
An Fe-doped Ni-based oxalate framework with a favorable electronic structure for electrocatalytic water and urea oxidation†
Chunzi
Yang
ac,
Ming
Zhao
a,
Chunmei
Zhang
a,
Shan
Zhang
*a,
Dongdong
Zhu
 *b and
Chunxian
Guo
*a
*b and
Chunxian
Guo
*a
aSchool of Materials Science and Engineering, Suzhou University of Science and Technology, Suzhou 215009, China. E-mail: cxguo@usts.edu.cn; zs@usts.edu.cn
bSchool of Chemistry and Materials Science, Institute of Advanced Materials and Flexible Electronics (IAMFE), Nanjing University of Information Science and Technology, Nanjing 210044, China. E-mail: dd.zhu@nuist.edu.cn
cSchool of Environmental Science and Engineering, Suzhou University of Science and Technology, Suzhou 215009, China
First published on 25th November 2024
Abstract
An Fe-doped Ni-based oxalate framework, synthesized via a facile co-precipitation method, is applied as an excellent bi-functional electrocatalyst for water and urea oxidation reactions. The obtained framework achieved a large current density of 100 mA cm−2 at 1.497 V and 1.375 V (vs. RHE) for the OER and UOR, highlighting its potential for practical hydrogen production.
The increasing energy demand and environmental issues call for clean energy carriers. Hydrogen attracts much interest due to its high energy density (142 MJ kg−1) and zero carbon dioxide emission.1 Electrocatalytic water splitting is recognized as a promising route to produce green hydrogen. Traditional water splitting involves the cathodic hydrogen evolution reaction (HER) and anodic oxygen evolution reaction (OER). Since the OER is a sluggish four-electron-transfer process,2 high potentials much more positive than the theoretical potential (1.23 V vs. RHE) are always required. Therefore, developing highly efficient OER electrocatalysts is essential for practical application of water electrolysis. Despite great achievements having been made in the design of advanced OER electrocatalysts, the energy consumption at the anode of water electrolysis is still large. Fortunately, the anodic OER can be replaced by some other oxidation reactions, which are thermodynamically more favourable.3,4 Among them, the urea oxidation reaction (UOR) receives great attention because of its low thermodynamic potential of only 0.37 V vs. RHE. Moreover, urea can be decomposed into non-toxic N2, O2 and H2O products via the UOR, which provides an environmentally friendly way to treat urea-containing industrial and sanitary wastewater. Coupling the more favored anodic UOR with the cathodic HER, a urea-assisted water electrolyzer enables energy-saving hydrogen production and wastewater treatment, simultaneously.5 However, the UOR still suffers from an intrinsically sluggish six-electron-transfer process, and highly active electrocatalysts for the UOR are always required. In this regard, it is urgent to construct highly active and robust bi-functional electrocatalysts for the OER and UOR to achieve efficient and stable hydrogen generation.6
Some noble metal-based materials are demonstrated as advanced electrocatalysts for the OER/UOR.7,8 However, the extraordinarily high price and low abundance of these noble metals severely limit their further application, thus making transition metal-based materials promising alternatives. Despite various transition metal-based materials having been reported for the OER,9,10 only Ni-based materials emerge as up-to-date active and most widely explored UOR electrocatalysts.11–14 The pre-oxidized NiOOH with Ni3+ species is identified as the real active species for catalyzing urea oxidation.15–19 Such in situ electrochemical surface reconstruction from Ni2+ to highly active Ni3+ species plays a crucial role in determining the UOR activity. To promote the formation of high-valence Ni3+ sites, heteroatom doping is reported as an effective strategy, as doped heteroatoms can modify the electronic structure of Ni cations.20 For instance, Fe doping can facilitate the generation of Ni3+ species, contributing to dramatically enhanced OER activity,21–23 though the exact mechanism of Fe doping is still under debate.24–26 It is also believed that Fe incorporation into NiOOH leads to improved electrical conductivity, which is highly desirable for electrochemical processes.27
Organic ligands with negative functional groups such as benzene dicarboxylic (BDC) are introduced to produce metal–organic frameworks (MOFs) as highly efficient UOR/OER electrocatalysts.28 Similarly, oxalate is also strongly electrophilic due to its two COO− groups.29,30 Taking advantage of the electro-withdrawing property, constructing oxalate-based catalysts can achieve a high percentage of active Ni3+ species, and improved OER/UOR performance can be expected.11 However, reports on oxalate-related materials as bi-functional OER and UOR electrocatalysts are still very limited. Taken together, it is proposed to combine Fe doping and using oxalate ligands in catalyst design to facilitate the generation of Ni3+ species, thus boosting the OER and UOR performance. Additionally, the typical solvothermal method to prepare MOFs with a BDC ligand often requires organic solvents such as triethylamine and N,N-dimethylformamide,31 and it is highly desirable to develop a facile and green strategy to obtain oxalate-based materials.
Herein, we report a series of Fe-doped Ni-based oxalate frameworks (NixFe2−xC2O4) with different ratios of Ni and Fe on nickel foam (NF) via a facile co-precipitation method (Scheme 1). Benefiting from the increased number of Ni3+ active sites caused by Fe doping and the oxalate ligand, the optimized Ni0.6Fe1.4C2O4 possesses superior catalytic activity towards the OER and UOR with small overpotentials and Tafel slopes. This work provides a low-cost and efficient bi-functional electrocatalyst for the OER and UOR.
 | ||
| Scheme 1 Schematic illustration of the synthetic route for NixFe2−xC2O4. | ||
XRD patterns of all NixFe2−xC2O4 samples (Fig. S1, ESI†) display similar diffraction peaks to those of Ni0.6Fe1.4C2O4 (Fig. 1a), which match well with NiC2O4·2H2O (JPCDS No. 25-0581) and α-FeC2O4·2H2O (JPCDS No. 23-0293). These results indicate the successful formation of Fe-doped Ni-oxalate, accompanying the color change of NF from metallic silver to greyish green (Fig. S2, ESI†). There is no noticeable disparity between diffraction peaks of Ni/Fe oxalates owing to the similar structure of Ni and Fe. The scanning electron microscopy (SEM) image of Ni0.6Fe1.4C2O4 in Fig. 1b shows that irregular nanocubes with a size of about 500 nm are stacked on the NF surface homogeneously. Compared to monometallic NiC2O4 and FeC2O4 samples (Fig. S3, ESI†), the deposition of Ni0.6Fe1.4C2O4 on NF is more compact and uniform. The transmission electron microscopy (TEM) image (Fig. 1c) reveals that the obtained irregular nanocubes are built by some nanoparticles. Furthermore, obvious lattice fringes of these nanoparticles can be clearly seen in high-resolution TEM images (Fig. 1d and Fig. S4, ESI†). The lattice spacing of 0.223 nm (inset in Fig. 1d) is well indexed to the (−314) plane of NiC2O4·2H2O (JPCDS No. 25-0581). The Ni, Fe, C and O elements are evenly distributed across the whole irregular nanocubes (Fig. 1e), implying the homogeneous incorporation of Fe in Ni0.6Fe1.4C2O4. All the results above demonstrate the successful preparation of the Ni–Fe-oxalate framework on NF.
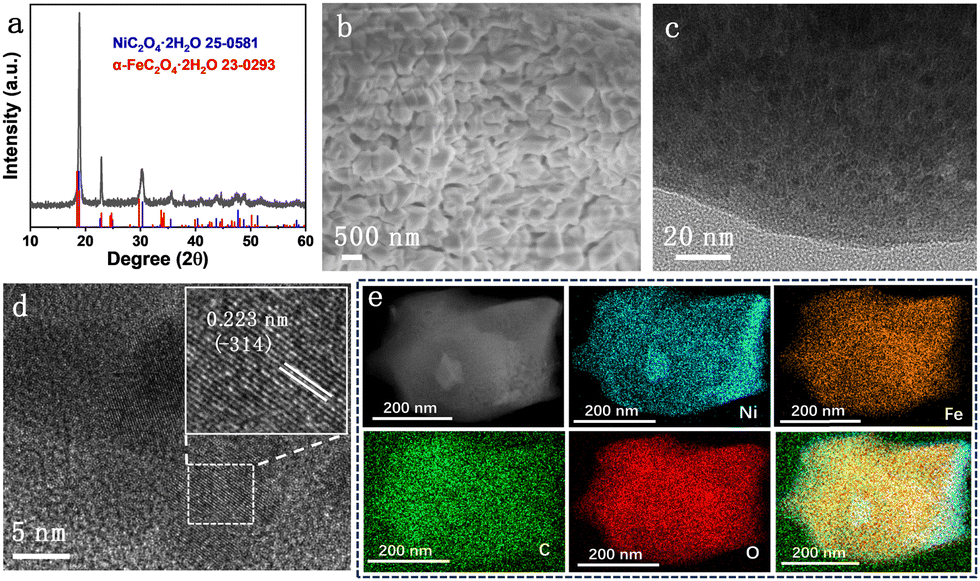 | ||
| Fig. 1 (a) XRD pattern of Ni0.6Fe1.4C2O4, (b) SEM, (c) TEM and (d) HRTEM images of Ni0.6Fe1.4C2O4, and (e) the corresponding EDX elemental mapping of Ni, Fe, C and O of Ni0.6Fe1.4C2O4. | ||
The surface composition and elemental valence state of Ni0.6Fe1.4C2O4 were determined by X-ray photoelectron spectroscopy (XPS). The high-resolution Ni 2p spectrum of Ni0.6Fe1.4C2O4 exhibits two different pairs of peaks (Fig. 2a), among which the peaks at 855.4 eV and 873.2 eV are attributed to 2p3/2 and 2p1/2 of Ni2+, respectively. While the other pair of peaks at 857.5 eV and 876.5 eV are assigned to Ni3+ 2p3/2 and 2p1/2, respectively.15 Moreover, the corresponding satellite peaks are observed at 861.1 eV and 880 eV.32 A small peak of 852.4 eV representing metallic Ni comes from the NF substrate.30,33 It is worth noting that the binding energy of Ni 2p in Ni0.6Fe1.4C2O4 has a positive shift of 0.36 eV compared to that of NiC2O4, indicating electron redistribution around Ni sites upon Fe doping. The content of active Ni3+ species in Ni0.6Fe1.4C2O4 is calculated to be 24.62%, while this value for NiC2O4 is only 7.99%, giving strong evidence that Fe doping increases the Ni3+ content in Ni0.6Fe1.4C2O4. Fig. 2b displays the Fe 2p spectra of the Ni0.6Fe1.4C2O4 and FeC2O4 samples. For Ni0.6Fe1.4C2O4, there are two peaks at around 712.16 eV and 724.34 eV, which belong to Fe2+ 2p3/2 and 2p1/2, respectively.34 Compared with the Fe 2p spectrum of FeC2O4, negative displacement of 0.4 eV for Ni0.6Fe1.4C2O4 can be observed. Considering the aforementioned binding energy change of Ni, it is suggested that electrons transfer from Ni to Fe, resulting in charge-redistribution around the active metal sites. Three peaks can be obtained in the C 1s spectrum (Fig. 2c), where 284.2 eV and 285.3 eV can be assigned to C–C and C–O bonds, respectively, while 288.9 eV corresponds to C2O42−.34 For the O 1s spectrum, as shown in Fig. 2d, three peaks can be obtained. The peaks at 531.3 eV and 533.4 eV can be well indexed to the metal–O (M–O) bond and C–O bond, respectively, while the peak located at 532.3 eV is ascribed to C2O42−, which is consistent with a previous report.30
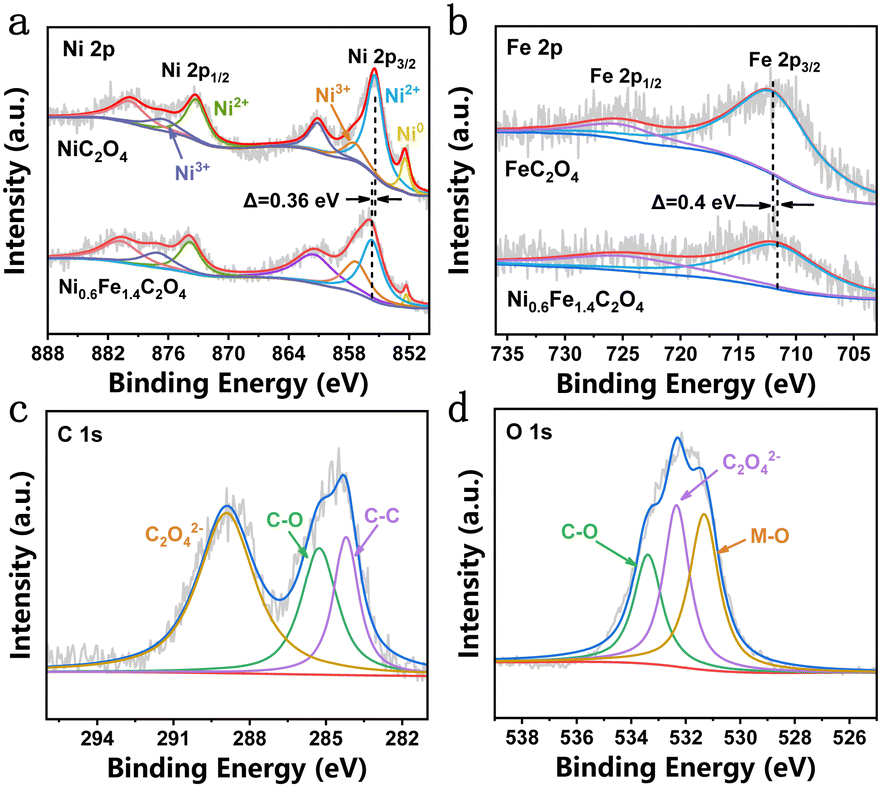 | ||
| Fig. 2 High-resolution XPS spectra of (a) Ni 2p of NiC2O4 and Ni0.6Fe1.4C2O4, (b) Fe 2p of FeC2O4 and Ni0.6Fe1.4C2O4, and (c) C 1s and (d) O 1s of Ni0.6Fe1.4C2O4. | ||
Thanks to the generation of desirable Ni3+ active sites, Ni0.6Fe1.4C2O4 can be utilized as a bi-functional electrocatalyst for both the OER and UOR. As can be seen from Fig. 3a, Ni0.6Fe1.4C2O4 shows excellent OER performance, surpassing other electrocatalysts with various compositions. The overpotentials of NixFe2−xC2O4 at the current density of 10 mA cm−2 (η10) and 100 mA cm−2 (η100) are collected in Fig. S5a (ESI†) for comparison. Of note, η10 is obtained from CV to avoid the influence of the pre-oxidation peak (Fig. S5b, ESI†). Ni0.6Fe1.4C2O4 displays the smallest η100 of 267 mV. In contrast, the single component NiC2O4 needs a large η100 of 461 mV. Such results indicate that Fe incorporation plays a pivotal role in boosting the OER performance. The superior OER activity of Ni0.6Fe1.4C2O4 is also demonstrated by a small Tafel slope of 22.63 mV dec−1 (Fig. 3b), implying its fast OER kinetics. The excellent OER performance of Ni0.6Fe1.4C2O4 makes it among the best-performing OER electrocatalysts (Table S1, ESI†). To further reveal the origin of the high OER activity of Ni0.6Fe1.4C2O4, the electrochemically active surface area (ECSA) and electrochemical impedance spectroscopy (EIS) were employed. As seen from Fig. S6 (ESI†), Ni0.6Fe1.4C2O4 possesses the largest Cdl of 10.7 mF cm−2, implying its largest ECSA with more active sites exposed. The intrinsic activity is further compared (Fig. S7a, ESI†), among which Ni0.6Fe1.4C2O4 displays the highest intrinsic OER activity. Moreover, the smallest charge transfer resistance of Ni0.6Fe1.4C2O4 in the Nyquist plots (Fig. S7b and inset, ESI†) suggests low charge transfer resistance at the electrode/electrolyte interface. As shown in the Bode diagrams (Fig. S8, ESI†), the peak of Ni0.6Fe1.4C2O4 disappears in the high frequency region at a voltage of 1.30 V, indicating that surface Ni2+ is completely reconstructed into a higher valence state, which is the actual active site for the OER process.35 The voltage of Ni0.6Fe1.4C2O4 is much lower than that of NiC2O4, implying that Fe incorporation benefits the formation of active NiOOH species. Chronoamperometric measurement of Ni0.6Fe1.4C2O4 is conducted to examine its OER durability. The current density displayed some loss during 50-h electrocatalysis, implying its moderate stability performance (Fig. 3c). Such substandard stability results may be attributed to its structural change during the OER process. To uncover the possible catalyst evolution, the morphology, composition and structure of Ni0.6Fe1.4C2O4 after the stability test were characterized. The nanocube morphology of Ni0.6Fe1.4C2O4 can still be observed, while some nanosheets also generate on the surface of the nanocubes (Fig. S9a and b, ESI†). Importantly, Ni(OH)2 species emerge as catalytically active sites for the OER (Fig. S9 and S10, ESI†).
 | ||
| Fig. 3 (a) LSV curves of NixFe2−xC2O4 for the OER in 1.0 M KOH, (b) the corresponding Tafel slopes, and (c) stability test of Ni0.6Fe1.4C2O4 for the OER. (d) LSV curves of NixFe2−xC2O4 for the UOR in 1.0 M KOH and 0.33 M urea, (e) the corresponding Tafel slopes, and (f) stability test of Ni0.6Fe1.4C2O4 for the UOR. | ||
Besides the OER, the as-prepared Ni0.6Fe1.4C2O4 is also performed for the UOR. Impressively, Ni0.6Fe1.4C2O4 displays greatly improved current densities when 0.33 M urea is added in 1.0 M KOH electrolyte (Fig. S11a, ESI†), and the potential to afford the UOR current density of 10 mA cm−2 is 1.36 V vs. RHE (Fig. 3d). The corresponding smallest Tafel slope of 15.12 mV dec−1 for Ni0.6Fe1.4C2O4 further implies its fast UOR kinetics (Fig. 3e). Furthermore, considering the overpotentials required at current densities of 10 and 100 mA cm−2 (Fig. S11b, ESI†), NixFe2−xC2O4 is most active towards the UOR. Specifically, Ni0.6Fe1.4C2O4 requires a potential of 1.375 V vs. RHE to reach 100 mA cm−2, while NiC2O4 and FeC2O4 require more positive potentials of 1.452 V and 1.418 V vs. RHE, respectively. For Ni0.6Fe1.4C2O4, both large ECSA with more exposed active sites, and higher intrinsic activity contribute to its excellent UOR performance (Fig. S12 and S13, ESI†). The smallest charge transfer resistance (Fig. S14, ESI†) of Ni0.6Fe1.4C2O4 further suggests the fast charge transfer at the electrode/electrolyte interface. As displayed in Table S2 (ESI†), Ni0.6Fe1.4C2O4 is one of the most efficient UOR electrocatalysts among these recently reported results. Bode diagrams (Fig. S15, ESI†) show that the peak of Ni0.6Fe1.4C2O4 in the high frequency region disappears at the voltage of 1.38 V, meaning that active NiOOH species are generated, which is earlier than that of NiC2O4 and FeC2O4. Moreover, Ni0.6Fe1.4C2O4 exhibits the smallest phase angle for the UOR, which reflects rapid dissociation kinetics of urea and strong electrooxidation ability of the reaction intermediates. Afterwards, chronoamperometric measurement of Ni0.6Fe1.4C2O4 was carried out in 1.0 M KOH and 0.33 M urea with refreshing the electrolyte three times (Fig. 3f). No obvious attenuation is observed over a continuous 40-h test, validating the excellent durability of Ni0.6Fe1.4C2O4 for the UOR. Post-characterization was conducted to investigate the morphology and structure change of Ni0.6Fe1.4C2O4. After the UOR stability test, the nanocube morphology of Ni0.6Fe1.4C2O4 is well retained, while some nanosheets can be observed on the surface of the nanocubes (Fig. S16a and b, ESI†). Similarly, Ni(OH)2 generates as real active species for the UOR (Fig. S16c and S17, ESI†). Though the original Ni0.6Fe1.4C2O4 is transferred to Fe-doped Ni(OH)2 during long-term electrolysis, Ni and Fe elements are still evenly distributed throughout the whole sample (Fig. S16d, ESI†).
In summary, an Fe doped Ni-based oxalate framework is facilely prepared through a one-step co-precipitation method, which is applied as a bi-functional electrocatalyst for the OER and UOR. Both Fe incorporation and introduction of electrophilic oxalate ligands facilitate the formation of highly desirable Ni3+ species. Therefore, the as-prepared Ni0.6Fe1.4C2O4 exhibits excellent performance towards the OER and UOR. Potentials of 1.497 V and 1.375 V vs. RHE are required to afford a large current density of 100 mA cm−2 for the OER and UOR, respectively. This work provides a dual-modulation strategy to construct Ni3+ rich electrocatalysts, which are promising for a variety of oxidation reactions beyond the OER and UOR.
The authors are grateful to the National Natural Science Foundation of China (22302140, 22372113), the Natural Science Foundation of Jiangsu Province (BK20220641), and the National Key Research and Development Program of China (No. 2021YFA0910403).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Wang, X. Cao and L. Jiao, eScience, 2021, 1, 69–74 CrossRef.
- B. Deng, G. Yu, W. Zhao, Y. Long, C. Yang, P. Du, X. He, Z. Zhang, K. Huang, X. Li and H. Wu, Energy Environ. Sci., 2023, 16, 5210–5219 RSC.
- Y. Sun, J. Wang, Y. Qi, W. Li and C. Wang, Adv. Sci., 2022, 9, e2200957 CrossRef.
- Y. Zhu, X. Zhu, L. Bu, Q. Shao, Y. Li, Z. Hu, C.-T. Chen, C.-W. Pao, S. Yang and X. Huang, Adv. Funct. Mater., 2020, 30, 2004310 CrossRef CAS.
- Y. Diao, Y. Liu, G. Hu, Y. Zhao, Y. Qian, H. Wang, Y. Shi and Z. Li, Biosens. Bioelectron., 2022, 211, 114380 CrossRef CAS PubMed.
- X. Jia, H. Kang, X. Yang, Y. Li, K. Cui, X. Wu, W. Qin and G. Wu, Appl. Catal., B, 2022, 312, 121389 CrossRef CAS.
- T. Kwon, H. Yang, M. Jun, T. Kim, J. Joo, J. Kim, H. Baik, J. Y. Kim and K. Lee, J. Mater. Chem. A, 2021, 9, 14352–14362 RSC.
- M. Zhong, M. Xu, S. Ren, W. Li, C. Wang, M. Gao and X. Lu, Energy Environ. Sci., 2024, 17, 1984–1996 RSC.
- X. Zhang, A. Wu, D. Wang, Y. Jiao, H. Yan, C. Jin, Y. Xie and C. Tian, Appl. Catal., B, 2023, 328, 122474 CrossRef CAS.
- C. Chen, M. Sun, F. Zhang, H. Li, M. Sun, P. Fang, T. Song, W. Chen, J. Dong, B. Rosen, P. Chen, B. Huang and Y. Li, Energy Environ. Sci., 2023, 4, 1685–1696 RSC.
- J. Kim, M. C. Kim, S. S. Han and K. Cho, Adv. Funct. Mater., 2024, 34, 2315625 CrossRef CAS.
- P. Wang, X. Bai, H. Jin, X. Gao, K. Davey, Y. Zheng, Y. Jiao and S. Z. Qiao, Adv. Funct. Mater., 2023, 33, 2300687 CrossRef CAS.
- M. Liu, W. Zou, J. Cong, N. Su, S. Qiu and L. Hou, Small, 2023, 19, 2302698 CrossRef CAS PubMed.
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustainability, 2021, 4, 868–876 CrossRef.
- L. Chen, L. Wang, J. T. Ren, H. Y. Wang, W. W. Tian, M. L. Sun and Z. Y. Yuan, Small Methods, 2024, 2400108, DOI:10.1002/smtd.202400108.
- Y. Zhu, C. Liu, S. Cui, Z. Lu, J. Ye, Y. Wen, W. Shi, X. Huang, L. Xue, J. Bian, Y. Li, Y. Xu and B. Zhang, Adv. Mater., 2023, 35, 2301549 CrossRef CAS.
- S. Zhou, H. He, J. Li, Z. Ye, Z. Liu, J. Shi, Y. Hu and W. Cai, Adv. Funct. Mater., 2023, 34, 2313770 CrossRef.
- L. Wang, Y. Zhu, Y. Wen, S. Li, C. Cui, F. Ni, Y. Liu, H. Lin, Y. Li, H. Peng and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10577–10582 CrossRef CAS PubMed.
- S. Zhou, S. Lv, J. Shi, L. Zhang, J. Li and W. Cai, Chem. Eng. J., 2024, 484, 149706 CrossRef CAS.
- Q. Zhang, Y. Hu, H. Wu, X. Zhao, M. Wang, S. Wang, R. Feng, Q. Chen, F. Song, M. Chen and P. Liu, ACS Nano, 2023, 17, 1485–1494 CrossRef CAS PubMed.
- K. Shen, Y. Tang, Q. Zhou, Y. Zhang, W. Ge, X. Shai, S. Deng, P. Yang, S. Deng and J. Wang, Chem. Eng. J., 2023, 471, 144827 CrossRef CAS.
- Y. Li, H. Guo, J. Zhao, Y. Zhang, L. Zhao and R. Song, Chem. Eng. J., 2023, 464, 142604 CrossRef CAS.
- C. Kou, J. Zhou, H. Wang, J. Han, M. Han, A. Vomiero, Y. Liu and H. Liang, Appl. Catal., B, 2023, 330, 122598 CrossRef CAS.
- P. Upale, S. Verma and S. B. Ogale, J. Mater. Chem. A, 2023, 11, 8972–8987 RSC.
- M.-L. Guo, Z.-Y. Wu, M.-M. Zhang, Z.-J. Huang, K.-X. Zhang, B.-R. Wang and J.-C. Tu, Rare Met., 2023, 42, 1847–1857 CrossRef CAS.
- M. Jiang, H. Zhai, L. Chen, L. Mei, P. Tan, K. Yang and J. Pan, Adv. Funct. Mater., 2023, 33, 2302621 CrossRef CAS.
- M. Cai, Q. Zhu, X. Wang, Z. Shao, L. Yao, H. Zeng, X. Wu, J. Chen, K. Huang and S. Feng, Adv. Mater., 2022, 35, 2209338 CrossRef PubMed.
- D. Zhu, C. Guo, J. Liu, L. Wang, Y. Du and S.-Z. Qiao, Chem. Commun., 2017, 53, 10906–10909 RSC.
- X. Gao, D. Chen, J. Qi, F. Li, Y. Song, W. Zhang and R. Cao, Small, 2019, 15, e1904579 CrossRef PubMed.
- J. Kim, M. C. Kim, S. S. Han and K. Cho, Adv. Funct. Mater., 2024, 34, 2315625 CrossRef CAS.
- K. Ge, S. Sun, Y. Zhao, K. Yang, S. Wang, Z. Zhang, J. Cao, Y. Yang, Y. Zhang, M. Pan and L. Zhu, Angew. Chem., Int. Ed., 2021, 60, 12097–12102 CrossRef CAS PubMed.
- X. Wang, Z. Li, S. Sun, H. Sun, C. Yang, Z. Cai, H. Zhang, M. Yue, M. Zhang, H. Wang, Y. Yao, Q. Liu, L. Li, W. Chu, J. Hu, X. Sun and B. Tang, J. Colloid Interface Sci., 2024, 662, 596–603 CrossRef CAS.
- H. Jin, X. Wang, C. Tang, A. Vasileff, L. Li, A. Slattery and S.-Z. Qiao, Adv. Mater., 2021, 33, 2007508 CrossRef CAS.
- X. Gao, D. Chen, J. Qi, F. Li, Y. Song, W. Zhang and R. Cao, Small, 2019, 15, 1904579 CrossRef CAS.
- H. Qin, Y. Ye, J. Li, W. Jia, S. Zheng, X. Cao, G. Lin and L. Jiao, Adv. Funct. Mater., 2022, 33, 2209698 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05385k |
| This journal is © The Royal Society of Chemistry 2025 |