
Fast, catalyst-free room temperature production of isocyanate-free polyurethane foams using aromatic thiols†
Maxime
Bourguignon
a,
Bruno
Grignard
ab and
Christophe
Detrembleur
 *ac
*ac
aCenter for Education and Research on Macromolecules (CERM), CESAM Research Unit, University of Liege, Sart-Tilman B6a, 4000 Liege, Belgium. E-mail: christophe.detrembleur@uliege.be
bFRITCO2T Platform, University of Liege, Sart-Tilman B6a, 4000 Liege, Belgium
cWEL Research Institute, avenue Pasteur, 6, 1300 Wavre, Belgique
First published on 26th November 2024
Abstract
Non-isocyanate polyurethane (NIPU) foams of the polyhydroxyurethane (PHU) type are promising greener alternatives to their conventional isocyanate-based polyurethane counterparts that dominate the foam market. Recently, concomitant organocatalyzed aminolysis and S-alkylation of the cyclic carbonates of PHU formulations offered a facile route to produce CO2 self-blown PHU foams. However, this process was limited to the production of foams of rather low Tg (commonly up to room temperature) and suffered from slow foaming (i.e. 30 min) at 120 °C, thus still far from the foaming timeframes (1–10 min) and room temperature (r.t.) needed for adaptation to industrial foaming equipment. In this work, we elaborate strategies to accelerate thiol-assisted NIPU foaming in order for it to be complete in 5–10 minutes from r.t. reactive formulations under catalyst-free conditions. This is achieved by substituting aliphatic thiols by more acidic aromatic ones, and by adding epoxides as heat release promotors that will accelerate both the foaming and the curing rates. Moreover, flexible, semi-rigid and rigid foams are easily accessible by the choice of the amine comonomer and epoxide additive. This work draws general and simple concepts for greatly speeding-up the self-blowing NIPU process, a crucial step toward decreasing the energetic and production costs, offering potential for retrofitting existing foam production plants.
1. Introduction
Polyurethane (PU) foams are essential materials of our modern life with multiple usages as insulation materials, items for comfort and furniture, seats, mattresses, etc.1,2 With an estimated market growth of 7.4% for the 2022–2027 period,3 the success of PU foams is due to their ease of fabrication from low cost isocyanates and polyols. Besides the formation of urethane linkages by polyaddition of the isocyanate with the polyol, the addition of some water within the reactive formulation causes hydrolysis of the isocyanate. This reaction furnishes an amine that participates in PU network construction via urea linkage formation, with CO2 acting as the blowing agent.4 These features make the foam production compatible with continuous slabstock foaming (e.g. for producing matresses) or reactive injection molding (RIM; e.g. for preparing foams of complex shapes such as car seats and decorative objects). Nevertheless, due to their acute toxicity,5,6 isocyanates face restrictions to their use by REACH.7 In addition, PU production also faces recent EU policies on decarbonization of the plastics sector that encourage the utilization of raw chemicals derived from bio-renewables and/or gaseous waste effluents such as CO2.8 Therefore, isocyanate-free pathways for the production of these irreplaceable products that will answer the sustainability goals of Europe are intensively sought. In the last decade, polyamine/poly(cyclic carbonate) copolymerization, leading to poly(hydroxyurethane)s (PHUs),9–12 has imposed itself as one of the main alternatives to conventional PU thermosets, particularly for producing foams, coatings and adhesives.13–19 The foaming of PHUs has been explored using a physical blowing agent such as Solkane20 – a fluorinated solvent that contributes to global warming – or supercritical CO2.21 Other recent concepts also use inorganic carbonate salts (K2CO3, NaHCO3) in synergy with acids (maleic or acetic acid) as the source of CO2 to formulate foamed materials from reactive formulations.22,23 The first self-blown PHU foams were described with Momentive MH15, a poly(methylhydrogensiloxane) reactive against amines, that releases in situ highly flammable H2 as the blowing agent.24–27 However, all these foaming approaches still face environmental or safety issues and are far from achieving a PU self-blowing technology for which one of the monomers, i.e. the isocyanate, serves both for the polymer chain construction and as a source of the blowing agent.Recently, our group developed two popular strategies for producing CO2-self-blown PHU foams. The most recent one involved the partial hydrolysis of 5-membered cyclic carbonates catalyzed by (organo)bases that initiated the foaming in a moderate time frame (about 30 min) at 100 °C.28,29 In the subsequent study, this process was improved for a rapid foaming (a few minutes) from room temperature formulations,30 reaching a foaming performance very close to industrial ones. This was achieved by adding some epoxide to the formulation that had a dual role, to create the additional exotherm needed to reach the foaming zone (85–100 °C) and to introduce additional crosslink nodes needed to rapidly fix the foam and avoid its collapse. The second method capitalized on the ambivalent reactivity of the cyclic carbonates. The nucleophilic attack of the carbonyl site by the amine of the formulation delivered hydroxyurethane linkages while a competitive S-alkylation reaction occurred via attack of the methylene site by (masked) thiols added to the formulation, leading to thioether bond formation and CO2 release promoting the self-blowing of the matrix.31–35 This strategy mimics the polyurethane chemistry as the reaction leading to the formation of the blowing agent also contributes to the foam cross-linking. Moreover, the structure of the thiol also influenced the foam properties as it was incorporated into the polymer network. However, the foaming conditions (several hours at 80–100 °C) were still far from the conventional PU ones (1–15 min) or the PHU ones produced by the optimized water-induced self-blowing procedure described above. Strategies to accelerate the PHU self-foaming promoted by thiols have primarily focused on modifying the process itself as exemplified by Torkelson.35 The decoupling of the gelation and foaming steps reduced the foaming time from hours to about 30 min, however a high foaming temperature (120 °C) was still required. This was achieved by a short pre-foaming step at 80 °C for 3–6 min to increase the formulation viscosity, followed by foaming for 20 min, at 120 °C. Further decreasing the foaming time while significantly decreasing the formulation temperature was not possible by this process. Recently, Verdejo reported the fast fabrication of thiol-induced CO2 self-blown polymers from r.t. reactive formulations by designing the partial carbonatation of epoxide (85% of cyclic carbonate and 15% of epoxide) to drive the foaming.36 According to the formulation composition presented in their article and FT-IR analysis, the foams contain a low hydroxyurethane content and are predominantly crosslinked by thioether bonds.
In this work, we address the challenge of rapidly producing PHU foams by the thiol-assisted self-foaming process from room temperature (r.t.) formulations. Herein, we explore the influence of the nature of the thiols on their ability to undergo S-alkylation by reaction with the cyclic carbonate, and thus the foaming. Model reactions carried out with aliphatic and aromatic thiols enable us to understand why the S-alkylation is facile with aromatic thiols under catalyst-free conditions. By utilizing some epoxide additives to the formulations, PHU foams were rapidly produced from r.t. formulations without the need for any catalyst. Ultimately, our evaluation of the foam properties revealed the system's versatility, as it allows the straightforward production of both flexible and rigid foams using aromatic thiols.
2. Experimental
2.1. Products
Trimethylol propane triglycidyl ether (TMPTE, Aldrich, technical grade), butanediol diglycidyl ether (BDGE, Aldrich >95%), DER 332™ (Aldrich), propylene carbonate (Pc, Aldrich, 98%), epoxidized soybean oil (ESBO, Vandeputte), m-xylylene diamine (XDA, Aldrich, 99%), 1,2-bis(2-aminoethoxy)ethane (EDR 148, TCI, >98%), tetrabutylammonium iodide (TBAI, Aldrich, 99%), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, Sigma Aldrich, 98%), hydrotalcite synthetic (HTC, Mg6Al2(CO3)(OH)16·4H2O, Aldrich), triethylamine (TEA, Aldrich, 99%), 1,3-bis(aminomethyl)cyclohexane (cis- and trans-mixture) (1,3 BAC, TCI, 98%), benzylamine (Aldrich, 99%), 2,2′-(ethylenedioxy)diethanethiol (T1, EDR-diSH, Aldrich, 95%), 1,3,4-thiadiazole-2,5-dithiol (T2, bismuthiol, ABCR, 95%), 4,4′ thiobisbenzenthiol (T3, Aldrich, 98%), thiophenol (T4, Alfa Aesar, >99%), and Portaflame SG63 (POR, Sibelco Europe MineralsPlus) were used.2.2. Method and characterization
The gel contents were measured in triplicate by incubating foam samples in 20 mL of THF for 24 h according to the following formula: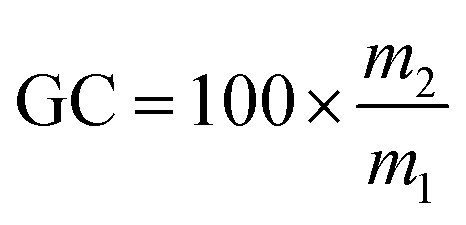 , where m1 and m2 are the mass before immersion and the mass after immersion and drying, respectively.
, where m1 and m2 are the mass before immersion and the mass after immersion and drying, respectively.
The foam density was assessed in triplicate by weighing the foam and dividing its mass by its volume.
The ATR spectra were recorded using a Nicolet IS5 spectrometer (Thermo Fisher Scientific) equipped with a transmission or a diamond attenuated transmission reflectance (ATR) device. Spectra were obtained in transmission or ATR mode as a result of 32 spectra in the range of 4000–500 cm−1 with a nominal resolution of 4 cm−1. Spectra were analyzed with ONIUMTM (Thermo Fisher Scientific) software. Analyses were carried out on the inner part of the foam after removing the foam skin.
Differential scanning calorimetry (DSC) was performed on a DSC250 TA Instruments calorimeter. The equilibrated Tg was measured on the first ramp of temperature (−40–80 °C, 10 °C min−1). Then, the DSC sample was allowed to stand at 80 °C for 2 h. A second cycle from −40 to 120 °C at 10 °C min−1 was then carried out to measure the Tg of the dried sample.
Scanning electron microscopy (SEM) was realized with a QUANTA 600 apparatus microscope from FEI. The cell size distribution was calculated by mean diameter measurements of at least 100 cells.
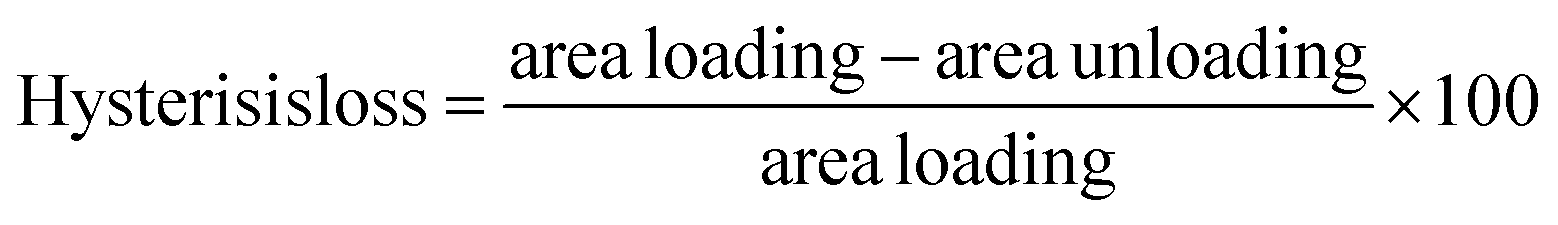 ,38 where area loading and area unloading are the air under the stress–strain curve until 50% deformation calculated using the “trapz” integration method of the scipy.integrate module of python3.12.
,38 where area loading and area unloading are the air under the stress–strain curve until 50% deformation calculated using the “trapz” integration method of the scipy.integrate module of python3.12.
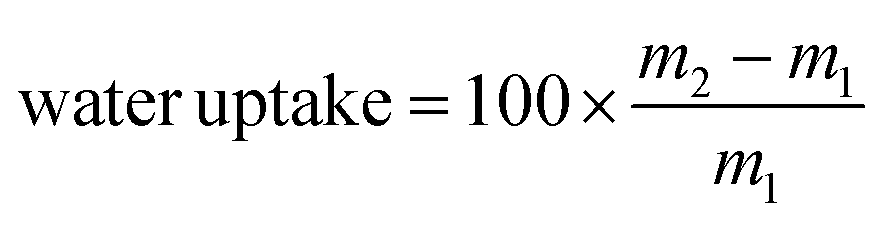 , where m1 and m2 are the mass of the dried sample and the mass after incubation at controlled humidity, respectively.
, where m1 and m2 are the mass of the dried sample and the mass after incubation at controlled humidity, respectively.
2.3. Procedure for thiol-induced foaming
3. Results and discussion
3.1. Aromatic thiol-induced self-blown PHU foams at 100 °C
Based on the hard and soft acid base theory of Pearson, the reactivity of S-nucleophiles is dictated by an interplay between the acidity and the charge delocalization of the sulfur electrons,39 and aromatic thiols should present higher reactivity vs. cyclic carbonates than aliphatic ones. To assert this, the widely used aliphatic, 2,2′-(ethylenedioxy)diethanethiol (T1; Fig. 1A),32,34,35,40 taken as a reference, was compared with an aromatic thiol (i.e. bismuthiol (T2)41 or 4,4′-thiobisbenzenthiol (T3)) in a typical formulation composed of a CO2-based tricyclic carbonate (TMPTC), a diamine (XDA) and DBU as the catalyst ([5CC]/[NH2]/[SH]/[DBU] in a ratio of 1/0.75/0.25/0.05). The three formulations were tested according to two different foaming procedures and validated for the water-based self-foaming process: (1) preheating the components at 100 °C before mixing them for foaming (fast foaming procedure), and (2) mixing the components at r.t., followed by heating the formulation at 100 °C (classic foaming process).28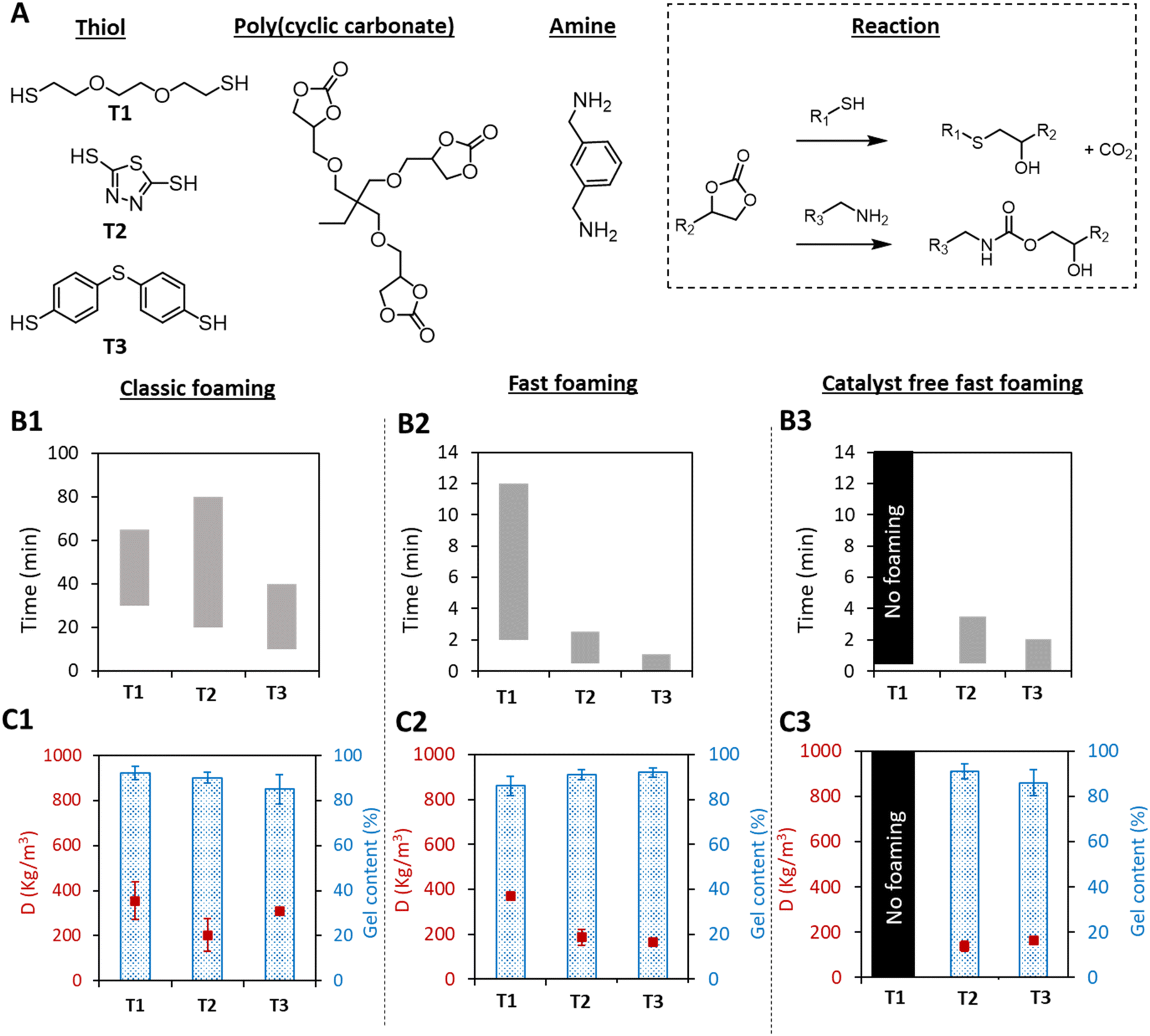 | ||
| Fig. 1 Thiol-induced self-blown PHU foaming. (A) Structure of the monomers and scheme of both reactions leading to PHU foams. (B) Expansion windows of the foams for the classic procedure (B1, mixing all components at room temperature before heating for 3 h at 100 °C), the fast procedure (B2, preheating the components to 100 °C before mixing them together and then curing for 30 min at 100 °C) and the fast foaming procedure under catalyst-free conditions (B3); T1, T2, and T3 refer to the thiols presented in A. (C). Densities and gel contents of foams obtained by the different foaming procedures (measured after 3 h for C1 and 30 min for C2 and C3). Conditions: TMPTC (5CC = 1 eq.), XDA (NH2 = 0.75 eq.), thiol (SH = 0.25 eq.), DBU (0.05 eq. for classic foaming and fast foaming 0 eq. for catalyst free fast foaming). Characteristics of the different foams are presented in Table S1.† 5CC, NH2 and SH state the number of cyclic carbonate, primary amine and thiol groups within the molecules. | ||
When using the classic foaming procedure, aliphatic thiol T1 showed a delayed foam expansion in comparison with the aromatic thiols with a time to initiate the expansion of 30 min for T1, 20 min for T2 and 10 min for T3 (Fig. 1B1). The foam density was also higher when using T1 (355 kg m−3 for T1vs. 203 and 310 kg m−3 for T2 and T3, respectively) with similar gel contents (GCT1 = 92%, GCT2 = 90%, GCT3 = 86%) (Fig. 1C1). The lower foam densities as well as the shorter foaming times obtained with the aromatic thiols were assumed to be the result of the faster S-alkylation of the cyclic carbonate, promoting faster production of the blowing agent together with faster cross-linking. Importantly, despite both aromatic thiols being solid compounds, T2 and T3 were easily dispersed in the PHU formulation and became soluble at the foaming temperature. Moreover, T2 is available on a large scale and at low cost. They are also more convenient to use as they are almost odorless.
Remarkably, when applying the fast foaming procedure, the foaming started after only 15 seconds with T2 and instantaneously with T3, and complete foam expansion was obtained after 2 min with T2 and 1 min with T3 (Fig. 1B2). PHU foams with a high gel content (>90%) and densities of 185 (with T2) and 166 kg m−3 (with T3) were obtained after 30 min of curing (Fig. 1C2). This is in sharp contrast to the same experiment carried out with the aliphatic thiol T1 where the foaming started after 2 min, with an expansion that lasted 10 min, and a final density of 372 kg m−3 (GC = 86%). Importantly, while the formulation containing the aliphatic thiol T1 did not provide any foam in the absence of the DBU catalyst, we discovered that PHU foams could be produced with T2 or T3 under catalyst-free conditions (Fig. 1B3 and C3). The expansion window (the time between the start and end of expansion) was only slightly extended by about 1 min, while similar foam densities and gel contents were obtained. We hypothesized that the diamine monomer (XDA) was sufficiently basic to activate the more acidic aromatic thiols T2 and T3, consequently accelerating the S-alkylation rate, and thus the CO2 formation and foam crosslinking.
To support this hypothesis, we monitored some model reactions by 1H-NMR spectroscopy (Fig. 2). The first one involved reacting a model cyclic carbonate (propylene carbonate, Pc) with a model aromatic thiol (T4, thiophenol) or an aliphatic one (T5, 1-butanethiol) in a stoichiometric ratio and under solvent- and catalyst-free conditions (after preheating the two components to mimic the fast foaming process). No reaction occurred after 5 min at 100 °C in the two cases. When triethylamine (1 eq. vs. propylene carbonate) was added to mimic the basicity of the diamine in the PHU formulation, S-alkylation was observed with thiophenol, with a propylene carbonate conversion of 53% after 5 min. No reaction was noted with 1-butanethiol in the presence of NEt3. This set of experiments supported the aromatic thiol being activated for the S-alkylation by the weak base. The basicity of the diamine comonomer in the PHU formulation was thus sufficient to catalyze the formation of the blowing agent in the presence of the aromatic thiols (T2 or T3).
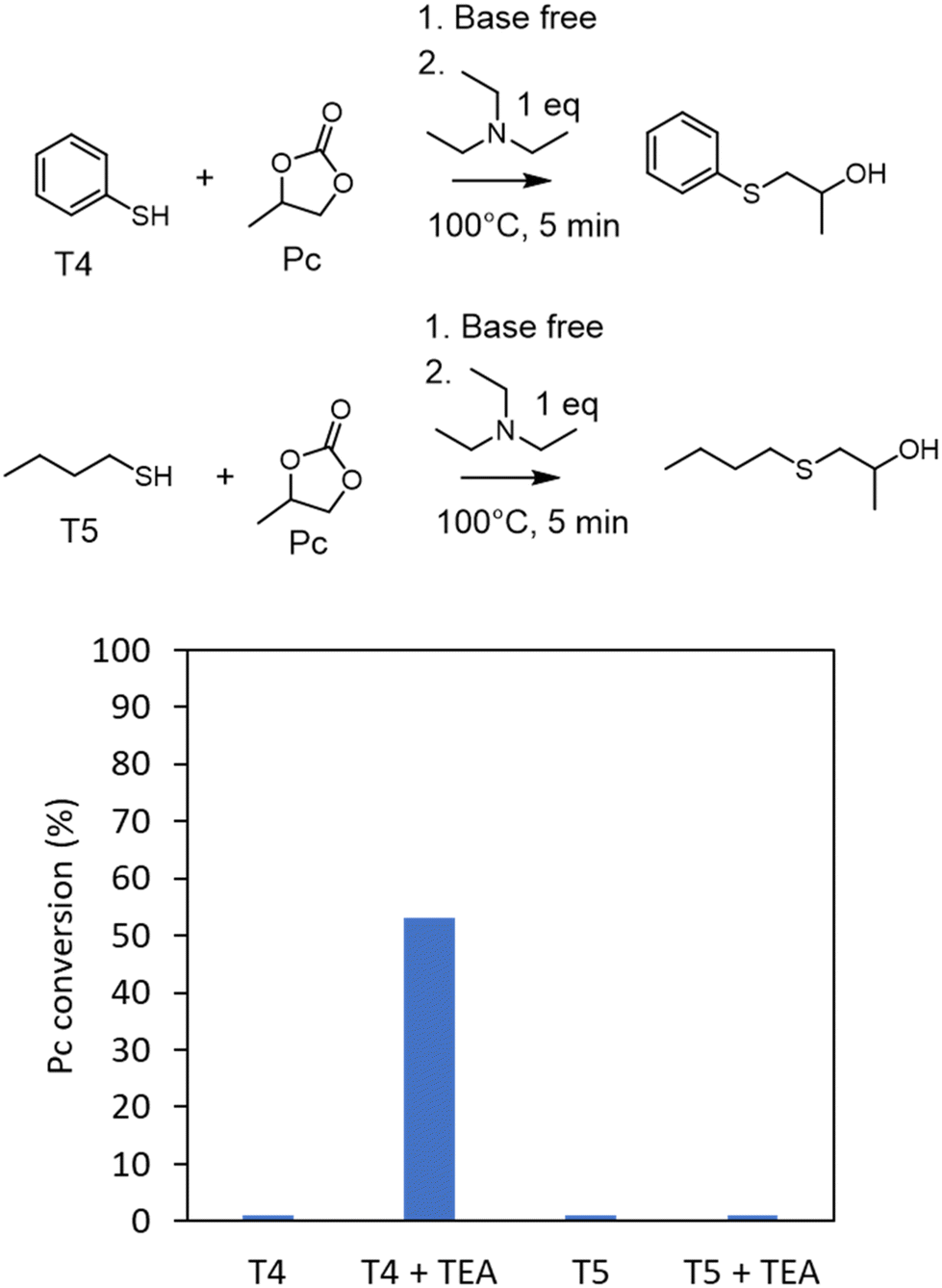 | ||
| Fig. 2 Propylene carbonate (Pc) conversion when reacted with thiophenol (T4) or 1-butanethiol (T5) in the presence or absence of triethylamine (TEA) determined by 1-H NMR spectroscopy (see Fig. S1† for NMR spectra). Conditions: propylene carbonate (5 mmol), thiol (5 mmol), TEA (5 mmol), preheating of the components at 100 °C before mixing them, followed by reaction at 100 °C for 5 min in a closed vial. | ||
Despite the gas generation being sufficiently fast to promote rapid expansion of the foam, we observed that 30 min of reaction was needed to reach high gel contents. Indeed, on using a standard formulation (TMPTC, XDA and T2; [5CC]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[NH2]:[SH] = 1:0.75:0.25) with the fast foaming process under catalyst-free conditions, the gel content values increased gradually with the reaction time, from 24% after 5 min to 91% after 30 min, with no significant difference in the foam density (142 kg m−3) (Fig. 3A). FT-IR analysis showed that the 5CC conversion was quite low after 5 min with an intense band of the carbonyl elongation at = 1800 cm−1 (Fig. 3B). Although the foam was fully expanded and did not collapse after 5 min, only partial conversion of the cyclic carbonate was noted, leading to a poorly cross-linked structure as evidenced by the low gel content.
:[NH2]:[SH] = 1:0.75:0.25) with the fast foaming process under catalyst-free conditions, the gel content values increased gradually with the reaction time, from 24% after 5 min to 91% after 30 min, with no significant difference in the foam density (142 kg m−3) (Fig. 3A). FT-IR analysis showed that the 5CC conversion was quite low after 5 min with an intense band of the carbonyl elongation at = 1800 cm−1 (Fig. 3B). Although the foam was fully expanded and did not collapse after 5 min, only partial conversion of the cyclic carbonate was noted, leading to a poorly cross-linked structure as evidenced by the low gel content.
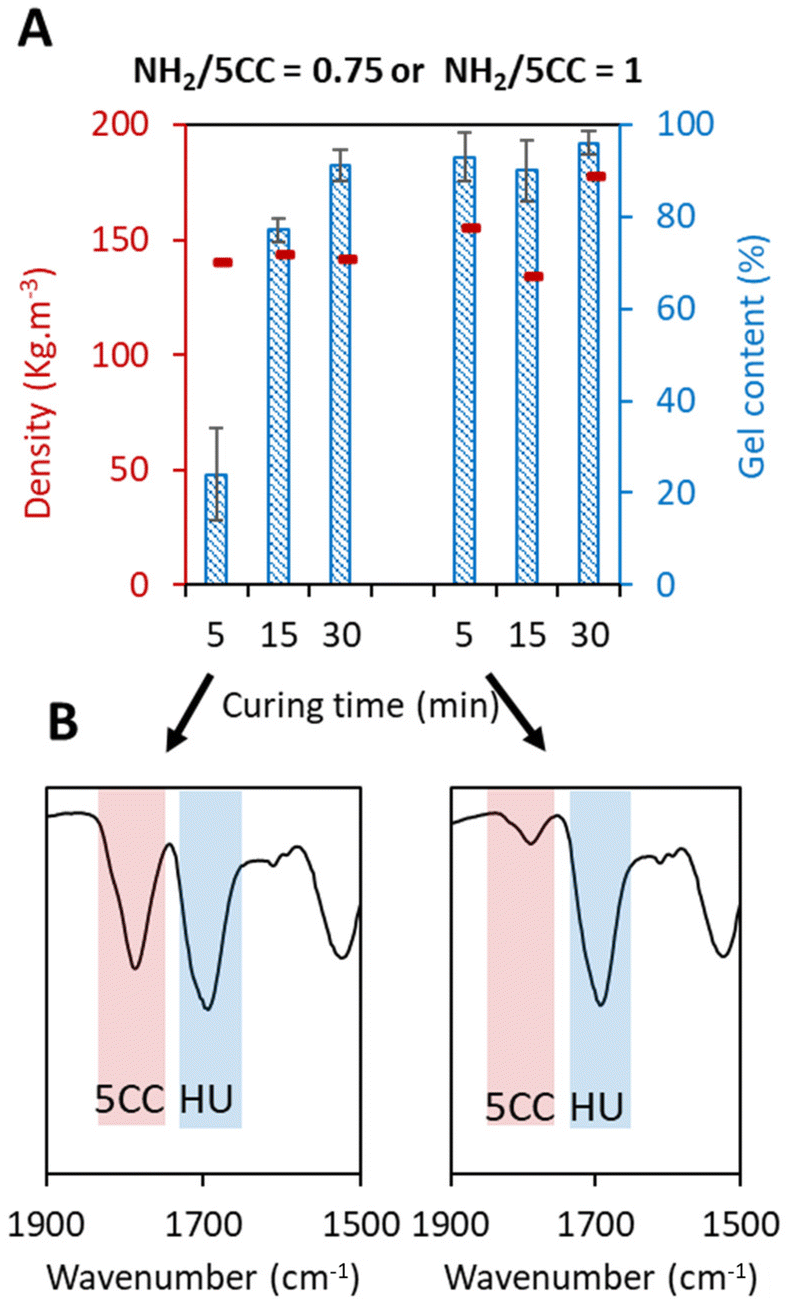 | ||
| Fig. 3 Comparison of foam properties (GC and density) after 5, 15, and 30 min with two different [NH2]:[5CC] ratios under catalyst-free fast foaming conditions. (A) Gel content and density values of the foams with different curing times. (B) Infrared spectroscopy after 5 min of curing (complete spectra of interest are depicted in Fig. S2†). Conditions: TMPTC, XDA, T2, catalyst-free, [5CC]/[NH2]/[SH] = 1/0.75/0.25 or 1/1/0.25, at 100 °C by the fast foaming process. | ||
To increase the gel content after 5 min of reaction, we slightly increased the amine content from 0.75 to 1 eq. Under these conditions ([5CC]:[NH2]:[SH] = 1:1:0.25), a gel content above 90% was reached after 5 min (Fig. 3A), together with the almost total consumption of 5CC groups as demonstrated by FT-IR (Fig. 3B and Fig. S2†). Importantly, the foam (155 kg m−3) was easily and directly demoldable after this short reaction time without the need to cool the whole system (foam and mold). This is particularly relevant for industrial applications where a high turnover is required during production, e.g. in the RIM process.
Based on this improved formulation ([5CC]/[NH2]/[SH] = 1/1/0.25), we produced a range of PHU foams under catalyst-free conditions in 5 min at 100 °C with two aromatic thiols (T2 and T3) or different diamines (XDA for F1 and F3 or EDR 148 for F2) (Fig. 4 and Table S2†). With T2, exchanging XDA (F2) and EDR 148 did not significantly influence the cell size (1.3 mm; Fig. 4A), the gel content (GC = 93%, Fig. 4C) or the density (155 vs. 156 kg m−3). As expected, the most significant difference was observed for the thermo-mechanical properties with a lower Tg (7.9 vs. 34 °C; Fig. 4D) and compression modulus (0.011 vs. 8.3 MPa; Table S3†) for the EDR-based foam compared to the XDA-based one. A flexible foam was thus obtained with EDR 148 and a rigid one with XDA. Note that for the XDA-based foam, we noticed the abrupt rupture of some rigid cells leading to stress release and giving a saw-tooth shape to the compression curve (Fig. 4B). This unusual shape of the stress/strain curve of F3 was certainly linked to the brittleness of the foam (see Fig. S10† for pictures of the foam before and after compression).
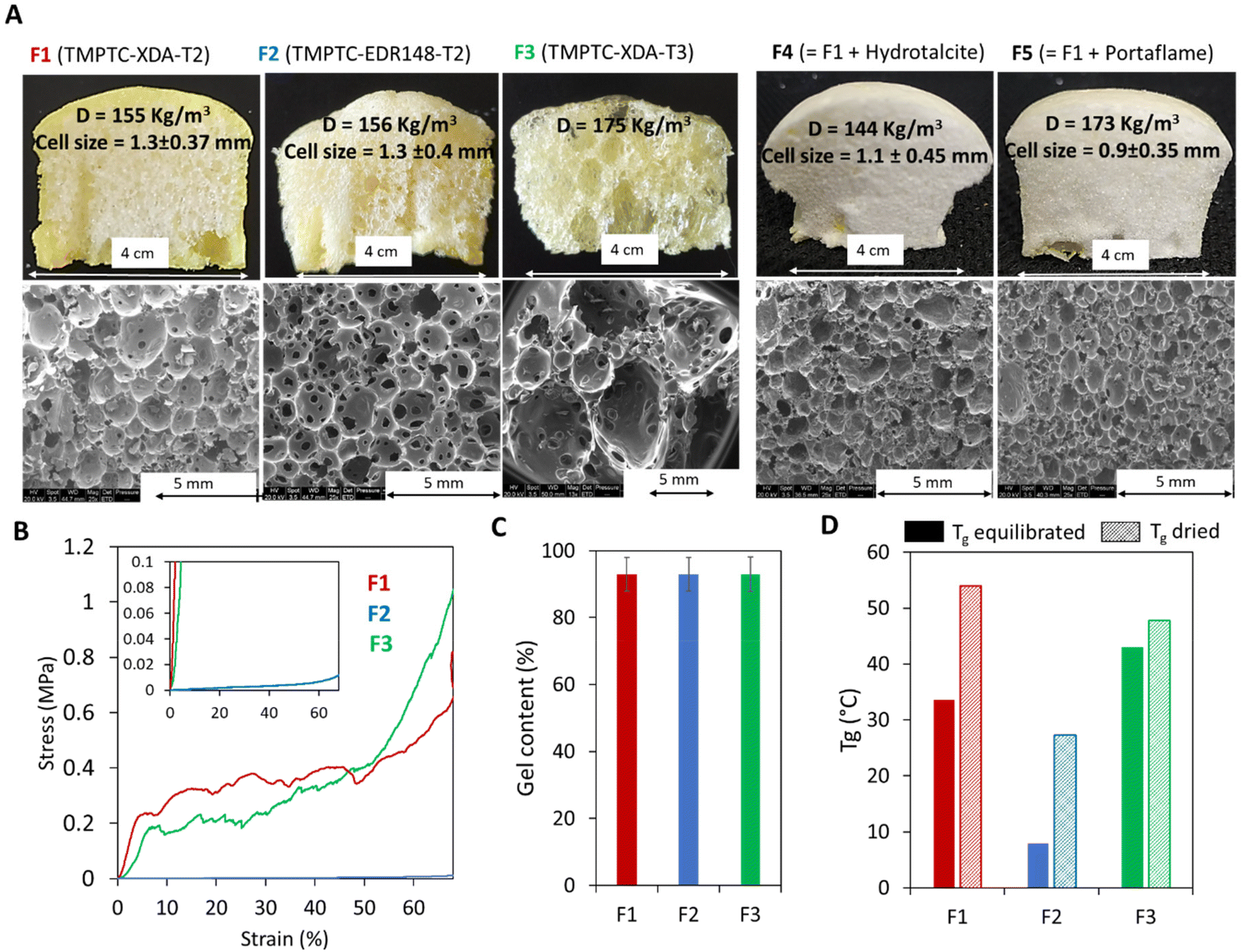 | ||
| Fig. 4 Characterization of the foams prepared from aromatic thiols. Synthesized with different thiols (T2 for F1, F2, F4, F5, or T3 for F3) or with different amines (XDA for F1, F3, F4, F5 or EDR 148 for F2), without (F1, F2, F3) or with a filler (F4 and F5). (A) Image and morphology of the foams. (B) Compressive curves and (C) gel contents of equilibrated additive-free foams, and (D) glass transition of additive-free foams equilibrated under ambient or dry conditions. Conditions: TMPTC, amine, thiol, catalyst-free, [5CC]/[NH2]/[SH] = 1/1/0.25, 5 min at 100 °C by the fast foaming process. DSC thermograms are presented in Fig. S3–S9.† | ||
Substituting T2 for T3 (formulation F3, Fig. 4A) delivered inhomogeneous foams with very large cells (d > 5 mm), coexisting with smaller ones (d < 1 mm). This is due to the higher reactivity of T3 (foam expansion is faster than that with T2 as illustrated in Fig. 5B1–B3) which promoted rapid production of the blowing agent when the matrix was still poorly cross-linked and exhibited an inappropriate viscosity. This could not avoid some coalescence of the growing cells within the matrix. As expected, this foam also showed a rigid character attested by a compression modulus of 2.97 MPa for 124 kg m−3 (due to foam heterogeneity, the density of the tested sample differs from the overall density) (F3, Fig. 4B). It is worth mentioning that as well as accelerating the foaming process and getting rid of any catalyst, the use of an aromatic thiol greatly broadened the variety of accessible foams, from flexible to rigid ones. This is an improvement compared to the previous foams obtained with aliphatic di- or poly-thiol32,34,35,40 that presented a flexible character with a low Tg (<10 °C), even when prepared from the rigid diamine XDA (entries 1 and 2, Table S1†). However, under more extreme humidity conditions (i.e. incubation for 48 h at 80% relative humidity), the use of aromatic thiols failed to significantly reduce the hydroplasticization of the PHU network as shown by the important drop of Tg by more than 30–40 °C in comparison with that under dry conditions (Tables S1 and S2†).
 | ||
| Fig. 5 Room temperature foaming using an epoxide additive for cascade exotherms. Conditions: TMPTC, BDGE, XDA, thiol, [5CC]:[epoxide]:[NH2 total]:[SH] = 1:1:1.5:0.5. | ||
The fast foaming process was also found to be compatible with the presence of different fillers (12 wt% vs. TMPTC). The addition of hydrotalcite or Portaflame® (a flame retardant) to the T2-based formulation (foam F4 and F5 in Fig. 4) allowed a decrease in the foam cell size and an increase in the homogeneity, as a result of the nucleating effect of the filler, without significantly affecting the GC and density (Table S2†).
3.2. Thiol-induced room temperature foaming
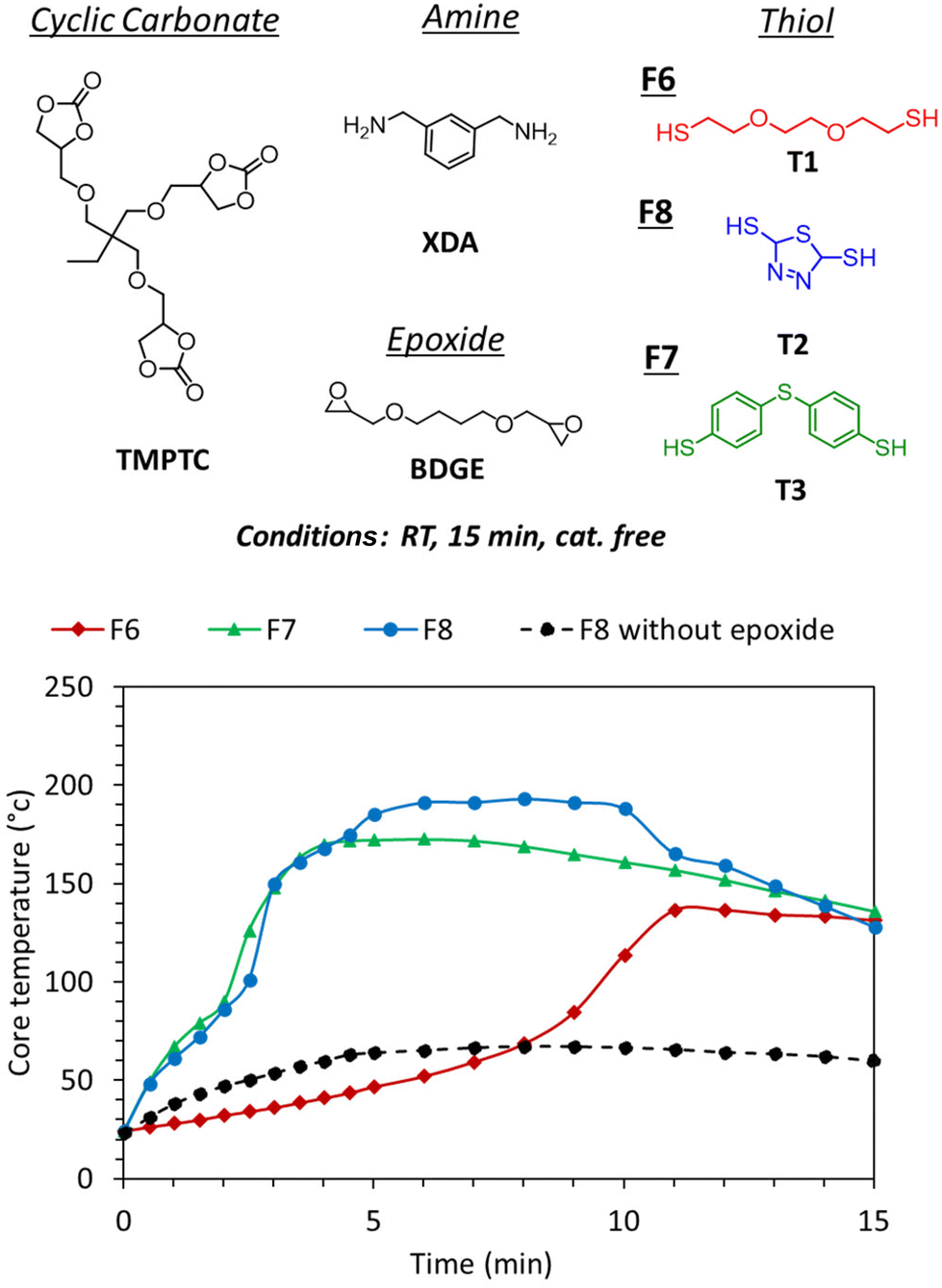
Despite increasing the kinetics of the gas generation, the reactivity of the aromatic thiol/amine was still not sufficient to induce foaming from room temperature reactive formulations. The examples in the previous section showed that the catalyst-free foaming induced by T2 demands temperatures of at least 100 °C and 80 °C when foaming with T3 (entry 7, Table S2†). As the PHU formation is known to be slightly exothermic, we monitored the exotherm of the polymerization when all components of the formulation were mixed at r.t. The goal was to evaluate how far the exotherm was from the foaming zone. Fig. 5 (F8 without epoxide, black curve) shows that the exotherm reached around 60 °C which was not sufficient for foaming.Recently, we presented the cascade exotherm strategy as a solution to improve the foaming of PHU-based formulations induced by hydrolysis.30 In this approach, epoxides were added to the formulation, leading to a rapid increase of the temperature to the foaming zone. The first fast exotherm generated by the aminolysis of the cyclic carbonates rapidly drove the highly exothermic aminolysis of the epoxides, thus leading to fast foaming and crosslinking. Here, we tested this strategy to induce fast foaming from r.t. formulations as the thiol–epoxy click reaction is known to be fast and exothermic.42–45 To do so, addition of the diepoxide BDGE (1 eq. of epoxide functional group vs. the 5CC functional group) to the previous formulation containing the aromatic thiol T2 or T3 promoted a rapid increase in the temperature in the corresponding formulations F8 and F7, respectively (up to 170 °C in less than 5 min, Fig. 5). This exotherm was accompanied by expansion of the matrix in less than 3 min (d = 229 kg m−3 for F8 and 324 kg m−3 for F7, Fig. 6) and cross-linking of the formulation (GC = 96% and 93%, Fig. 6). The infrared spectra of the different foams confirmed the PHU nature of the foam with almost complete disappearance of the carbonyl elongation of 5CC moieties at 1800 cm−1 and the appearance of a strongly intense and new signal of hydroxyurethane functions at 1700 cm−1 (Fig. S11–S16†). Similar to the observation linked to Fig. 4A, a foam with a rather larger cell size was obtained with T3 (F7) in comparison with T2 (F8) (Fig. 7). Nevertheless, in each case, we noticed a decrease in the cell size in comparison with the foam obtained without epoxide. This is rationalized by faster and denser cross-linking in the presence of the epoxide that stabilized the cell walls during expansion, limiting their coalescence.
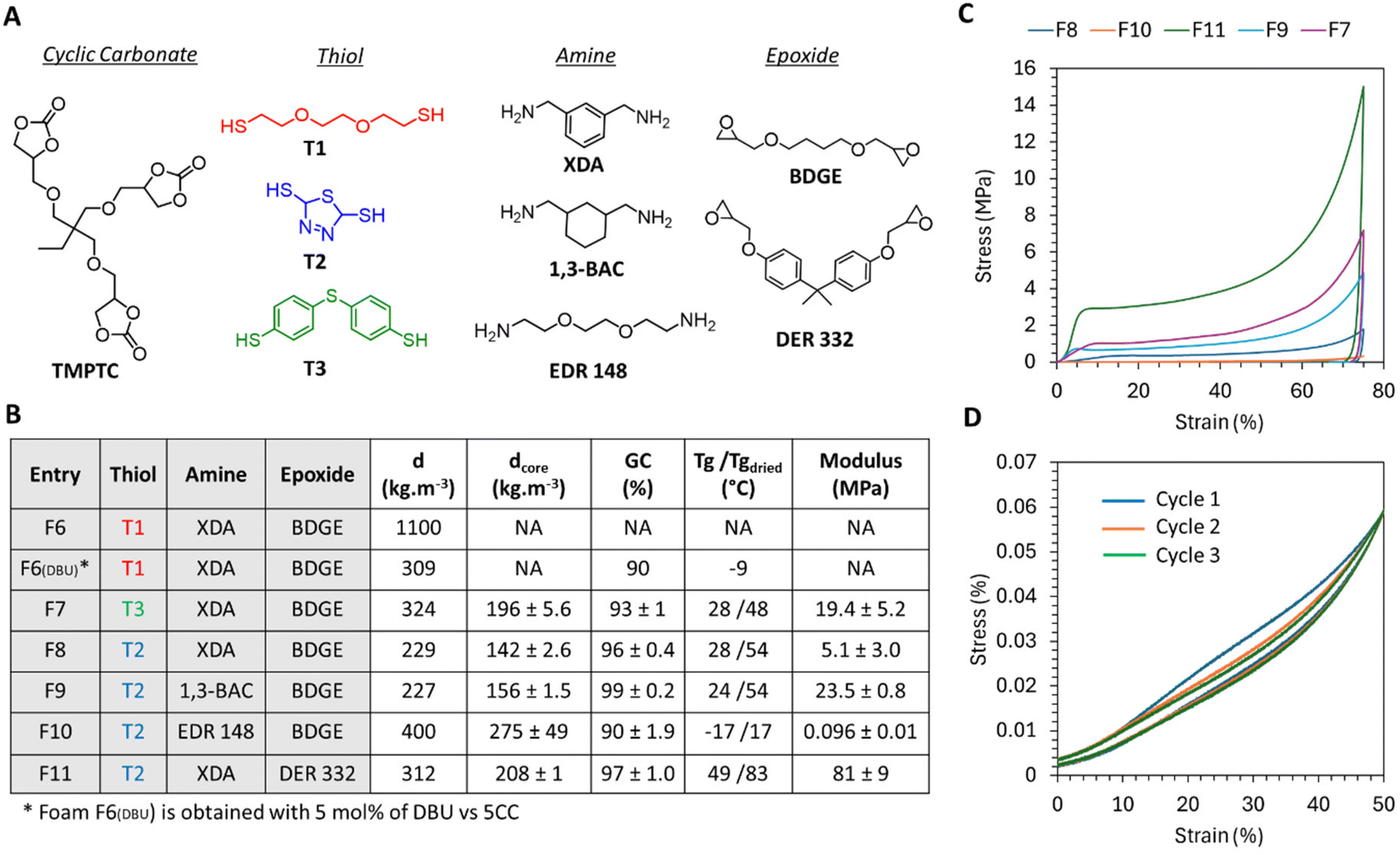 | ||
| Fig. 6 Foaming from r.t. formulations under catalyst free conditions and foam properties. (A) Structure of the component involved in the different formulations. (B) Composition of the formulations and properties of the foams. (C) Compressive curve of the foams synthesized with T2. (D) Three consecutive loading–unloading cycles of F10 at an initial compressive rate = 0.0025 s−1. DSC thermograms are presented in Fig. S17–S22 in the ESI.† Conditions: TMPTC, epoxide, amine, thiol, [5CC]:[epoxide]:[NH2 total]:[SH] = 1:1:1.5:0.5. d states for the density measured on the whole foam, and dcore states for the density measured at the center of the foam. | ||
 | ||
| Fig. 7 Images and SEM of the different foams obtained from r.t. formulations under catalyst free conditions. Conditions: TMPTC, epoxide, amine, thiol with [5CC]:[epoxide]:[NH2 total]:[SH] = 1:1:1.5:0.5, foaming RT. | ||
Importantly, during the foaming of r.t. formulations, we observed the formation of some aggregates when the aromatic thiols were involved, but not with the aliphatic one. The solid compounds were rapidly formed when all formulation components were mixed at r.t., and they then solubilized when the temperature reached 80–100 °C. This was rapidly followed by foaming. It was hypothesized that these solid compounds originated from the acid–base reaction between the thiol and the amine, leading to the thiol-amine salt as already observed elsewhere.46 This was further supported by mixing benzylamine with T2, T3 or thiophenol (T4) without any epoxide, with the formation of a solid compound that precipitated, together with a rapid increase in the temperature despite the low quantity of the reagent (2–3 g scale owing to safety concerns) (Fig. 8B). This was in sharp contrast to the results obtained with the aliphatic thiol T1 that did not show any precipitate nor any exotherm when mixed with the amine (Fig. 8B). This directly explains the important delay observed between the exotherm with F7 or F8 (with aromatic thiol) and F6 (with aliphatic thiol), the latter requiring three times more time to reach 100 °C (∼9 min vs. ∼3 min) during the foaming procedure (Fig. 5). This difference between the various thiols was explained by the higher acidity of the aromatic thiols vs. the aliphatic one.
 | ||
| Fig. 8 Model reaction of a thiol with an epoxide and/or amine. (A) Scheme of the three considered reactions. (B) Exotherm for the three reactions. (C) Epoxide conversion after the reaction of the model epoxide with different thiols and the thiol amine salt. Conditions: acid–base reaction [epoxide]/[NH2] = 1:1, thiol-epoxy reaction:[epoxide]/[SH] = 1:1, and thiolate salt-epoxy reaction [epoxide]/[SH]/[NH2] = 1:1:3. 1H-NMR spectra are presented in Fig. S23–S27 in the ESI.† | ||
Thiols are also known to react with epoxides, and this thiol epoxy-click reaction might be involved in the curing process. This reaction is however generally efficient at a temperature higher than 60 °C.42,43 We decided to test for the occurrence of this reactionusing model 1,2-epoxydodecane with the aliphatic thiol T1, the aromatic thiol T3 or the model thiophenol (T4) at r.t. In all cases, no exotherm was observed (Fig. 8B, blue curves) and the 1H-NMR analysis of the reaction medium after 15 min at r.t. showed no epoxide consumption (Fig. 8C; T3 was not tested due to its poor dispersibility in the epoxide). On the other hand, the reaction proceeded well when catalyzed by DBU (0.05 mol%) and was faster with aromatic thiols (96% conversion for T4 instead of only 28% for T1; Fig. 8C). Surprisingly, the reaction of bismuthiol (T2) with the epoxide occurred rapidly from r.t. with an important exotherm (more than 100 °C) being reached after 4 min in the absence of any catalyst (Fig. 8B and C). The 1H-NMR analysis of the model reaction confirmed 70% epoxide conversion after 15 min of reaction with the formation of the corresponding adduct (Fig. S26†). However, under the foaming conditions, bismuthiol (T2) was not able to directly react with the epoxide because the thiol/amine acido–basic reaction took place very rapidly, leading to the corresponding salt, which was unable to react with the epoxide. This was proved by first reacting bismuthiol (T2) with an excess of amine (SH/NH2 = 1/3, a similar composition to that in the foam formulation) before adding the epoxide. In these conditions, no exotherm (green curve in Fig. 8B for T2) was observed and almost no conversion of the epoxide by the thiol was observed by 1H-NMR spectroscopy (Fig. 8C, T2 amine-salt) at r.t. Whereas thiols are able to oxidize and form disulfide bonds under basic conditions, model reaction studies carried out in the presence of amines did not show the occurrence of this reaction under the foaming conditions (see section 2.4 for a detailed discussion, and Fig. S28 and S29† for 1H-NMR studies).
Despite the difficulty of fully determining the relative kinetics of all possible reactions, we hypothesized that the thiol-amine acid–base reaction was the first to take place, thus promoting the first exotherm needed to initiate the aminolysis of the epoxide. The stable thiol-amine salt limited the reaction of the thiol with the epoxide and 5CC, until the temperature reached 80–100 °C at which salt dissociation was expected to occur. Beyond 100 °C, the thiol reacted with 5CC to generate CO2 and with the epoxide.
We then investigated the versatility of the process. Through careful selection of the amine and epoxide components in the formulation, it was possible to achieve both flexible and rigid foams. As expected, the most rigid foam was obtained by combining the most rigid amine (XDA) with the aromatic epoxide monomer (DER 332), resulting in a foam with a Tg of 49 °C (F11, Fig. 6B). The compressive curve of this foam exhibited plastic deformation behaviour,47,48 with a modulus of 81 MPa, a yield stress of approximately 3 MPa, and an almost zero plateau beyond the yield stress (green curve, Fig. 6C). Substituting the aromatic epoxide in this formulation with an aliphatic one (BDGE) significantly altered the characteristics of the foam. The resulting foam (F8) exhibited a Tg value of 28 °C and a compressive modulus of 5 MPa, consistent with the properties of a semi-rigid foam (Fig. 6). Additionally, replacing XDA with the cyclo-aliphatic amine produced a foam (F9) with a similar Tg value but with an enhanced compressive modulus (23.5 MPa) for a similar density (Fig. 6). The properties of foam F7 obtained from the other aromatic thiol T3 are in line with these values (a Tg value of 28 °C and a compressive modulus of 19.4 MPa; Fig. 6). We observed that aromatic thiols played a crucial role in achieving rigid or semi-rigid foams. For the sake of comparison, the foam F6DBU produced from the aliphatic thiol (T1), XDA, and BDGE in the presence of DBU exhibited a low Tg value of −9 °C together with a low modulus of 0.0192 MPa, resulting in a foam that was significantly more flexible than the ones discussed in this section (Fig. S30†). Note that in all cases, foams were hydroplasticized by moisture, a common feature for PHU.13,28,29,31,49,50
We also investigated the influence of the epoxide structure on foaming. For this purpose, we substituted BDGE with epoxidized soybean oil (ESBO) featuring internal epoxides instead of external ones. Despite being of great interest due to the good availability and biobased origin of such internal epoxides, the low reactivity of the internal epoxides did not allow generation of a large exotherm and thus, induction of foaming. Indeed, only a temperature of 42.9 °C was reached when using ESBO instead of BDGE (Fig. S31C†) under similar foaming conditions. The reaction of the thiol with the internal epoxides was also tested using a model reaction between bismuthiol and ESBO. Bismuthiol reacted slowly with ESBO with the formation of 17% thioether linkage (Fig. S31A†) after 15 min without any catalyst at room temperature with a very slight exotherm (Fig. S31B†) compared to 70% thioether bonding when reacting with 1,2-epoxydodecane (Fig. 8C).
Finally, the combination of both an aliphatic amine (EDR 148) and an aliphatic epoxide (BDGE) resulted in the flexible foam F10, as evidenced by a Tg value below room temperature (−17 °C) and a low modulus of 0.096 MPa (Fig. 6). The compressive curve in Fig. 6C shows a constant plateau modulus beyond the elastic region and the foam was able to recover its shape after compression with a small amount of hysteresis loss (hysteresis loss = 17% for 50% compression strain; Fig. 6D). By repeating the compression cycle 2 more times, we observed that the modulus during loading decreased from 0.1071 to 0.088 MPa between the first and second cycle and there was an almost full recovery of the modulus between cycles 2 and 3 (0.0844 MPa). In contrast, the unloading curves superimposed quite well and then showed hysteresis losses of 12.8 and 11.5% during unloading respectively for cycles 2 and 3.
4. Conclusions
Thiol-induced self-blown non-isocyanate polyurethane (NIPU) foams are promising CO2-sourced alternatives to conventional isocyanate-based foams. However, their production is still slow (i.e. 30 min at 120 °C), far from the 1–10 minutes required for many industrial foaming processes. Herein, we investigated the reasons for these slow processes, and developed optimized strategies for the production of NIPU foams in short time frames (5 minutes) notably by replacing aliphatic thiols with aromatic ones. Remarkably, as aromatic thiols are more acidic than aliphatic ones, the amine comonomer was basic enough to catalyze the S-alkylation, thus leading to fast foaming (5 min) at 100 °C without requiring an organobase catalyst (DBU) as is commonly used in the original process. Since the system was not sufficiently reactive for room temperature foaming, the cascade exotherm strategy employed for the water-induced self-foaming process was then implemented to rapidly raise the formulation temperature to the foaming zone in minutes. Model reactions showed that the higher acidity of the aromatic thiol led to a strong acid–base reaction with the amine comonomer. This first exothermic reaction allowed initiation of the other ones (i.e. the aminolysis of the cyclic carbonates and epoxides). This cascade exotherm enabled the foaming zone to be reached very quickly, delivering stable crosslinked NIPU foams in 5 minutes under catalyst-free conditions from r.t. formulations. With aliphatic thiols, a strong organobase catalyst (DBU) was needed to achieve such a foaming performance. More than accelerating the foaming, aromatic thiols allowed much more freedom to prepare foams of very distinct properties. Indeed, by the choice of the amine comonomer and epoxide, flexible, semi-rigid or rigid foams presenting Tg values from −17 to 49 °C were easily accessible, such versatility not being possible with the aliphatic thiol.This work shows that, by a careful understanding of the foaming process, simple strategies can be implemented to tackle the low reactivity of cyclic carbonates for the fast self-blowing of solvent-free NIPU formulations, offering potential retrofitting of existing PU foam production plants.
Author contributions
Maxime Bourguignon carried out the experimental design, data analysis and manuscript writing. Bruno Grignard synthesized the cyclic carbonate, and participated in result discussions and manuscript correction. Christophe Detrembleur obtained the funding resources, supervised the project, and participated in result discussions and manuscript correction.Data availability
Supplementary data for this article, including DSC thermograms, infrared spectra, 1H NMR spectra, supplementary syntheses, and cyclic carbonate synthesis, are present in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Region Wallonne for funding the Win2Wal project “ECOFOAM” (convention 2010130). They also thank the company NMC sa (Belgium) for their support in this research. The authors are very grateful to Grégory Cartigny for technical assistance in the preparation of the formulations and the foams. Christophe Detrembleur is the FNRS Research Director and thanks the Fonds de la Recherche Scientifique (F.R.S.-FNRS) for funding. Christophe Detrembleur is particularly grateful to the Region Wallonne for funding the FRFS-WEL-T project Chemistry (convention WEL-T-CR-2023 A).References
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 CAS.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CAS.
- https://www.imarcgroup.com/polyurethane-foam-market .
- J. Peyrton and L. Avérous, Mater. Sci. Eng., R, 2021, 145, 100608 Search PubMed.
- M. H. Karol and J. A. Kramarik, Toxicol. Lett., 1996, 89, 139–146 CrossRef CAS.
- D. Bello, C. A. Herrick, T. J. Smith, S. R. Woskie, R. P. Streicher, M. R. Cullen, Y. Liu and C. A. Redlich, Environ. Health Perspect., 2007, 115, 328–335 CrossRef CAS PubMed.
- S. Merenyi, REACH: Regulation (EC) No 1907/2006: Consolidated version (June 2012) with an introduction and future prospects regarding the area of Chemicals legislation (Vol. 2), GRIN Verlag Search PubMed.
- European Commisson, Communication from the commission to the European Parliament and the council, Sustainable carbon cycles, Brussels, 2021 Search PubMed.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 Search PubMed.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 CAS.
- F. D. Bobbink, A. P. van Muyden and P. J. Dyson, Chem. Commun., 2019, 55, 1360–1373 CAS.
- T. Theerathanagorn, T. Kessaratikoon, H. U. Rehman, V. D'Elia and D. Crespy, Chin. J. Chem., 2024, 42, 652–685 CrossRef CAS.
- P. Sen Choong, N. X. Chong, E. K. Wai Tam, A. M. Seayad, J. Seayad and S. Jana, ACS Macro Lett., 2021, 10, 635–641 CrossRef PubMed.
- G. Seychal, P. Nickmilder, V. Lemaur, C. Ocando, B. Grignard, P. Leclère, C. Detrembleur, R. Lazzaroni, H. Sardon, N. Aranburu and J.-M. Raquez, Composites, Part A, 2024, 185, 108311 CrossRef CAS.
- G. Seychal, C. Ocando, L. Bonnaud, J. De Winter, B. Grignard, C. Detrembleur, H. Sardon, N. Aramburu and J.-M. Raquez, ACS Appl. Polym. Mater., 2023, 5, 5567–5581 CrossRef CAS.
- P. Helbling, F. Hermant, M. Petit, T. Tassaing, T. Vidil and H. Cramail, Polym. Chem., 2023, 14, 500–513 CAS.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 CAS.
- A. Gomez-Lopez, S. Panchireddy, B. Grignard, I. Calvo, C. Jerome, C. Detrembleur and H. Sardon, ACS Sustainable Chem. Eng., 2021, 9, 9541–9562 CAS.
- F. Magliozzi, A. Scali, G. Chollet, D. Montarnal, E. Grau and H. Cramail, ACS Sustainable Chem. Eng., 2020, 8, 9125–9135 CrossRef CAS.
- H. Blattmann, M. Lauth and R. Mülhaupt, Macromol. Mater. Eng., 2016, 301, 944–952 CrossRef CAS.
- B. Grignard, J.-M. Thomassin, S. Gennen, L. Poussard, L. Bonnaud, J.-M. Raquez, P. Dubois, M.-P. Tran, C. B. Park, C. Jerome and C. Detrembleur, Green Chem., 2016, 18, 2206–2215 CAS.
- T. Dong, E. Dheressa, M. Wiatrowski, A. P. Pereira, A. Zeller, L. M. L. Laurens and P. T. Pienkos, ACS Sustainable Chem. Eng., 2021, 9, 12858–12869 CAS.
- C. Amezúa-Arranz, M. Santiago-Calvo and M.-Á. Rodríguez-Pérez, Eur. Polym. J., 2023, 197, 112366 Search PubMed.
- A. Cornille, S. Dworakowska, D. Bogdal, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 66, 129–138 CAS.
- A. Cornille, C. Guillet, S. Benyahya, C. Negrell, B. Boutevin and S. Caillol, Eur. Polym. J., 2016, 84, 873–888 CAS.
- G. Coste, M. Denis, R. Sonnier, S. Caillol and C. Negrell, Polym. Degrad. Stab., 2022, 202, 110031 CAS.
- G. Coste, C. Negrell, L. Averous and S. Caillol, ACS Sustainable Chem. Eng., 2022, 10, 8549–8558 CAS.
- M. Bourguignon, B. Grignard and C. Detrembleur, Angew. Chem., Int. Ed., 2022, 61, e202213422 CrossRef CAS.
- D. Trojanowska, F. Monie, G. Perotto, A. Athanassiou, B. Grignard, E. Grau, T. Vidil, H. Cramail and C. Detrembleur, Green Chem., 2024, 26, 8383–8394 RSC.
- M. Bourguignon, B. Grignard and C. Detrembleur, J. Am. Chem. Soc., 2024, 146, 988–1000 CrossRef CAS.
- F. Monie, B. Grignard and C. Detrembleur, ACS Macro Lett., 2022, 11, 236–242 CrossRef CAS PubMed.
- F. Monie, B. Grignard, J.-M. Thomassin, R. Mereau, T. Tassaing, C. Jerome and C. Detrembleur, Angew. Chem., Int. Ed., 2020, 59, 17033–17041 CAS.
- G. Coste, C. Negrell and S. Caillol, Macromol. Rapid Commun., 2022, 43, 2100833 CAS.
- N. S. Purwanto, Y. Chen and J. M. Torkelson, ACS Appl. Polym. Mater., 2023, 5, 6651–6661 CAS.
- N. S. Purwanto, Y. Chen, T. Wang and J. M. Torkelson, Polymer, 2023, 272, 125858 CAS.
- M. Chaib, S. El Khezraji, S. Thakur, H. Ben Youcef, M. Lahcini and R. Verdejo, React. Funct. Polym., 2024, 200, 105924 CAS.
- B. Li and R. M. Aspden, J. Bone Miner. Res., 1997, 12, 641–651 CrossRef CAS PubMed.
- S. Wang, W. Liu, D. Yang and X. Qiu, Ind. Eng. Chem. Res., 2019, 58, 496–504 CrossRef CAS.
- B. Grignard, P. Mampuys, J. Escudero, D. Masullo, F. Lemière, B. U. W. Maes and C. Detrembleur, Polym. Chem., 2022, 13, 6599–6605 RSC.
- N. S. Purwanto, Y. Chen and J. M. Torkelson, Eur. Polym. J., 2024, 206, 112775 CAS.
- Y. Hu, C.-Y. Li, X.-M. Wang, Y.-H. Yang and H.-L. Zhu, Chem. Rev., 2014, 114, 5572–5610 CAS.
- Z. Yao, B. Dai, Y. Yu, H. Ji, L. Zhou and K. Cao, RSC Adv., 2017, 7, 10881–10884 CAS.
- K. Jin, W. H. Heath and J. M. Torkelson, Polymer, 2015, 81, 70–78 CAS.
- A. O. Konuray, X. Fernández-Francos and X. Ramis, Polymer, 2017, 116, 191–203 CAS.
- T. H. Lee, Y. Il Park, S. M. Noh and J. C. Kim, Prog. Org. Coat., 2017, 104, 20–27 CrossRef CAS.
- T. Habets, F. Siragusa, A. J. Müller, Q. Grossman, D. Ruffoni, B. Grignard and C. Detrembleur, Polym. Chem., 2022, 13, 3076–3090 RSC.
- Z. H. Tu, V. P. W. Shim and C. T. Lim, Int. J. Solids Struct., 2001, 38, 9267–9279 CrossRef.
- F. Rahimidehgolan and W. Altenhof, Composites, Part B, 2023, 253, 110513 CrossRef CAS.
- C. Pronoitis, M. Hakkarainen and K. Odelius, ACS Sustainable Chem. Eng., 2022, 10, 2522–2531 CrossRef CAS.
- I. Łukaszewska, A. Bukowczan, K. N. Raftopoulos and K. Pielichowski, Polymer, 2024, 302, 127060 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00971a |
| This journal is © The Royal Society of Chemistry 2025 |