
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanogels with tailored hydrophobicity and their behavior at air/water interfaces†
Ruiguang
Cui‡
a,
Maret
Ickler‡
b,
Johannes
Menath
 b,
Nicolas
Vogel
*b and
Daniel
Klinger
*a
b,
Nicolas
Vogel
*b and
Daniel
Klinger
*a
aInstitute of Pharmacy, Freie Universität Berlin, Königin-Luise-Str. 2-4, 14197 Berlin, Germany. E-mail: daniel.klinger@fu-berlin.de
bInstitute of Particle Technology, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany. E-mail: nicolas.vogel@fau.de
First published on 27th November 2024
Abstract
The interfacial behavior of micro-/nanogels is governed to a large extent by the hydrophobicity of their polymeric network. Prevailing studies to examine this influence mostly rely on external stimuli like temperature or pH to modulate the particle hydrophobicity. Here, a sudden transition between hydrophilic and hydrophobic state prevents systematic and gradual modulation of hydrophobicity. This limits detailed correlations between interfacial behavior and network hydrophobicity. To address this challenge, we introduce a nanogel platform that allows accurate tuning of hydrophobicity on a molecular level. For this, via post-functionalization of active ester-based particles, we prepare poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA) nanogels as a hydrophilic benchmark and introduce gradually varied amounts of hydrophobic propyl or dodecyl moieties to increase the nanogel hydrophobicity. We study the deformation and arrangement of these particles at an air/water interface and correlate the results with quantitative measures for nanogel hydrophobicity. We observe that increasing hydrophobicity of nanogels, either by increasing the hydrophobic moiety ratio or the alkyl chain length, leads to decreased particle deformability and aggregation of an interfacially-adsorbed monolayer. Contrary to what may be intuitively assumed, these changes are not gradual, but rather occur suddenly above a threshold in hydrophobicity. Our study further shows that the effect of hydrophobicity affects the nanogel properties differently in bulk and when adsorbed at liquid interfaces. Thus, this study establishes the transition of interfacial behavior between soft gel-like particles to a solid spherical morphology triggered by the increase in hydrophobicity.
Introduction
Nano-/microgels are crosslinked polymer networks that can be swollen by solvents1 and are potentially useful as drug delivery systems,2 microreactors,3 biosensors,4,5 or emulsion stabilizers.6–8 Nano-/microgels are well known to adsorb efficiently to air/water interfaces9,10 and serve as interesting model systems to study the effect of particle softness on interfacial adsorption11,12 and self-assembly.13 Especially, the assembly of the nano-/microgels at interfaces can be used to stabilize emulsions and foams14–20 or to transfer a particle monolayer to a substrate for surface patterning.21–25The interfacial morphology of such soft particles is mostly governed by two important parameters: the network structure and the chemical composition of the polymers forming the network. The network structure depends to a large extent on the colloidal preparation method and the crosslinking density. When using conventional batch precipitation reactions for nano-/microgel preparation, a core–corona morphology is often observed as the result of a cross-linking gradient, because the crosslinker reacts faster than the monomer.13,26 Nano/microgels adsorb to the liquid interfaces as their constituent polymer chains are typically surface active. The particles subsequently stretch out at the interface under the influence of surface tension.10,13 In particular, the less crosslinked outer region spreads at the interface, while the more crosslinked core deforms less, leading to a “fried-egg” or core–corona structure.27–29 Especially nano-/microgels with a lower crosslinking density become softer and deform more at the interface due to long dangling chains and less resistance of the network to elongation, forming a characteristic expanded corona structure.29–32
In addition, the nano-/microgels’ interfacial behavior depends on the chemical composition of the network polymer or copolymer.33 Especially the corresponding particle hydrophobicity governs their interaction with the polar water phase and the non-polar air. This parameter is mostly controlled through external stimuli that change the network hydrophobicity, like by variations in temperature34 or pH.35 For example, poly(N-isopropylacrylamide) (PNIPAm) nano-/microgels can transition from a swollen state at low temperatures to a collapsed state above their volume phase transition temperature (VPTT) around 32 °C.9 This behavior results from the inherent amphiphilicity of PNIPAm's repeat units. In bulk water, the contraction of PNIPAm at high temperatures is driven by aggregation of pendant hydrophobic isopropyl groups, which leads to an entropy gain. At lower temperatures, hydrogen bonding of water is energetically preferable and leads to swelling of the polymer network.36–38 Conversely, at the water surface, only the submerged sections of the nano-/microgels shrink above the VPTT, whereas the chains at the interface remain non-responsive.27,34,39,40 This loss of responsiveness at the air/water interface is due to the orientation of hydrophobic moieties towards the air phase. Here, the absence of surrounding water molecules diminishes the entropic effect.27 This pronounced discrepancy between the behavior of collapsed microgels in the bulk and at interfaces underlines the importance of studying the effects of hydrophobicity within soft, water-swellable nano-/microgels.
Up to now, the majority of studies on nano-/microgels at air/water interfaces have focused on such temperature-responsive PNIPAm systems.27,34 However, in these cases, the transition between hydrophilic and hydrophobic state occurs suddenly, i.e., upon crossing the VPTT. This “all or nothing” behavior prevents a gradual modulation of hydrophobicity, thus hindering a systematic correlation between network hydrophobicity and the interfacial properties. Consequently, an experimentally more direct approach is required to systematically vary the colloidal hydrophobicity. This could be realized through controlling the chemical composition of the network copolymer. One approach is varying the number and length of hydrophobic side chains within the network,41,42 which, however, is experimentally difficult. In conventional precipitation polymerizations, the composition of the network can only be varied through the used (co-)monomers, which requires the copolymerization of hydrophilic and hydrophobic monomers.43 However, this may change the polymerization kinetics, can produce gradients, and may affect crosslinker distribution.44–48 Another approach is to introduce hydrophobic monomers to hydrophilic nano-/microgels resulting in nano-/microgels infiltrated with hard nanoparticles via seeded emulsion polymerization, which can lead to more inhomogeneous internal particle architectures.49–51 As a result of both approaches, the hydrophobicity of the network is coupled to other nano-/microgel features and different microgels are difficult to compare.
To overcome this challenge and provide nanogels (NGs) with comparable architecture but changing hydrophobicity, we control the amount of hydrophilic and hydrophobic groups after particle synthesis. For this, we use reactive precursor colloidal particles, whose internal network can be functionalized with the desired side groups via active ester chemistry. This post-functionalization protocol guarantees control over the chemical composition of the network while keeping the colloidal features of the produced nanogels similar to the precursor particles.52,53 In our former work, we have shown that in such nanogels, hydrophobic moieties such as cholesteryl or dodecyl groups can form ordered internal hydrophobic nanodomains53 and exhibit higher Young's moduli.54 Based on these studies, we suggest that similar particles represent suitable platforms to investigate the effects of hydrophobicity on the nanogels’ behavior at air/water interface.
Using our synthetic platform, we synthesize a library of nanogels with varied hydrophobicity but consistent colloidal features. We systematically vary the hydrophobicity of the nanogels by tuning the ratio between hydrophilic 2-hydroxyl propyl groups and different hydrophobic groups, i.e., either short propyl groups or long dodecyl chains. For the resulting nanogels, we investigate the position and deformation at the air/water interface as a function of hydrophobicity. We use in situ gel-trapping and ex situ atomic force microscopy to analyze the morphologies. We correlate the observed differences in interfacial properties with the nanogels’ compressibility at the interface, studied by compression curves on a Langmuir trough. Furthermore, we show that hydrophobicity changes the surface patterns that are created when interfacially-adsorbed particles are transferred to a solid substrate.
Materials and methods
Materials
Amino-2-propanol (HPA), acryloyl chloride and ethylene glycol dimethacrylate (EGDMA) were obtained from Sigma Aldrich. Acryloyl chloride was purified via vacuum distillation. EGDMA was filtered through a short Al2O3 column to remove the inhibitor prior to use. 2,6-Lutidine, ammonium persulfate (APS) and sodium dodecyl sulfate (SDS) were purchased from Merck. Pentafluorophenol (PFP) was purchased from fluorochem. Triethylamine (TEA) was purchased from abcr GmbH. Propylamine (PROPA) was purchased from TCI. Dodecyl amine (DODA) was purchased from Thermo Scientific. Kelcogel® gellan gum was provided by CPKelco and sodium chloride purchased from Honeywell chemicals. Sylgard 184 (polydimethylsiloxane (PDMS) gel along with its curing agent) was purchased from Dow Corning. All chemicals were used as received unless noted otherwise. De-ionized (DI) water was obtained from LaboStar TWF systems (18.2 MΩ cm, Siemens Ultrapure Water Systems).Synthesis of PROPA and DODA nanogels
We synthesized PROPA and DODA nanogels through the functionalization of preformed precursor particles.52,54,55 (1) The monomer pentafluorophenyl methacrylate (PFPMA) was synthesized according to a literature procedure.56 (2) We synthesized the precursor nanoparticles (PPFPMA) via emulsion polymerization according to modified literature procedures.6,52 Typically, 256.8 mg (0.890 mmol) SDS as surfactant were added into a 250 mL flask, followed by adding a mixture of 21 g monomer PFPMA (83.3 mmol) and 153.4 μL (0.083 mmol) crosslinker EGDMA. Then 210 mL water were added to the flask to obtain the final SDS concentration of 4 mM. The flask was placed in a 60 °C oil bath and deoxygenated by bubbling with nitrogen for 90 min. After that, 2 mL deoxygenated aqueous solution of 126.6 mg mL−1 APS (1.1 mmol in total), were added to the system to initiate the reaction, which was allowed to proceed for 5 days. Then PPFPMA particles were purified by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min and washing with water, repeated 4 times. Finally, the water dispersion with precursor particles was freeze-dried to give a white powder with a yield of 75% (16 g). (3) PROPA and DODA nanogels were prepared by post-functionalizing PPFPMA particles according to the literature.52 Typically, 2 g (7.9 mmol PFPMA units) PPFPMA precursor powder were dispersed in 150 mL DMF using a ultrasonication bath. Then, a defined amount of propylamine (PROPA) or dodecyl amine (DODA) was dissolved in 25 mL DMF in a vial, followed by adding the corresponding amount of 2-hydroxylpropyl amine (HPA), as summarized in Tables 1 and 2. The amine mixture was then added into the flask with the PPFPMA precursor dispersion, after which 3.310 mL TEA (23.64 mmol) were added. The mixture was left to react for 6 days under stirring at 40 °C (PROPA), and at 60 °C (DODA).
000 rpm for 20 min and washing with water, repeated 4 times. Finally, the water dispersion with precursor particles was freeze-dried to give a white powder with a yield of 75% (16 g). (3) PROPA and DODA nanogels were prepared by post-functionalizing PPFPMA particles according to the literature.52 Typically, 2 g (7.9 mmol PFPMA units) PPFPMA precursor powder were dispersed in 150 mL DMF using a ultrasonication bath. Then, a defined amount of propylamine (PROPA) or dodecyl amine (DODA) was dissolved in 25 mL DMF in a vial, followed by adding the corresponding amount of 2-hydroxylpropyl amine (HPA), as summarized in Tables 1 and 2. The amine mixture was then added into the flask with the PPFPMA precursor dispersion, after which 3.310 mL TEA (23.64 mmol) were added. The mixture was left to react for 6 days under stirring at 40 °C (PROPA), and at 60 °C (DODA).
| HPA | Propylamine | |||||
|---|---|---|---|---|---|---|
| Volume [mL] | Mass [g] | n [mmol] | Volume [mL] | Mass [g] | n [mmol] | |
| PROPA0 | 1.873 | 1.787 | 23.8 | 0 | 0 | 0 |
| PROPA10 | 1.686 | 1.608 | 21.4 | 0.196 | 0.141 | 2.4 |
| PROPA20 | 1.498 | 1.429 | 19.0 | 0.393 | 0.283 | 4.8 |
| PROPA30 | 1.311 | 1.251 | 16.7 | 0.589 | 0.424 | 7.1 |
| PROPA40 | 1.124 | 1.072 | 14.3 | 0.786 | 0.565 | 9.5 |
| HPA | Dodecyl amine | ||||
|---|---|---|---|---|---|
| Volume [mL] | Mass [g] | n [mmol] | Mass [g] | n [mmol] | |
| DODA10 | 1.686 | 1.608 | 21.4 | 0.438 | 2.4 |
| DODA40 | 1.124 | 1.072 | 14.3 | 1.753 | 9.5 |
The resulting nanogels were purified by dialysis against DMF, changing solvent every two days for five times in total and subsequently against water, changing the water phase three times a day for another week. The nanogel dispersion was concentrated by centrifugation at 10000 rpm for 2 hours and redispersion in water. The final concentration was gravimetrically determined by lyophilizing aliquots of the nanogel dispersion. The final required concentration was then adjusted by dilution with water. Before usage for air/water interfacial behavior studies, the nanogels were purified by washing 3 times via centrifugation and redispersion. Here, for washing 1:1 water/ethanol mixture was used for PROPA0–PROPA40 and DMF for DODA10 and DODA40. In the final cycle, all particles were washed and redispersed in water.
Synthesis of PROPA and DODA polymers
To prepare samples for the contact angle measurements and solubility measurements, we synthesized the respective non-crosslinked PROPA and DODA polymers. For these polymers, we first synthesized the PFPMA precursor particles without crosslinker while keeping other synthetic parameters the same. Afterwards, the same feed ratios of reagents and the same reaction conditions as for the crosslinked nanogels were used to prepare the linear PROPA and DODA polymers.Solubility of PROPA polymers
The solubility of the respective PROPA polymers in water was measured as such: 50 mg PROPA10 to PROPA40 polymers were added to individual centrifuge tubes. Subsequently, 1 mL water was added to each tube, and the samples were stirred for one day. The solubility of polymers was measured after centrifugation of the samples at 5000 rpm for 10 minutes to separate the supernatant and the possible sediment. The supernatants and the sediments were then freeze-dried to gravimetrically determine the solubility of polymers in water.Calculation of Flory–Huggins parameters of polymers in water and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P values
P values
Flory–Huggins parameters of polymers and water indicate the affinity between polymers and water and were calculated according to previously developed methods as described in the literature.6
The logP value is the decadic logarithm of the partition coefficient (P) between water and hydrophobic 1-octanol and can indicate the hydrophobicity of a compound. Herein, we calculated logP values with the software MOE 2022.02 (Molecular Operating Environment; Chemical Computing Group ULC, Montreal, QC, Canada).57,58 As input, logP(o/w) values of the corresponding DODA or PROPA polymers with 10 repeating units were predicted with the ‘Calculate Descriptors’ function (class: 2D, code: logP(o/w)).
Gel trapping experiments
To enhance the observation of nanogel deformation and positioning at the air/water interface, we employed the gel trapping technique that immobilizes the nanogels within a gel matrix, modifying a method described by Paunov et al. in the literature.59 In brief, 0.1 wt% gellan gum was dissolved in water at 80 °C and mixed with 0.3 wt% sodium chloride. Subsequently, a nanogel dispersion in a 1:1 volume ratio water/ethanol mixture was added to the surface of this solution by a pipette. As the solution cooled down to room temperature, the water phase transformed into a gel. Subsequently, an approximately 5 mm thick PDMS gel layer was created by blending PDMS base and curing agent in a 10:1 weight ratio and gently pouring it over the gellated gellan gum layer. This assembly was then left in a fume hood for 48 hours. Afterwards, the PDMS gel was carefully removed, and any residual gellan gum was washed off with hot water. The initial positions of the nanogels in the water phase, showing up as protrusions from the PDMS were determined using Atomic Force Microscopy (AFM) on the PDMS gel surface. The initial positions in air were identified by the cavities left within the PDMS gel after the nanogels were removed via tape peeling.
Characterization
The hydrodynamic diameters of the nanogels were measured using dynamic light scattering (DLS) with laser wavelength of 638 nm at the scattering angle of 90° (Nicomp Nano Z3000 Gauss system). Transmission electron microscopy (TEM) images were obtained by the scanning transmission electron microscopy (STEM) mode with the acceleration voltage of 30 kV and current of 10 μA (Hitachi FE-SEM SU8030). Scanning electron microscopy (SEM) images were measured using a Zeiss Gemini 500 and a voltage of 1 kV. Fourier-transform infrared spectroscopy (FT-IR) spectra were measured over the range of 4000 to 650 cm−1 (Shimadzu IRSpirit QATR-S ATR FT-IR system). Atomic force microscopy (AFM) images were obtained by using a JPK Nano Wizard and Anfatec NSC 18 cantilevers. The contact angle measurements were conducted with the Dataphysics OCA 20 instrument using the sessile drop method, where a 5 μL water droplet was deposited to the polymer film and was allowed to equilibrate to obtain the contact angle.Interfacial characterization
We applied the simultaneous compression and deposition method as described by Rey et al.31,60,61 We used a Teflon Langmuir–Blodgett trough (KSV NIMA) (area = 243 cm2, width = 7.5 cm) with Delrin barriers. We filled the trough with Milli-Q water, cleaned the water surface by suction with a peristaltic pump and measured the surface pressure using a Wilhelmy plate. The nanogel dispersion in water was mixed with an equal volume of ethanol before spreading. The dispersion was spread dropwise using a 100 μL pipette and equilibrated for 20 min. Upon start of the experiment, the barriers were closed at a speed of 4 mm min−1. The monolayers were deposited with a dipping speed of 0.8 mm min−1 to 5 × 1 cm2 N-type silicon wafers (LG Silicon Inc.), which were previously cleaned with ethanol and oxygen plasma (Diener). The substrate was mounted to the dipper at a 45° angle. The AFM measurements for nanogels at low surface pressure were conducted on dried particles that were transferred from the air/water interface at surface pressure of around 5 mN m−1 onto a solid substrate. The nearest neighbor distance between nanogels were obtained by analyzing the SEM images with over 100 nanogels.Results and discussion
Synthesis and characterization of nanogels: influence of network composition on hydrophobicity
To study the influence of nanogel hydrophobicity on their interfacial properties, we first prepared a library of nanogels with varying hydrophobicity, namely, PROPA nanogels and DODA nanogels (Fig. 1a). The nanogel networks are composed of amphiphilic copolymers featuring hydrophilic moieties (2-hydroxyl propyl amide groups—HPA) and hydrophobic moieties (propyl amides for PROPA nanogels and dodecyl amides for DODA nanogels). Thus, we suggest that the hydrophobicity of the nanogels is defined by the molar ratio of the hydrophobic moieties. This is represented by the number in the sample name, e.g., PROPA10 nanogels contain 10 mol% propyl amide side groups and 90 mol% 2-hydroxy propyl amide side groups. Importantly, PROPA0 nanogels do not contain any hydrophobic PROPA or DODA moieties, thus the polymer backbone structure equals the well-known hydrophilic polymer poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA). We utilize PHPMA as a benchmark to assess the influence of hydrophobic moieties, PROPA or DODA, on the nanogel properties. In the following, we will refer to these PHPMA nanogels as PROPA0 or DODA0 to simplify the comparison with the nanogels with various amount of PROPA or DODA moieties.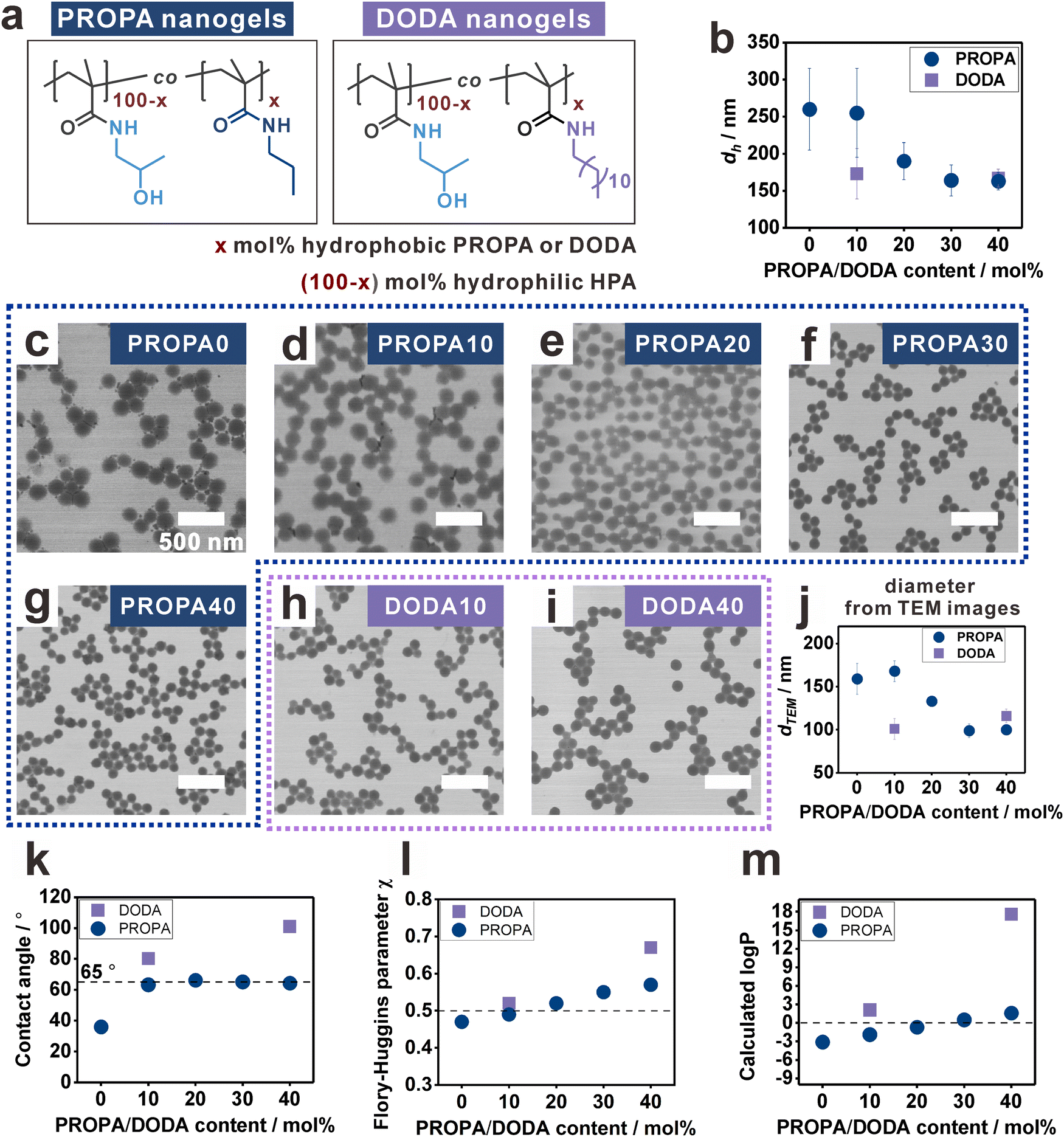 | ||
| Fig. 1 Functionalization of precursor particles gives access to well-defined nanogels with narrow size distribution, homogeneous morphology and controlled hydrophobicity. (a) Nanogel hydrophobicity can be varied by changing the content of hydrophobic PROPA and DODA groups in the amphiphilic network copolymers. Note that for our nomenclature, PROPA0 equals DODA0, because the polymer backbone structure of PROPA0 or DODA0 is the same as the hydrophilic polymer poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA). (b) Hydrodynamic diameters of the nanogels decrease with an increasing network hydrophobicity, i.e., an increasing PROPA/DODA content. (c)–(i) TEM images of PROPA and DODA nanogels show well-defined particles with homogenous spherical morphology and narrow size distribution. The scale bar is 500 nm for all images. (j) Diameters of the nanogels calculated from TEM images decrease with increasing PROPA/DODA content, thus suggesting a decreasing network flexibility with increasing hydrophobicity. (k)–(m) Network hydrophobicity can be controlled by the PROPA/DODA content. An increasing amount of hydrophobic groups increases the hydrophobicity as shown by: (k) contact angles of films from PROPA/DODA polymers, (l) calculated Flory–Huggins parameters between PROPA/DODA polymers and water, and (m) calculated logP values of the PROPA/DODA polymers. | ||
All nanogels were synthesized via a post-functionalization protocol,52,54i.e., reactive pentafluorophenyl ester moieties in the precursor particles were replaced by hydrophilic and hydrophobic amines in defined molar ratios (Fig. S1, ESI†).55 The complete substitution of reactive groups by amines was verified by FT-IR spectra (Fig. S1, ESI†). This post-functionalization protocol thus provides a library of nanogels with varying hydrophobicity while keeping similar colloidal features such as crosslinking density and size distribution. Notably, this post-functionalization method has been proven to be effective and quantitative, resulting in nanogel compositions that match the input ratios.6,52
All PROPA and DODA nanogels exhibit a homogenous spherical morphology with narrowly dispersed sizes. This is evidenced by their hydrodynamic diameters (dh) (Fig. 1b) and their TEM images (Fig. 1c–i). For PROPA nanogels, dh decreases with increasing amount of hydrophobic PROPA moieties (Fig. 1b). Notably, this decrease is not linear: while PROPA0 and PROPA10 show comparable hydrodynamic diameters around 250 nm (Fig. 1b), dh of PROPA20 decreases to 190 nm, and PROPA30 and PROPA40 show a further decrease in diameter to 160 nm (Fig. 1b). This value is close to the diameter of the precursor particles (156 ± 12 nm), thus indicating that these more hydrophobic particles do not significantly swell in bulk water. For DODA-functionalized nanogels, the decrease in hydrodynamic diameter is more pronounced and occurs more rapidly: DODA10 already shows a much smaller diameter (173 ± 34 nm) than PROPA10, and dh does not change significantly for increasing the DODA content to DODA40 (167 ± 12 nm). Thus, both DODA nanogels swell as little as the most hydrophobic PROPA-based nanogels (PROPA30–40).
In addition to their size in aqueous dispersion, we calculated the diameters of the dry nanogels by TEM image analysis (Fig. 1j and Fig. S2, ESI†). It can be seen that these values follow a similar trend as the hydrodynamic diameters. PROPA0–PROPA10 are in the range of 160 nm, PROPA20 around 130 nm, and PROPA30–PROPA40 around 100 nm. Again, the sizes of both DODA nanogels are similar to the more hydrophobic PROPA30 and PROPA40 nanogels, with DODA10 at around 100 nm and DODA40 at around 115 nm. The larger size of more hydrophilic nanogels from TEM measurement should be due to their more flexible network and flattening on the TEM grids.54 Overall, the decrease of nanogel size and dh suggests that the degree of swelling and the flexibility of the network depend on the amount and length of hydrophobic alkyl side chains., i.e., on the network hydrophobicity. However, clear differences between the PROPA and the DODA nanogels are visible: for PROPA-based nanogels, a rather sudden change in hydrophilicity is observed between PROPA10 and PROPA30. In contrast, DODA-based nanogels already exhibit a hydrophobic, collapsed character with the lowest amount of DODA (10%).
To systematically examine these influences, the hydrophobicity of the PROPA and DODA nanogels should be quantified, i.e., connecting a specific network composition to a numerical measure for hydrophobicity. For this, we focused on static contact angles as a measure for hydrophobicity (Fig. 1k). Since direct measurement on nanogel films could lead to artifacts due to surface roughness,62 here we prepared spin-cast films from linear PROPA and DODA polymers as non-crosslinked analogues to the nanogel networks (see Materials and methods section for synthetic details). The contact angle of PROPA0 is 36 ± 0°, indicating that PROPA0 is hydrophilic. With an increasing amount of propyl chains, the contact angle increases to 63 ± 1° for PROPA10 but keeps almost constant from PROPA10 to PROPA40 with a value near 65°. In contrast, introduction of dodecyl groups has a more pronounced influence on the hydrophobicity, with contact angles of 80 ± 1° for DODA10 and 101 ± 1° for DODA40.
To interpret the contact angle values, it can be stated that, generally, a surface with a contact angle above 90° is defined as hydrophobic.63–65 This would mean that only the DODA40 sample is actually hydrophobic. However, the small hydrodynamic diameters of DODA10, PROPA20, and PROPA30 are in the same range as the dh of DODA40. Apparently, in these nanogels the hydrophobic character already dominates as well. This can be explained by the “Berg limit”, which refers to a contact angle of around 65° that has also been discussed as the threshold for hydrophobicity in biological systems.66,67 In this context, the contact angles of PROPA10 to PROPA40 polymer films all reside near the “Berg limit”, thus suggesting a hydrophobic character of all macroscopic PROPA films. In contrast, the bulk swelling behavior of the nanogels shows a clear difference between PROPA10 and PROPA40. These differences underline the different effect of hydrophobic groups in a hydrophobic environment (i.e. air) and a hydrophilic environment (i.e. water).
Since the contact angles of PROPA10 and PROPA40 do not sufficiently differentiate their hydrophobicity, we conducted a solubility evaluation of PROPA10 to PROPA40 polymers in water to further study the hydrophobicity differences. We added 1 mL water to 50 mg of the corresponding PROPA polymers and stirred them for one day. From the optical images (Fig. S3, ESI†), we observed that the PROPA10 polymer dispersion is completely transparent, indicating its complete dissolution in water, while PROPA20 dispersion is opaque and only slightly turbid. In contrast, PROPA30 and PROPA40 polymer dispersions show very high turbidity. The quantitative solubility in water verifies the observed trend, with the values of solubility decreasing from PROPA10 (>50 mg) to PROPA20 (23 mg mL−1) and finally to almost zero for PROPA30 (1 mg mL−1) and PROPA40 (2 mg mL−1). These solubility measurements clearly indicate the transition of hydrophobicity between PROPA10 and PROPA30.
Moreover, we calculated the Flory–Huggins parameters (FHP) between polymers and water to assess the hydrophobicity. Generally, the FHP between polymer and a solvent indicates their affinity, where a value smaller than 0.5 signifies that the polymer is soluble, while a value larger than 0.5 suggests incompatibility (i.e. that the polymer is insoluble). Our calculated FHP between PROPA/DODA polymers and water increase with the propyl and dodecyl content (Fig. 1l). The results suggest that PROPA0 is hydrophilic with FHP < 0.5, while PROPA30, PROPA40 and DODA40 are hydrophobic with FHP > 0.5. PROPA10, PROPA20 and DODA10 are at the boundary of transition from hydrophilic to hydrophobic.
As an alternative measure, we also calculated the logP values of all polymers (Fig. 1m). Generally, the logP value is the logarithm of the partition coefficient (P) of a compound in a hydrophobic solvent and water. If the value is smaller than 0, the compound is hydrophilic. If it is larger than 0, it is hydrophobic. Our calculated logP values indicate that PROPA0 and PROPA10 are hydrophilic, while PROPA40, DODA10, and DODA40 are hydrophobic. PROPA20 and PROPA30 are transitioning from hydrophilic to hydrophobic.
The slight difference between the FHP and logP results arise from their different calculation methods and considerations. Note that the slopes for FHP and logP vs. PROPA/DODA content are steeper for the DODA samples than for the PROPA samples, thus suggesting a stronger influence of the longer dodecyl chains on the hydrophobicity. The slower increase in the slopes of the FHP and logP curves against propyl content suggests that propyl chains exert a subtle and consistently gradual control over the hydrophobicity of the polymers.
Overall, we showed that the hydrophobicity of the nanogels can be controlled by changing the molecular structure of the network, i.e., the copolymer composition. Here, the longer dodecyl side chains strongly influence nanogel hydrophobicity. Already small molar ratios of 10 mol% DODA lead to hydrophobic characteristics, as evidenced by all analysis methods shown in Fig. 1. For the shorter propyl side chains, our analyses suggest that the transition from hydrophilic to more hydrophobic characteristics occurs more gradually and can be observed between PROPA10 and PROPA30. PROPA20 seems to be in between these two extremes.
Deformation of nanogels on a surface: influence of network composition on elasticity
Having established a correlation between network composition and particle hydrophobicity, we examine the structural properties of the nanogels as these govern the nanogels deformability. This becomes important at liquid/liquid and liquid/air interfaces, where the stretching of nanogel networks is determined by the interplay between surface tension and their bulk elasticity.30,68,69 We assume that the elasticity strongly depends on the hydrophobic groups in the nanogels’ network. This assumption is based on our previous work, where we showed that in an aqueous dispersion, pendant hydrophobic groups form hydrophobic nanodomains in the nanogels’ interior.53 It is known that such domains act as additional physical crosslinks with an increasing number of hydrophobic interactions leading to more rigid nanogels due to a higher overall crosslinking density.41,42,53,54 To examine this proposed effect, we determine the deformation of PROPA and DODA nanogels via AFM. These experiments were performed on dried particles transferred from the air/water interface onto a solid substrate (see the interfacial characterization part in the Materials and methods section for Experimental details).Representative AFM height images are shown in Fig. 2a–f for PROPA0–PROPA40 and DODA10. To determine the deformation, we quantify both width and height from AFM line scans and average 10 nanogels for each curve (Fig. 2g). This analysis shows that nanogels with low PROPA contents exhibit a large deformation: PROPA0 and PROPA10 have small heights of around 30 nm and large widths of 275 nm. Increasing the PROPA content leads to an increasing height and a slightly smaller width, e.g., PROPA20 exhibits a height of 43 ± 3 nm and a width of 261 ± 8 nm. Further increasing the PROPA content causes a sudden change in height and width: PROPA30 and PROPA40 both exhibit a much larger height of 103 ± 6 nm and 95 ± 6 nm, respectively, and a corresponding width of 167 ± 10 nm and 160 ± 14 nm. Thus, we conclude that upon increasing the PROPA content from PROPA10 to PROPA30, the nanogels transit from a flexible and highly deformed state to a more rigid and spherical morphology. This transition range correlates with the change of dh observed in the bulk dispersion and the macroscopic contact angles (Fig. 1).
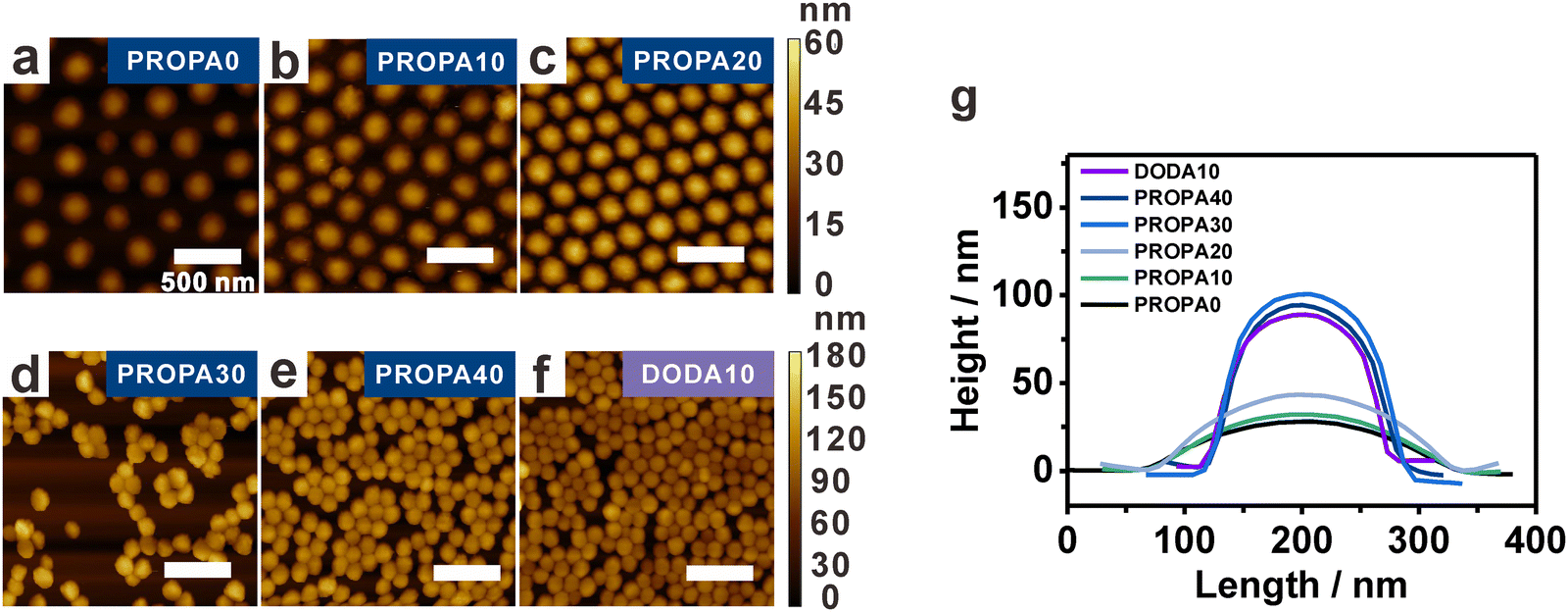 | ||
| Fig. 2 AFM height measurements of nanogels on a solid substrate show that nanogel deformability depends on their hydrophobicity, i.e., the PROPA and DODA content. (a)–(f) AFM height images of PROPA0–PROPA40 and DODA10 show differences in particle deformation. Images were taken on nanogels that were transferred to a silicon wafer from the air/water interface at low surface pressures. (g) AFM height profiles of the respective nanogels show that nanogel deformability decreases with increasing PROPA and DODA content. All scale bars are 500 nm. | ||
For DODA nanogels, even with a low DODA content of 10 mol% (DODA10), the nanogel profile already suggests a high rigidity with a height of 89 ± 5 nm and width of 156 ± 8 nm. These values are similar to the most hydrophobic PROPA30 and PROPA40 nanogels, corroborating the bulk properties which show a small hydrodynamic diameter. Clearly, the dodecyl chains are efficient in rendering the nanogel hydrophobicity, thus causing additional physical crosslinking that limits swelling and deformation. This stronger influence on the nanogel's rigidity from dodecyl side chains than from propyl side chains is consistent with the corresponding higher contact angles, larger FHP values and larger logP values as discussed above.
Gel-trapping to examine the 3D deformation of nanogels at the air/water interface
The results above describe the deformation of the nanogels on a substrate, thus giving access to the elasticity of the individual nanogels. On the air/water interface, however, the elasticity competes with the interfacial tension, which can lead to a different deformation behavior. Therefore, to visualize the position and shape of the different particles directly at the air/water interface, we used the gel-trapping technique,59 as illustrated in Fig. 3a. This technique involves spreading the nanogel dispersion in a water/ethanol mixture on the surface of a gellan gum aqueous solution. This aqueous solution was liquid upon spreading, and thus resembled a pure water phase.59 However, upon cooling from 80 °C to room temperature, the solution solidified, trapping the nanogels on the surface of a gel matrix. Subsequently, a polydimethylsiloxane (PDMS) precursor was carefully added on top of the gelled water phase and hardened by crosslinking. Finally, the gellan gum gel was removed, leaving the interfacial nanogel layer adsorbed within the PDMS film. Thus, using AFM imaging, the initial interfacial position of the nanogels can be characterized. | ||
| Fig. 3 Gel trapping experiments show that the hydrophobicity of nanogels determines their deformation at the air/water interface. (a) Schematic of the gel trapping protocol: 1st, preparation of a gellan aqueous solution. 2nd, spreading nanogel dispersion onto the surface. 3rd, solidification of gellan into a gel matrix. 4th, application and solidification of PDMS on the surface. 5th, AFM measurement of nanogels after cleaning the PDMS surface. (b) Representative height profiles of PROPA0–PROPA40 show that protrusion into the water phase increases with nanogel hydrophobicity, with the exception of PROPA0, which also shows a pronounced protrusion due to increased swelling. (c) Height-to-length ratio of PROPA nanogels extracted from the AFM measurements show that nanogel deformation decreases with increasing PROPA content. (d) Illustration of different deformation of PROPA0 vs. PROPA40: more hydrophobic nanogels deform less than hydrophilic nanogels at air/water interface. | ||
AFM height images taken on the PDMS side show the protrusion of the interfacial nanogels (PROPA0–PROPA40) into the original water phase (Fig. S4a–e, ESI†). AFM images of the cavities in PDMS after the removal of nanogels provide information on the protrusion of nanogels in the air phase (Fig. S4f–j, ESI†). Fig. 3b shows the resulting height profiles of interfacial nanogels that represent their protrusion into the air and water phase. These data reveal two trends. First, the purely hydrophilic PROPA0 shows a large penetration into the water phase, thus suggesting pronounced swelling of these nanogels in the water subphase. Second, for nanogels containing hydrophobic PROPA groups, the protrusion into the water phase increases with their hydrophobicity. This trend is especially visible when comparing PROPA10 to PROPA40. These findings may seem contradictory as the more hydrophobic nanogels clearly swell less in water compared to their hydrophilic analogues (Fig. 1), and therefore suggest that with increasing hydrophobicity the decreasing nanogel elasticity governs their protrusion into the water rather than their swelling characteristics.
To provide a more coherent picture, we exclude effects from size deviations when comparing the different samples. For this, we quantified the particle deformation by calculating height-to-length ratios from the AFM height profiles in Fig. 3b. The height-to-length ratios are shown in Fig. 3c as a function of PROPA contents. Here, the height includes both the height in water and in air. Note that for an ideal rigid round particle, the height-to-length ratio is 1 and a low height-to-length ratio indicates a deformed morphology. The more hydrophilic nanogels, PROPA0 and PROPA10, exhibit similar small height-to-length ratios. This indicates pronounced deformation at the interface, which qualitatively agrees with the AFM results from the dry state (Fig. 2g). We assume that the high deformation results from the swollen nature and high elasticity of the hydrophilic nanogels, in agreement with the larger bulk hydrodynamic radius, the FHP, and the logP evaluation (Fig. 1). In contrast, more hydrophobic nanogels, PROPA30–PROPA40, show a higher height-to-length ratio, and thus a less deformed morphology. Notably, even the most hydrophobic samples have a height-to-length ratio much smaller than 1 (0.23 for PROPA40), showing that these hydrophobic nanogels are still significantly deformed at the interface. This is surprising as the bulk properties of such hydrophobic nanogels do not indicate pronounced swelling (Fig. 1). These results underline that hydrophobic properties have different effects at the interface and within the bulk. The observed trend of less deformation for increasingly hydrophobic nanogels is in agreement with the AFM measurements of Fig. 2. However, the sudden change in height up to approximately 100 nm for hydrophobic nanogels is not reproduced by gel-trapping. We assume that when drying on a silicon wafer the adhesion of hydrophobic particles to the substrate is low. As a result, these particles can recover their original shape to a certain extent, which is not possible when trapped in a gel-matrix.
Compression curves depend on the PROPA and DODA content
We have established that hydrophobicity and corresponding elasticity of our PROPA and DODA nanogels span a broad range between purely hydrophilic (PROPA0 and PROPA10) and rather hydrophobic (PROPA40 and DODA40). Therefore, our library bridges the worlds of hydrophilic soft microgels, like PNIPAm, and hydrophobic hard particles, such as polystyrene or silica. Since these two contrasting types of conventional particles show tremendously different behavior at the air/water interface, we examine the behavior of our nanogels in comparison and assume that their behavior lies in-between these extremes.To test this assumption, we investigated the behavior of nanogel ensembles adsorbed to the air/water interface under interfacial compression. We use a Langmuir trough and record the change in surface tension for the different nanogels via compression curves.32 To compare the compression curves of different nanogels, we normalized the isotherms at 5 mN m−1, which is the point at which the surface pressure visibly increases and the formation of a monolayer of nanogels covering the interface can be expected. Fig. 4a shows the representative compression curves for all PROPA samples. We observe a pronounced difference between PROPA0–PROPA30 and PROPA40.
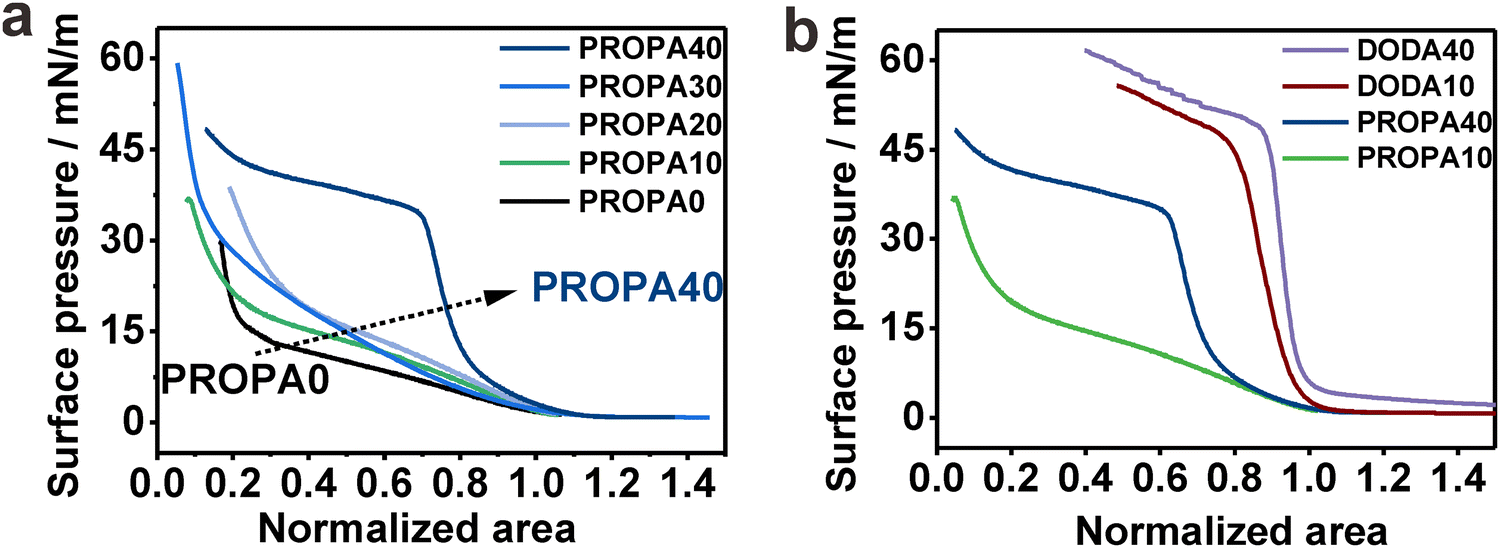 | ||
| Fig. 4 Interfacial properties under surface pressure show that PROPA and DODA nanogel ensembles combine properties of soft hydrophilic microgels and hard hydrophobic particles. This is demonstrated by representative surface pressure–normalized area isotherms of (a) PROPA nanogels of different hydrophobicity, and (b) comparison of PROPA nanogels and DODA nanogels. | ||
For PROPA0–PROPA30, the surface pressure increases slowly over a wide range of interfacial area and rises steeply at higher compressions. This behavior combines typical elements from soft hydrophilic microgels and incompressible hydrophobic particles: at large surface areas, the behavior resembles conventional microgels with a core–corona structure. For such particles, an increase in surface pressure at large areas is caused by the compression of loose dangling chains that constitute the corona.70,71 Upon compression, this is then followed by a broad plateau where the surface pressure does not significantly rise, but the individual particles can be compressed and deformed further.31,34 In our case, the PROPA0–PROA30 nanogels show a comparable increase in surface pressure at large surface areas, albeit this increase is less pronounced than in typical nanogels formed by precipitation polymerization. At small surface areas, a sudden rise of the surface pressure in our samples indicates the formation of a close-packed layer. This resembles the case of incompressible particles which typically show a negligible rise in surface pressure over most of the compression, until a monolayer covers the complete interface, at which point the surface pressure rises rapidly.72–74 The hybrid behavior of our nanogels can be rationalized by their internal structure. The slow increase in the compression curve relates to their compressibility, arising from the deformation at the liquid interface (Fig. 3). In contrast to conventional microgels, the nanogels are prepared by post-functionalization of hydrophobic precursor particles that come from an emulsion polymerization. We hypothesize that during this formation of precursor particles, the crosslinker distributes more evenly inside the monomer-swollen particles and thus produces a more uniform crosslinking structure compared to microgels synthesized via conventional precipitation polymerization. This, in turn, would lead to a less pronounced corona of dangling chains compared to conventional microgels and thus a lower degree of compressibility.
For the most hydrophobic PROPA sample (PROPA40), we observed a profile that rather resembles an incompressible material: this sample shows a steep rise in the compression curve, but lacks the soft part where the surface pressure rises slowly at larger available areas. For the more hydrophobic DODA samples, this behavior becomes even more pronounced (Fig. 4b). In this case, the increase in surface pressure is steeper, indicating even less compressible nanogels. The pseudo-plateau after the steep increase in surface pressure can be assigned to buckling of the formed interfacial colloidal monolayer, which we observed as a whitish and wrinkling appearance of the monolayer for the three most hydrophobic samples (PROPA40, DODA10 and DODA40).
Summarizing these experiments, we observed a sudden transition from a compression behavior typically associated with soft materials to a behavior that is typically associated with incompressible particles between PROPA30 and PROPA40. Interestingly, in these experiments, PROPA30 shows an interfacial behavior that resembles more the hydrophilic samples, while both bulk properties (Fig. 1) and investigation of individual particles at the interface (Fig. 2 and 3) indicate that this sample behaves more like the hydrophobic sample PROPA40.
Structural evolution of nanogel monolayers under compression—interparticle interactions are governed by hydrophobicity and deformability
Varying the nanogel hydrophobicity has a pronounced effect on their interfacial behavior as demonstrated by the different compression curves. It is known that such a compression of interfacially adsorbed nanogel layers changes the ordering of the nanogels with respect to each other. To examine this effect in detail, we now investigate the structure of colloidal monolayers as a function of nanogel hydrophobicity and corresponding deformability (Fig. 5). For this, we used the simultaneous compression–deposition technique.13,70 In this technique, the interfacial monolayers were transferred to a solid substrate at specific interfacial compressions. This enabled us to investigate the evolution of monolayer structure as a function of the available area per particle by SEM (Fig. 5a–p and Fig. S7, ESI†). Note that in contrast to larger colloidal particles,71,75 the small size of the nanogels prevent a direct observation at the interface. We therefore resorted to this indirect way of assessing the structure via the described transfer to a solid substrate and note that capillary forces acting during drying can have a significant effect on the resultant morphology.22,71 We therefore discuss the resultant morphologies after drying as a function of the particle hydrophobicity. | ||
| Fig. 5 Nanogel hydrophobicity governs their interfacial arrangement under compression. This was examined on nanogel monolayers after transfer to a solid substrate from the air/water interface. (a)–(p) Representative SEM images show the structure of nanogel monolayers under various surface pressures. Hydrophilic nanogels show the compression of close packed hexagonal structures: (a)–(d) PROPA0, (e)–(h) PROPA10, and (i)–(l) PROPA20. In contrast, hydrophobic nanogels show agglomeration into chain- and island-like structures: (m)–(p) PROPA40. All scale bars are 1 μm. (q) and (r) Statistical evaluation of the formed nanogel patterns reveal distinct differences between hydrophilic and hydrophobic nanogels. Analysis was performed on monolayers of nanogels with different hydrophobicity after transfer to a solid surface at low surface pressure (∼5 mN m−1) and include: (q) area per particle within the transferred layer; (r) nearest neighbor distance between the nanogels. | ||
At low surface pressure, PROPA0, PROPA10, and PROPA20 form a hexagonal monolayer with apparently a small distance between the nanogels (Fig. 5a, e and i). Upon closer inspection via phase image AFM, one can identify a small corona surrounding these particles (Fig. S6, ESI†), indicating that the particles in this phase are in corona–corona contact. With increasing compression, PROPA0 and PROPA10 show a continuous decrease in interparticle distance (Fig. 5a–h and Fig. S5, ESI†) although small clusters of collapsed particles are observed. These resemble “flower-like” structures of 4–5 nanogels aggregated around a central particle (e.g.Fig. 5b, c, f and g). Upon closer inspection, it seems that the central particles are often smaller nanogels. These flower structures likely arise from defects in the hexagonal lattices. Smaller nanogels tend to induce defects that facilitate a collapse of neighboring particles, a phenomenon previously observed for PNIPAM microgels.61 Finally, at higher surface pressures the nanogels arrange into a closed packed structure with core–core contacts (Fig. 5d, h and l). In contrast to monolayers of conventional PNIPAM microgels, which show a pronounced transition from non-close packed (corona–corona contact) to close packed (core–core contact) arrangements upon compression,31,60,70 this transition is much less pronounced in our particles. Generally, the particle packing density is higher for PROPA20 (Fig. 5i–k) than for the samples with lower PROPA content. We assume that this is a consequence of PROPA20's lower deformability. Overall, PROPA20 shows a similar pattern evolution as PROPA0 and PROPA10. In all cases, a decrease in interparticle distance upon compression is clearly visible. However, in contrast to PROPA0 and PROPA10, one can see particles in close contact already at lower pressures for PROPA20.
PROPA40 monolayers show a different structural evolution than the other PROPA nanogels (Fig. 5m–p). At a low surface pressure (5.4 mN m−1), all particles are already agglomerated and form island- and chain-like structures on the silicon substrate. Upon compression, these initial agglomerates are continuously pushed together, leading to a growth in agglomerate size at the expense of uncovered substrate regions (Fig. 5n and o). Notably, even at maximum compression (PI = 47.2 mN m−1), PROPA40 nanogels did not form a complete, hexagonal monolayer (Fig. 5p). We suggest that the formed network becomes so rigid that it cannot further relax into a complete monolayer. PROPA30 transferred to a solid substrate shows a similar behavior to PROPA40 (Fig. S7a–d, ESI†).
We further quantified the preferred interfacial morphology of the different particles, i.e., in a state without significant compression (at low surface pressure of around 5 mN m−1) (Fig. 5a, e, i and m). From the SEM images shown in Fig. 5a, e, i and m, we quantified the average area that one nanogel occupies within such a monolayer (Fig. 5q) and statistically evaluated the nearest neighbor distances (NND) between neighboring nanogels within this monolayer (Fig. 5r). Importantly, the area per particle not only includes the area of one nanogel, but also includes the space between cores. Fig. 5q shows that a single PROPA0 or PROPA10 nanogel occupies around 0.1 μm2, which is approximately twice the area occupied by a PROPA20 or PROPA30 nanogel, and almost four times the area of a PROPA40 nanogel. This increased footprint of the more hydrophilic particles is a direct consequence of their larger deformability, corroborating our AFM investigations (Fig. 2 and 3).
PROPA0 and PROPA10 nanogels show a single peak in their NNDs at approx. 350 nm, corresponding to the separated particle cores visible in the SEM image of the hexagonal monolayer (Fig. 5a and e). PROPA20 nanogels also exhibit a single peak in the NND distribution, but at a smaller distance of 220 nm. The larger distance between PROPA0 or PROPA10 nanogels is consistent with the larger area per particle (Fig. 5q). In contrast, PROPA30 and PROPA40 show two peaks in the NND distributions, one main peak at 130 nm, and a broader peak at 210 nm, indicating two distinct nearest neighbor distances. These peaks correspond to particles within chains and in between chains in the aggregated state these particles assume after transfer even at low compression. Overall, these statistical investigations show characteristic differences between the more hydrophilic samples (PROPA0–PROPA10), and the more hydrophobic samples (PROPA30–PROPA40). The PROPA20 sample shows characteristics in between these two extremes. This behavior corroborates the investigations on the single particle level discussed above.
The morphology of the transferred monolayers of DODA-functionalized particles closely resembles the most hydrophobic PROPA sample (Fig. S7e–h and i–l, ESI†). As an example, a DODA10 nanogel monolayer already shows a percolated morphology in the absence of compression. Then upon compression, the percolated network is continuously pushed together. Similar to the case of PROPA40, the hydrophobic and rigid character of the network prevents a complete consolidation into a well-ordered, hexagonally close packed monolayer even at highest compression.
The differences of these hydrophobic nanogel monolayers compared to their hydrophilic analogues can be either a direct agglomeration at the interface, or an increased affinity upon drying. It is known that surfaces with hydrophobicity above the “Berg limit” show long-range attractive forces. While the origin of the attractive force is still under debate,66,67 a limited repulsion by extended chains at the interface, and a higher affinity to other apolar surfaces76 can facilitate the formation of agglomerated networks at the air/water interface. The stiffer interfacial properties upon compression (Fig. 4) could be a consequence of this agglomeration. On the other hand, the AFM investigations on a solid substrate (Fig. 2) showed an increased height, and the ability to collapse into a more spherical shape on the substrate. Both factors can collude to increase collapse of neighboring particles under the influence of immersion capillary forces upon drying.
Conclusions
In conclusion, the hydrophobicity of nanogels significantly influences their behavior at the air/water interface. In this study, we have developed a library of nanogels with different levels of hydrophobicity but comparable colloidal structure through a post-functionalization technique. This allowed us to isolate effects of hydrophobicity on interfacial morphology of individual particles and the structure of self-assembled monolayer of such particles.With increasing hydrophobicity, nanogels are more rigid and less deformed at the interface. We suggest that this is the result of increased hydrophobic interactions between their polymer chains, thus giving additional physical crosslinks. Surprisingly, while the more hydrophobic nanogels do not swell in bulk water, they still experience a significant interfacial deformation. When compressed, the hydrophilic particles show a more continuous transition of surface pressure, indicative of a soft behavior. More hydrophobic particles instead show a more sudden increase in surface pressure, indicative of a more rigid particle network. The hydrophobicity differences also lead to differences in the morphology of an interfacial layer upon compression: softer particles form seemingly non-close packed morphologies which can be compressed to heal out defects and form a homogeneous monolayer. Particles with hydrophobicity above the “Berg limit” agglomerate upon compression and transferring to a solid substrate afterwards, forming a rigid network that cannot be easily compressed into a homogeneous, dense layer.
Using well-defined nanogel model systems, we have bridged the two worlds of hydrophilic soft microgels and hydrophobic incompressible particles. As a consequence, our study sheds light on the critical role of varying hydrophobicity on the behavior of particles in the bulk and when adsorbed to liquid interfaces. It demonstrates that the effect of hydrophobicity can affect the nanogel properties differently in bulk and when adsorbed at liquid interfaces. The systematic variation of hydrophobicity in otherwise similar nanogels thus provides a model system to facilitate understanding of hydrophobic effects for interface-bound macromolecules.
Author contributions
RC, MI and JM have performed the experiments, where RC synthesized the particles and provided the data for Fig. 1, MI for Fig. 3 and JM for Fig. 2, 4 and 5. NC and DK supervised the project. All authors have contributed to the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† The raw data of the measurements are available under the following https://doi.org/10.5281/zenodo.14065518.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
NV acknowledges support from the Deutsche Forschungsgemeinschaft (DFG) under grant number VO 1824/13-1. RC acknowledges China Scholarship Council (CSC) for financial support under grant number 202008320344. In addition, we would like to thank the Core Facility BioSupraMol supported by the DFG for aiding the TEM measurements.References
- B. R. Saunders and B. Vincent, Adv. Colloid Interface Sci., 1999, 80, 1–25 CrossRef CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS.
- M. Vatankhah-Varnoosfaderani, M. Ina, H. Adelnia, Q. Li, A. P. Zhushma, L. J. Hall and S. S. Sheiko, Macromolecules, 2016, 49, 7204–7210 CrossRef CAS.
- A. Fernández-Barbero, I. J. Suárez, B. Sierra-Martín, A. Fernández-Nieves, F. J. de las Nieves, M. Marquez, J. Rubio-Retama and E. López-Cabarcos, Adv. Colloid Interface Sci., 2009, 147–148, 88–108 CrossRef.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- R. Cui, M. Ickler, A. Markovina, S. Kanwal, N. Vogel and D. Klinger, ACS Nano, 2024, 18, 25499–25511 CrossRef CAS PubMed.
- M. Destribats, V. Lapeyre, M. Wolfs, E. Sellier, F. Leal-Calderon, V. Ravaine and V. Schmitt, Soft Matter, 2011, 7, 7689–7698 RSC.
- V. Schmitt and V. Ravaine, Curr. Opin. Colloid Interface Sci., 2013, 18, 532–541 CrossRef CAS.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS PubMed.
- M. Karg, A. Pich, T. Hellweg, T. Hoare, L. A. Lyon, J. J. Crassous, D. Suzuki, R. A. Gumerov, S. Schneider, I. I. Potemkin and W. Richtering, Langmuir, 2019, 35, 6231–6255 CrossRef CAS PubMed.
- H. Minato, M. Murai, T. Watanabe, S. Matsui, M. Takizawa, T. Kureha and D. Suzuki, Chem. Commun., 2018, 54, 932–935 RSC.
- M. C. Tatry, E. Laurichesse, A. Perro, V. Ravaine and V. Schmitt, J. Colloid Interface Sci., 2019, 548, 1–11 CrossRef CAS PubMed.
- M. Rey, M. A. Fernandez-Rodriguez, M. Karg, L. Isa and N. Vogel, Acc. Chem. Res., 2020, 53, 414–424 CrossRef CAS.
- Y. Chevalier and M. A. Bolzinger, Colloids Surf., A, 2013, 439, 23–34 CrossRef CAS.
- O. S. Deshmukh, D. van den Ende, M. C. Stuart, F. Mugele and M. H. G. Duits, Adv. Colloid Interface Sci., 2015, 222, 215–227 CrossRef CAS.
- M. A. Fernandez-Rodriguez, A. Martin-Molina and J. Maldonado-Valderrama, Adv. Colloid Interface Sci., 2021, 288, 102350 CrossRef CAS PubMed.
- S. Lam, K. P. Velikov and O. D. Velev, Curr. Opin. Colloid Interface Sci., 2014, 19, 490–500 CrossRef CAS.
- M. Kuhnhammer, K. Graff, E. Loran, O. Soltwedel, O. Lohmann, H. Frielinghaus and R. von Klitzing, Soft Matter, 2022, 18, 9249–9262 RSC.
- B. Brugger, B. A. Rosen and W. Richtering, Langmuir, 2008, 24, 12202–12208 CrossRef CAS.
- M. Rey, J. Kolker, J. A. Richards, I. Malhotra, T. S. Glen, N. Y. D. Li, F. H. J. Laidlaw, D. Renggli, J. Vermant, A. B. Schofield, S. Fujii, H. Lowen and P. S. Clegg, Nat. Commun., 2023, 14, 6723 CrossRef CAS PubMed.
- I. N. Arrebola, L. Billon and G. Aguirre, Adv. Colloid Interface Sci., 2021, 287, 102333 CrossRef.
- K. Volk, F. Deissenbeck, S. Mandal, H. Lowen and M. Karg, Phys. Chem. Chem. Phys., 2019, 21, 19153–19162 RSC.
- F. Grillo, M. A. Fernandez-Rodriguez, M.-N. Antonopoulou, D. Gerber and L. Isa, Nature, 2020, 582, 219–224 CrossRef CAS.
- J. Menath, J. Eatson, R. Brilmayer, A. Andrieu-Brunsen, D. M. A. Buzza and N. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2113394118 CrossRef CAS PubMed.
- D. Suzuki, Langmuir, 2023, 39, 7525–7529 CrossRef CAS.
- M. Stieger, W. Richtering, J. S. Pedersen and P. Lindner, J. Chem. Phys., 2004, 120, 6197–6206 CrossRef CAS PubMed.
- J. Harrer, M. Rey, S. Ciarella, H. Löwen, L. M. C. Janssen and N. Vogel, Langmuir, 2019, 35, 10512–10521 CrossRef CAS.
- K. Geisel, L. Isa and W. Richtering, Langmuir, 2012, 28, 15770–15776 CrossRef CAS PubMed.
- K. Zielińska, H. Sun, R. A. Campbell, A. Zarbakhsh and M. Resmini, Nanoscale, 2016, 8, 4951–4960 RSC.
- F. Camerin, M. Á. Fernández-Rodríguez, L. Rovigatti, M.-N. Antonopoulou, N. Gnan, A. Ninarello, L. Isa and E. Zaccarelli, ACS Nano, 2019, 13, 4548–4559 CrossRef CAS PubMed.
- M. Rey, X. A. Hou, J. S. J. Tang and N. Vogel, Soft Matter, 2017, 13, 8717–8727 RSC.
- A. Scotti, M. F. Schulte, C. G. Lopez, J. J. Crassous, S. Bochenek and W. Richtering, Chem. Rev., 2022, 122, 11675–11700 CrossRef CAS.
- Y. D. Wu, S. Wiese, A. Balaceanu, W. Richtering and A. Pich, Langmuir, 2014, 30, 7660–7669 CrossRef CAS PubMed.
- S. Bochenek, A. Scotti, W. Ogieglo, M. A. N. Fernández-Rodríguez, M. F. Schulte, R. A. Gumerov, N. V. Bushuev, I. I. Potemkin, M. Wessling and L. Isa, Langmuir, 2019, 35, 16780–16792 CrossRef CAS.
- S. Schmidt, T. Hellweg and R. von Klitzing, Langmuir, 2008, 24, 12595–12602 CrossRef CAS PubMed.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- J. Vialetto, S. N. Ramakrishna and L. Isa, Sci. Adv., 2022, 8, eabq2019 CrossRef CAS PubMed.
- S. Bochenek, F. Camerin, E. Zaccarelli, A. Maestro, M. M. Schmidt, W. Richtering and A. Scotti, Nat. Commun., 2022, 13, 3744 CrossRef CAS PubMed.
- J. Hao and R. A. Weiss, Macromolecules, 2011, 44, 9390–9398 CrossRef CAS.
- S. Abdurrahmanoglu, V. Can and O. Okay, Polymer, 2009, 50, 5449–5455 CrossRef CAS.
- C. Biglione, T. M. P. Neumann-Tran, S. Kanwal and D. Klinger, J. Polym. Sci., 2021, 59, 2665–2703 CrossRef CAS.
- R. Acciaro, T. Gilányi and I. Varga, Langmuir, 2011, 27, 7917–7925 CrossRef CAS PubMed.
- X. Wu, R. H. Pelton, A. E. Hamielec, D. R. Woods and W. McPhee, Colloid Polym. Sci., 1994, 272, 467–477 CrossRef CAS.
- P. Liu, C. M. Pearce, R.-M. Anastasiadi, M. Resmini and A. M. Castilla, Polymers, 2019, 11, 353 CrossRef PubMed.
- T. Hoare and D. McLean, Macromol. Theory Simul., 2006, 15, 619–632 CrossRef CAS.
- T. Hoare and R. Pelton, J. Phys. Chem. B, 2007, 111, 11895–11906 CrossRef CAS.
- T. Watanabe, M. Takizawa, H. Jiang, T. Ngai and D. Suzuki, Chem. Commun., 2019, 55, 5990–5993 RSC.
- T. Watanabe, Y. Nishizawa, H. Minato, C. Song, K. Murata and D. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 8849–8853 CrossRef CAS PubMed.
- Y. Nishizawa, T. Watanabe, T. Noguchi, M. Takizawa, C. Song, K. Murata, H. Minato and D. Suzuki, Chem. Commun., 2022, 58, 12927–12930 RSC.
- A. Gruber, D. Isik, B. B. Fontanezi, C. Bottcher, M. Schafer-Korting and D. Klinger, Polym. Chem., 2018, 9, 5553 RSC.
- A. F. Thunemann, A. Gruber and D. Klinger, Langmuir, 2020, 36, 10979–10988 CrossRef PubMed.
- A. Gruber, A. A. Joshi, P. Graff, J. L. Cuellar-Camacho, S. Hedtrich and D. Klinger, Biomacromolecules, 2022, 23, 112–127 CrossRef CAS PubMed.
- A. Gruber, L. Navarro and D. Klinger, Adv. Mater. Interfaces, 2019, 7, 1901676 CrossRef.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS.
- S. A. Wildman and G. M. Crippen, J. Chem. Inf. Comput. Sci., 1999, 39, 868–873 CrossRef CAS.
- P. Labute, J. Mol. Graphics Modell., 2000, 18, 464–477 CrossRef CAS PubMed.
- V. N. Paunov, Langmuir, 2003, 19, 7970–7976 CrossRef CAS.
- A. Rauh, M. Rey, L. Barbera, M. Zanini, M. Karg and L. Isa, Soft Matter, 2016, 13, 158–169 RSC.
- J. Harrer, S. Ciarella, M. Rey, H. Löwen, L. M. Janssen and N. Vogel, Soft Matter, 2021, 17, 4504–4516 RSC.
- S. Staufenbiel, M. Merino, W. Li, M.-D. Huang, S. Baudis, A. Lendlein, R. H. Müller and C. Wischke, Int. J. Pharm., 2015, 485, 87–96 CrossRef CAS PubMed.
- K.-Y. Law, J. Phys. Chem. Lett., 2014, 5, 686–688 CrossRef CAS PubMed.
- J. Bico, C. Marzolin and D. Quéré, Europhys. Lett., 1999, 47, 220 CrossRef CAS.
- N. A. Patankar, Langmuir, 2003, 19, 1249–1253 CrossRef CAS.
- M. Kanduč, E. Schneck and R. R. Netz, Phys. Chem. Chem. Phys., 2024, 26, 713–723 RSC.
- E. A. Vogler, Adv. Colloid Interface Sci., 1998, 74, 69–117 CrossRef CAS PubMed.
- R. W. Style, L. Isa and E. R. Dufresne, Soft Matter, 2015, 11, 7412–7419 RSC.
- H. Mehrabian, J. Harting and J. H. Snoeijer, Soft Matter, 2016, 12, 1062–1073 RSC.
- M. Rey, M. Á. Fernández-Rodríguez, M. Steinacher, L. Scheidegger, K. Geisel, W. Richtering, T. M. Squires and L. Isa, Soft Matter, 2016, 12, 3545–3557 RSC.
- K. Kuk, V. Abgarjan, L. Gregel, Y. Zhou, V. Carrasco Fadanelli, I. Buttinoni and M. Karg, Soft Matter, 2023, 19, 175–188 RSC.
- N. Vogel, L. de Viguerie, U. Jonas, C. K. Weiss and K. Landfester, Adv. Funct. Mater., 2011, 21, 3064–3073 CrossRef CAS.
- E. Santini, J. Krägel, F. Ravera, L. Liggieri and R. Miller, Colloids Surf., A, 2011, 382, 186–191 CrossRef CAS.
- J. Kumaki, Macromolecules, 1988, 21, 749–755 CrossRef CAS.
- M. Rey, A. D. Law, D. M. A. Buzza and N. Vogel, J. Am. Chem. Soc., 2017, 139, 17464–17473 CrossRef CAS PubMed.
- M. Kanduč, E. Schneck and R. R. Netz, Chem. Phys. Lett., 2014, 610–611, 375–380 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01186d |
| ‡ Co-first authorship. |
| This journal is © The Royal Society of Chemistry 2025 |