 4.05 g of BHET/BHEI (61 mol% yield) and the remaining water solution was stirred with the above prepared β-CD/TFTN particles (200 mg). After 30 seconds, the suspension was filtered, and the retained particles were separately filtered with methanol (5 mL) and the solution was dried under reduced pressure at 60 °C. The BHET/BHEI aqueous solution was again stirred with the particles for 30 seconds, filtered, and again recovered in methanol; the process was repeated a total of 4 times to afford 190, 161, 150, and 151 mg of BHET/BHEI (95, 80, 75, and 76 wt% of particle mass, respectively).Conclusions
As part of a broader effort to make polyester recycling more efficient, an investigation was conducted to determine the utility of crosslinked cyclodextrin adsorbents to selectively extract terephthalate monomers from depolymerized PET. Binding constant measurements indicate a favourable supramolecular interaction between cyclodextrin with terephthalate monomers over isophthalate isomers. When crosslinked with urethane or aryl linkers, the cyclodextrin particle were effective at extracting and recovering the organic monomers from solution, but with a loss of competitive binding between isomers. Nonetheless, the heterogeneous particles were effective at recovering pure monomers from the aqueous filtrate from glycolysis depolymerization. Through an iterative filter/rinse process with 4 wt% of particles, the monomer recovery was increased from 61 to 72%. Optimization of the continuous extraction process and the investigation of other host systems through a combination of computational and experimental means is warranted, to further develop this paradigm of supramolecular purification of recycled feedstocks. It may also be possible to improve the heterogenization chemistry to retain the cyclodextrin selectivity and further advance the paradigm. In summary, crosslinked cyclodextrin adsorbents may be a means of boosting atom recovery from chemical recycling methods.Data availability
The materials, instruments, synthetic procedures, elemental analysis measurements, spectral data, regression fits, formula derivation, and supplemental references supporting this article have been included as part of the ESI.†Author contributions
GWL curated data, formal analysis, method development, project administration, supervised, validated, visualized, and wrote the manuscript. TP contributed to method development, and data validation. JME conceptualized, curated data, analysis, provided resources, acquired funding, wrote the manuscript and contributed project administration and supervision. All authors contributed to investigation and validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Center for Tire Research (CenTiRe) and the National Science Foundation IUCRC (Award #2137261).Notes and references
- Modern Polyesters: Chemistry and Technology of Polyesters and Copolyesters, ed. J. Scheirs and T. E. Long, Wiley, 2004 Search PubMed.
- S. Deng, R. Chen, S. Duan, Q. Jia, X. Hao and L. Zhang, SusMat, 2023, 3, 581–608 CrossRef.
- J. C. Tapia-Picazo, J. G. Luna-Bárcenas, A. García-Chávez, R. Gonzalez-Nuñez, A. Bonilla-Petriciolet and A. Alvarez-Castillo, Fibers Polym., 2014, 15, 547–552 CrossRef.
- L. Wu, G. Lienhart, S. Basak, S. C. Jana, K. A. Cavicchi and J. M. Eagan, Tire Sci. Technol., 2023 DOI:10.2346/tire.23.22020.
- P. Benyathiar, P. Kumar, G. Carpenter, J. Brace and D. K. Mishra, Polymers, 2022, 14, 2366 CrossRef.
- Y. Qin, M. Qu, J. Kaschta, V. Allen and D. W. Schubert, Studies on Recycled Polyester, 2020, pp. 29–67 Search PubMed.
- H. Abedsoltan, Polym. Eng. Sci., 2023, 63, 2651–2674 CrossRef.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- A. Bohre, P. R. Jadhao, K. Tripathi, K. K. Pant, B. Likozar and B. Saha, ChemSusChem, 2023, 16, e202300142 CrossRef PubMed.
- Eastman Provides Updates on Massive PET Recycling Plant, https://resource-recycling.com/plastics/2023/08/01/eastman-provides-updates-on-massive-pet-recycling-plant/, accessed August 2024.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. Mcneil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef.
- C. Y. Chen, C. C. Chen and Y. C. Chung, Bioresour. Technol., 2007, 98, 2578–2583 CrossRef PubMed.
- G. Crini, S. Bertini, G. Torri, A. Naggi, D. Sforzini, C. Vecchi, L. Janus, Y. Lekchiri and M. Morcellet, J. Appl. Polym. Sci., 1998, 68, 1973–1978 CrossRef.
- S. Murai, S. Imajo, Y. Takasu, K. Takahashi and K. Hattori, Environ. Sci. Technol., 1998, 32, 782–787 CrossRef.
- M. H. Mohamed, L. D. Wilson, D. Y. Pratt, R. Guo, C. Wu and J. V. Headley, Carbohydr. Polym., 2012, 87, 1241–1248 CrossRef.
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef PubMed.
- J. Szejtli, J. Mater. Chem., 1997, 7, 575–587 RSC.
- J. Szejtli, Pure Appl. Chem., 2004, 76, 1825–1845 CrossRef.
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef PubMed.
- M. J. Klemes, Y. Ling, M. Chiapasco, A. Alsbaiee, D. E. Helbling and W. R. Dichtel, Chem. Sci., 2018, 9, 8883–8889 RSC.
- L. D. Wilson and R. Guo, J. Colloid Interface Sci., 2012, 387, 250–261 CrossRef.
- N. E. A. Abu Rahim, N. I. Wan Azelee, S. F. Z. Mohd Fuzi, N. Masngut, Z. A. Zakaria, A. Zulkharnain, R. M. Illias and N. H. Abdul Manas, Curr. Pollut. Rep., 2023, 9, 680–693 CrossRef.
- S. Kawano, T. Kida, K. Miyawaki, Y. Fukuda, E. Kato, T. Nakano and M. Akashi, Polym. J., 2015, 47, 443–448 CrossRef.
- L. Xiao, C. Ching, Y. Ling, M. Nasiri, M. J. Klemes, T. M. Reineke, D. E. Helbling and W. R. Dichtel, Macromolecules, 2019, 52, 3747–3752 CrossRef CAS.
- H. Ritter, Prog. Polym. Sci., 2002, 27, 1713–1720 CrossRef CAS.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 8350–8369 CrossRef PubMed.
- A. di Marino and F. Mendicuti, Appl. Spectrosc., 2004, 58, 823–830 CrossRef.
- M. Y. Khaled and H. M. McNair, J. High Resolut. Chromatogr., 1996, 19, 143–150 CrossRef.
- S. Kawano, T. Kida, K. Miyawaki, Y. Noguchi, E. Kato, T. Nakano and M. Akashi, Environ. Sci. Technol., 2014, 48, 8094–8100 CrossRef PubMed.
- G. Crini and M. Morcellet, J. Sep. Sci., 2002, 25, 789–813 CrossRef.
- L. D. Wilson, M. H. Mohamed and C. L. Berhaut, Materials, 2011, 4, 1528–1542 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The spectral data, regression fits, formula derivation, and synthetic procedures supporting this article. See DOI: https://doi.org/10.1039/d4su00330f |
| This journal is © The Royal Society of Chemistry 2024 |