
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the dynamic CO2 capture performance of MgO promoted with molten salts and CaCO3via fixed bed reactor experiments†
Theodoros
Papalas
 *ab,
Andy N.
Antzaras
a and
Angeliki A.
Lemonidou
*ac
*ab,
Andy N.
Antzaras
a and
Angeliki A.
Lemonidou
*ac
aDepartment of Chemical Engineering, Aristotle University of Thessaloniki, University Campus, 54124 Thessaloniki, Greece. E-mail: alemonidou@cheng.auth.gr
bDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, CB3 0AS Cambridge, UK. E-mail: tp557@cam.ac.uk
cChemical process & Energy Resource Institute, CPERI/CERTH, 57001 Thermi, Thessaloniki, Greece
First published on 23rd October 2024
Abstract
Carbonate looping using MgO-based materials has recently ignited scientific interest for CO2 capture at intermediate temperatures (275–375 °C), with the main limitation being the slow carbonation kinetics of MgO. Molten alkali nitrates and metal carbonates have been identified as promoters that provide an alternative reaction mechanism for an enhanced carbonation rate. However, the evaluation of the ability of these materials to effectively remove CO2 from a gas feed under realistic reactor configurations is still required. This study investigated the CO2 capture performance of magnesite-derived MgO promoted with limestone and molten Li, Na and K nitrates under carbonate looping conditions in a fixed bed reactor. The CO2 capture efficiency was enhanced in the presence of H2O, by increasing the gas–solid contact time and by decreasing the carbonation temperature. The evaluation demonstrated that ∼75% CO2 stripping of a gas feed with 30% CO2 concentration at 275 °C and a space velocity of 300 h−1 is possible, a performance that highlights and expands the potential and possible applications of MgO-based materials.
1. Introduction
The heavy reliance on fossil fuels to satisfy the global energy requirements has led to a significant increase in CO2 emissions and severe environmental problems. The mitigation of emissions can be attained by adopting renewable resources, increasing the efficiency of current energy technologies and implementing CO2 capture, utilisation and storage (CCUS).1,2 Despite the notable progress in renewable energy technologies, their high costs and low maturity, along with the varying availability across regions and the periodic nature of renewable resources, suggest that the global economy will continue to rely on fossil fuels for the foreseeable future. In contrast, CCUS can be applied by retrofitting fossil fuel-dependent energy systems, such as the heavy industry (cement, steel, and chemicals production), which accounts for 20% of industrial emissions.3 This approach can help carbon-intensive industries meet the requirements of the energy transition period and operate sustainably in the future. Hence, research should aim to find viable CCUS solutions.Schemes based on reversible gas–solid reactions between CO2 and metal oxides, such as carbonate looping, comprise a promising carbon capture technology. CO2 is captured via the carbonation of the metal oxide, while the formed carbonate is transferred and decomposed to a second reactor operating at higher temperatures, enabling the reuse of the metal oxide for carbonation. CaO has been favoured for this technology due to its high CO2 capture capacity (17.8 mmol CO2 per g) and fast carbonation kinetics at 550–650 °C.4–6 Such attributes have enabled the pilot-scale investigation of calcium looping, reaching a technology readiness level of 6–7.7,8 However, harsh decarbonation temperatures (≥900 °C) are detrimental to the CaO stability and process energy demand, while they make retrofitting of existing industries less flexible, as flue gas is typically available at lower temperatures.9,10
MgO has recently gained interest due to the favourable thermodynamics (Fig. 1) for carbonation (eqn (1)) at intermediate temperatures (275–375 °C). With a high theoretical CO2 capture capacity (24.8 mmol CO2 per g) and mild decarbonation conditions (400–450 °C), MgO presents a sustainable alternative for flexible CO2 capture applications.4,11,12 The main limitation comprises the slow carbonation kinetics, arising from the high lattice energy of MgO and the formation of a non-crystalline carbonate layer of 7 Å thickness in contact with CO2,13 resulting in an uptake of less than 0.5 mmol CO2 per g after 1 h exposure to CO2.14,15
| MgO(s) + CO2(g) ⇄ MgCO3(s), ΔH°298K = ±118 kJ molCO2−1 | (1) |
 | ||
| Fig. 1 Equilibrium curves of the carbonation of pure MgO (solid line) and MgO–CaCO3 (dashed line) with a CaCO3 to MgO molar ratio of 0.05 at 1 bar (recreated from ref. 16). In the figure, A refers to MgCO3, B to CaMg(CO3)2, C to MgO and D to CaCO3. | ||
Many researchers have also investigated various binary and ternary salt mixtures except for NaNO3 to evaluate the effect of the composition of the molten phase on the CO2 capture activity.19,21,24–26 Addition of alkali nitrates with an alkali of small ionic radius can enhance the CO2 solubility and thus the CO2 uptake of the material, while nitrates with an alkali of large ionic radius are resistant to decomposition, thereby retaining their beneficial effect on multiple carbonation and decarbonation cycles.12,21 Beyond alkali nitrates, there is high interest in using alkali or alkaline earth carbonate promoters (e.g. Na2CO3, K2CO3, CaCO3, BaCO3) to improve the MgO carbonation rate.27–30 Carbonates serve as nucleation seeds, which are beneficial due to the autocatalytic behaviour of the nuclei formation and growth mechanism. Moreover, they may also form mixed carbonates with MgO, whose formation is thermodynamically feasible at lower partial pressures of CO2 compared to MgCO3,13,31,32 as illustrated in Fig. 1 for the case of a mixture of MgO and CaCO3. Our previous work examined MgO-based materials derived from magnesite and promoted with limestone and a mixture of Li, Na and K nitrates (Li/Na/K = 30/18/52). Limestone enabled the formation of CaMg(CO3)2 and a fast nucleation rate, when applying a CaCO3 to MgO molar ratio of 0.05. Increasing the alkali nitrate content accelerated the generation of carbonates, while the optimal molar ratio of alkali nitrates to MgO was equal to 0.20. The optimum CaCO3 and alkali salt contents resulted in an uptake of 7.2 mmol CO2 per g (30% CO2, 300 °C, 30 min) and minimal activity loss (∼6%) after 50 cycles in a thermogravimetric analyser.16,33
Despite extensive research on molten salt-promoted MgO, the evaluation of CO2 capture activity has mainly been conducted with pure CO2 gas feed and long carbonation durations in thermogravimetric analysers, thereby not reflecting realistic operating conditions. Thermogravimetric analysis can efficiently determine the CO2 uptake capacity of such materials; however, it is essential to also examine the CO2 stripping efficiency from the gas feed.12,34 Chen et al. evaluated MgO-based materials promoted with various alkali nitrates and carbonates (LiNO3, KNO3, Na2CO3, K2CO3) in a fixed bed reactor. They found a CO2 capture efficiency less than 10% when carbonation was conducted at 325 °C even with the use of a gas feed with unrealistically high CO2 concentration (80% CO2) for either pre- or post-combustion CO2 capture applications.35 Experiments should be performed under real flow reaction conditions in bench-scale units, with such studies being currently scarce in the literature,35–40 combined with continuous reactor gas outlet monitoring.
This study builds on our previous work16,33 and investigates the developed MgO-based material promoted with CaCO3 and alkali nitrates under carbonate looping conditions via fixed bed reactor experiments. The ability to remove CO2 from a gas feed was evaluated by varying temperature, CO2 concentration, H2O presence and space velocity. The results of this study reveal the potential and limitations of dynamic CO2 capture at intermediate temperatures and provide insight on the possible industrial applications of the MgO-based materials.
2. Materials and methods
2.1. Preparation of the material
A MgO-based material was prepared via a two-step protocol, initiated by wet mixing of calcined magnesite and calcined limestone in a 12 vol% acetic acid solution and followed by wet impregnation of the resulting solids with Li, Na and K nitrates (with an atomic ratio of Li/Na/K equal to 30/18/52). The prepared material had nominal molar ratios of CaCO3 and alkali salts to MgO of 0.05 and 0.20, respectively, and was designated as MgCa0.05A0.20. The detailed preparation protocol and characterisation results, including ex situ and in situ X-ray diffraction, N2 adsorption, and scanning electron microscopy, for both fresh and used materials subjected to carbonate looping conditions can be found elsewhere.16,332.2. Evaluation of the MgO-based material under carbonate looping conditions
Thermogravimetric analysis (TGA, TG 209 F3 Tarsus, NETZSCH) was utilised to explore the effect of pelletisation on the CO2 capture activity of the MgCa0.05A0.20 material. After the final calcination step of the preparation protocol at 450 °C, the material powders were compressed into small discs with a manual hydraulic press. The discs were then crushed and sieved to obtain a particle size range of 106 to 212 μm. After loading ∼10 mg of the material in an Al2O3-based crucible, the temperature was increased to 450 °C under a 100% N2 flow to remove any CO2 or H2O captured from the atmosphere. The TGA chamber was cooled down to 300 °C with a rate of 50 °C min−1 and the gas feed was switched to a 30% CO2/N2 flow for 30 min to conduct the carbonation stage. Afterwards, the sample was heated up to 450 °C at 10 °C min−1 to decompose the formed carbonates under a 100% N2 flow. A total of 50 carbonation and decarbonation cycles were performed, while the same experiment was conducted also with a non-pelletised material for comparison.The performance of the MgCa0.05A0.20 material was investigated by performing experiments in a continuous flow unit equipped with a fixed bed reactor. A schematic illustration of the unit is available in the ESI† (Fig. S1). The reactor was loaded with 0.5, 2, or 4 g of material with a particle diameter below 106 μm obtained by sieving the material without prior pelletisation. Solids were pre-treated by increasing the temperature to 450 °C under a 100% He flow to release any captured CO2 or H2O. The reactor was then cooled down to 275, 300 or 325 °C and the gas feed was switched to 30% CO2/He or 15% CO2/He (50 mL min−1) to conduct the carbonation stage. The effect of steam was also examined in an experiment, in which the gas feed passed through a steam saturator to attain a 5% H2O content. The composition of the carbonation gas feed was selected to replicate the flue gas of the cement industry.41,42 Carbonation was followed by the increase of temperature at 450 °C and the change of gas feed to 100% He (120 mL min−1) to carry out the decarbonation stage. The carbonation and decarbonation stages were repeated for a total of 10 cycles. The duration of the carbonation stage was equal to 30 min, except from the 1st and 10th cycle, where the duration was extended to 60–180 min. The prolonged carbonation stages aimed to fully detect the transition of the kinetically controlled stage to the CO2 diffusion through the formed carbonate layers, which was different depending on the remaining operating conditions (temperature, CO2 content in the gas feed, GHSV). On the other hand, decarbonation lasted until the full conversion of carbonates formed in the preceding carbonation stage. In addition to the experiments described, blank experiments were conducted under the same operating conditions with inert SiO2 that helped to analyse the results. All experiments were conducted at atmospheric pressure, while pressure drop did not display an increase higher than 15% of inlet pressure, satisfying the general rule of thumb for isobaric conditions at laboratory-scale fixed bed reactor experiments. The detailed methodology followed for analysing the results of TGA and reactor experiments is available in the ESI.†
3. Results and discussion
3.1. Evaluation of the effect of pelletisation with thermogravimetric analysis
Prior to the fixed bed experiments, the performance of the MgCa0.05A0.20 material was investigated in TGA apparatus to check the effect of pelletisation on the performance of the material to be used in the bench-scale unit. The MgO carbonation conversion and CO2 capture capacity of the material is presented in Fig. 2. The material sieved to obtain a particle size lower than 106 μm without prior pelletisation displayed an initial CO2 capture uptake of 7.2 mmol CO2 per g of material and a high stability, with a minimal 6% activity loss over 50 consecutive carbonation and decarbonation cycles. On the other hand, the material with a particle size between 106 and 212 μm obtained after pelletisation displayed a ∼70% lower initial uptake than the non-pelletised material. However, the material exhibited a continuous self-activation, reaching up to 5.4 mmol CO2 per g of material, while the lower initial activity, coupled with the simultaneous self-activation, agrees with previously reported results in the literature for pelletised MgO.35,43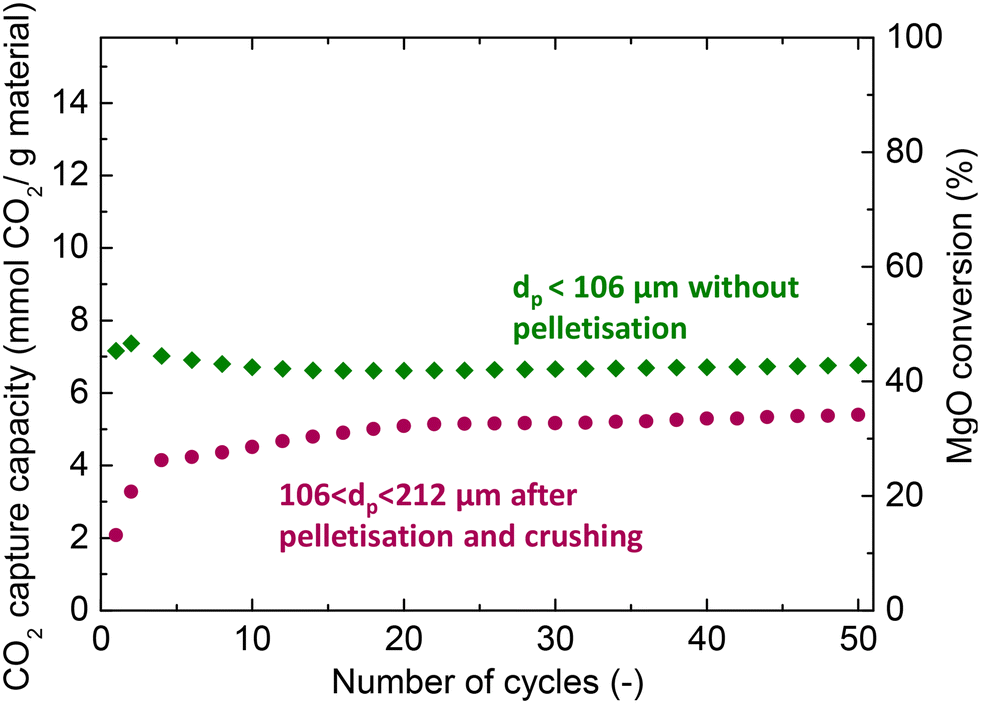 | ||
| Fig. 2 CO2 capture efficiency and MgO conversion for the MgCa0.05A0.20 material obtained with and without pelletisation when investigated via carbonate looping experiments in a thermogravimetric analyser (carbonation stage: 30% CO2/N2, 300 °C; decarbonation stage: 450 °C, 100% N2). | ||
The activity and stability of molten salt-promoted MgO have been linked with various phenomena. While molten salts enhance the CO2 capture activity, they also dissolve solid components and promote their densification, and thus the annihilation of the pore network, known as liquid phase sintering.33,44 The cyclic performance is also affected by changes in the distribution of molten salts,25 as a result of the lower affinity of the molten phase with MgCO3 compared to MgO.19,32 The re-distribution can be beneficial for the CO2 capture activity under certain operating conditions and mask the extent of sintering,33,45 ultimately being the reason behind the stable performance of the non-pelletised material.
Pelletisation caused a decrease of the surface area (from 10.1 to 5.3 m2 g−1) and pore volume (from 0.08 to 0.03 cm3 g−1), as found by characterising the materials with N2 adsorption analysis. The inferior textural properties prevent the efficient distribution and exploitation of molten alkali salts, leading to a lower initial CO2 uptake.35 However, Rekhtina et al. noted that alkali nitrates can gradually decompose and react with CO2 to form carbonates, which, as mentioned before, act as nucleation seeds and promote carbonation. They further suggested that this effect accounts for the self-activation phenomenon.31
In general, granulation of MgO-based CO2 capture materials is essential for their practical applications. However, current efforts in this area and existing granulation techniques are limited.38,40,43 Future research should prioritise the development of MgO granules that exhibit strong mechanical properties and high CO2 capture ability. A key consideration for any granulation method is its scalability for industrial use, since large quantities of MgO are required for practical looping systems. Additionally, the cost-effectiveness of the manufacturing process is another critical factor.12 In any case, finding an efficient granulation method was rendered out of the scope of this study and thus all experiments described below were performed using a non-pelletised material with a particle size below 106 μm to evaluate the full potential for stripping CO2 from a gas feedstock.
3.2. CO2 capture and release efficiency in a fixed bed reactor
As described before, evaluating the CO2 capture capacity with TGA does not provide information on the CO2 capture efficiency from the gas phase, an important parameter for the application of the proposed MgO-based materials. Thus, this section focuses on the performance evaluation under carbonate looping conditions in a fixed bed reactor. Fig. 3a presents the evolution of the CO2 concentration over time at the exit of the reactor when loaded either with the reactive MgCa0.05A0.20 or an inert SiO2 material (blank experiment). The CO2 signal was initially zero, reflecting the time needed for the flow to reach the analyser and for the latter to respond. In the blank experiment with SiO2, the signal gradually increased and stabilised at 30%, the inlet CO2 concentration. In contrast, the CO2 signal was noticeably lower in the experiment with MgCa0.05A0.20, demonstrating the effective CO2 capture from the gas phase. It was possible to divide the profile into three regimes, as inferred from other studies.21,35,40,46 There is an initial period (1st regime) that refers to the time needed for MgO, CaCO3 and CO2 to be dissolved in the molten salts till saturation and enables the precipitation of the first MgCO3 and CaMg(CO3)2 nuclei. During this period, CO2 also reacted with uncoated MgO to form unidentate or bidentate carbonates and this may be the reason behind the extended induction period in the experiment with MgCa0.05A0.20 compared to the inert SiO2 material, as also stated by Zheng et al.40 After some point, the CO2 capture performance accelerated (2nd regime), since the molten phase remains oversaturated with dissolved reactants, leading to the gradual growth of the size of the nuclei, a faster step compared to their initial formation and the saturation of the molten phase. After some time, the CO2 diffusion through the formed carbonate layers becomes the kinetically controlled stage, which is slower compared to the previous stages, leading to the gradual reduction of the attained CO2 capture over time (3rd regime). From an industrial viewpoint, it is preferred to operate before the 3rd regime, where the CO2 capture is high.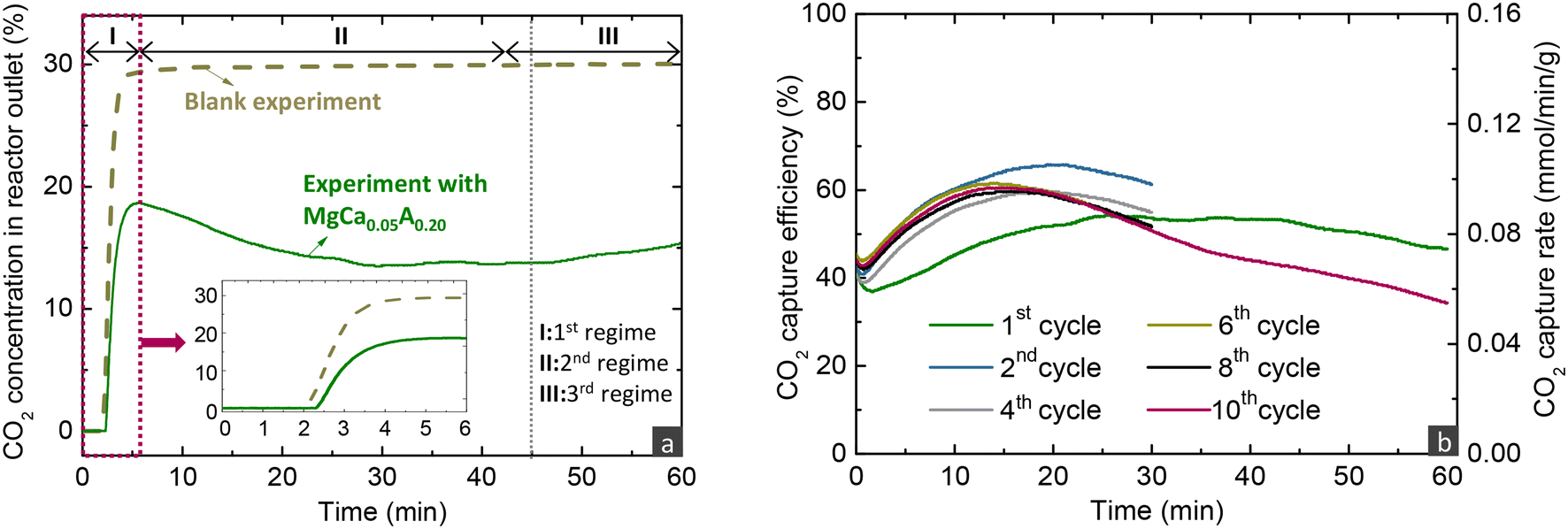 | ||
| Fig. 3 (a) Outlet CO2 concentration over time for the blank experiment with SiO2 and the experiment with the MgCa0.05A0.20 material and (b) CO2 capture efficiency and rate over time for the MgCa0.05A0.20 material during the carbonation stage of different cycles when investigated via carbonate looping experiments in a fixed bed reactor (carbonation stage: 30% CO2/He, 300 °C, 300 h−1; decarbonation stage: 450 °C, 100% He, 720 h−1). The inset in figure (a) is the magnification for the first 6 min. | ||
The recorded CO2 concentration enabled the calculation of the CO2 capture efficiency and rate, as described in the methodology in the ESI.†Fig. 3b illustrates the results from the investigation of the MgCa0.05A0.20 material in 10 consecutive carbonation and decarbonation cycles in a fixed bed reactor containing 4 g of the material. The first and last cycles featured an extended carbonation duration of 60 minutes, while the intermediate cycles lasted 30 minutes each. For simplicity, the data from the first 4 minutes of exposure to CO2 are excluded from the figure. With the exception of the initial cycle, the CO2 capture efficiency and rate in the first and second regimes remained consistent throughout the subsequent cycles, attaining a CO2 capture efficiency and rate of ∼60% and ∼0.09 mmol min−1 g−1, respectively, when exposed to a gas stream with 30% CO2 concentration at 300 °C. The slightly reduced CO2 capture performance in the first cycle indicates a lower initial capture capacity; while in subsequent cycles, the material displayed some self-activation, a phenomenon also noted in the literature.32,33,44
Fig. 4a displays the outlet CO2 concentration over time during the decarbonation stage conducted under a 100% He flow, while increasing the temperature to 450 °C. In realistic applications, decarbonation would occur in a CO2 atmosphere to yield a high-purity CO2 stream, suitable for sequestration or further use. The performance of the studied material under such conditions was investigated in our previous studies.16,33 In the current work, the CO2 release lasted ∼9 min for all cycles, with the duration being prolonged only in the 1st and 10th cycle, due to the extended preceding carbonation. In contrast to MgO carbonation, decarbonation was very fast, due to the applied temperature (450 °C), which surpasses the minimum temperatures that thermodynamically allow the full conversion of MgCO3 or CaMg(CO3)2 compounds (404 and 424 °C as seen in Fig. 1). This implies an enhanced kinetic driving force, which is defined as the difference between the equilibrium CO2 partial pressure and the actual CO2 partial pressure (PCO2,eq − PCO2). Several researchers have mentioned that the molten phase can have a positive effect on the dissociation of carbonate ions and rapid decarbonation of formed carbonates,21,24,25 while may also initiate at lower temperatures compared to non-promoted MgO.32,44
 | ||
| Fig. 4 (a) Outlet CO2 concentration during the decarbonation stage of different cycles and (b) MgO and MgCO3/CaMg(CO3)2 conversions over cycles for the MgCa0.05A0.20 material when investigated via carbonate looping experiments in a fixed bed reactor (carbonation stage: 30% CO2/He, 300 °C, 300 h−1; decarbonation stage: 450 °C, 100% He, 720 h−1). | ||
Fig. 4b depicts the MgO and MgCO3/CaMg(CO3)2 conversions as determined from the carbonation and decarbonation stages, respectively, following the methodology described in eqn S5 and S6 of the ESI.† Similar carbonation and decarbonation conversions were found, indicating that all the CO2 captured in each carbonation stage was completely released in the subsequent decarbonation stage. For cycles with a carbonation duration of 30 min, the MgO conversion reached 20%, while the transition to the 3rd regime had taken place before the end of the carbonation stage. The low conversion suggests that applying MgO-based materials in a realistic process would require a reactor with high solids inventory to attain efficient CO2 capture since after surpassing 20% MgO conversion, the slow CO2 diffusion controls the reaction rate.
It is important to note that the material displayed high stability over cycles. Even though the solids underwent sintering during cycling operation, as verified by employing X-ray diffraction and by comparing the MgO crystal size between fresh and used materials (Fig. S2 and Table S1 in the ESI†), the high stability is, as mentioned before, a result of a possible alkali salt redistribution in the surface of MgO.24,33
3.3. Parametric evaluation for the carbonation stage operation
The effect of the different operating parameters was evaluated by conducting experiments of 10 consecutive carbonation and decarbonation cycles. The figures presented below that illustrate the CO2 capture efficiency and rate over time profiles contain two curves for each parameter investigated, referring to the first and last carbonation stages of each experiment. Even though the first and last cycles were run for an extended duration (60–180 min) compared to other cycles (30 min), the CO2 capture capacity as a function of cycles presented below refer to the CO2 uptake calculated only from the first 30 min of the carbonation stage. | ||
| Fig. 5 (a) CO2 capture efficiency during the carbonation stage of the 1st and 10th cycles and (b) CO2 capture capacity and MgO conversion over cycles for the MgCa0.05A0.20 material when investigated via carbonate looping experiments in a fixed bed reactor with different space velocities for the carbonation stage (carbonation stage: 30% CO2/He, 300 °C; decarbonation stage: 450 °C, 100% He, 720 h−1). | ||
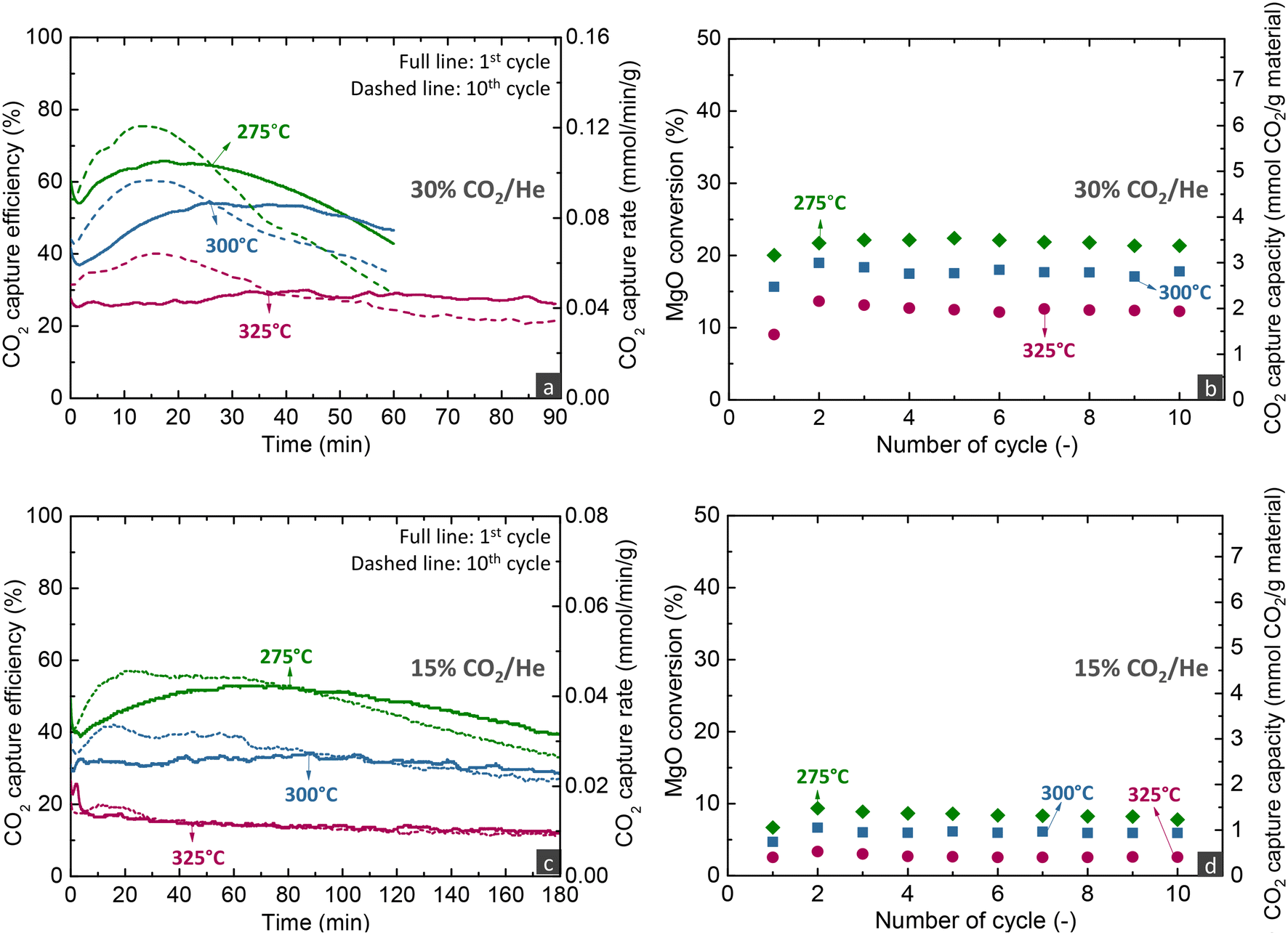 | ||
| Fig. 6 (a) and (c) CO2 capture efficiency and rate during the carbonation stage of the 1st and 10th cycles and (b) and (d) CO2 capture capacity and MgO conversion over cycles for the MgCa0.05A0.20 material when investigated via carbonate looping experiments in a fixed bed reactor with different temperatures and gas feeds for the carbonation stage (carbonation stage: 300 h−1; decarbonation stage: 450 °C, 100% He, 720 h−1). | ||
Temperature has multiple effects on the carbonation mechanism, as discussed in our previous studies,16,33 while the current results of fixed bed reactor experiments confirm our initial findings. Increasing the temperature contributes beneficially to the Arrhenius parameter of the reaction rate, but is detrimental to the driving force of carbonation due to the decrease of the PCO2,eq term. Moreover, temperature affects the solubility of all reactants in the molten phase, with the CO2 solubility decreasing and the MgO and CaCO3 solubilities increasing at higher temperatures.21,47 Nonetheless, MgO and CaCO3 exhibit different relationships between their solubility and temperature, with the solubility of MgO promoted at lower temperatures, while the solubility of CaCO3 increases at higher temperatures. Specifically at 275 °C, due to the lower CaCO3 solubility, the CaMg(CO3)2 phase does not crystallise.16 Thus, at 275 °C, CaCO3 contributed to the faster kinetics only by acting as a carbonate seed, promoting the formation of the first nuclei. It is worth to mention that following the transition to the 3rd regime, the CO2 capture efficiency decreased faster over time at lower temperatures, due to the lower diffusivity of CO2, a phenomenon also seen in CaO-based materials for high-temperature CO2 capture.48
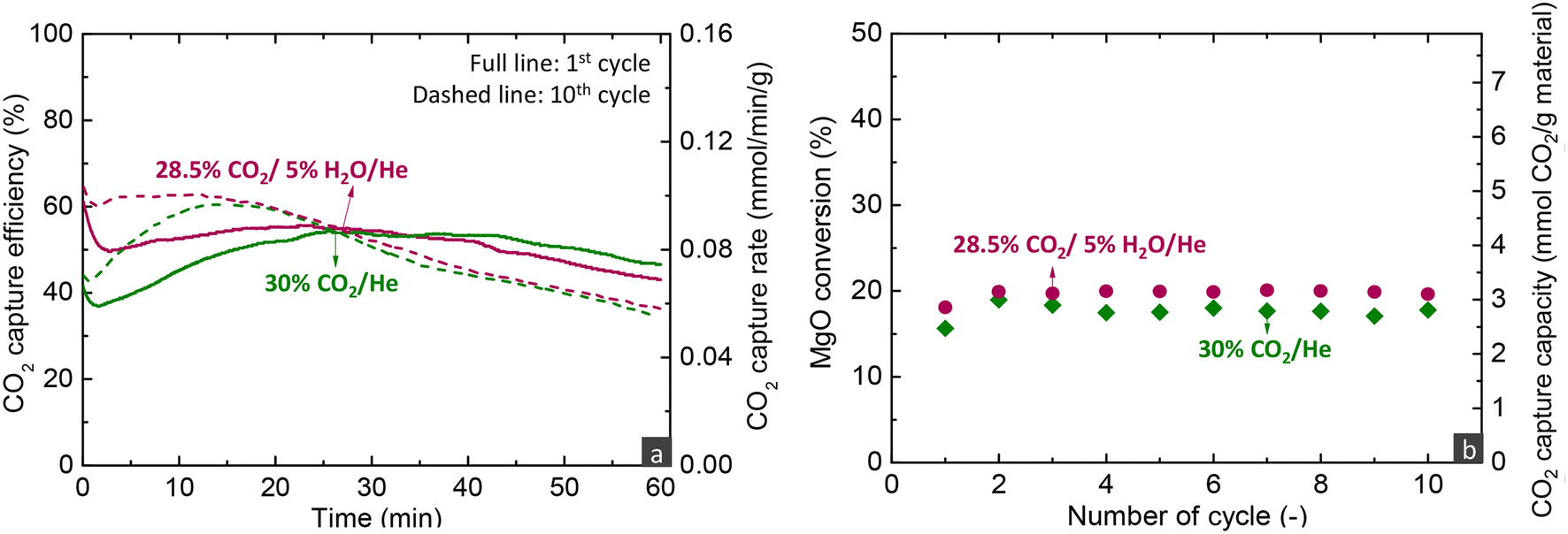 | ||
| Fig. 7 (a) CO2 capture efficiency and rate during the carbonation stage of the 1st and 10th cycles and (b) CO2 capture capacity and MgO conversion over cycles for the MgCa0.05A0.20 material when investigated via carbonate looping experiments in a fixed bed reactor under dry and wet operating conditions for the carbonation stage (carbonation stage: 300 h−1; decarbonation stage: 450 °C, 100% He, 720 h−1). | ||
Overall, the parametric analysis on the carbonation stage revealed that the MgCa0.05A0.20 material can attain 75% CO2 capture efficiency from a gas feed with 30% CO2 at a significantly low temperature of 275 °C, for low MgO conversions (<20%) in the kinetically controlled regime. However, this result was observed with a quite low space velocity (300 h−1), a performance which contrasts sharply the behaviour of CaO-based materials used for CO2 capture at high temperatures (600–750 °C). Our previous studies demonstrated that such materials can attain ∼90% capture from a gas feed with 12% CO2 at 650 °C, while the transition of the kinetically controlled stage to the slow CO2 diffusions occurs after reaching 70% CaO conversion.5,6 Moreover, the CaO-based materials attained this activity in shorter contact times (GHSV > 5000 h−1) than MgO-based materials (300 h−1).
Even though these discrepancies can be attributed to the different reaction mechanisms of the two materials and the faster carbonation kinetics of CaO mostly because of the higher temperature used, they highlight that CaO-based materials exhibit better CO2 stripping efficiency compared to MgO-based materials, which indicates that the former materials are perhaps more suitable for post-combustion CO2 capture applications. However, the promising activity of MgO-based materials and their ability to react with CO2 at intermediate temperatures merit further investigation, particularly in higher operating pressure applications and the intensification of H2-producing technologies operating at similar temperatures to MgO, such as sorption-enhanced water gas shift and steam reforming of methanol.51,52
4. Conclusions
MgO has emerged as a promising material for CO2 capture at intermediate temperatures (275–375 °C). However, its broad application has been limited by its slow carbonation kinetics. Recent research has focused on developing MgO-based materials promoted with alkali salts, which enhance the carbonation kinetics via an alternative reaction mechanism. Despite extensive evaluations of the CO2 capture activity of these materials with thermogravimetry, there has been a lack of consideration for the efficiency of stripping CO2 from a gas feedstock in realistic reactor configurations. In this work, we evaluated a mineral MgO-based material promoted with limestone and Li/Na/K nitrates with molar ratios of CaCO3 and alkali salts to MgO equal to 0.05 and 0.20, respectively, by conducting carbonate looping experiments in a fixed bed reactor. The results showed that carbonation initiates with a high CO2 capture efficiency and rate, followed by a gradual decrease of performance due to the slow diffusion of CO2 though the formed carbonate products. The transition between the fast and slow carbonation was found to occur at very low MgO conversions (<20%), dependent on the CO2 content of the gas feed. The parametric analysis for the carbonation stage revealed that lowering the operating temperature to 275 °C enables ∼75% and ∼60% CO2 stripping efficiency from a 30% and 15% CO2 feed, respectively, while the increase of space velocity and the presence of H2O promote the carbonation rate. Overall, even though the activity of MgO-based materials for post-combustion CO2 capture may be inferior to CaO-based materials, the results of this study highlight the potential of MgO-based materials for CO2 capture and process intensification purposes.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Theodoros Papalas: conceptualisation, formal analysis, investigation, methodology, validation, visualisation, and writing – original draft. Andy N. Antzaras: validation, supervision, and writing – review & editing. Angeliki A. Lemonidou: supervision, writing – review & editing, resources, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been co-financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code: T1EDK-01532).References
- S. Fawzy, A. I. Osman, J. Doran and D. W. Rooney, Environ. Chem. Lett., 2020, 18, 2069–2094 CrossRef CAS.
- IEA, Net zero by 2050: A roadmap for the global energy sector, 2021.
- IEA, Energy Technology Perspectives 2020 – Special Report on Carbon Capture Utilisation and Storage, 2020.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed.
- T. Papalas, A. N. Antzaras and A. A. Lemonidou, Ind. Eng. Chem. Res., 2020, 59, 9926–9938 CrossRef CAS.
- A. N. Antzara, A. Arregi, E. Heracleous and A. A. Lemonidou, Chem. Eng. J., 2018, 333, 697–711 CrossRef CAS.
- J. A. Garcia, M. Villen-Guzman, J. M. Rodriguez-Maroto and J. M. Paz-Garcia, J. Environ. Chem. Eng., 2022, 10, 108470 CrossRef CAS.
- S. A. Theofanidis, A. N. Antzaras and A. A. Lemonidou, Curr. Opin. Chem. Eng., 2023, 39, 1–10 Search PubMed.
- R. Han, Y. Wang, S. Xing, C. Pang, Y. Hao, C. Song and Q. Liu, Chem. Eng. J., 2022, 450, 137952 CrossRef CAS.
- J. Chen, L. Duan and Z. Sun, Energy Fuels, 2020, 34, 7806–7836 CrossRef CAS.
- A. H. Ruhaimi, M. A. A. Aziz and A. A. Jalil, J. CO2 Util., 2020, 43, 101357 CrossRef.
- Y. Hu, Y. Guo, J. Sun, H. Li and W. Liu, J. Mater. Chem. A, 2019, 7, 20103–20120 RSC.
- A. H. Bork, M. Rekhtina, E. Willinger, P. Castro-Fernández, J. Drnec, P. M. Abdala and C. R. Müller, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2103971118 CrossRef CAS.
- K. K. Han, Y. Zhou, W. G. Lin and J. H. Zhu, Microporous Mesoporous Mater., 2013, 169, 112–119 CrossRef CAS.
- M. Bhagiyalakshmi, J. Y. Lee and H. T. Jang, Int. J. Greenhouse Gas Control, 2010, 4, 51–56 CrossRef CAS.
- T. Papalas, I. Polychronidis, A. N. Antzaras and A. A. Lemonidou, J. CO2 Util., 2021, 50, 101605 CrossRef CAS.
- K. Zhang, X. S. Li, Y. Duan, D. L. King, P. Singh and L. Li, Int. J. Greenhouse Gas Control, 2013, 12, 351–358 CrossRef CAS.
- K. Zhang, X. S. Li, W. Z. Li, A. Rohatgi, Y. Duan, P. Singh, L. Li and D. L. King, Adv. Mater. Interfaces, 2014, 1, 1400030 CrossRef.
- A. Dal Pozzo, A. Armutlulu, M. Rekhtina, P. M. Abdala and C. R. Müller, ACS Appl. Energy Mater., 2019, 2, 1295–1307 CrossRef.
- W. Gao, J. Xiao, Q. Wang, S. Li, M. A. Vasiliades, L. Huang, Y. Gao, Q. Jiang, Y. Niu, B. Zhang, Y. Liu, H. He and A. M. Efstathiou, Adv. Mater., 2021, 34, 2106677 CrossRef.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Chem. Mater., 2015, 27, 1943–1949 CrossRef CAS.
- H. A. Lara-García, W. Gao, A. Gómez-Cortés, G. Diaz, H. Pfeiffer and Q. Wang, Ind. Eng. Chem. Res., 2019, 58, 5501–5509 CrossRef.
- A. Landuyt, P. V. Kumar, J. A. Yuwono, A. H. Bork, F. Donat, P. M. Abdala and C. R. Müller, JACS Au, 2022, 2, 2731–2741 CrossRef CAS.
- H. Jeon, M. L. T. Triviño, S. Hwang, J. H. Moon, J. Yoo and J. G. Seo, J. CO2 Util., 2020, 39, 101153 CrossRef CAS.
- H. Lee, M. L. T. Trivinìo, S. Hwang, S. H. Kwon, S. G. Lee, J. H. Moon, J. Yoo and J. G. Seo, ACS Appl. Mater. Interfaces, 2018, 10, 2414–2422 CrossRef CAS.
- X. Zhao, G. Ji, W. Liu, X. He, E. J. Anthony and M. Zhao, Chem. Eng. J., 2018, 332, 216–226 CrossRef CAS.
- A. T. Vu, K. Ho, S. Jin and C. H. Lee, Chem. Eng. J., 2016, 291, 161–173 CrossRef CAS.
- H. Cui, Q. Zhang, Y. Hu, C. Peng, X. Fang, Z. Cheng, V. V. Galvita and Z. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 20611–20620 CrossRef CAS.
- H. Cui, Z. Cheng and Z. Zhou, J. Mater. Chem. A, 2020, 8, 18280–18291 RSC.
- L. Wang, Z. Zhou, Y. Hu, Z. Cheng and X. Fang, Ind. Eng. Chem. Res., 2017, 56, 5802–5812 CrossRef CAS.
- M. Rekhtina, M. Krödel, Y. H. Wu, A. Kierzkowska, F. Donat, P. M. Abdala and C. R. Müller, Sci. Adv., 2023, 9, eadg5690 CrossRef CAS PubMed.
- S. I. Jo, Y. I. An, K. Y. Kim, S. Y. Choi, J. S. Kwak, K. R. Oh and Y. U. Kwon, Phys. Chem. Chem. Phys., 2017, 17, 6224–6232 RSC.
- T. Papalas, A. N. Antzaras and A. A. Lemonidou, J. CO2 Util., 2021, 53, 101725 CrossRef CAS.
- F. Donat and C. R. Müller, Curr. Opin. Green Sustainable Chem., 2022, 36, 100645 CrossRef CAS.
- J. Chen, L. Duan, F. Donat and C. R. Müller, ACS Sustainable Chem. Eng., 2021, 9, 6659–6672 CrossRef CAS.
- P. Teixeira, P. Correia and C. I. C. Pinheiro, Chem. Eng. Sci., 2024, 289, 119856 CrossRef CAS.
- G. Bang, K.-M. Kim, S. Jin and C.-H. Lee, Chem. Eng. J., 2022, 433, 134607 CrossRef CAS.
- Y. Cai, W. Liu, Z. Sun, Y. Yang and P. Li, J. CO2 Util., 2022, 61, 102047 CrossRef CAS.
- K. H. Chai, L. K. Leong, S. Sethupathi, K. C. Chong, T. C. K. Yang, S. P. Ong and Y. H. Yap, Chem. Eng. J. Adv., 2024, 17, 100578 CrossRef CAS.
- Y. Zheng, J. Wu, L. Zhang, Y. Guo, Z. Xu, Y. Huang, P. Huang, J. Zhang and C. Zhao, Chem. Eng. J., 2022, 450, 137944 CrossRef CAS.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- D. Ipsakis, G. Varvoutis, A. Lampropoulos, S. Papaefthimiou, G. E. Marnellos and M. Konsolakis, Renewable Energy, 2021, 179, 1884–1896 CrossRef CAS.
- Y. Hu, X. Liu, Z. Zhou, W. Liu and M. Xu, Fuel, 2017, 187, 328–337 CrossRef CAS.
- S. Jin, K. Ho, A. T. Vu and C. H. Lee, Energy Fuels, 2017, 31, 9725–9735 CrossRef CAS.
- X. Ma, H. Cui, Z. Cheng and Z. Zhou, AIChE J., 2023, 69, e18146 CrossRef CAS.
- W. Gao, M. A. Vasiliades, C. M. Damaskinos, M. Zhao, W. Fan, Q. Wang, T. R. Reina and A. M. Efstathiou, Environ. Sci. Technol., 2021, 55, 4513–4521 CrossRef CAS.
- T. Harada and T. A. Hatton, Chem. Mater., 2015, 27, 8153–8161 CrossRef CAS.
- Y. A. Criado, B. Arias and J. C. Abanades, Ind. Eng. Chem. Res., 2018, 57, 12595–12599 CrossRef CAS.
- S. Jin, K. Ho and C. H. Lee, Chem. Eng. J., 2018, 333, 697–711 CrossRef.
- W. Gao, L. Sun and Q. Wang, in Pre-combustion Carbon Dioxide Capture Materials, ed. Q. Wang, Royal Society of Chemistry, 2018, pp. 61–143 Search PubMed.
- Y. Hu, H. Cui, Z. Cheng and Z. Zhou, Chem. Eng. J., 2018, 377, 119823 CrossRef.
- H. Li, H. Tian, S. Chen, Z. Sun, T. Liu, R. Liu, S. Assabumrungrat, J. Saupsor, R. Mu, C. Pei and J. Gong, Appl. Catal., B, 2020, 276, 119052 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00432a |
| This journal is © The Royal Society of Chemistry 2025 |