
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecyclable iridium-containing copolymers for homogeneous photoredox catalysis†
Shweta
Gaikwad
a,
Argha
Bhattacharjee
a,
Stanley
Baldwin
a,
Steven
Huss
a,
Anna
Griggs
a,
Michael
Spicuzza
a and
Elizabeth
Elacqua
 *ab
*ab
aDepartment of Chemistry, The Pennsylvania State University, University Park, Pennsylvania 16802, USA. E-mail: elizabeth.elacqua@psu.edu
bMaterials Research Institute, The Pennsylvania State University, University Park, Pennsylvania 16802, USA
First published on 12th March 2025
Abstract
Iridium-based polypyridyl complexes have gained recent attention in photoredox catalysis, with the potential for enhanced efficiency across a variety of applications. However, challenges such as poor recyclability, catalyst loss, and high costs can hinder their practical utility. Herein, we report the synthesis and characterization of three poly(methyl methacrylate)s featuring pendant cyclometalated iridium complexes based upon [Ir(df(CF3)ppy)2(dmbpy)](PF6), [Ir(ppy)2(dmbpy)](PF6), and fac-Ir(ppy)3. Our homogeneous polymer catalysts exhibited exceptional performance across a diverse array of photoredox-mediated transformations, including Birch-type photo-reduction, cycloaddition reactions, dual catalytic cross-coupling systems, and regioselective alkene functionalizations, while closely mimicking the catalytic and photophysical properties of their small-molecule counterparts. The new polymer photocatalyst maintained fidelity during reactions, enabling it to be recycled and reused up to at least five cycles. This approach combines high performance with sustainability, offering a promising pathway toward greener photoredox catalysts while broadening the practical applications of iridium-based systems.
Introduction
Transition metal polypyridyl complexes constitute an important class of coordination complexes that are widely used across organic and organometallic synthesis, supramolecular chemistry, polymer chemistry, and materials science.1–6 Interest in these structures over the past several decades has centered around those which possess visible charge-transfer absorption features; ruthenium and iridium polypyridyl complexes, for example, have photophysical and redox properties7–14 which have been leveraged to design new materials and molecules for widespread use in applications ranging from bio-sensing and therapeutics to solid-state materials and catalysis.A particularly potent application for these complexes lies in their use as photosensitizers to catalyze small-molecule organic transformations, with polypyridyl compounds based on Ru(II) or Ir(III) comprising the vast majority of the catalysts studied owing to their ability to absorb visible light, with the light selectively targeting the catalyst over any organic/organometallic complexes. Additionally, they access long-lived stable excited states owing to metal-to-ligand charge transfer (MLCT) events followed by intersystem crossing.14,15 Moreover, the polypyridyl chromophores can be easily modified to tune their intrinsic physical and photophysical properties; thus, a wide range of redox properties can be accessed by these light-absorbing species.
Although their efficacy in synthetic and light-harvesting applications is well-documented, the high cost of these precious metals has made their applicability beyond the microscale often less practical. Moreover, the potential to recycle and reuse these potent catalysts has been less studied with molecular species. One particular challenge with these charged transition metal polypyridyl complexes is that their solubility is often best in high dielectric polar aprotic solvents, such as DMF and DMSO,16 both of which make biphasic separations that are achievable with some molecular systems less practical. Moreover, the intrinsic photophysical and electrochemical properties are known to be solvent dependent with solvent polarity having an influence on the photocatalyst's photooxidant/photoreductant potential.17 For example, a change from THF to MeCN for the well-studied [Ir(df(CF3)ppy)2(dtbbpy)]PF6 complex results in a concomitant change in the excited state redox potential so significantly that it can be the difference between seeing no reaction and the desired chemistry. Thus, efforts to make these reactions even more ‘green’ have a higher chance of being less successful when compared to organic photocatalysts. Lastly, although this is underexplored, the ‘fate’ of iridium photocatalysts during/after reaction is less straightforward,18 with side reactions that compromise the fidelity of the polypyridyl ligands being present when there are sacrificial electron donors involved, evidenced by both drastic decreases in emission intensity and substantial hypsochromic shifts in emission being observed.
Thus, amongst the growing literature on iridium-based photoredox catalysis, only few reports concern the recycling of commercial catalysts. In one report, fac-Ir(ppy)3 enabled a photoinduced controlled radical polymerization wherein water-induced phase separation parsed the catalyst into the organic phase, while the polymer was in the water phase.19 Key to this method was the use of water-soluble monomers that could be suitably solubilized and polymerized in an ethanol/dichlorobenzene system. In another study, the photocatalyzed (E) to (Z) isomerization of stilbene was investigated, wherein [Ir(ppy)2(bpy)](PF6) was immobilized in the ionic liquid phase of a toluene/[bmim/BF4] solvent system, leading to easy separation.20 More recently, the fac-Ir(ppy)3 catalyzed α-amino arylation was accomplished using greener solvents and with the ability to recycle and reuse the commercial photocatalyst.21
The need to generate more sustainable forms of photoredox catalysts has been recognized by the materials community, wherein these photoactive complexes have been integrated into metal–organic frameworks (MOFs), covalent–organic frameworks (COFs), and polymers,2,12,22–31 wherein the resultant material generally performs as well as the molecular catalyst. Many of these frameworks have utilized the principles of confinement to accelerate catalysis.32–36 From a green chemistry perspective, the use of macromolecular and/or reticular frameworks could mitigate the high cost of the complex by promoting both facile removal and recycling of the metal polypyridyl catalysts. As such, ruthenium- and iridium-based polypyridyl complexes have been integrated into MOFs, COFs, and/or polymers in an effort to harness their properties. For example, the high photocatalytic activity of [Ru(bpy)3]2+ has led to incorporating the complex into the cavities of MOFs, resulting in dual emission composite materials for applications such as sensing.37 Similarly, [Ru(bpy)3]2+ has been featured as pendant units on monomers,2,12,38 affording a wide range of polymers that function in several applications including as light-emitting or light-harvesting devices, and homogeneous photocatalysts. The well-known photophysics of ruthenium has rendered it a staple in both small molecule and materials chemistry.
The photophysics of iridium polypyridyl complexes are well-documented;13 yet their translation to the materials community has only recently emerged in the past decade or so, with the main application targeted being photoredox catalysis. A library of ligands with different photosensitizing and electrochemical properties have been developed for iridium polypyridyl complexes, allowing for the design of tunable platforms for catalysis. Indeed, both MOFs and COFs have been leveraged for catalysis owing to the promise of recyclability while enabling two cooperative catalysts (such as Ir and Ni) to be embedded in a highly periodic structure for heterogeneous catalysis. In a pioneering example, a zirconocene-based MOF studded with 2,2′-bipyridine and 2-phenylpyridine struts allowed Ir- and Ni-based catalytic components to be proximally installed, thus facilitating electron and radical transfers that enabled more efficient photoredox catalysis.34 In this seminal study, the dual catalytic MOF could be reused for up to five additional cycles without loss of catalyst activity. Other subsequent reticular materials based on iridium have demonstrated a far larger capacity to recycle and reuse iridium photosensitizers for 5–10 cycles;28,32,39,40 in some cases, performance dropped by 15–20% in latter rounds of recycling. While it is not abundantly clear why the performance decreases over time, many of the COFs investigated are for dual catalysis; thus, if metal leaching of the cocatalyst occurs, the performance of the system itself would be expected to decrease.
Recently, several macromolecular systems including porous organic polymers have been used for heterogeneous catalysis.26,28,31,41–44 Such frameworks are versatile and efficient in facilitating chemical reactions while being recyclable, making them valuable tools in catalysis. Many have been found to promote enhanced catalysis when compared to molecular counterparts.45 While macromolecular scaffolds comprised of iridium2 have been investigated mainly for emerging applications in organic electronics, such as light-emitting diodes, other flexible polymers containing iridium have been used to promote photoredox catalysis46,47 resulting in recyclable scaffolds. Given the relative ease in incorporating N-heterocycles as side chains in polymers, we sought to develop a modular platform to investigate homogeneous iridium photocatalysis. Installing iridium complexes as side chains on a polymer could enable different solubility, selectivity, and efficiency when compared to other catalysts. Moreover, environmental factors such as solvent selection are known to alter photophysical properties;1 thus, the polymer design and microenvironment around the metal species may impact MLCT properties.
Herein, we present the synthesis and characterization of three distinct poly (methyl methacrylate)s featuring pendant iridium complexes based on 2,2′-bipyridine (bpy) and 2-phenylpyridine (ppy) ligands: [Ir(df(CF3)ppy)2(dmbpy)]PF6, [Ir(ppy)2(dmbpy)]PF6, and fac-Ir(ppy)3. The polymers, obtained from reversible addition–fragmentation chain-transfer (RAFT) polymerization, were applied successfully as homogeneous photoredox catalysts for a variety of organic transformations, with low catalyst loadings, while achieving yields that were on par with the analogous molecular cyclometalated iridium complexes. We also demonstrate that each catalyst is recyclable and reusable over at least five cycles.
Results and discussion
Synthesis and characterization
We envisioned developing homogeneous polypyridyl ligand-based polymers as a modular means to coordinate different metal-based photocatalysts. Methyl methacrylate (MMA) was selected as the polymer backbone for its ease of incorporation with the targeted catalysts and chemical inertness, thus enabling properties similar to those of both small molecule and other macromolecular/reticular-based iridium catalysts. A library of iridium(III)-based cyclometalated compounds have demonstrated effective photoredox over the past decade, each exhibiting distinct photoredox properties.7Our studies began with targeting a modular platform based on a 4,4′-dimethyl-2,2′-bipyridine (dmbpy)-containing methacrylate, wherein a rich variety of photocatalytically-active polymers could be obtained through the addition of phosphorescent cyclometalated iridium(III) dimers. To accomplish this, the ligand (4′-methyl-[2,2′-bipyridine]-4-yl)methanol (dmbpy-OH) was converted to the methacrylate. Subsequently, RAFT polymerization with MMA was conducted in the presence of 2,2′-azobis(2-methylpropionitrile) (AIBN) as the initiator and 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid as the chain transfer agent. The corresponding polymer dmbpy-co-MMA was analyzed using 1H NMR spectroscopy to determine the amount of dmbpy incorporated, while molecular weight was analyzed through GPC (see ESI†). The dmbpy-co-MMA was then treated with an iridium dimer ([(dF(CF3)ppy)2-Ir-µ-Cl]2) to coordinate the catalyst to the polymer backbone. 1H NMR spectroscopy confirmed 50% incorporation of iridium onto the polymer backbone. While encouraged by the result, the uncoordinated dmbpy ligands could impact the targeted organic transformations, particularly those with either coordinative additives and/or acid present. This was confirmed when we tested the polymer-based iridium catalyst in a MacMillan-type decarboxylation reaction of N-Boc proline with 4-bromoacetophenone,48 wherein the results obtained were suboptimal. Consequently, we reevaluated our synthetic strategy and targeted iridium-containing methacrylates to incorporate directly into the backbone using RAFT (Scheme 1). We targeted three monomers featuring widely used catalysts based on dmbpy and/or ppy ligands: [Ir(df(CF3)ppy)2(dmbpy)]PF6, (IrCat1), [Ir(ppy)2(dmbpy)]PF6, (IrCat2), and fac-Ir(ppy)3 (IrCat3).
 | ||
| Scheme 1 RAFT polymerization of MMA-based monomers toward IrCat polymers. | ||
Thus, the hydroxyl-terminated ligands dmbpy-OH and (2-phenylpyridine-4-yl)methanol (ppy-OH) were separately added into methacryloyl chloride, furnishing the monomers. The dmbpy monomer was then separately treated with iridium dimers ([(dF(CF3)ppy)2-Ir-µ-Cl]2) and ([(ppy)2-Ir-µ-Cl]2), affording IrCat1 and IrCat2, respectively. The dimer ([(ppy)2-Ir-µ-Cl]2) was also treated with the ppy monomer, yielding IrCat3. The amount of iridium incorporated within the polymer was analyzed using 1H NMR spectroscopy. Generally, the broad resonances between 7.0 and 9.0 ppm, assigned to the aromatic protons of the polypyridyl ligands, were compared to the MMA ester resonance at 3.5 ppm. In the case of IrCat1, approximately 8% of iridium photocatalyst had been incorporated (see ESI† for details on all polymers).
The bulkiness and charged nature of the iridium complexes made GPC analysis a less reliable technique to garner precise molecular weight of the polymer catalysts. Consequently, molecular weight was determined through end-group analysis.49–51 For this process, the terminal carboxylic acids were coupled with 3-trimethylsilylpropargyl alcohol in the presence of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride,12 wherein the –SiMe3 resonance would be distinguishable from the broader polymer resonances in a corresponding 1H NMR spectrum. After purification, a new resonance at 0.13 ppm appeared, indicating successful functionalization with trimethylsilyl propargyl groups. By correlating this shift with the integration of the resonances indicated above, the molecular weights of the copolymers were around 22–29 kDa (see ESI†).
Photophysical characterization
The series of iridium copolymers were characterized using UV-Vis absorption spectroscopy and compared with the corresponding small molecule analogue. Generally, iridium(III) complexes with polypyridyl ligands (e.g., ppy and/or bpy) exhibit a combination of ligand-centered (LC) and metal-to-ligand charge transfer (MLCT) transitions that are observed as at least two prominent bands in the absorption spectrum,6,52–55 the exact positioning of which differs based on electronics of the ligand and solvent choice. We chose to study the optical properties in a variety of common organic solvents of various dielectric constants given the polymer assembly in each solvent might impact the properties.In the case of IrCat1, two intense bands appeared in the ultraviolet region (λabs = 265 and 305–320 nm), along with one weaker and broader peak centered around 384–390 nm with the exact values being slightly influenced by the solvent polarity (see ESI†). These are attributed to two LC transitions and one MLCT transition, consistent with that observed in the seminal reports on [IrdF(CF3)ppy)2(dtbbpy)]PF6.54 IrCat2 comprises a mixture of ppy and bpy-based ligands, thus exhibiting an absorption spectrum with more LC transitions. Similar to IrCat1, there are some small solvent-related effects (see ESI†), with the ultraviolet region showing the most intense bands centered at approximately 255–260 nm and 310–315 nm corresponding to LC transitions. Additional broad features between 385–405 nm and 410–420 nm are ascribed to MLCT transitions, in line with similar reports on monomeric [Ir(ppy)2bpy]1+ complexes.52 Lastly, IrCat3, effectively a polymeric form of fac-Ir(ppy)3, exhibits similar LC transitions in the ultraviolet region below 300 nm arising from the cyclometalated ppy species, along with a broad unresolved band ranging from 320–400 nm, which is assigned to both LC and MLCT transitions.55 The collective observations are consistent with the reported small molecule photocatalysts, thus confirming the photophysical properties aren't markedly different when attached to a polymer backbone.
Transition metal polypyridyl complexes display both MLCT and triplet charge transfer emission that is sensitive to the local environments of the metal complexes.1,8 For photoredox processes, solvents that accomplish the transformations are typically limited to the catalyst dissolution profiles.16 This, combined with the polymer's added potential to solubilize the iridium polypyridyl species while influencing the microenvironment led us to further investigate the emission properties in the aforementioned solvents.
The collective polymers emit at different regions of the visible range and in some cases, exhibit solvent dependent properties (Fig. 1). IrCat1 emitted in the green (∼500 nm, with a shoulder centered around 470–480 nm) in the visible spectrum measured using DCM or THF. The dual bands are suggestive of more 3π–π* character being observed in the mixed-ligand transition metal complex.53,56 Performing measurements in solvents with higher dielectrics, such as MeCN and DMSO, did not drastically alter the position of the emission profile. In contrast, using a solvent with a lower dielectric constant (i.e., toluene) exhibited an emission maximum at 477 nm, with a shoulder around 500 nm. The subtle changes may result from the polymer's solubility in the different solvents.
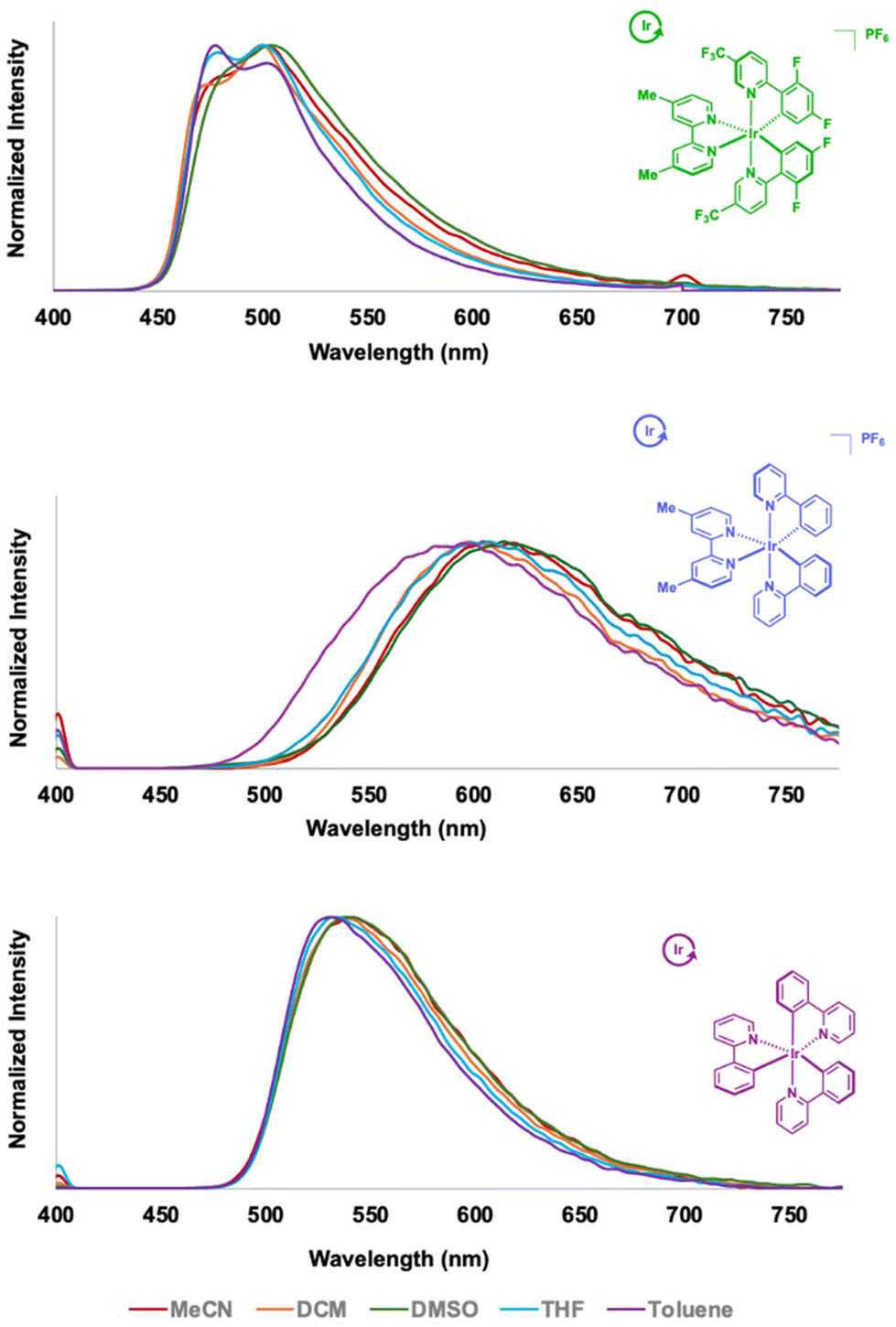 | ||
| Fig. 1 Emission spectral overlays obtained for IrCat1 (top), IrCat2 (middle) and IrCat3 (bottom) in toluene, THF, DCM, MeCN, and DMSO. IrCat1 was excited at 350 nm while IrCat2 and IrCat3 were excited at 400 nm. | ||
IrCat2 exhibited a broad and featureless emission profile centered around 606 nm in DCM akin to the reported molecular analogue,52 with slight red shifting observed when using THF (611 nm) or MeCN (616 nm). Similar measurements in toluene revealed a hypsochromic shift of ca. 20 nm in the MLCT emission band. IrCat2 exhibited a bathochromic shift to λEM = 626 nm when measured in DMSO. The lower energy transitions were consistent with increases in the dielectric constant (and thus, polarity), which was indicative of the dipolar nature and solvent-sensitivity of the excited state, as observed in other cyclometalated iridium complexes.53,57 IrCat3 emitted in the green region (∼540 nm) when measured in DMSO, DCM, or MeCN and featured a broad emission at ambient temperature, akin to molecular fac-Ir(ppy)3.55 We note slight blue shifting to ca. 530 nm in less polar solvents such as THF and toluene.
Synthetic applications
Among the synthesized iridium-based linear photocatalysts, IrCat1 exhibited structural and functional parallels to small molecules of fluorinated heteroleptic iridium photocatalysts. Extant literature underscores that such catalysts have been exceptionally proficient as potent oxidizing photocatalysts, a property largely attributed to the incorporation of fluoro and trifluoromethyl substituents on the phenylpyridine ligands.54 In light of this, we hypothesized that IrCat1, despite its primary function as a photosensitizer or energy-transfer catalyst, can efficiently facilitate single-electron transfer (SET) oxidations and support a broad range of chemical transformations. We investigated five diverse reactions to evaluate the catalytic efficiency of IrCat1, alongside [Ir(df(CF3)ppy)2(dmbpy)]PF6 to confirm the viability of our polymer-based homogeneous photoredox catalyst.In 2019, König reported an efficient Birch-type photo-reduction of planar aromatic substrates to their two-electron reduced forms using [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as an electron-transfer and energy-transfer catalyst.58 Encouragingly, under similar conditions, IrCat1 demonstrated nearly complete conversion of anthracene to 9,10-dihydroanthracene within 24 hours, with [Ir(df(CF3)ppy)2(dmbpy)]PF6 yielding comparable results (Fig. 2a). To further investigate IrCat1's versatility, it was employed in an intermolecular alkyne–alkene [2 + 2]-cycloaddition between diphenylacetylene and N-methylmaleimide, targeting the synthetically valuable cyclobutene ring structure under conditions reported by Park.59
 | ||
| Fig. 2 Summary of organic transformations investigated using (a–e) polymeric IrCat1, (f–i) IrCat2, and (j–n) IrCat3 with yields achieved compared to small molecule analogues using the same conditions. Yields are reported based on NMR spectroscopic analysis in the presence of trimethoxybenzene as an internal standard (see ESI† for details). | ||
Notably, IrCat1 delivered a higher yield of product (71%), compared to [Ir(df(CF3)ppy)2(dmbpy)]PF6 (64%) (Fig. 2b). Additionally, IrCat1 was investigated as an energy-transfer catalyst for a dearomative [4 + 2]-cycloaddition, following the methodology by Maji,60 between 2-acetyl naphthalene and 1-chloro-4-vinylbenzene to access the synthetically challenging sp3-rich bicyclo[2.2.2]octa-2,5-diene 3D scaffold. Here, IrCat1 achieved a 55% yield of the product, compared to a 60% yield when using [Ir(df(CF3)ppy)2(dmbpy)]PF6 (Fig. 2c).
To broaden the scope of IrCat1 within a dual catalysis framework that synergistically combines photoredox and transition metal catalysis, two distinct cross-coupling reactions were explored. In 2014, MacMillan reported a direct decarboxylative sp3–sp2 cross-coupling of amino acids with aryl halides, employing [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as a photoredox catalyst in conjunction with an organometallic nickel(II) catalyst.48 Later, the same group utilized this system for cross-electrophile reactions, efficiently coupling alkyl bromides with aryl or heteroaryl bromides.61 Encouragingly, our results demonstrate that IrCat1, in combination with a nickel(II) catalyst, achieved a 60% yield for the decarboxylative cross-coupling between N-Boc-protected proline and methyl-4-bromoacetophenone (Fig. 2d). Moreover, a dehalogenative cross-coupling between bromocyclohexane and methyl-4-bromobenzoate delivered a moderate yield of 62% (Fig. 2e).
IrCat2 closely resembled the structure and functionality of heteroleptic iridium small molecule photocatalysts.52 Studies emphasized the high versatility of these catalysts, which can serve as both strong photooxidants as well as photoreductants depending on reaction conditions, due to the synergistic interactions between the phenylpyridine and bipyridine ligands.2,6,13,14,52,53,55 Like IrCat1, IrCat2 is hypothesized to efficiently undergo both photosensitization and SET to mediate a broad range of redox transformations. To validate the catalytic performance of IrCat2, we investigated four distinct reactions and compared the observed results with the corresponding [Ir(ppy)2(dmbpy)]PF6 small molecule to assess the potential of this polymeric system in photoredox catalysis.
To begin, the aza-Henry reaction was selected as the initial model system, employing Stephenson's protocol due to its simplicity and operational efficiency.62 Under visible-light irradiation, IrCat2 enabled near-quantitative oxidative Henry coupling of nitromethane with N-aryltetrahydroisoquinolines without external oxidants, highlighting the photocatalyst's intrinsic oxidative capacity (Fig. 2f). Further exploration of IrCat2's utility in tertiary amine functionalization through photocatalytic oxidation of sp3 α-C–H bonds adjacent to nitrogen. This was conducted using a Ugi-type multicomponent reaction wherein N,N-dimethylaniline functioned as the iminium ion precursor, p-toluenesulfonylmethyl isocyanide as the main reactant, and water as the nucleophile.63 Despite the inherent challenges associated with this ‘one-pot’ multicomponent methodology, the reaction delivered the desired α-amino amides in a satisfactory yield of 57% (Fig. 2g).
The aforementioned reactions unequivocally demonstrated a pivotal step wherein IrCat2 undergoes reductive quenching in the presence of tertiary amines. Capitalizing on this mechanistic insight, the direct oxidative hydroxylation of phenylboronic acid under aerobic conditions was also explored using diisopropylethylamine as a sacrificial electron and proton donor.31 This approach yielded phenol in an excellent yield of 84% (Fig. 2h). Yang recently demonstrated the sp3–sp3 homocoupling of benzyl pyridyl ethers, utilizing the synergistic combination of Ir-based photocatalysts and tertiary amines as co-catalysts.64 Mechanistically, the reaction's challenge lies in the generation of neutral radicals from C(sp3)–O bond homolysis, which are susceptible to side reactions. Notably, under analogous conditions, IrCat2 achieved the desired bibenzyl product in 62% yield, demonstrating its distinct efficacy similar [Ir(ppy)2(dtbbpy)]PF6 (Fig. 2i).
Within our series of polymeric iridium-based linear photocatalysts, IrCat3 exhibited structural and functional resemblance to small-molecule homoleptic iridium complexes. Existing studies suggested that this class of catalysts is highly effective as photoreductants, characterized by uniform cyclometalated phenylpyridine ligands.2,6,11,13,14 Accordingly, we predicted that our newly developed IrCat3, acting as a photoreductant, can efficiently engage in SET reductions for a wide range of substrates, thereby enabling an oxidative quenching mechanism. We thus examined the catalytic efficiency of IrCat3 across five diverse array reaction types and provided a comparative analysis with its small-molecule counterpart fac-Ir(ppy)3 to validate the utility of this polymer-based model.
In 2011, MacMillan and colleagues introduced an innovative, one-step α-functionalization strategy for amines, enabling the construction of versatile benzyl amine motifs – a key structural component present in eight of the top 100 selling pharmaceuticals, using fac-Ir(ppy)3 as a photoredox catalyst.65 Notably, under similar conditions, our newly developed IrCat3 also facilitated nearly complete formation of 4-(1-phenylpyrrolidin-2-yl)benzonitrile though direct decyanative cross-coupling between 1,4-dicyanobenzene with N-phenylpyrrolidine (Fig. 2j). In addition to its efficacy in sp3–sp2 cross-coupling, IrCat3 has also been applied to an another cross-coupling reaction, involving regioselective C(sp2)–H activation of the pharmaceutically-relevant imidazo[1,2-a]pyridine scaffold, differing from previous approaches reliant on C(sp3)–H activation.66 Utilizing bromoacetonitrile as the coupling partner, this transformation achieved a commendable 71% yield of C3-substituted imidazo[1,2-a]pyridine derivatives (Fig. 2k). Notably, IrCat3 performed comparably to fac-Ir(ppy)3, further underscoring its versatility and potential for applications in pharmaceutical synthesis.
Additionally, IrCat3 has been examined in a synthetically challenging ‘one-pot’ three-component regioselective alkene difuntionalization reaction utilizing Umemoto's reagent for oxytrifluoromethylation of styrene and benzylic C–O substitution using D2O as O-nucleophile, under conditions similar to those reported by Akita.67 Encouragingly, this reaction also resulted in the complete formation of the desired product (Fig. 2l). IrCat3 was applied for another regioselective alkene functionalization, specifically facilitating the oxidative coupling of 4-tert-butylstyrene with ethyl-2-bromopropanoate to produce a γ-ketoester derivative—a valuable synthetic building block.68 This reaction resulted in a moderate 45% yield of the target product with IrCat3, comparable to that obtained with fac-Ir(ppy)3 (Fig. 2m).
Finally, IrCat3 was evaluated within a dual catalysis framework for cross-coupling, following a slightly modified method reported by MacMillan48 and later by Seeberger.69 Mechanistically, this approach uses the iridium catalyst as a photosensitizer, while an in situ generated organometallic nickel(II) catalyst in its excited state facilitates the dehalogenative coupling of N-Boc-proline and methyl-4-bromobenzoate, efficiently yielding O-aryl ethers. Remarkably, this dual catalytic system was highly compatible with IrCat3, achieving a 71% yield of the desired product (Fig. 2n), demonstrating its adaptability and efficiency in advanced catalytic systems.
Recyclability studies
Given iridium is one of the rarest and most precious metals, we were prompted to explore the recyclability of our newly developed polymeric catalysts, with the goal of improving sustainability in catalytic processes. To comprehensively evaluate this, we selected a representative reaction for each of the disclosed polymeric iridium catalysts, ensuring comparable reaction conditions (such as time, yield, light source, and additives) across all systems, and studied the ability to recycle and reuse the polymer catalyst over five cycles (Fig. 3). For IrCat1, we chose the alkyne–alkene [2 + 2]-cycloaddition, a key reaction in organic synthesis, photochemistry, and the development of bioactive molecules and advanced materials. In the first cycle, using diphenylacetylene and N-methylmaleimide, we achieved a 71% yield of the cycloadduct.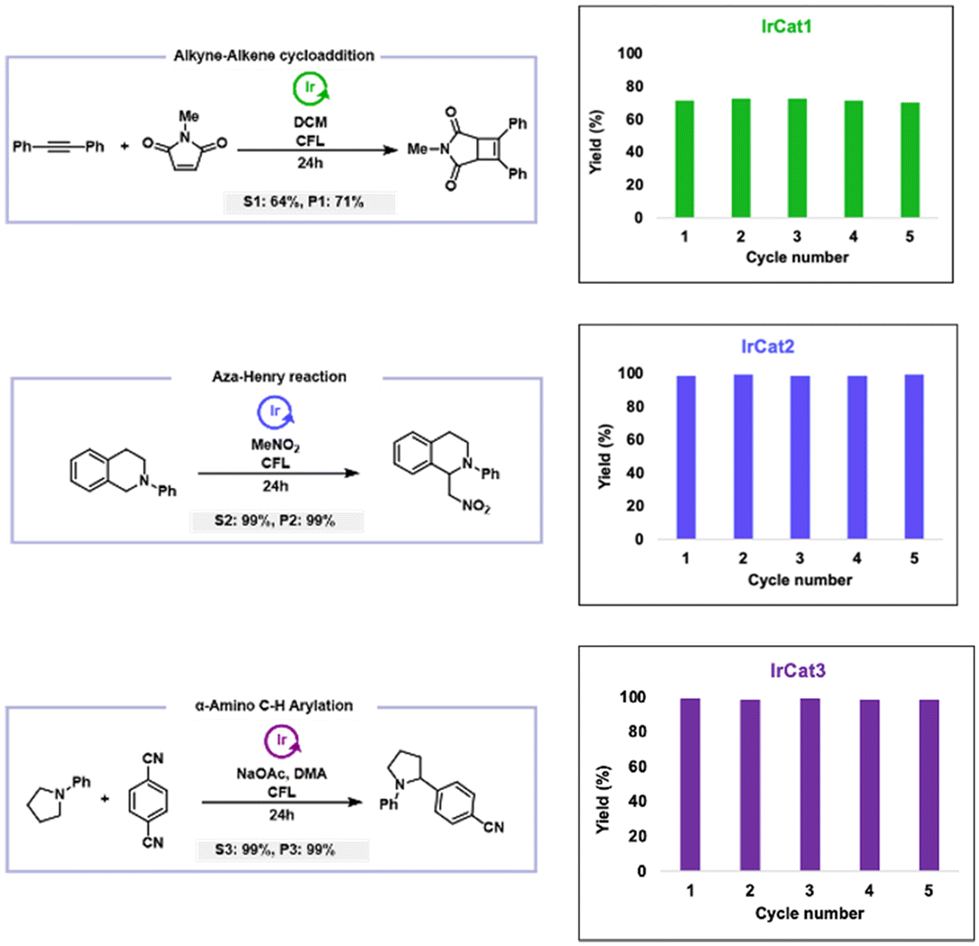 | ||
| Fig. 3 Recyclability of IrCat1 (top), IrCat2 (middle) and IrCat3 (bottom) in the alkyne–alkene cycloaddition, aza-Henry, and C–H arylation reactions, respectively. After reaction, the respective IrCat is recycled from the reaction mixture through multiple reprecipitations into methanol, followed by centrifugation, drying, and reuse in a fresh reaction. | ||
It is worth noting that the highly electron-deficient phenylpyridine ligands of IrCat1 can limit the overall efficiency of the reaction, as intermediate carbon-centered radicals may undergo direct addition to the electrophilic arenes, reducing selectivity and impacting the resultant MLCT properties. Given this possibility, we sought to investigate the catalyst fidelity over the course of the reaction. Thus, throughout the reaction of diphenylacetylene and N-methylmaleimide, aliquots were taken with their emission profile recorded (Fig. 4). While we observed minor changes in intensity as the reaction progressed (likely owing to precision of the aliquot volume), we observed that data collected prior to irradiation (0 hour trace) and upon completion of the reaction (24 hours trace) was nearly identical in intensity. This suggested that the cyclometalated iridium complex maintained high fidelity throughout the duration of the reaction. This particular conclusion was further manifested as IrCat1 demonstrated excellent recyclability, maintaining 72% yield in the second cycle, with yields of 70–72% being demonstrated in the subsequent three cycles.70 Nevertheless, despite the challenges, IrCat1 remained catalytically active through five cycles, showcasing its potential as a reusable homogeneous catalyst.
 | ||
| Fig. 4 Emission spectral overlay of aliquots drawn during the alkynealkene cycloaddition, catalyzed by IrCat1. For each sample, 100 μL of the reaction mixture was diluted to 500 μL in DCM, followed by recording a spectrum. While some small changes to the emission intensity are noted, the intensity prior to the reaction (0 hour trace) and at the conclusion of the reaction (24 hour trace) are nearly identical, suggesting the photocatalyst maintains high fidelity throughout the course of the reaction. | ||
For IrCat2, we turned to the aza-Henry reaction, capitalizing on its ability to selectively activate C–H bonds across diverse substrates, thus enhancing synthetic versatility. When N-phenyl tetrahydroisoquinoline was treated with CH3NO2 in the presence of IrCat2, the reaction yielded 1-(nitromethyl)-2-phenyl-1,2,3,4-tetrahydroisoquinoline with an impressive 99% yield in the first cycle. Similar to IrCat1, we monitored the emission profile during the reaction to gain insights into the fidelity of the photocatalyst. While we observed the emission intensity increasing throughout the duration of the reaction, we did not see any signs of degradation or catalyst deactivation (see Fig. S57†). This observation was validated as IrCat2 exhibited outstanding recyclability, maintaining yields of 99% in the second through fifth cycles,70 highlighting its robustness and efficiency for repetitive use in synthetic applications. For IrCat3, we selected the α-amino C–H arylation reaction, specifically the direct decyanative cross-coupling of 1,4-dicyanobenzene with N-phenylpyrrolidine, which produced 4-(1-phenylpyrrolidin-2-yl) benzonitrile with a 99% yield in the first cycle. When monitoring the emission profile during the reaction, no notable decreases in intensity were observed (see Fig. S58†). While the emission maxima exhibited a hypsochromic shift of 20 nm from the period of irradiation onward, IrCat3, demonstrated impressive recyclability and maintained high yields of 98–100% across all five cycles.70 This suggested that the observed alteration in the emission profile during the reaction did not impact the catalyst activity.
Conclusions
In summary, we have developed three homogeneous polymeric catalysts based on photoactive cyclometalated iridium complexes. The three catalysts – IrCat1, IrCat2, and IrCat3 – demonstrated efficient homogeneous photoredox chemistry across a broad swath of organic transformations, including both oxidative and reductive couplings, bimolecular cycloadditions, and alkene functionalization reactions, highlighting their versatility. Each IrCat exhibits remarkable recyclability, maintaining high catalytic activity through (at least) five reaction cycles. Moreover, for the reactions studied for recyclability, each IrCat preserved the original emission behavior after reaction, suggesting This highlights the potential of polymeric iridium-based homogeneous catalysts in sustainable, scalable processes in organic synthesis.When comparing these polymeric catalysts with their small-molecule counterparts, it is evident that both enable comparative reactivity across multiple challenging organic transformations. The polymeric nature of IrCat1, IrCat2, and IrCat3, however, also allows for efficient recycling with high durability over multiple cycles, while providing a clear advantage in terms of sustainability and long-term performance. Our findings suggest that polymeric iridium-based catalysts represent a promising alternative to traditional small-molecule catalysts, providing an exciting pathway toward more efficient and environmentally friendly catalytic processes.
Data availability
All experimental data are provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation Early CAREER program (CHE-2046470) and Research Experience for Undergraduates Program (CHE-2050927). The authors also acknowledge support from the Alfred P. Sloan Foundation (FG-2021-15490) and start-up funds generously provided by the Pennsylvania State University. We also acknowledge Dr Daniela Arias-Rotondo of Kalamazoo College, and thank her for several enlightening discussions on the photophysics of photoredox catalysts and fruit flies.References
- K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 Search PubMed.
- V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 618–635 RSC.
- S. Telitel, F. Dumur, S. Telitel, O. Soppera, M. Lepeltier, Y. Guillaneuf, J. Poly, F. Morlet-Savary, P. Fioux, J.-P. Fouassier, D. Gigmes and J. Lalevée, Polym. Chem., 2015, 6, 613–624 RSC.
- D. R. Martir and E. Zysman-Colman, Coord. Chem. Rev., 2018, 364, 86–117 CrossRef.
- K. P. S. Zanoni, B. K. Kariyazaki, A. Ito, M. K. Brennaman, T. J. Meyer and N. Y. Murakami Iha, Inorg. Chem., 2014, 53, 4089–4099 CrossRef CAS PubMed.
- S. Conejero, C. Maya, M. Paneque, A. Petronilho, M. L. Poveda, F. Vattier, E. Álvarez, E. Carmona, A. Laguna and O. Crespo, Dalton Trans., 2012, 41, 14126–14136 RSC.
- R. Bevernaegie, S. A. M. Wehlin, B. Elias and L. Troian-Gautier, ChemPhotoChem, 2021, 5, 217–234 Search PubMed.
- K. Y. Zhang, H.-W. Liu, T. T.-H. Fong, X.-G. Chen and K. K.-W. Lo, Inorg. Chem., 2010, 49, 5432–5443 Search PubMed.
- S. Chen, Y. Zhou and X. Ma, Dalton Trans., 2024, 53, 2731–2740 RSC.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- S. Huss, A. R. Walsh, A. Griggs, D. A. Rodriguez-Acevedo, D. M. Arias-Rotondo and E. Elacqua, Polym. Chem., 2023, 14, 4560–4568 Search PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 Search PubMed.
- D. Jespersen, B. Keen, J. I. Day, A. Singh, J. Briles, D. Mullins and J. D. Weaver III, Org. Process Res. Dev., 2019, 23, 1087–1095 CrossRef CAS PubMed.
- M. A. Bryden, F. Millward, O. S. Lee, L. Cork, M. C. Gather, A. Steffen and E. Zysman-Colman, Chem. Sci., 2024, 15, 3741–3757 RSC.
- J. C. Bawden, P. S. Francis, S. DiLuzio, D. J. Hayne, E. H. Doeven, J. Truong, R. Alexander, L. C. Henderson, D. E. Gómez, M. Massi, B. I. Armstrong, F. A. Draper, S. Bernhard and T. U. Connell, J. Am. Chem. Soc., 2022, 144, 11189–11202 CrossRef CAS PubMed.
- X. Liu, Y. Ni, J. Wu, H. Jiang, Z. Zhang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2018, 9, 584–592 Search PubMed.
- D. C. Fabry, M. A. Ronge and M. Rueping, Chem. – Eur. J., 2015, 21, 5350–5354 Search PubMed.
- A. Quintavalla, D. Carboni, C. Sepe, L. Mummolo, N. Zaccheroni and M. Lombardo, Adv. Synth. Catal., 2023, 365, 252–262 CrossRef CAS.
- Y. Quan, Y. Song, W. Shi, Z. Xu, J. S. Chen, X. Jiang, C. Wang and W. Lin, J. Am. Chem. Soc., 2020, 142, 8602–8607 CrossRef CAS PubMed.
- J.-L. Wang, C. Wang and W. Lin, ACS Catal., 2012, 2, 2630–2640 Search PubMed.
- T. Liu, C. Deng, D. Meng, Y. Zhang, R. Duan, H. Ji, H. Sheng, J. Li, C. Chen, J. Zhao and W. Song, ACS Appl. Mater. Interfaces, 2023, 15, 5139–5147 CrossRef CAS PubMed.
- X. Yu and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 12320–12323 CrossRef CAS.
- T. Kajiwara, M. Fujii, M. Tsujimoto, K. Kobayashi, M. Higuchi, K. Tanaka and S. Kitagawa, Angew. Chem., Int. Ed., 2016, 55, 2697–2700 CrossRef CAS PubMed.
- X. Yu, L. Wang and S. M. Cohen, CrystEngComm, 2017, 19, 4126–4136 RSC.
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed.
- A. López-Magano, B. Ortín-Rubio, I. Imaz, D. Maspoch, J. Alemán and R. Mas-Ballesté, ACS Catal., 2021, 11, 12344–12354 CrossRef PubMed.
- M. Traxler, S. Reischauer, S. Vogl, J. Roeser, J. Rabeah, C. Penschke, P. Saalfrank, B. Pieber and A. Thomas, Chem. – Eur. J., 2023, 29, e202202967 CrossRef CAS PubMed.
- Z.-Y. Xu, Y. Luo, D.-W. Zhang, H. Wang, X.-W. Sun and Z.-T. Li, Green Chem., 2020, 22, 136–143 RSC.
- Z. Wang, P. Yeary, Y. Fan, C. Deng and W. Lin, ACS Catal., 2024, 14, 9217–9223 CrossRef CAS.
- Y. Hao, Y.-L. Lu, Z. Jiao and C.-Y. Su, Angew. Chem., Int. Ed., 2024, 63, e202317808 CrossRef CAS PubMed.
- Y.-Y. Zhu, G. Lan, Y. Fan, S. S. Veroneau, Y. Song, D. Micheroni and W. Lin, Angew. Chem., Int. Ed., 2018, 57, 14090–14094 Search PubMed.
- M. Spicuzza, S. P. Gaikwad, S. Huss, A. A. Lee, C. V. Craescu, A. Griggs, J. Joseph, M. Puthenpurayil, W. Lin, C. Matarazzo, S. Baldwin, V. Perez, D. A. Rodriguez-Acevedo, J. R. Swierk and E. Elacqua, Polym. Chem., 2024, 15, 1833–1838 RSC.
- J. J. Piane, L. E. Chamberlain, S. Huss, L. T. Alameda, A. C. Hoover and E. Elacqua, ACS Catal., 2020, 10, 13251–13256 CrossRef CAS.
- Y. Isaka, Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Catal. Sci. Technol., 2019, 9, 1511–1517 RSC.
- K. L. Metera and H. Sleiman, Macromolecules, 2007, 40, 3733–3738 CrossRef CAS.
- A. Jati, A. K. Mahato, D. Chanda, P. Kumar, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2024, 146, 23923–23932 CrossRef CAS PubMed.
- Y.-L. Li, F. Wang, J. J. Vittal, P. Jin, S.-L. Huang and G.-Y. Yang, J. Mater. Chem. A, 2024, 12, 9164–9172 RSC.
- V. Mouarrawis, R. Plessius, J. I. van der Vlugt and J. N. H. Reek, Front. Chem., 2018, 6, 623 CrossRef CAS.
- S. Gisbertz, S. Reischauer and B. Pieber, Nat. Catal., 2020, 3, 611–620 CrossRef CAS.
- X. Zhao, C. Deng, D. Meng, H. Ji, C. Chen, W. Song and J. Zhao, ACS Catal., 2020, 10, 15178–15185 CrossRef CAS.
- A. Vijeta, C. Casadevall and E. Reisner, Angew. Chem., Int. Ed., 2022, 61, e202203176 CrossRef CAS PubMed.
- E. Tsuchida and H. Nishide, Adv. Polym. Sci., 1977, 1–87, DOI:10.1007/3-540-08124-0_1.
- D. Rackl, P. Kreitmeier and O. Reiser, Green Chem., 2016, 18, 214–219 RSC.
- Y. Pan, N. Zhang, C.-H. Liu, S. Fan, S. Guo, Z.-M. Zhang and Y.-Y. Zhu, ACS Catal., 2020, 10, 11758–11767 CrossRef CAS.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed.
- J. F. R. Van Guyse, Y. Bernhard, A. Podevyn and R. Hoogenboom, Angew. Chem., Int. Ed., 2023, 62, e202303841 CrossRef CAS PubMed.
- D. J. Lunn, E. H. Discekici, J. Read de Alaniz, W. R. Gutekunst and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2903–2914 CrossRef CAS.
- F. O. Garces, K. A. King and R. J. Watts, Inorg. Chem., 1988, 27, 3464–3471 Search PubMed.
- M. G. Colombo, A. Hauser and H. U. Guedel, Inorg. Chem., 1993, 32, 3088–3092 CrossRef CAS.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290–9299 CrossRef CAS.
- M. S. Lowry, W. R. Hudson, R. A. Pascal and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS.
- R. A. Smith, E. C. Stokes, E. E. Langdon-Jones, J. A. Platts, B. M. Kariuki, A. J. Hallett and S. J. A. Pope, Dalton Trans., 2013, 42, 10347–10357 RSC.
- A. Chatterjee and B. König, Angew. Chem., Int. Ed., 2019, 58, 14289–14294 CrossRef CAS.
- S. Ha, Y. Lee, Y. Kwak, A. Mishra, E. Yu, B. Ryou and C.-M. Park, Nat. Commun., 2020, 11, 2509 CrossRef CAS.
- P. Rai, K. Maji, S. K. Jana and B. Maji, Chem. Sci., 2022, 13, 12503–12510 RSC.
- P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed.
- M. Rueping and C. Vila, Org. Lett., 2013, 15, 2092–2095 CrossRef CAS PubMed.
- J. Chen, F. Wang, X. Li, L. Wang, W. Yu, K. Sun and J. Yang, Chem. Commun., 2023, 59, 11409–11412 RSC.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed.
- Q. Chang, Z. Liu, P. Liu, L. Yu and P. Sun, J. Org. Chem., 2017, 82, 5391–5397 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS PubMed.
- X.-X. Fang, P.-F. Wang, W. Yi, W. Chen, S.-C. Lou and G.-Q. Liu, J. Org. Chem., 2019, 84, 15677–15684 CrossRef CAS PubMed.
- J. A. Malik, A. Madani, B. Pieber and P. H. Seeberger, J. Am. Chem. Soc., 2020, 142, 11042–11049 CrossRef CAS PubMed.
- We note that, at the scale used, a few mg of polymer are lost on recycling. Instead of increasing the reaction duration to account for the lower amount of catalyst, we adjusted the limiting reagent accordingly to match the optimal conditions of the first cycle and mmol of catalyst recovered.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures for monomers and polymers and characterization. See DOI: https://doi.org/10.1039/d4py01391c |
| This journal is © The Royal Society of Chemistry 2025 |