
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H functionalization reactions enabled by hydrogen atom transfer to carbon-centered radicals
Sumon
Sarkar†
 ,
Kelvin Pak Shing
Cheung†
and
Vladimir
Gevorgyan
*
,
Kelvin Pak Shing
Cheung†
and
Vladimir
Gevorgyan
*
Department of Chemistry and Biochemistry, University of Texas at Dallas, 800 W Campbell Rd, Richardson, Texas 75080, USA. E-mail: vlad@utdallas.edu
First published on 16th November 2020
Abstract
Selective functionalization of ubiquitous unactivated C–H bonds is a continuous quest for synthetic organic chemists. In addition to transition metal catalysis, which typically operates under a two-electron manifold, a recent renaissance in the radical approach relying on the hydrogen atom transfer (HAT) process has led to tremendous growth in the area. Despite several challenges, protocols proceeding via HAT are highly sought after as they allow for relatively easy activation of inert C–H bonds under mild conditions leading to a broader scope and higher functional group tolerance and sometimes complementary reactivity over methods relying on traditional transition metal catalysis. A number of methods operating via heteroatom-based HAT have been extensively reported over the past few years, while methods employing more challenging carbon analogues have been less explored. Recent developments of mild methodologies for generation of various carbon-centered radical species enabled their utilization in the HAT process, which, in turn, led to the development of remote C(sp3)–H functionalization reactions of alcohols, amines, amides and related compounds. This review covers mostly recent advances in C–H functionalization reactions involving the HAT step to carbon-centered radicals.
Introduction
Selective functionalization of C–H bonds in organic molecules is highly desirable as this atom- and step-economic approach allows for late stage functionalization and diversification of complex molecules, and sometimes previously unattainable unique retrosynthetic disconnections.1 However, selective and efficient functionalization of ubiquitous C–H bonds is a highly challenging task due to the inertness of these bonds, and also lack of control for activation of a specific C–H site among other similar C–H bonds available.2Over the past decades, transition metal-catalyzed C–H activation and carbene/nitrene insertion reactions have been developed tremendously (Scheme 1a).3 However, these methods typically require high temperature conditions, as well as directing groups, some of which are not removable. The selectivity of C–H activation mostly depends on thermodynamic stability of the resultant carbon–metal species. Thus, primary C–H sites are easier to activate compared to their secondary counterparts, while activation of tertiary C–H bonds is rare. On the other hand, carbene and nitrene insertion reactions occur preferentially at the most electron-rich C–H bonds. Nonetheless, steric influence can be the dominant factor in some cases. Moreover, these processes usually employ diazo compounds or amides to access the corresponding metal carbene and metal nitrene species. An electron withdrawing group such as an ester (i.e. α-diazo ester) is often required, thus limiting the choice of reaction substrates.
 | ||
| Scheme 1 (a) Different modes of remote C–H functionalization. (b) Guiding factors for HAT. (c) Remote C–H functionalization via HAT to C-centered radicals. | ||
In contrast, hydrogen atom transfer (HAT)4 represents an alternative approach toward C–H functionalization. Since HAT is highly influenced by bond dissociation energy (BDE), for a given system, HAT from tertiary C–H sites is faster than that from secondary and primary positions. Thus, methods relying on HAT exhibit a selectivity trend complementary to that for two-electron transition metal-catalyzed approaches.5
The HAT process has long been recognized as a versatile tool for C–H functionalization due to its high reactivity and regioselectivity. Historic precedence of 1,5-HAT initiated by nitrogen- and oxygen-centered radicals was showcased in the Hofmann–Löffler–Freytag reaction and Barton's nitrite photolysis.6 This radical approach discovered in the 19th century, despite being highly regioselective and exergonic, did not make an appearance among the mainstream C–H functionalization methods until mild generation of radicals was developed.7 Likewise, carbon-centered radical-mediated HAT was first reported in 1954 (ref. 8) and was not utilized in organic synthesis until the late 1980's;9 however, it has enjoyed rapid employment in synthesis in the past decade.
The rate of HAT is mostly controlled by enthalpic factors; however, the Thorpe–Ingold effect also plays an important role in the case of intramolecular HAT.10 With few exceptions,11 HAT proceeds via an early transition state due to its exothermic nature. Usually aryl, vinyl, or primary alkyl radicals undergo HAT at alkyl C–H sites due to their higher parent C–H BDEs (Scheme 1b(i)). In the case where these BDE differences are comparable, the process can potentially be reversible;12 thus a selective functionalization becomes more challenging. Another important factor governing the efficiency and selectivity of HAT is radical philicity (polarity).13 The radical philicity, electrophilicity, or nucleophilicity index can be quantified from the ionization potential (IP) and electron affinity (EA) of free radicals. Given similar BDE differences, an electrophilic radical is expected to undergo HAT preferentially from a more hydridic C–H site and vice versa (Scheme 1b(ii)).4b,14 In case of polarity mismatch, another HAT agent can be used for polarity reversal catalysis (Scheme 1b(iii)).15
C-Centered radicals can abstract hydrogen via intramolecular 1,n-HAT where n = 4–8, or even higher (Scheme 1b(iv)).7a,b,d–g The predictable site-selective HAT relies on optimal orientation (linear to quasi-linear: ≤35°) and distance (≤3 Å) between the radical and the H-atom at the abstraction site.16 Higher activation energy for other intramolecular HATs can be attributed to increased C–H–C strain for 1,3- and 1,4-HAT, or entropic barrier for higher order HATs. However, incorporation of heteroatoms can alter the preferred geometry and thus the site of HAT. For example, a silicon-based tether displays a preference for 1,6-HAT, while an arylsulfonyl tether is predisposed to 1,7-HAT (see below).
Common generation of O- and N-centered radicals for HAT involves single electron reduction of very weak oxygen- or nitrogen-heteroatom bonds or strong oxidants required to oxidize nitrogen or oxygen centers. In contrast, generation of C-centered radicals can now be done under a variety of benign conditions using organic halides, redox-active esters, triflates, and diazonium salts. However, there are a few challenges associated with the C–H functionalization via HAT of C-centered radicals (Scheme 1c). Since both the initial and translocated radicals are very similar in nature, there is a possibility of a premature coupling process. However, this problem can be addressed by using radicals of different radical philicities and by matching the polarity of the reagent and translocated radical.17 In addition, due to lower BDE differences, the rate of HAT to the C-centered radical from the alkyl C–H site can be low, thus competing with other HAT processes, including an unwanted intermolecular HAT process with solvent giving rise to reduction byproducts.
This review covers C–H functionalizations involving HAT to a C-centered radical, with emphasis placed on the theoretical background and challenges of HAT involving C-centered radicals. It should be noted that this topic has been partially covered in several excellent reviews;5,7a–d however, since then an increasing number of reports have appeared in the literature. Hence, this review summarizes the progress in this emerging area of HAT, organized by the distance of HAT and type of C-centered radical.
HAT involving alkyl radicals
Intramolecular HAT
 | ||
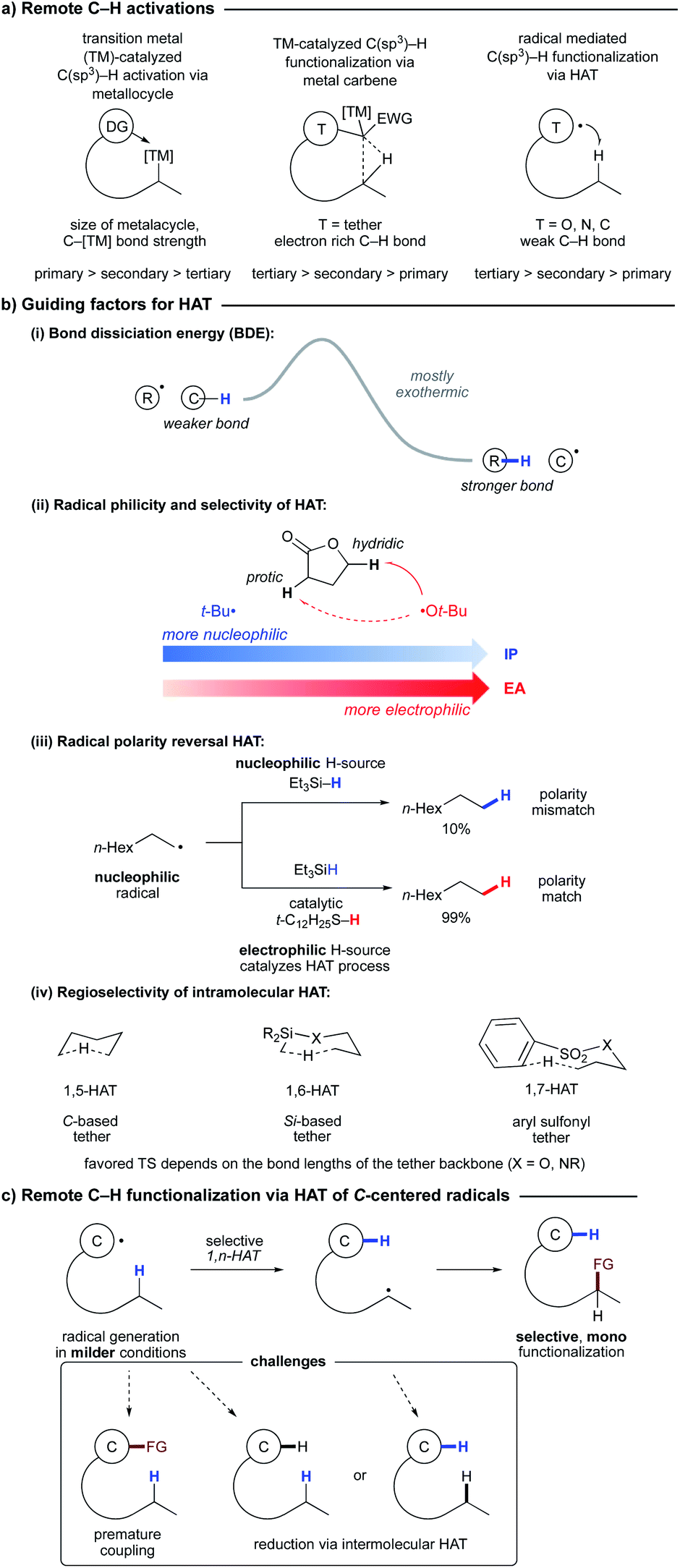
| Scheme 2 1,4-HAT in synthetic studies toward gelsemine. | ||
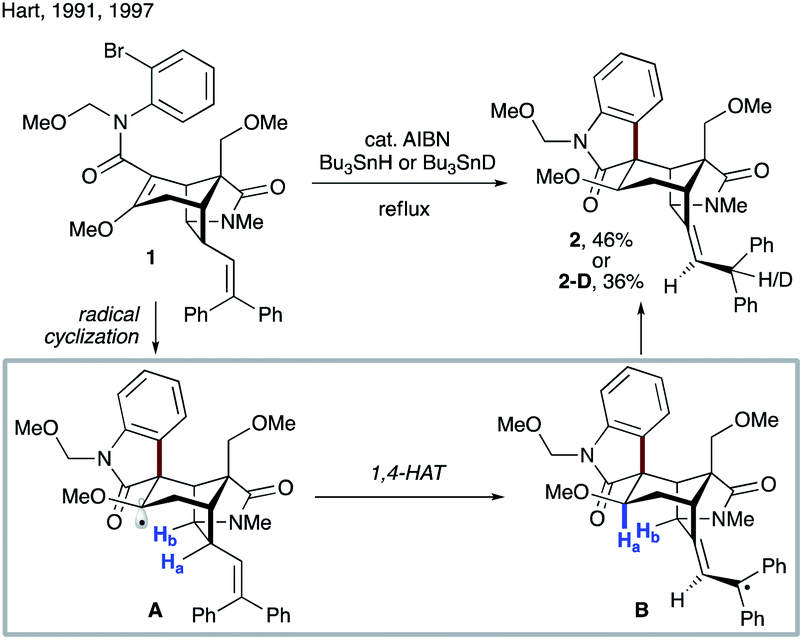
Crich and co-workers showed that 1,4-HAT could be a competitive pathway during the inversion of α- to β-pyranosides via 1,5-HAT.19 As depicted in Scheme 3a, reaction of 3 with Bu3SnH in the presence of AIBN leads to radical A, which can undergo 1,5-HAT, followed by axial HAT, to afford the desired β-mannoside 4 after workup. Alternatively, competing 1,4-HAT can occur, resulting in intermediate B. The latter may afford α-mannoside 5 or glucoside 6via HAT, or ketone 7 upon β-scission. The product distribution clearly shows that 1,4-HAT is not only viable but indeed the major pathway in this rigid system. In addition, exclusive formation of ketone 7 was observed when glucoside 8 was used (Scheme 3b).
 | ||
| Scheme 3 (a) 1,4-HAT triggered by radical A originating from bromoethyl mannoside 3. (b) Synthesis of ketone 7 by 1,4-HAT/fragmentation of bromoethyl glucoside 8. | ||
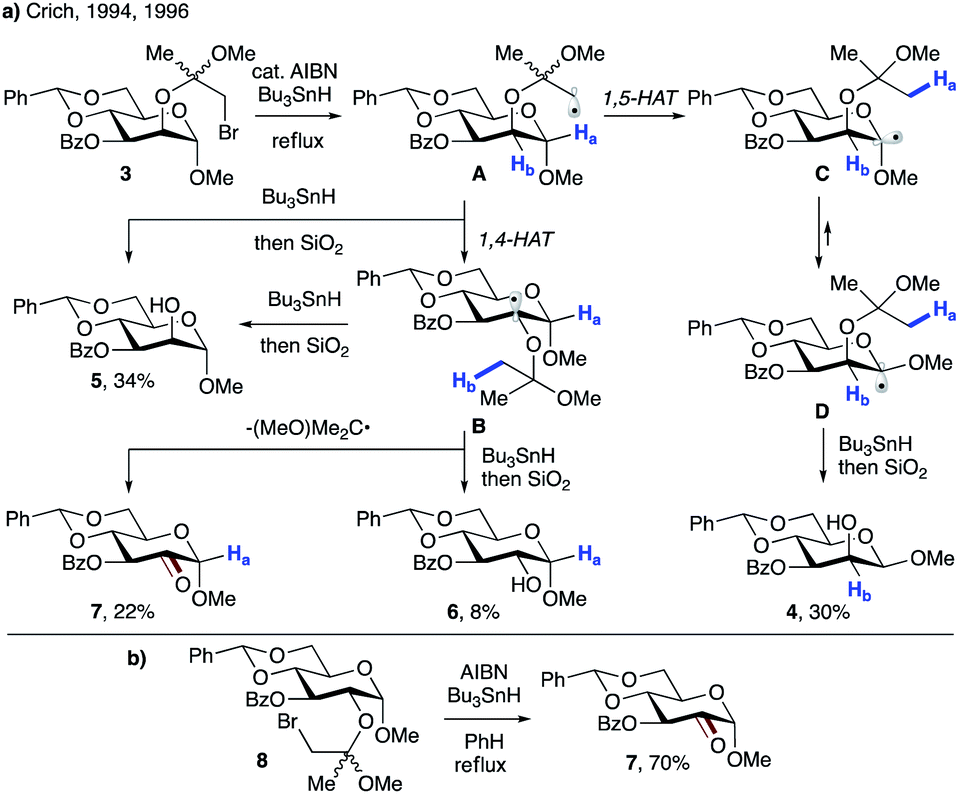
In 2001, Zard and Cassayre reported a short synthesis of (−)-dendrobine,20 in which the pyrrolidine core was thought to be constructed via radical cyclization of the corresponding trichloroacetamide (9 → 10, Scheme 4a). However, upon treatment of model substrate 9 with nickel and acetic acid in the presence of diphenyl diselenide, enamide 11 was obtained instead of the anticipated bicyclic lactam 10. This suggests that 1,4-HAT toward stable allylic radical B occurred preferentially over the 5-exo-trig cyclization. Bertrand, Nechab and co-workers provided a plausible explanation based on the conformational analysis of conformers A-1 and A-2 leading to 1,4-HAT and 5-exo-trig processes, respectively (Scheme 4b).21
 | ||
| Scheme 4 (a) Formation of cyclic enamide 11via a 1,4-HAT/arylselenation cascade. (b) Conformational analysis of the 1,4-HAT process vs. 5-exo-trig cyclization. | ||
Ryu and co-workers observed formation of three different products while investigating the reactivity of α-ketenyl radicals with an internal amino group (Scheme 5a).22a The reaction begins with radical addition of the tributyltin radical to 12, followed by trapping of carbon monoxide to give α-ketenyl radical A, which then engages in nucleophilic cyclization with a tethered amino group. A subsequent proton transfer affords hydroxylallyl radical B, which may either be oxidized to lactam 13, convert to reduced lactam 14 upon intermolecular HAT with Bu3SnH, or perform 1,4-HAT to produce intermediate C. The latter then extrudes the tributyltin radical to furnish 15. Alternatively, 15 could be obtained via protodestannylation of 13, as exemplified in the one-pot two-step transformation of 12c to 15c. The product distribution varies with the lactam ring size, with larger rings favoring 1,4-HAT over the other two pathways. This was later rationalized by DFT calculations, which showed that despite all being highly exothermic, the activation barrier decreased with ring size (17.0 kcal mol−1 for n = 4 vs. 25.3 kcal mol−1 for n = 1).22b It was also found that in the case of phenethylamines 16, ejection of the phenethyl radical preceded proton transfer and hence bypassed 1,4-HAT, resulting in intermediate A, which was exclusively converted to B, which upon treatment with TMSCl afforded lactam 17 (Scheme 5b).
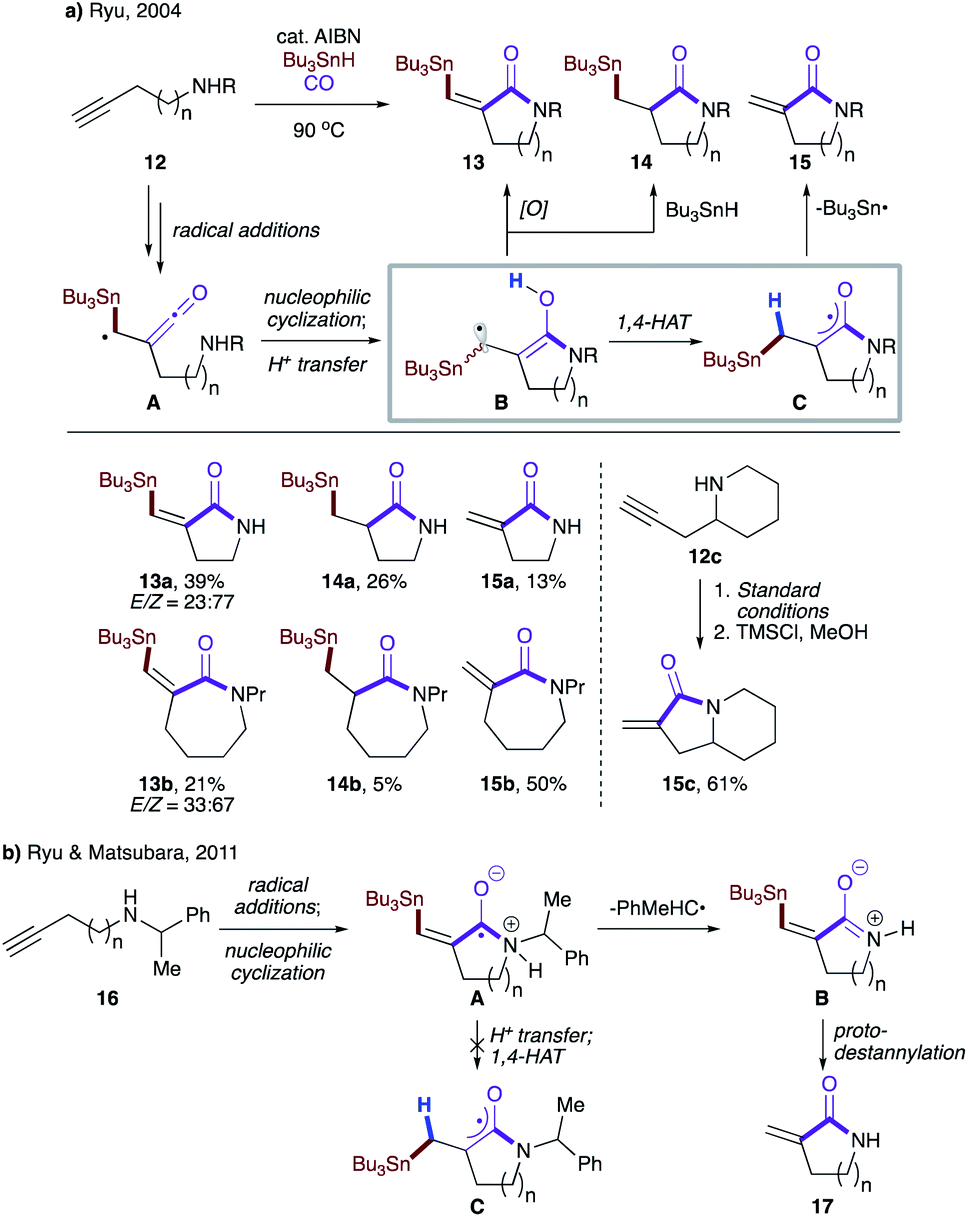 | ||
| Scheme 5 (a) Rationale for the formation of three products. (b) Phenethyl radical as a leaving group. | ||
In 2006 and 2008, Dake and co-workers disclosed synthetic routes toward nitiol derivatives 20 (ref. 23) and fusicoccane A–B ring fragment 21 (ref. 24) involving tandem Norrish type 1 fragmentation/1,4-HAT (Scheme 6). First, upon UV irradiation, a photoexcited ketone 18 undergoes fragmentation to form biradical A, which after intersystem crossing (ISC) undergoes 1,4-HAT to afford ketene intermediate B. Nucleophilic trapping with methanol solvent leads to the key intermediate, ester 19.
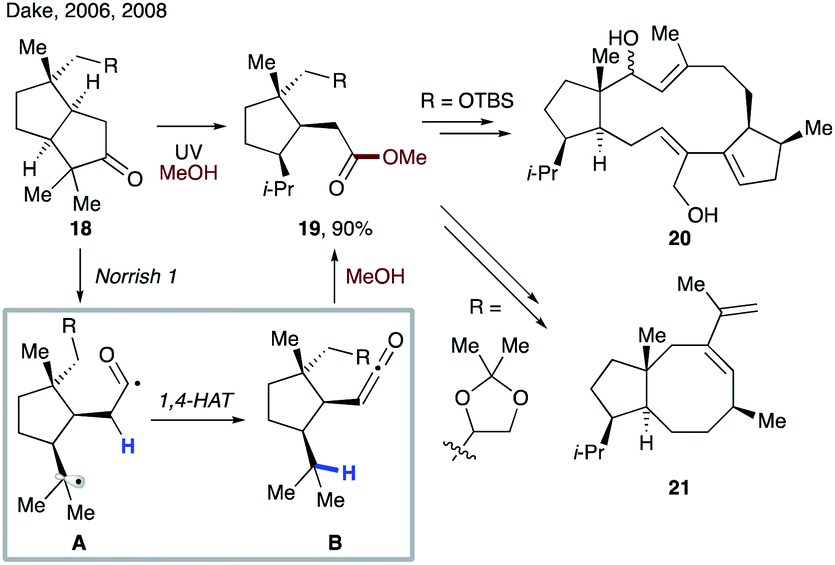 | ||
| Scheme 6 Synthesis of natural product derivatives 20 and 21via the 1,4-HAT process. | ||
In 2012, the group of Li and Shi reported the synthesis of bicyclo[3.1.0]hexane derivatives 22via radical reaction between vinylidene cyclopropanes and diphenyl diselenide.25 The vinylcyclopropane moiety of 22 undergoes thermally induced ring-opening, followed by 1,4-HAT, to give biradical C, which produces cyclohexene derivatives 23 or 24 (Scheme 7a). In all cases, excellent stereoselectivity was observed, which could be explained by unfavourable formation of the conformer B. Involvement of 1,4-HAT in this thermal rearrangement was supported by the reaction of D-labeled substrate 22c, as well as by DFT calculations (Scheme 7b).
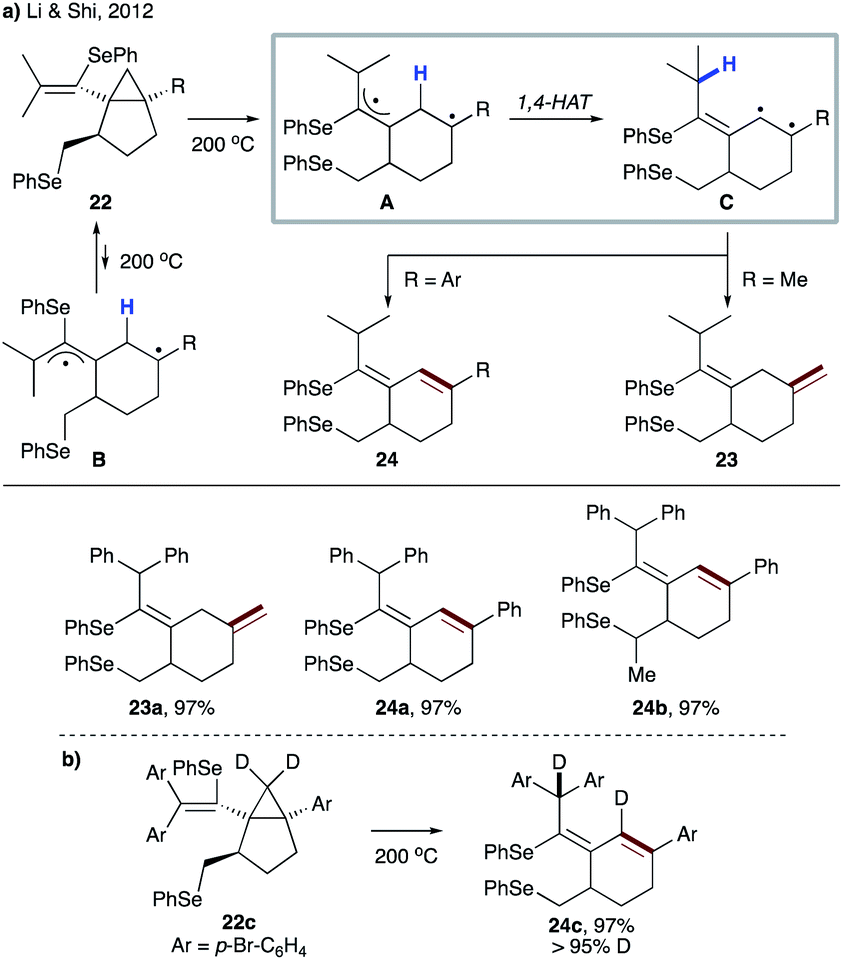 | ||
| Scheme 7 (a) Thermally induced ring-opening/1,4-HAT cascade transformation of 22. (b) Evidence for the 1,4-HAT process from a deuterium-labelling experiment. | ||
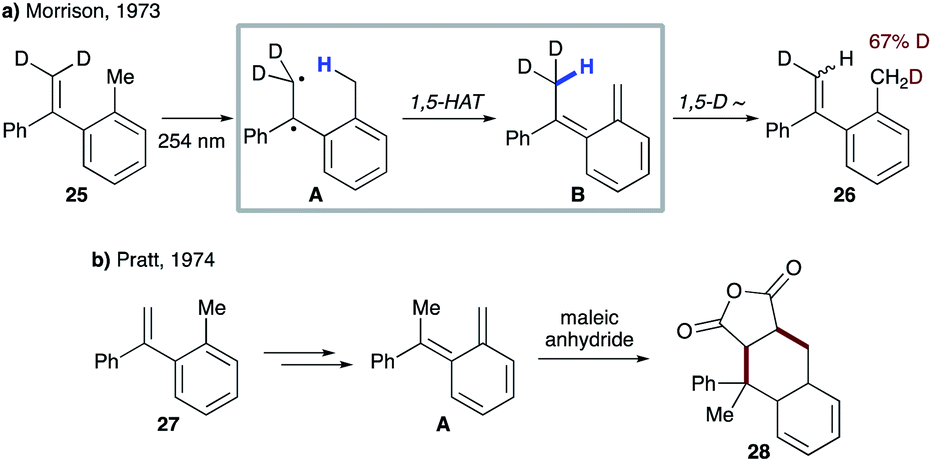 | ||
| Scheme 8 (a) Photoinduced internal H/D exchange of 25. (b) Trapping of the o-xylylene intermediate with maleic anhydride. | ||
Very recently, Koert's group reported an interesting example of naphthocyclobutene synthesis from methyl (α-napthyl) acrylates 29.29 1,2-Biradical A, formed by energy transfer (EnT) from a photoexcited iridium catalyst, underwent 1,5-HAT to produce intermediate B. In contrast to the above cases, instead of the formation of o-xylylene, cyclobutane 30 is furnished after ISC/recombination of radicals. Thus, this transformation represents a carbon analogue of Norrish–Yang reaction. The reaction performed with a deuterium-labelled substrate provided evidence for 1,5-HAT, as well as it being the rate-determining step (30e) (Scheme 9).
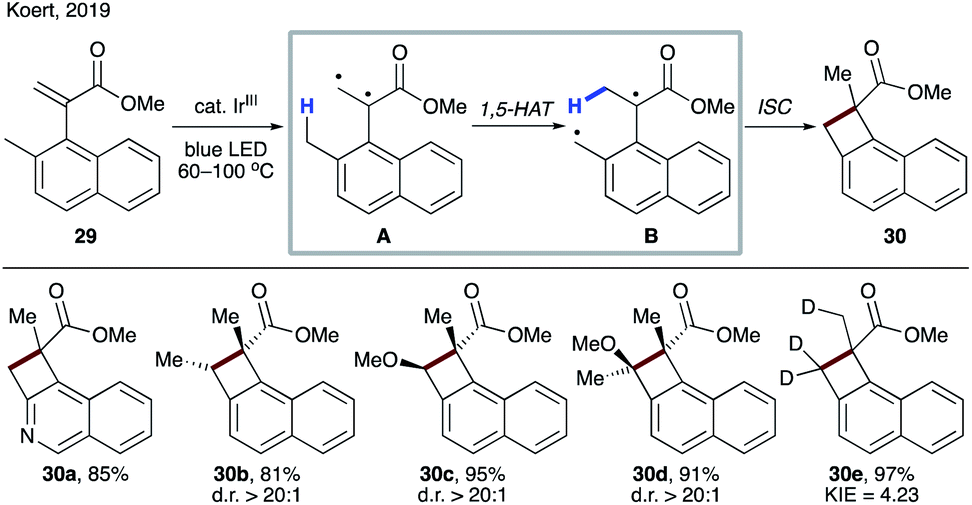 | ||
| Scheme 9 Synthesis of cyclobutanes 30via EnT-induced 1,5-HAT to a 1,2-biradical. | ||
In 2014, Liu, Tan, and co-workers reported enantioselective C–H alkoxylation involving a 1,5-HAT process (Scheme 10a).30a The trifluoromethyl radical, formed by single electron reduction of Togni's reagent by a copper(I) catalyst, undergoes radical addition to substrate 31. A subsequent 1,5-HAT occurs to form more stabilized α-amido radical B, followed by a single electron oxidation to produce imine intermediate C. Finally, the nucleophilic attack of alcohol in the presence of chiral phosphoric acid (CPA) furnishes enantioenriched product 32. The authors also demonstrated the feasibility of HAT with an aliphatic amide (32d). Later, a related transformation that also relied on 1,5-HAT was disclosed (Scheme 10a).30b In this case, a catalytic amount of 1,2-bis(diphenylphosphino)benzene (dppBz) serves as a single electron reductant and generates the trifluoromethyl radical with Togni II reagent under thermal conditions. Similarly, radical addition to 33 leads to the more stabilized radical B. Electron transfer to another equivalent of Togni II reagent, followed by a proton loss leads to, in contrast to an imine in the first report, enamide C, which is susceptible to another radical addition. Single electron oxidation followed by proton loss delivers the reaction product 34 and regenerates the phosphine catalyst, respectively. A one-pot procedure was also developed for the synthesis of various 5-(trifluoromethyl)oxazoles such as 35 (Scheme 10b).
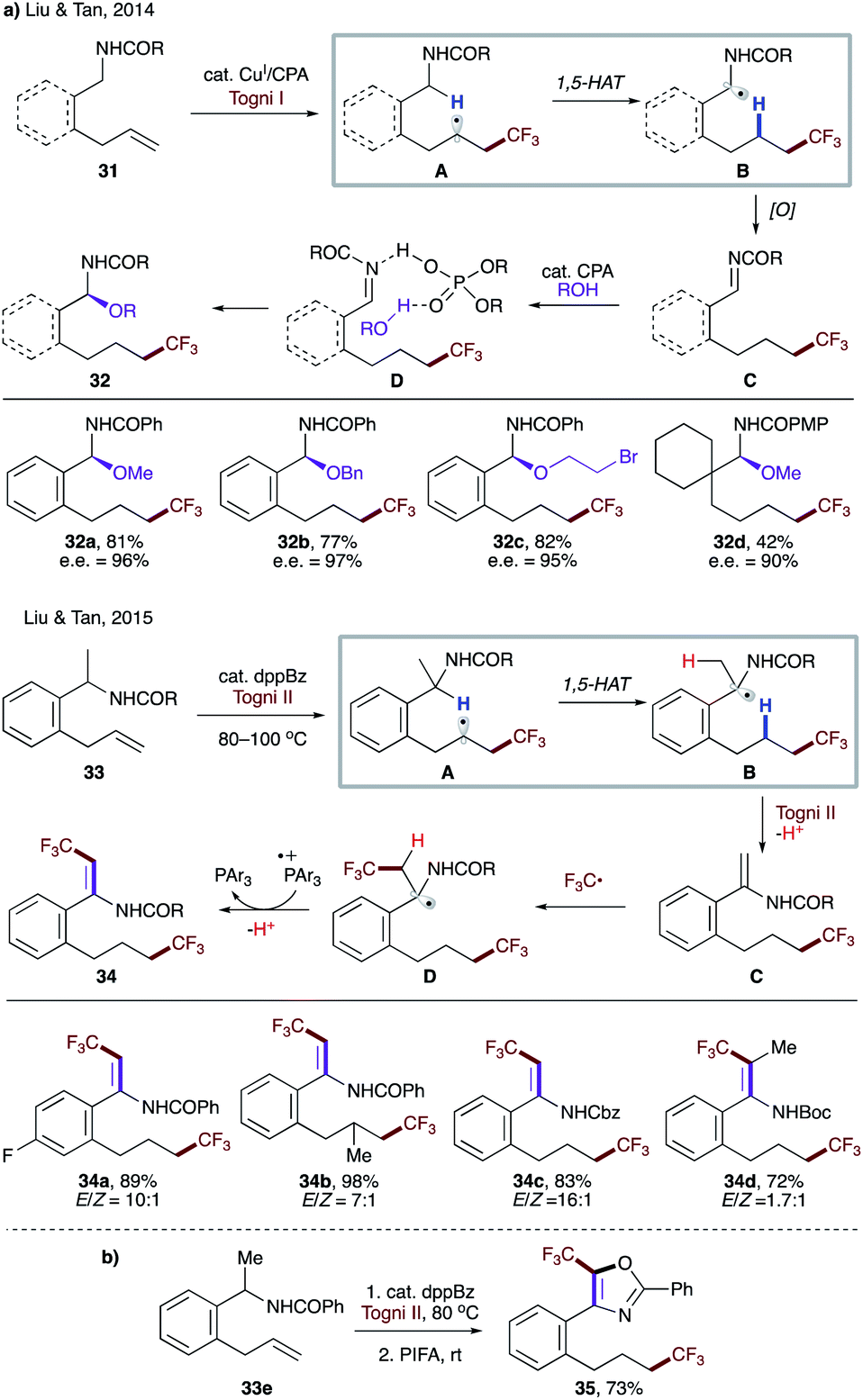 | ||
| Scheme 10 (a) Radical addition-triggered 1,5-HAT. (b) One-pot synthesis of 5-(trifluoromethyl)oxazole 35. | ||
A reaction mode where 1,5-HAT is induced by radical addition to a tethered double bond has proven to be efficient and general. Based on this strategy, transformations targeting weak C–H bonds, including benzylic (Scheme 11a),31 α-carbonyl (Scheme 11b),32 α-hydroxyl (Scheme 11c),31a,33 and formyl (Scheme 11d),34 have been developed. Notably, these substrates are also amenable to entropically less favored 1,6- and 1,7-HAT due to the presence of radical-stabilizing groups.
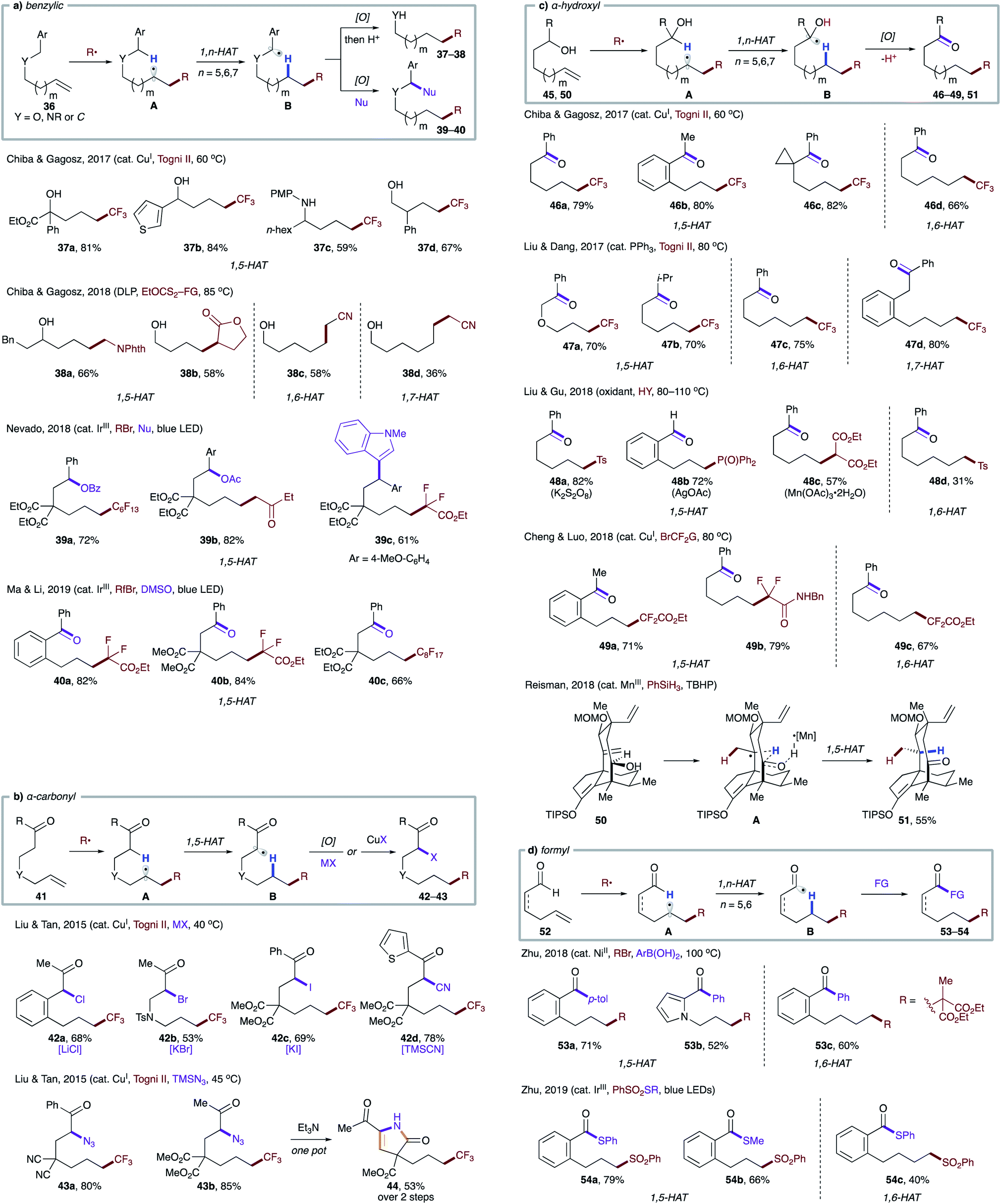 | ||
| Scheme 11 Intramolecular HAT from activated C–H bonds initiated by radical addition to a tethered olefin. (a) Benzylic C–H. (b) α-Carbonyl C–H. (c) α-Hydroxyl C–H. (d) Formyl C–H. | ||
Very recently, Wang and co-workers reported iron-catalyzed remote functionalization that operates in a similar fashion.35 Remarkably, the catalytic system could be applied not only to activated systems, but also to unactivated ones (Scheme 12). Notably, substrate bearing secondary C–H sites could also be employed (56c).
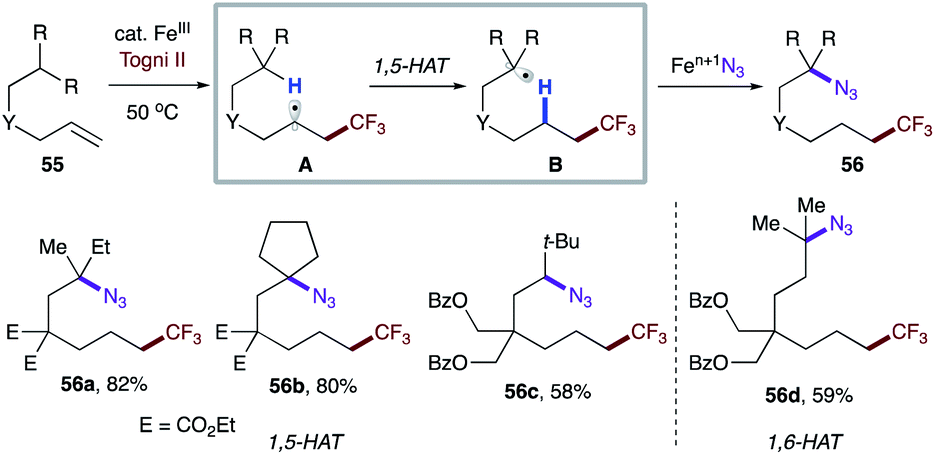 | ||
| Scheme 12 Intramolecular HAT from unactivated C–H bonds. | ||
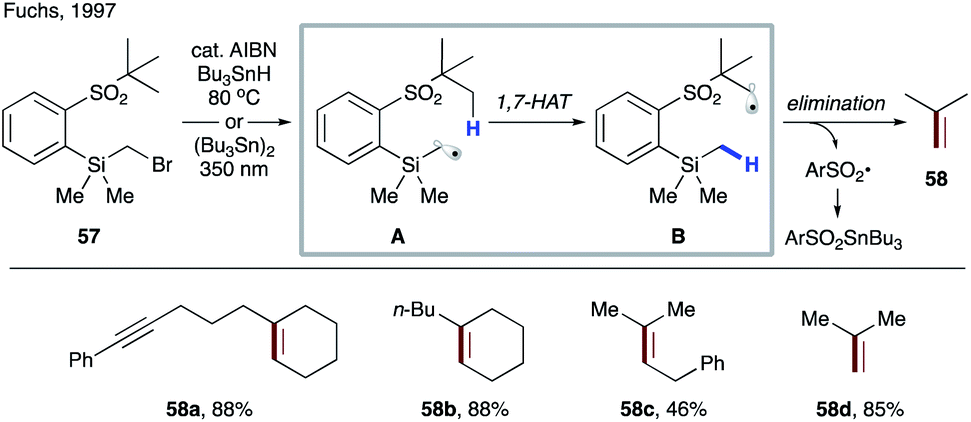 | ||
| Scheme 13 Formation of an alkene via 1,7-HAT/β-elimination of 57. | ||
In 2013, Beau, Urban and co-workers reported selective mono-debenzylation of benzyl-protected carbohydrate templates 59 that involved a key 1,7-HAT step triggered by a silylmethyl radical (Scheme 14a).37 The substrates were readily converted to the corresponding xanthate methylsilyl ethers 60via a sequential silylation and substitution by xanthate. Thermal decomposition of dilauroyl peroxide (DLP) initiates the process to produce electrophilic silylmethyl radical intermediate A, which undergoes 1,7-HAT to furnish benzylic radical B. Oxidation of the latter by DLP generates a stable benzylic cation, which is trapped in the form of acyl acetal C. A subsequent hydrolysis and desilylation delivers the reaction product, diol 61. In the case of β anomers such as 60a, an alternative path could be operational, which involves a successive 1,5-HAT from anomeric C–H and then benzylic C–H sites (Scheme 13b). However, the authors discounted this possibility as α anomer 60b, which does not possess suitable hydrogen at the anomeric site, reacted smoothly to give 61b with similar efficiency (Scheme 13c).
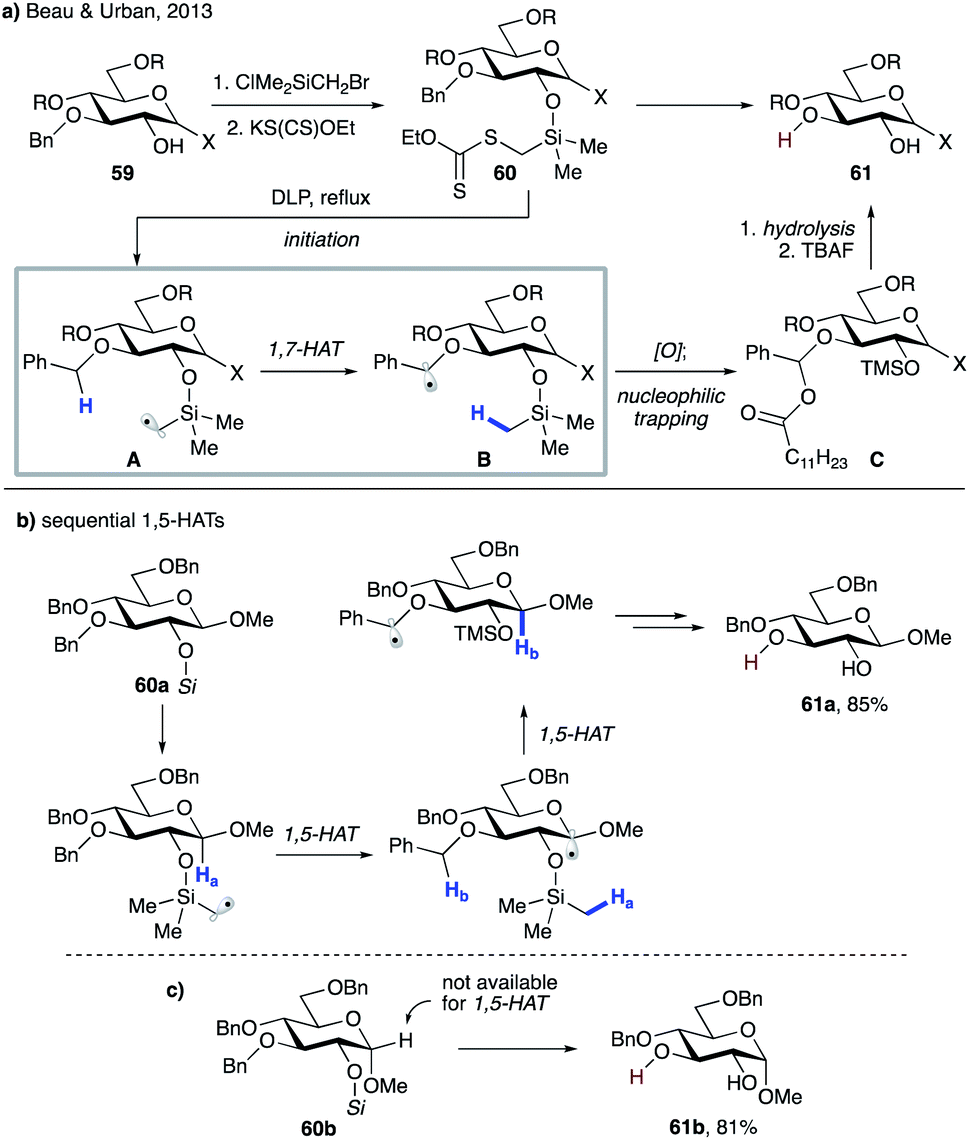 | ||
| Scheme 14 (a) Selective mono-debenzylation of 59. (b) Plausible mechanism for sequential 1,5-HATs leading to the same product. (c) Validation of 1,7-HAT based on efficient debenzylation of 60b. | ||
Later in 2015, the group of Zeng, Ren and Wu utilized the 1,7-HAT process in the construction of an isocopalane skeleton (Scheme 15).38 Radical precursor 63 was readily prepared from naturally abundant andrographolide 62. Upon heating, 63 undergoes homolytic cleavage of the S–Cl bond and SO2 extrusion leading to primary alkyl radical A. 1,7-HAT then follows, resulting in the more stable α-acyloxy- or α-alkoxy radical B. Finally, fragmentation of B affords the reaction product, aldehyde 64. Deuterium labelling studies confirmed the migration of the hydrogen atom, thus ruling out a sequential 1,5-HAT (vide supra). Furthermore, catalytic amounts of benzoyl peroxide accelerated the reaction while TEMPO shut down the reaction, suggesting a radical pathway for this transformation. Several protecting groups R were examined, which led to moderate to good yields (64a–d), with the exception of the benzoylated substrate (64e). Interestingly, methyl ether was a viable substrate (64f).
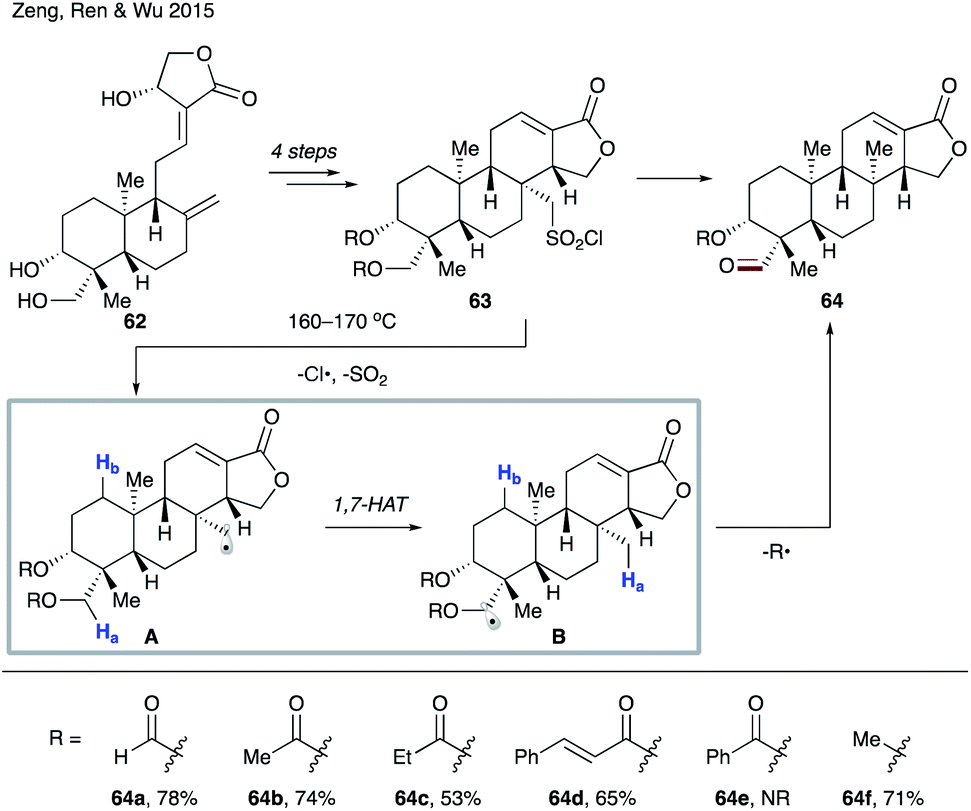 | ||
| Scheme 15 Construction of isocopalane framework 64 involving 1,7-HAT. | ||
Intramolecular HAT at competing sites
In 2017, Gevorgyan and co-workers demonstrated the ability of hybrid alkyl palladium-radical species to undergo selective intramolecular HAT at unactivated C(sp3)–H sites, featuring an easily installable/removable silicon-based auxiliary (Scheme 16).39 Substrate 65, readily accessible via routine silyl protection of the corresponding alcohol, engages in a single electron transfer (SET) with a photoexcited Pd(0) catalyst to form hybrid radical A, which undergoes rate-limiting site-selective 1,n-HAT to produce translocated radical B. The latter may either recombine with Pd(I) followed by β-hydride elimination, or undergo direct HAT with Pd(I) to furnish remote desaturation product 66 after desilylation with TBAF. A side hydrodehalogenation process, which would produce reduced product 67, was suppressed under optimized conditions. This protocol allows for activation of both secondary and tertiary C(sp3)–H bonds at β-, γ-, and δ sites (66a–c). Remarkably, a high level of site-selectivity was maintained even with substrates bearing competitive sites with similar BDEs (66d–f). Kinetic studies suggested the selectivity preference to be 1,6-HAT (γ) > 1,5-HAT (β) > 1,7-HAT (δ). The preference of 1,6-HAT over the more common 1,5-HAT can be attributed to the increased tether length and flexibility of the tether possessing silicon atom.40 This method represents the first practical catalytic desaturation of aliphatic alcohols under mild, auxiliary-enabled, visible light-induced conditions.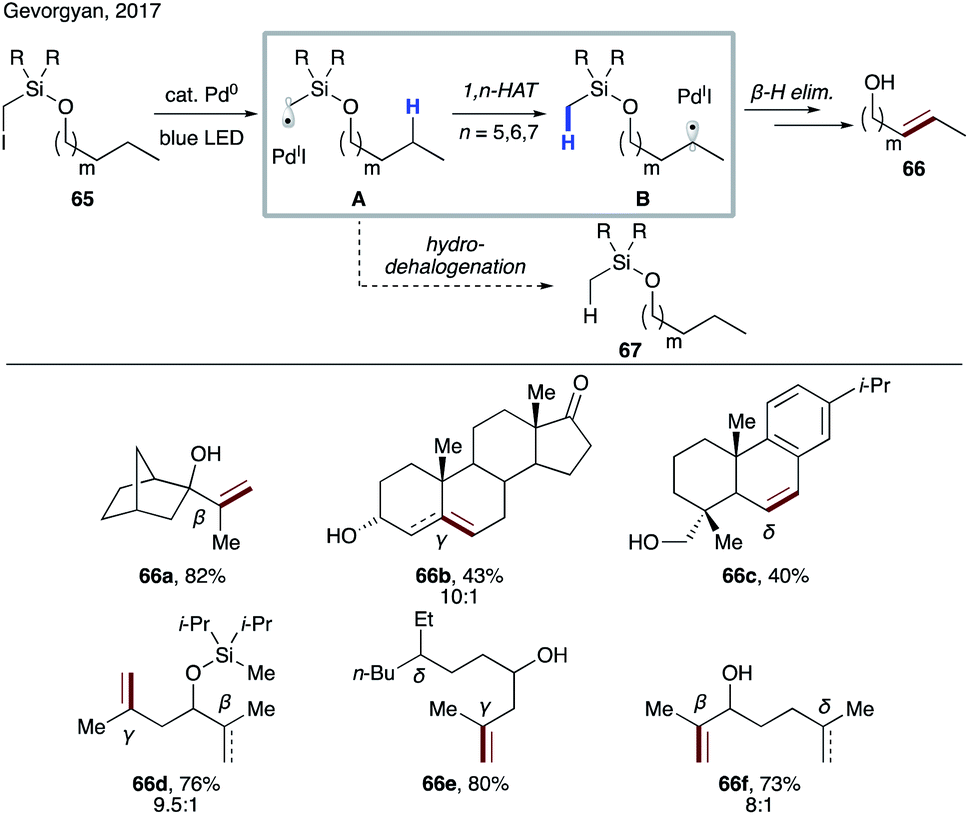 | ||
| Scheme 16 Remote desaturation of aliphatic alcohols via selective 1,n-HAT enabled by an iodomethylsilyl auxiliary. | ||
Photoexcited palladium catalysis has been developed for alkyl Heck reaction and C–H activation,41 which had not been utilized synergistically to achieve more complex transformations. Very recently, Gevorgyan's group disclosed site-selective radical relay Heck reaction of aliphatic alcohol derivatives 65 with various alkenes 68 to achieve alkenylation at remote unactivated C(sp3)–H sites (Scheme 17).42 Potential side reactions, such as premature Heck reaction (70) and desaturation (71), were suppressed under optimized conditions. Interestingly, iodide C was observed over the course of reaction, which was shown to produce alkenol 69 under reaction conditions, thus suggesting its intermediacy in this transformation. As in the previous case (Scheme 16),39 this reaction exhibits the same site-selectivity: γ > β > δ.
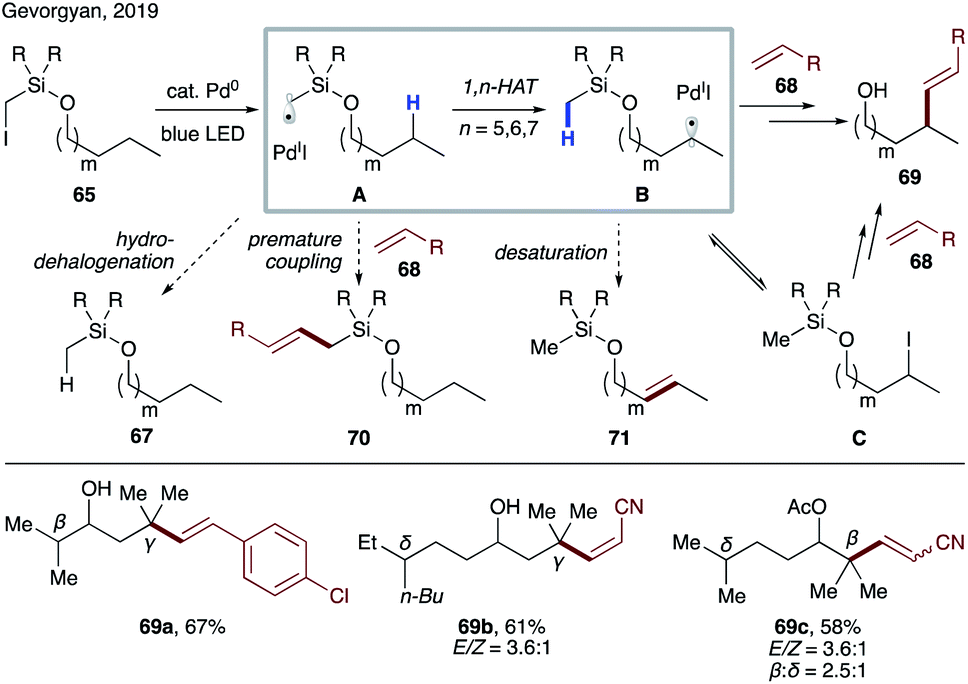 | ||
| Scheme 17 Radical relay Heck reaction of aliphatic alcohols. | ||
In continuation of employing silicon-based tethers for remote transformations, Gevorgyan's group reported a transition metal- and light-free directed amination of 65 (Scheme 18).16 This method represents the first general approach for selective amination of inert C(sp3)–H bonds at β-, δ-, and γ sites. In the presence of lithium formate, readily available aryl diazonium salt 72 is reduced to generate an aryl radical, which abstracts an iodine atom from the substrate. The resultant silyl methyl radical A is electrophilic and thus not predisposed to premature coupling with 72. As such, the desired 1,n-HAT occurs, leading to a transposed nucleophilic radical B, which undergoes facile radical addition to 72 to furnish diazenylation product 73. It is noteworthy that the primary C–H site is also reactive under the conditions of this method (73d). In addition, protected amino alcohols 74 can be obtained in reasonable yields via a one-pot three-step protocol.
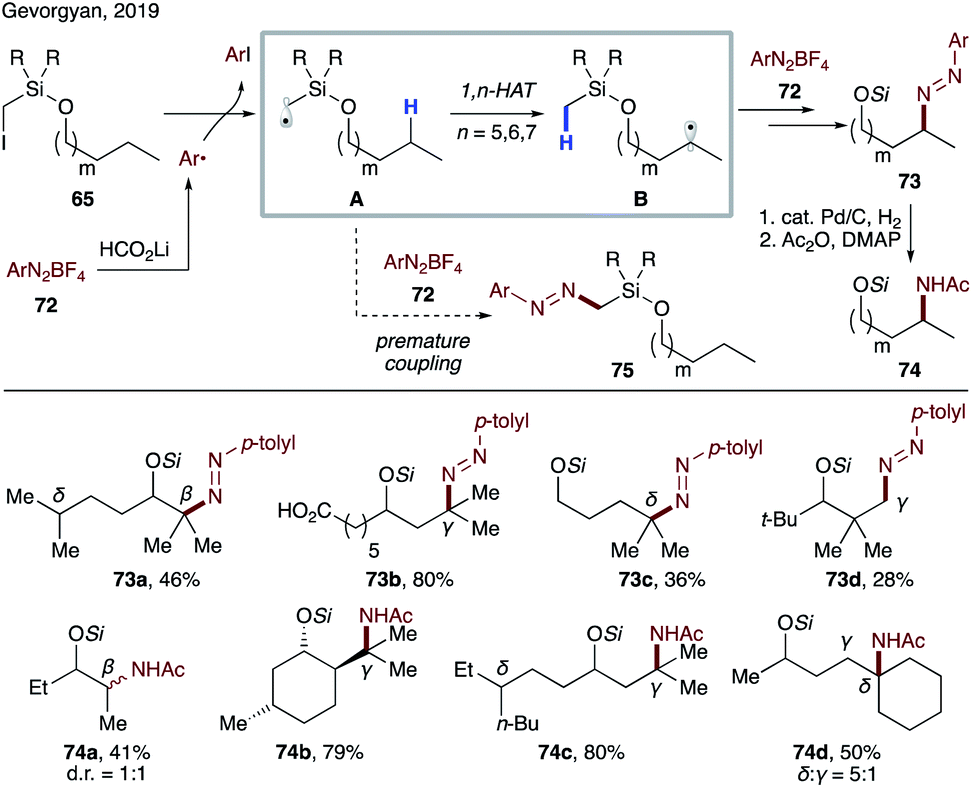 | ||
| Scheme 18 Transition metal- and light-free remote amination of aliphatic alcohols. | ||
Intermolecular HAT
Efficient transformations involving intermolecular HAT mediated by sp3-hybridized C-centered radicals are rare due to a smaller thermodynamic driving force, as compared to those mediated by C(sp2)-, N- or O-centered radicals. In 2002, Tomioka's group utilized the instability of the methyl radical to achieve intermolecular radical addition of ethers to aldimines (Scheme 19a).43 Dimethylzinc and oxygen initiate the reaction by producing a methyl radical, which abstracts activated α-hydrogen from ethereal solvent 76 to generate nucleophilic radical A. The latter then adds to aldimine 77 to give aminyl radical B, which is reduced by dimethylzinc to furnish product 78 after aqueous workup. Meanwhile, the methyl radical is regenerated to close the radical chain cycle. Both cyclic and linear ethers are suitable substrates, albeit with moderate diastereoselectivity in most cases. Employment of diethylzinc and diisopropylzinc led to a drastic decrease in product yield due to significant formation of alkyl adduct 79 and/or reduction product 80, highlighting the pivotal role of methyl radical in efficient intermolecular HAT (Scheme 19b). Direct HAT between 76 and N-centered radical B is unlikely, as the reaction requires a super-stoichiometric amount of zinc reagent. Later in 2003, the same group developed chemoselective addition of the THF radical to aldehydes under Et3B/air conditions.44 This initiator-dependence was also applied to three-component reaction of aldehyde, amine and THF.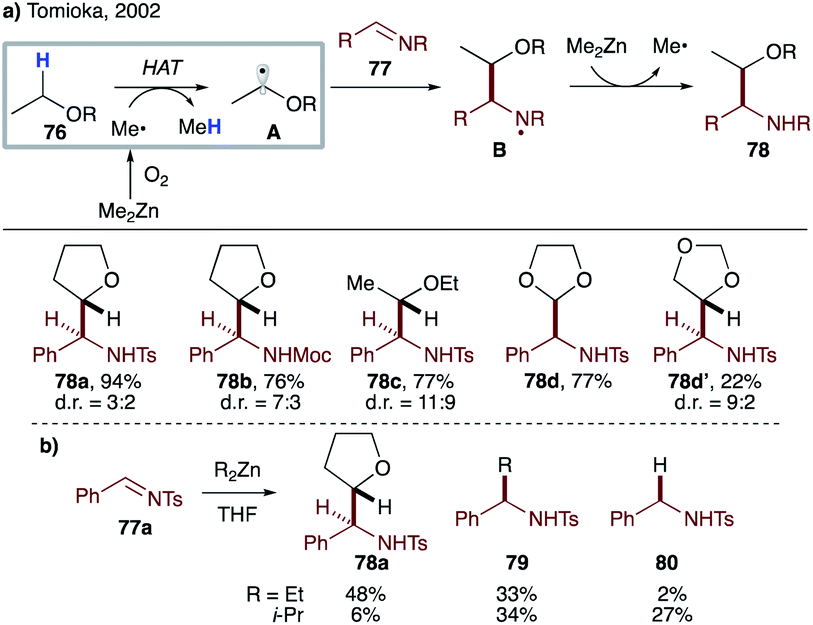 | ||
| Scheme 19 (a) Intermolecular HAT from 76 to the methyl radical. (b) Superiority of the methyl radical vs. ethyl- and isopropyl radicals. | ||
Given the relatively small thermodynamic driving force for the HAT step to the C-based radicals, the polarity match becomes an increasingly important factor. In 2015, the Li group reported visible light-promoted transesterification and debenzylation reactions of alkyl benzyl ethers 81 (Scheme 20).45a The trichloromethyl radical is first generated via a single electron reduction of bromotrichloromethane by a photoexcited iridium catalyst.46 A polarity-matched intermolecular HAT between this electrophilic radical and the electron-rich C–H bond in 81 occurs, resulting in nucleophilic radical A. A subsequent halogen atom transfer (XAT) produces α-bromoether B and a trichloromethyl radical, thereby propagating the radical chain process. Intermediate B can either be hydrolysed in methanol in a one-pot two-step manner to the debenzylation products 82 or undergo in situ transesterification to afford esters 83.
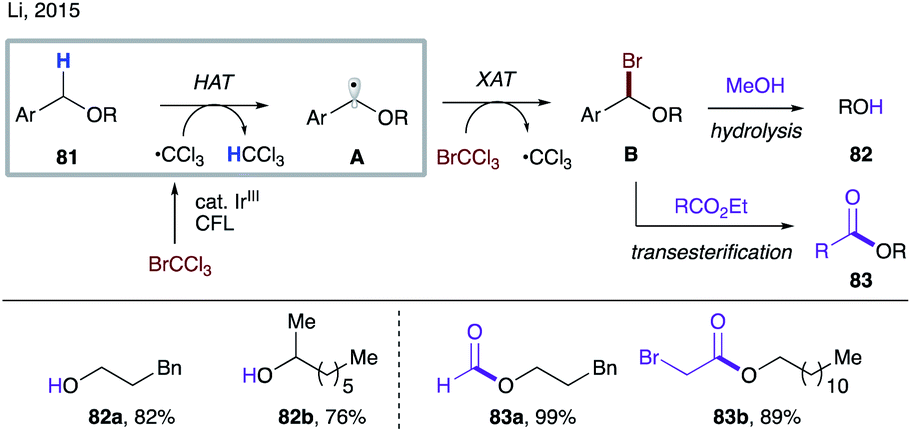 | ||
| Scheme 20 Employment of the trichloromethyl radical in intermolecular HAT. | ||
Later, Li's group showed that substrates possessing secondary benzylic C–H bonds (84) are also competent for analogous bromination, thus affording benzyl bromides 85 in good to excellent yields (Scheme 21a).45b In addition, a one-pot synthesis of benzyl amines 86 was developed, albeit a huge excess of amine was required to achieve good yields. In 2019, Kappe and co-workers disclosed the same process for toluene derivatives 87 under a continuous flow reaction setup using benzophenone as a photocatalyst (Scheme 21b).45c Moderate yields of benzylamines 89 were obtained when 87 was susceptible to dibromination (89a).
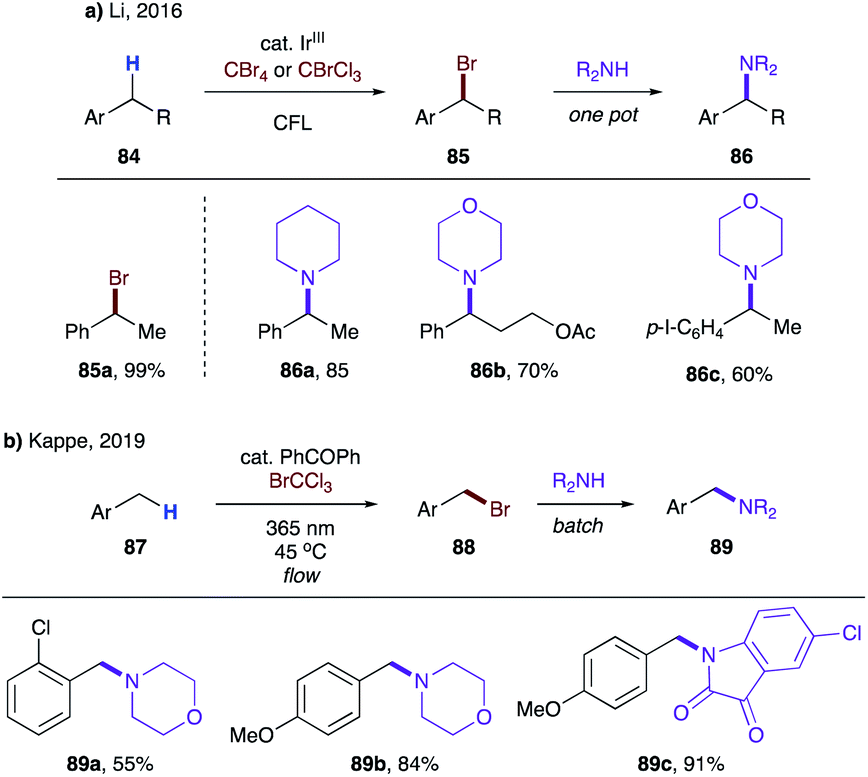 | ||
| Scheme 21 Benzylic C–H bromination en route to benzyl amines via intermolecular HAT. (a) Secondary benzylic C–H. (b) Primary benzylic C–H. | ||
HAT involving vinyl radicals
A larger BDE difference between olefinic C–H (∼110 kcal mol−1) and aliphatic C–H (∼96–100 kcal mol−1) bonds makes the 1,n-HAT process more efficient compared to that by an alkyl radical.47 According to studies by Gilbert and co-workers, the rate of 1,5-HAT at the activated C–H site by a vinyl radical is more than 105 s−1.48 After the HAT process, usually the alkene moiety serves as a radical acceptor for the translocated radical, leading to the formation of cyclopentane or cyclohexane derivatives.49 In general, a vinyl radical is generated by halogen abstraction/single electron reduction of the corresponding vinyl halides or via radical addition to alkynes.50 The second approach is more attractive from a practical standpoint, since the first protocol requires synthesis of vinyl halides. However, despite being exothermic in nature, the intermolecular radical addition to an alkyne is a relatively slow process. The feasibility of HAT by vinyl radicals was first reported in 1967 by Heiba and Dessau in the novel radical cascade involving addition of ˙CCl3 across a triple bond that triggered 1,5-HAT.50b However, it was not until 1988 when the first systematic studies on 1,5-HAT of vinyl radicals were reported by Parsons.9bIntramolecular HAT
In 2016, Du Bois and co-workers achieved concise asymmetric synthesis of the natural (−) and non-natural (+) antipodes of batrachotoxin, as well as both enantiomers of a C-20 benzoate-modified derivative (Scheme 22).52 The key radical cascade sequence involving 1,4-HAT of a vinyl radical formed in situ is highly efficient and selective. The process commences with addition of a tributyl tin radical at the terminal alkyne of 90, followed by sequential 6-endo-trig/5-exo-dig cyclizations to produce reactive intermediate A. This electrophilic radical then undergoes 1,4-HAT to form stabilized nucleophilic allylic radical species B, which is quenched by Bu3SnH to deliver 91 in 75% yield.
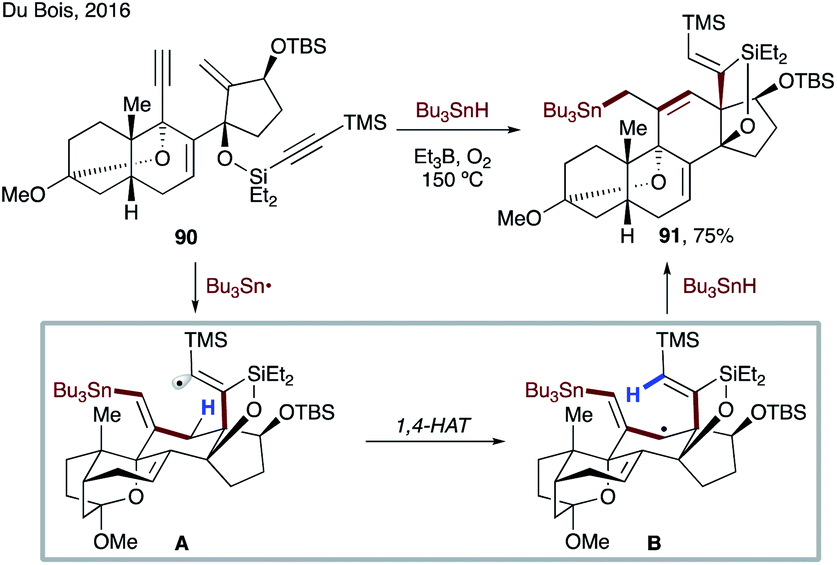 | ||
| Scheme 22 Synthesis of the steroidal core of batrachotoxin via 1,4-HAT of a vinyl radical. | ||
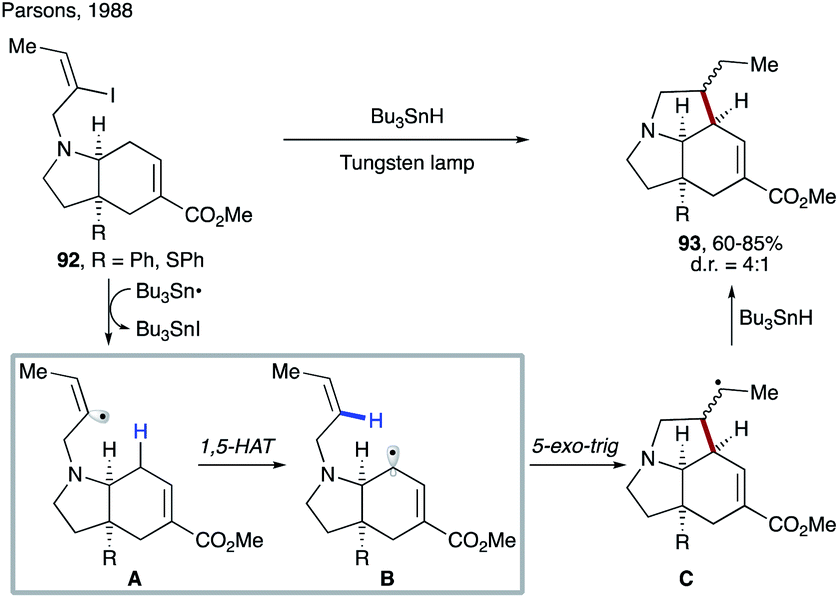 | ||
| Scheme 23 1,5-HAT triggered by a vinyl radical in the synthesis of a pyrrolizidine ring system. | ||
In their seminal work, Curran's group reported translocation of vinyl radicals via 1,5-HAT (Scheme 24a).9a Realising that C–H bonds, the most attractive and simplest ‘radical precursor’, could not be activated via intermolecular HAT by a tin radical due to its significantly endothermic nature, they utilized Bu3SnH as an atom transfer agent to access vinyl radical A from 94. Efficient 1,5-HAT occurs to form translocated alkyl radical B, which is cyclized to produce cyclopentane derivatives 96 after reduction. A premature reduction of the vinyl radical via intermolecular HAT leads to the acyclic alkene byproduct 95. In 1993, Curran's group disclosed the substituent effect on these types of 1,5-HAT transformations.53 Although a few exceptions exist, the authors found a correlation between the rate of 1,5-HAT and the BDE of the C(sp3)–H bond. They also found that the overall geometry of the moiety undergoing HAT was likely to have a greater impact than the particular substituent at the target C–H site.
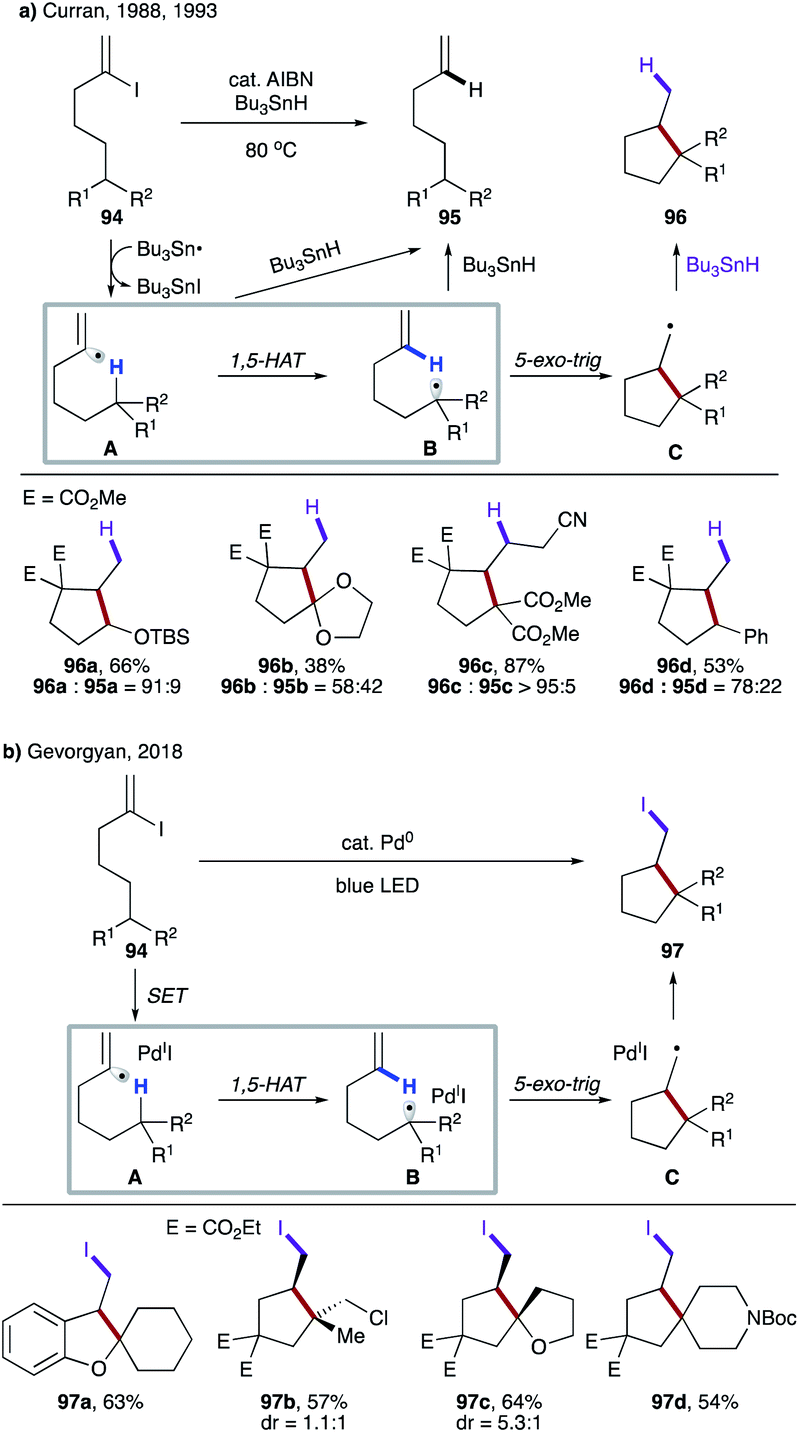 | ||
| Scheme 24 (a) Free radical translocation via 1,5-HAT. (b) 1,5-HAT of the hybrid vinyl palladium radical intermediate. | ||
In 2018, Gevorgyan's group reported photoinduced palladium-catalyzed non-chain atom transfer radical cyclization (ATRC) at remote unactivated aliphatic C–H sites with similar types of substrates (Scheme 24b).54 The hybrid vinyl palladium-radical A undergoes a 1,5-HAT process to generate the translocated radical B, which upon 5-exo-trig cyclization leads to the intermediate C. Finally, iodine atom transfer from the putative Pd(I)I affords iodomethyl carbo- and heterocyclic motifs 97, thus contrasting with the reductive transformation by Curran. Notably, no formation of Heck-type products via β-hydrogen elimination of the radical intermediate C was observed.
As discussed above, the alternative way to form vinyl radicals involves addition of radicals to alkynes. In 1967, Heiba and Dessau observed 1,5-HAT of a vinyl radical during addition of polyhalomethanes to terminal alkynes.55 In 1993, Bachi and co-workers reported the formation of vinyl radical Avia addition of the tributyltin radical to an alkyne moiety of 98. A subsequent 1,5-HAT, followed by 6-endo-trig cyclization and β-tin elimination produces β-lactam C (Scheme 25).56 Upon DBU-catalyzed isomerization of the double bond, methyl 1-oxahomoceph-4-em-5-carboxylate 100 was obtained in 40% yield, along with 25% of reduction byproduct 99.
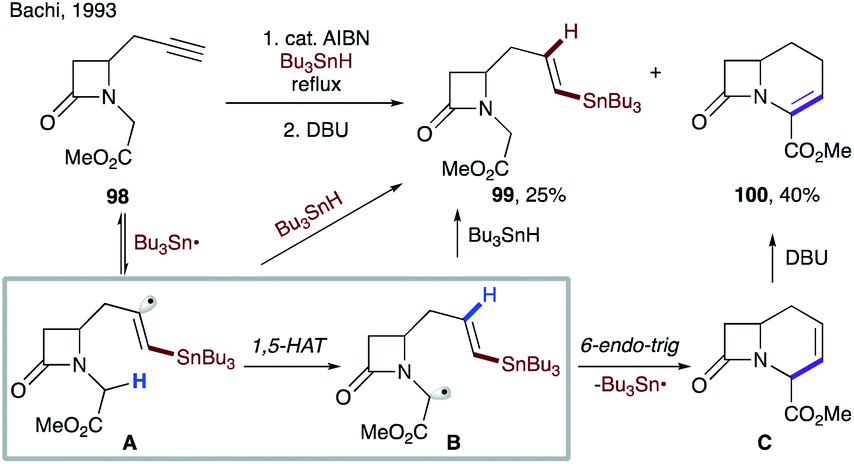 | ||
| Scheme 25 1,5-HAT of a vinyl radical in the synthesis of bicyclic β-lactam. | ||
Few years later, Malacria's group reported an efficient radical cascade reaction for diastereoselective synthesis of highly functionalized cyclopentanes (Scheme 26a).57 In this transformation, substrate 101 forms an α-silyl radical upon XAT, which then cyclizes to give vinyl radical species A. 1,3-Allylic interaction in E-A and eclipsed interaction between methyl and isopropyl groups in Z-A2 render them less favourable conformers than Z-A1. The latter undergoes highly regioselective 1,5-HAT at the unactivated C(sp3)–H site, leading to primary alkyl radical intermediate B. A following rarely observed 5-endo-trig cyclization constructs the cyclopentane moiety. Finally, the cleavage of the siloxy ring with MeLi affords product 102 in impressive 74% overall yield.
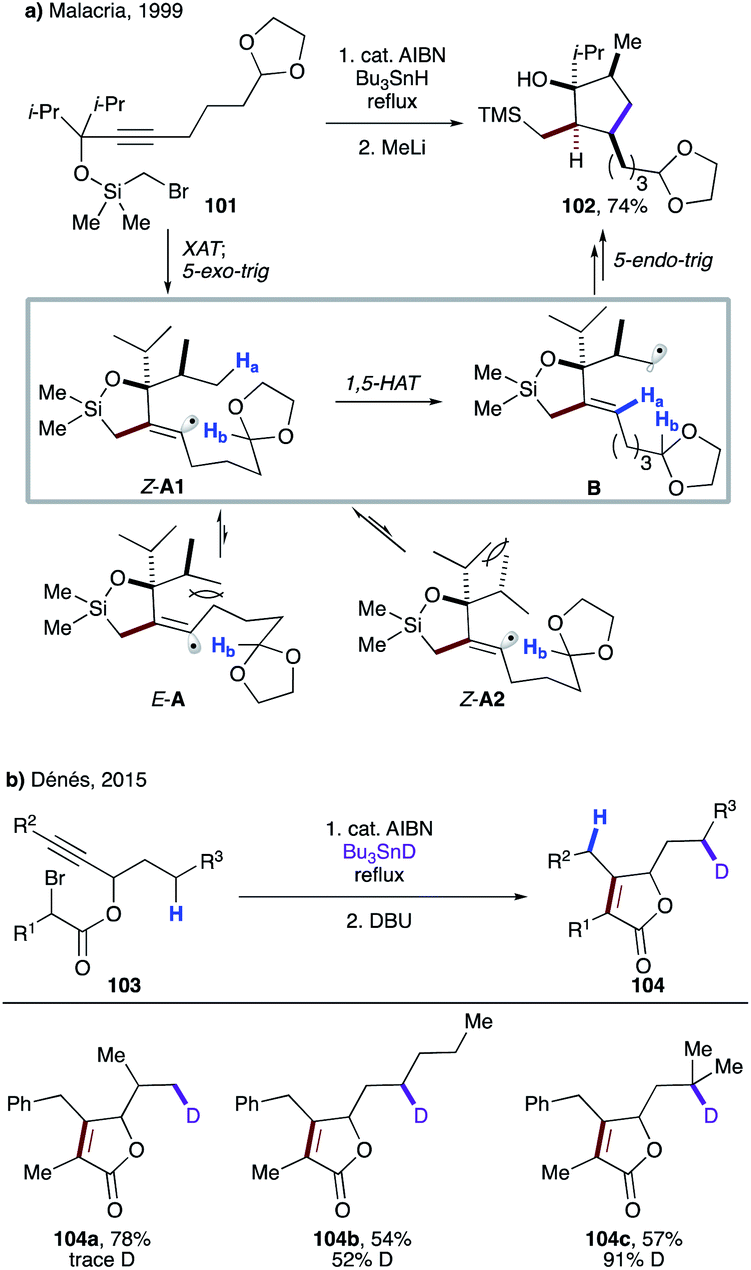 | ||
| Scheme 26 (a) Diastereoselective 1,5-HAT event triggered by 5-exo-dig cyclization, followed by 5-endo-dig cyclization. (b) 5-Exo-dig cyclization followed by “invisible” 1,5-HAT. | ||
Recently, Dénés' group disclosed the involvement of 1,5-HAT in a similar type of reaction sequence during the synthesis of polysubstituted γ-butenolides (Scheme 26b).58 The deuterium labelling experiments revealed an “invisible” 1,5-HAT to the formed vinyl radical from the aliphatic side chain. Expectedly, due to the greater stability of the tertiary radical, the higher deuterium incorporation was observed in 104c.
In 2019, Zhu and co-workers reported mild, photocatalytic methods for activation of δ C(sp3)–H bonds via formation of vinyl radicals (Scheme 27a).59 In both cases, the authors synthesized the tertiary propargyl alcohol derivatives 107via addition of acetylide 106 at ketone 105. Under the reaction conditions, the excited photocatalyst undergoes SET to generate electrophilic radical Y˙, which adds to an alkyne to trigger a 1,5-HAT event. The translocated radical species B undergoes a sequence of 5-exo-trig cyclization, β-elimination and oxidation by a photocatalyst to afford the final product 108–109. Using this protocol, valuable, complex fluoroalkylated alkenes 108a–c,59a and sulfonylvinylation products 109a–c (ref. 59b) were easily accessed.
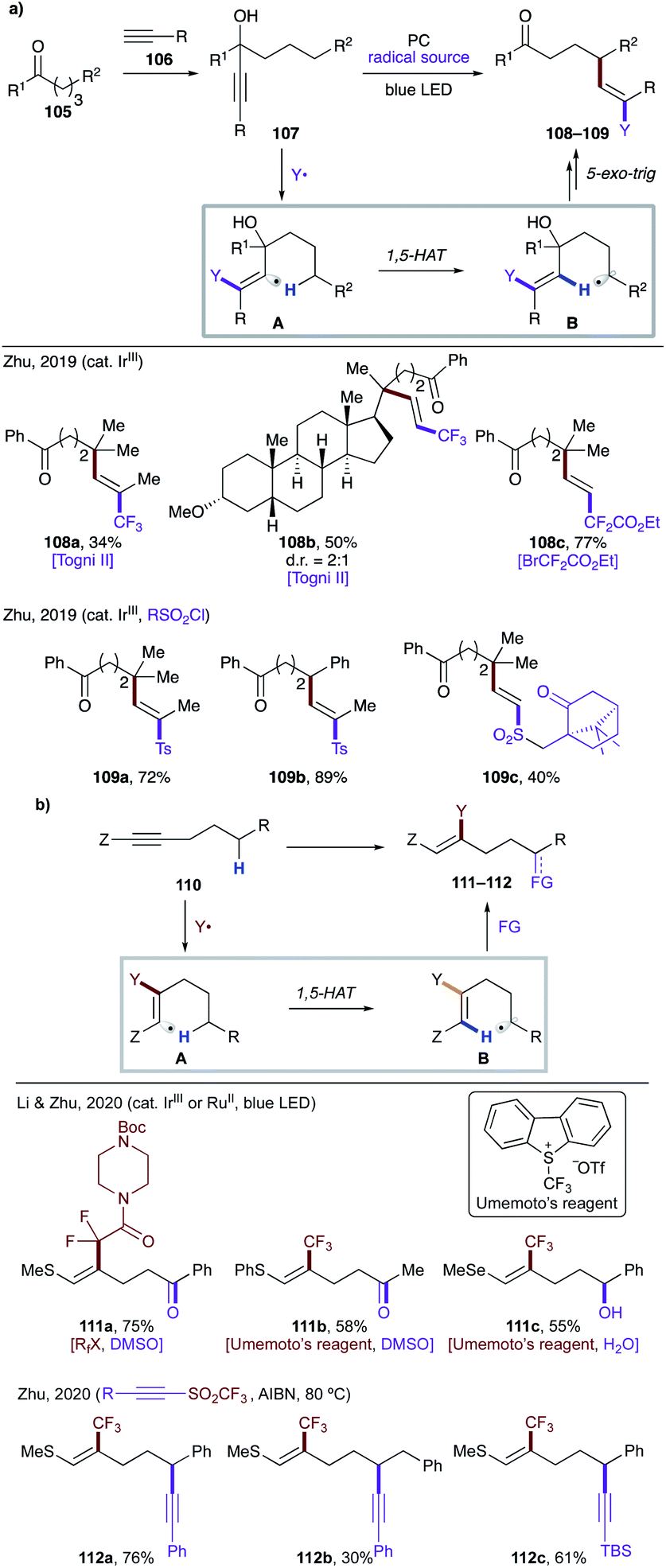 | ||
| Scheme 27 (a) Regioselective vinylation via 1,5-HAT, triggered by radical addition to an alkyne. (b) Remote C–H functionalizations of an alkyne via 1,5-HAT of a vinyl radical formed by β-radical addition. | ||
Recently, Li, Zhu and co-workers reported remote functionalization of heteroalkyne 110via HAT of a vinyl radical formed by β-radical addition (Scheme 27b).60 The method involves addition of a perfluoroalkyl radical to an alkyne to generate vinyl radical A, which undergoes 1,5-HAT to produce nucleophilic alkyl or benzyl radicals B. In the first scenario, the translocated radical B is oxidized selectively to ketone or alcohol 111a–c, while in the second case, the radical B is trapped by an electrophilic alkyne to afford remote alkynylation products 112a–c.
Very recently, Chu's group developed photoredox/nickel dual-catalyzed conditions for sequential deoxygenative vinylation/C–H arylation of tertiary oxalates (Scheme 28).61 This multicomponent cascade is initiated by a single electron oxidation of oxalate 113 by a photoexcited iridium catalyst, which results in decarboxylation, followed by addition of the formed tertiary alkyl radical to alkyne 114 to form vinyl radical A. The succeeding 1,5-HAT affords a translocated secondary alkyl radical B, which is captured by Ni(0) to form alkyl Ni(I) species C. The reaction product 116 is obtained upon nickel-mediated cross-coupling with an aryl halide.
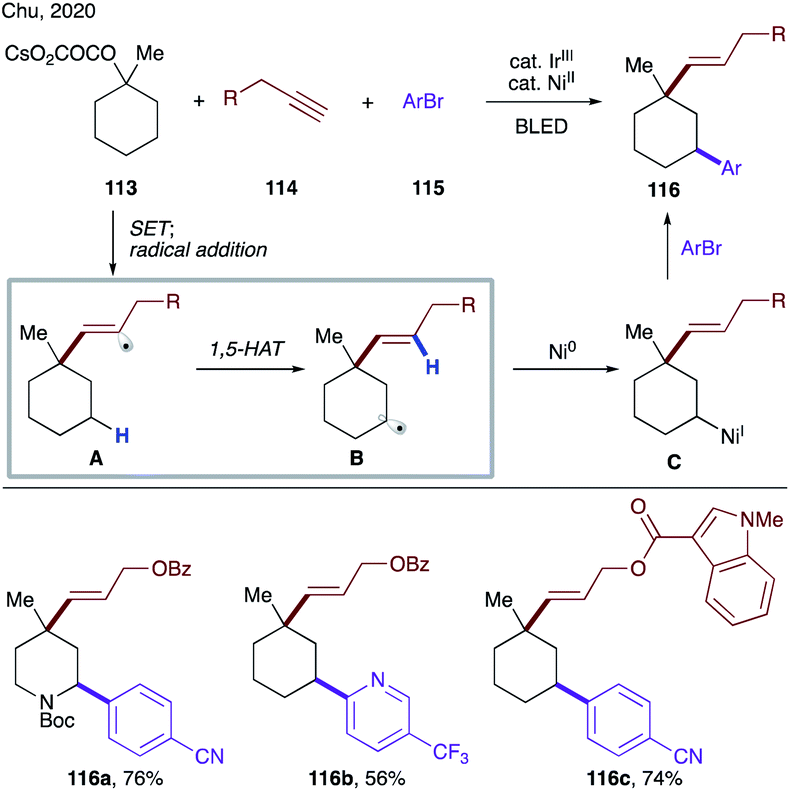 | ||
| Scheme 28 Photoredox/nickel dual-catalyzed radical cascade involving 1,5-HAT of vinyl radicals. | ||
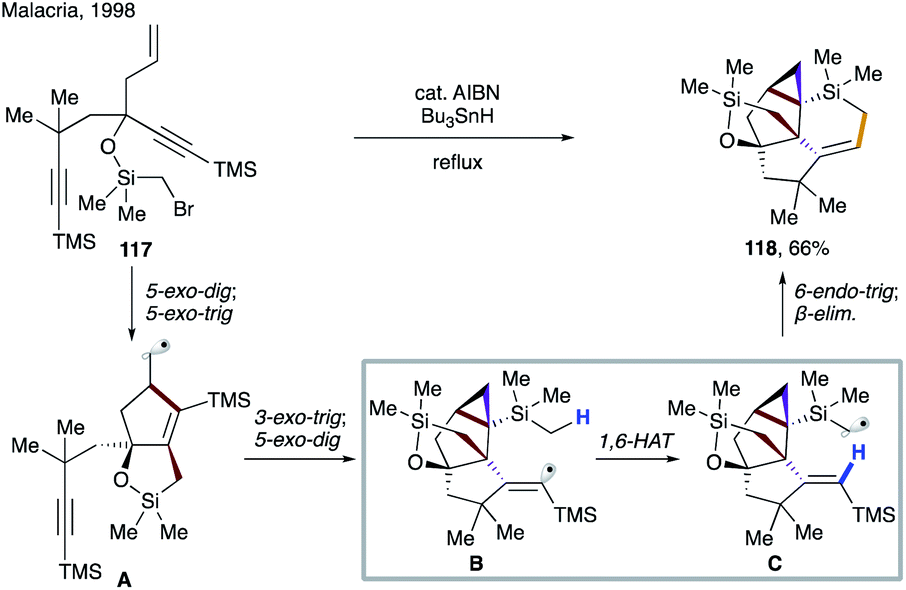 | ||
| Scheme 29 Highly diastereoselective 1,6-HAT in the synthesis of triquinane derivatives. | ||
Several examples of radical cascade reactions showcased the formation of electrophilic vinyl radical A generated from 119, which undergoes 1,6-HAT to form nucleophilic α-heteroatomic radical B. A subsequent 5-exo-trig cyclization, followed by reduction of the forming alkyl radical affords the cyclic product 120 (Scheme 30). In 2002, Parsons and co-workers utilized this strategy to synthesize 122, the precursor of mitomycin ring systems.63 Few years later, Reiser's group reported the synthesis of 2,3-disubstituted dihydrobenzofurans 125a–d involving a similar type of 1,6-HAT process.64 In 2017, the same group employed photoredox catalysis to synthesize 2,3-disubstituted indolines 127 using a similar strategy.65 Beaudry and co-workers also utilized this 1,6-radical translocation strategy to construct the central indoline core of several natural products (129, 130).66 In 2007, Yoshimura, Takahata, and co-workers reported the synthesis of 6,5′-C-cyclouridines 132 and 133, in which the tris(trimethylsilyl)silyl (supersilyl) radical initiates the process by selenium abstraction in 131.67
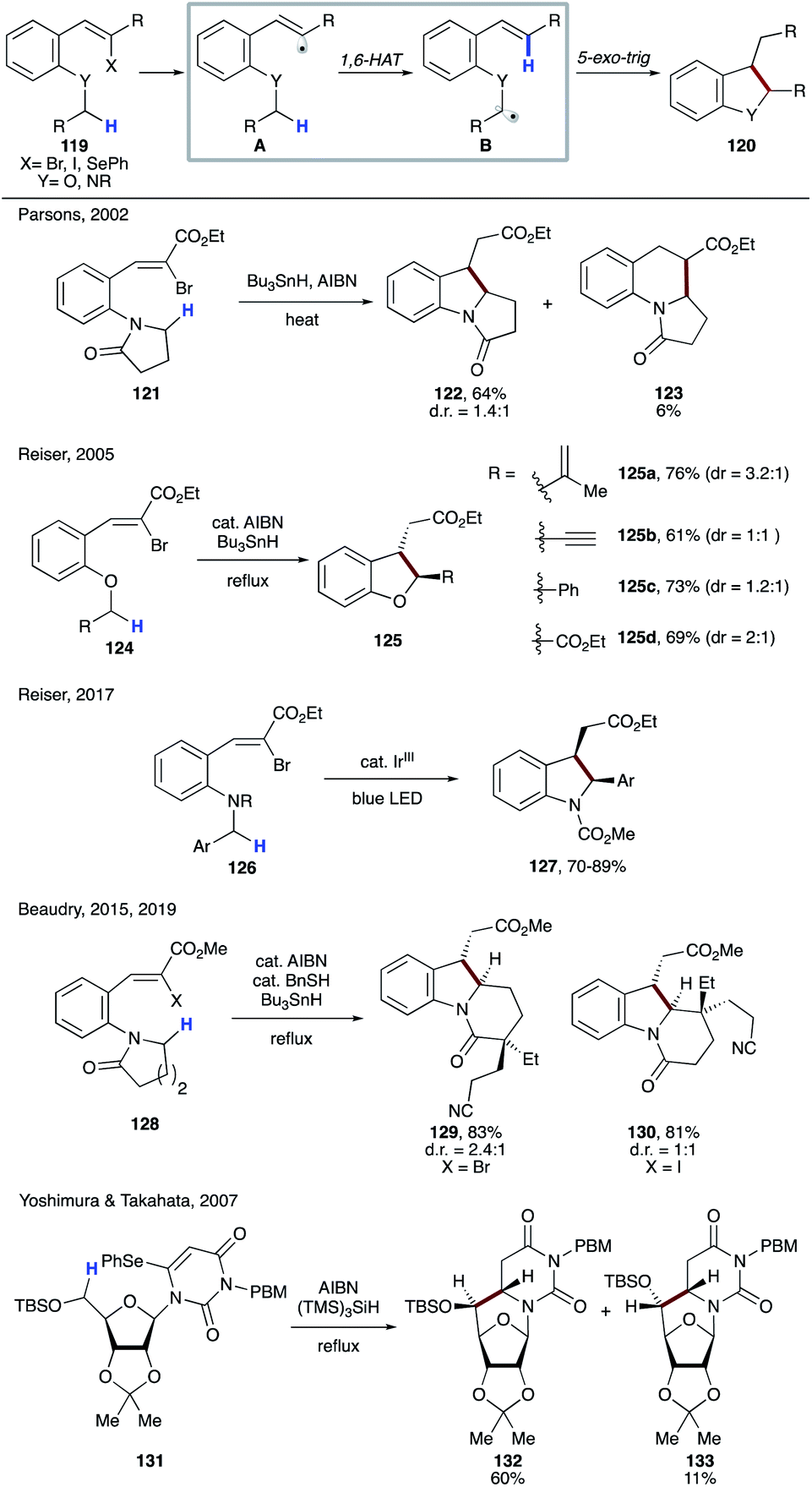 | ||
| Scheme 30 Cascade radical reactions of vinyl radicals involving 1,6-HAT at C(sp3)–H sites α to the heteroatom. | ||
HAT involving aryl radicals
Aryl radicals are extremely reactive species. Similarly to HAT to vinyl radicals, HAT to aryl radicals from aliphatic C–H sites is energetically favourable due to the high BDE difference between aryl C–H bonds (Ph–H, BDE = 112 kcal mol−1) and aliphatic C–H bonds (BDE ∼ 96–100 kcal mol−1).47 In 1954, Hey and co-workers recognized intramolecular 1,5-HAT during the decomposition of o-(dimethylaminocarbonyl)aryl or (N-aryl-N-methylaminocarbonyl)aryl diazonium salts.8 Later, Cohen and co-workers also showed that aryl radicals could undergo 1,5-HAT during copper-catalyzed decomposition of diazonium salts, which was supported by extensive mechanistic studies.68 The deuterium labelling experiments suggested that the rate of 1,5-HAT is much faster than the rotation around the amide C–N bond but slower than that of the methyl C(sp3)–N bond, presumably due to the partial double bond character of the former. In 1978, Pines and co-workers reported the intramolecular HAT process of aryl radicals generated from different ortho-substituted aryl diazonium salts. Furthermore, intramolecular 1,5-, 1,6-, and 1,7-HATs were observed during copper-catalyzed decomposition of o-di-n-propylaminosulfonylbenzenediazonium salts.69Intramolecular HAT
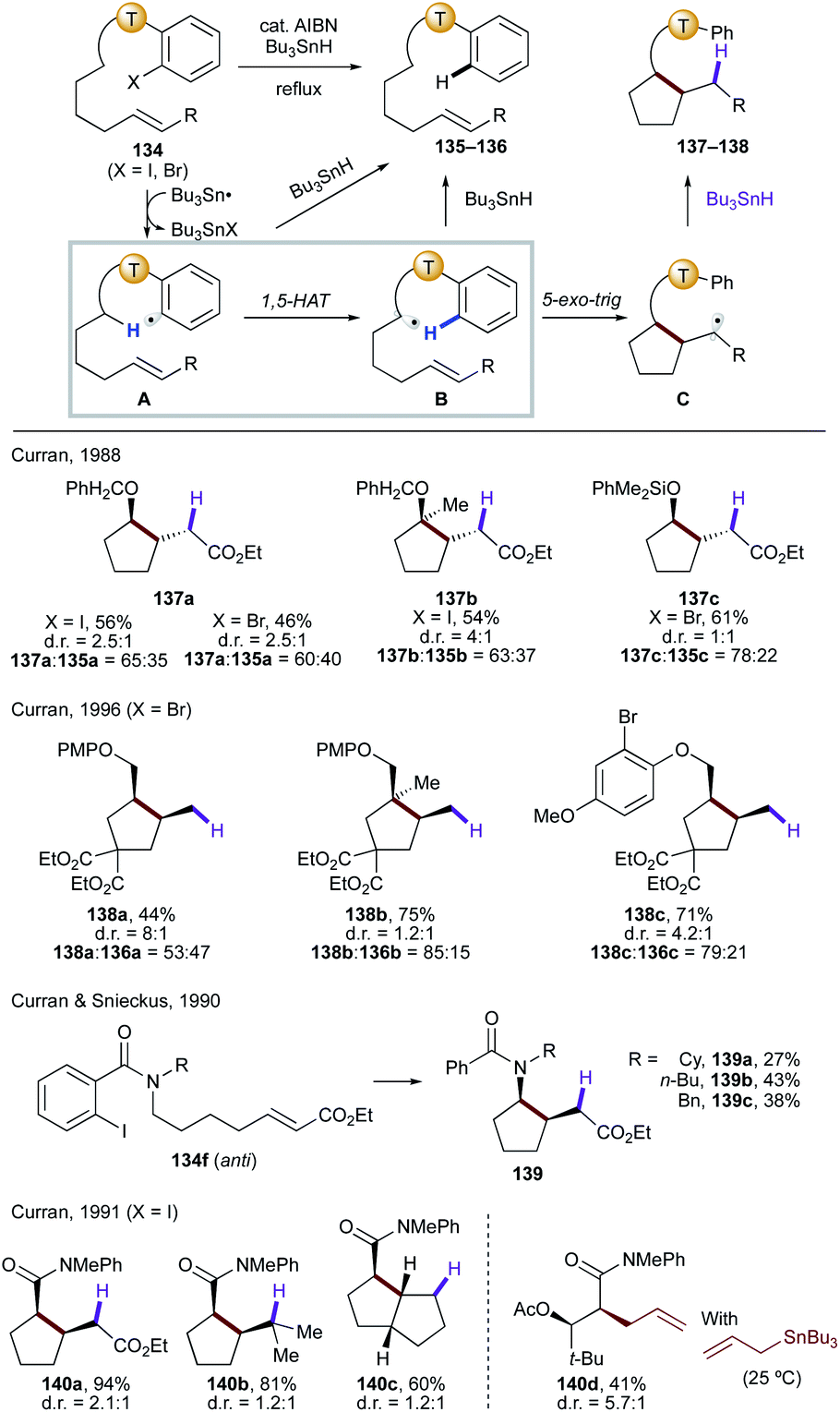 | ||
| Scheme 31 Translocation of radicals from C(sp2) to C(sp3) sites via 1,5-HAT, followed by cyclization to afford cyclopentane derivatives. | ||
Similarly, amides have also been utilized in the related process involving the 1,5-HAT step.72 In the case of o-iodobenzamides,72a,b only the anti-rotamer of 134f will lead to the desired cyclic product (139a–c). Thus, the reaction outcome is highly dependent on the R group as it affects the anti/syn ratio of the amide. On the other hand, employment of o-iodoanilides allows for the direct α C–H functionalization of carbonyl compounds without enol or enolate formation.72c,d The reaction is shown to be effective for intermolecular radical trapping of the translocated radical with deuterium or an allylic group (140d).
In 2016, Gevorgyan's group introduced a 1,5-HAT event of hybrid palladium-radical species (Scheme 32).73 Aryl halides are well-known substrates in palladium catalysis, typically involved in a two-electron oxidative addition process. But in this case the authors have discovered that irradiation with blue light switched the reaction pathway leading to an SET process to generate hybrid palladium-radical species A. Due to the radical nature of this intermediate, it undergoes a 1,5-HAT process to generate translocated radical species B, which upon β-hydrogen loss delivers silyl enol ethers 142a–d. In this light-induced protocol, palladium plays a double role by harvesting light and catalyzing the bond breaking/forming process. Notably, this mild method for dehydrogenation of silyl-protected alcohols to silyl enol ethers proceeds at room temperature without use of exogenous photosensitisers or oxidants.
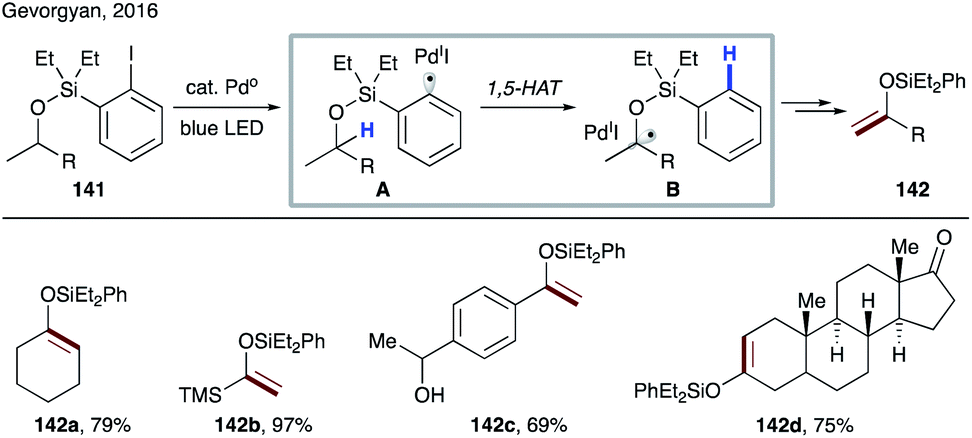 | ||
| Scheme 32 HAT event of hybrid Pd-radical species. | ||
In 1994, Weinreb's group utilized copper catalyzed diazotation of o-aminobenzamides 143 for oxidation of amides, which can also be translated to more complex systems like 144d, used in the synthesis of antibiotic anisomycin (Scheme 33).74 In this case, in situ diazotization of o-aminobenzamide 143, followed by reduction via SET from a copper catalyst generates aryl radical A. 1,5-HAT of the latter affords nucleophilic radical B, which then via the copper-catalyzed radical-polar crossover process gets oxidized to an α-amino carbocation that is trapped by methanol to deliver products 144a–d. Recently, Qi and Zhang's group reported photocatalytic, metal-free methods with similar types of substrates to achieve α-amino C(sp3)–H functionalization.75 Likewise, single electron reduction of the in situ formed diazonium salt by an excited Eosin Y photocatalyst leads to an aryl radical intermediate, which eventually affords α-alkoxybenzamides (145a–d) in moderate to good yields.
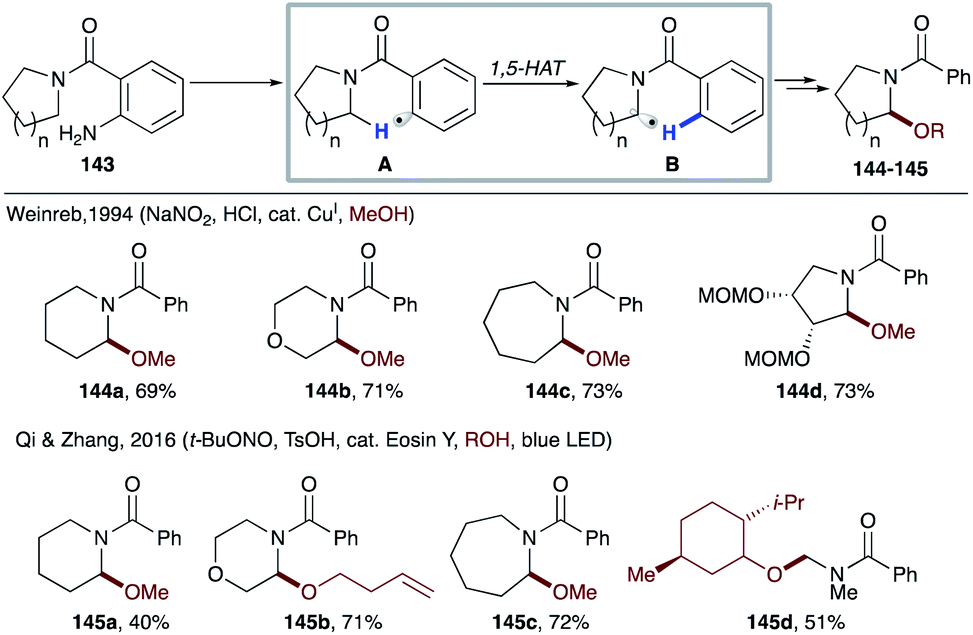 | ||
| Scheme 33 Oxidation of amides via 1,5-HAT. | ||
At the same time, Zeng and co-workers reported site-selective silylation of the α-amino C(sp3)–H bond (Scheme 34).76 The use of Grignard reagent enables generation of N-magnesiated aryl radical A from the corresponding fluoride 146. This method represents the first example of α C–H silylation of amides.
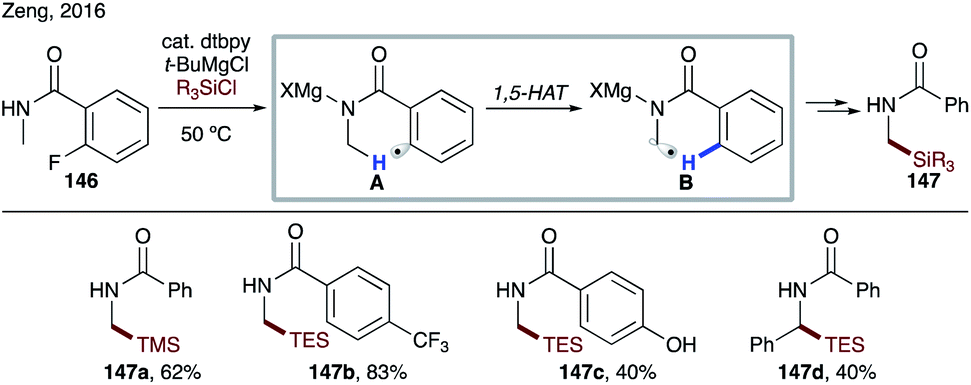 | ||
| Scheme 34 Silylation of benzamides via 1,5-HAT. | ||
It has been shown that in the absence of a tethered alkene as in the previous examples (Scheme 31), the translocated radical B can undergo radical addition to an arene, thus affording cyclization products 149–152 after rearomatization (Scheme 35). In 2013, Kalyani and co-workers reported the use of a nickel or phenanthroline catalyst for aryl chlorides and bromides to afford various isoindolin-1-ones 149a–d.77 Shortly after, Kumar's group developed metal-free conditions targeting bromides and iodides to deliver similar products (150a–d).78 In 2016, Xu's group employed iridium photocatalysis to achieve similar transformations (148 → 151) with iodides and its application to the formal synthesis of (±)-coerulescine and (±)-physovenine.79 More recently, Gevorgyan and co-workers achieved the first photoinduced palladium-catalyzed C(sp2)–O bond activation of an aryl triflate to form a hybrid aryl palladium-radical intermediate,80 which underwent the same sequence of 1,5-HAT/radical cyclization to afford oxyindole and isoindolin-1-ones 152a–d.
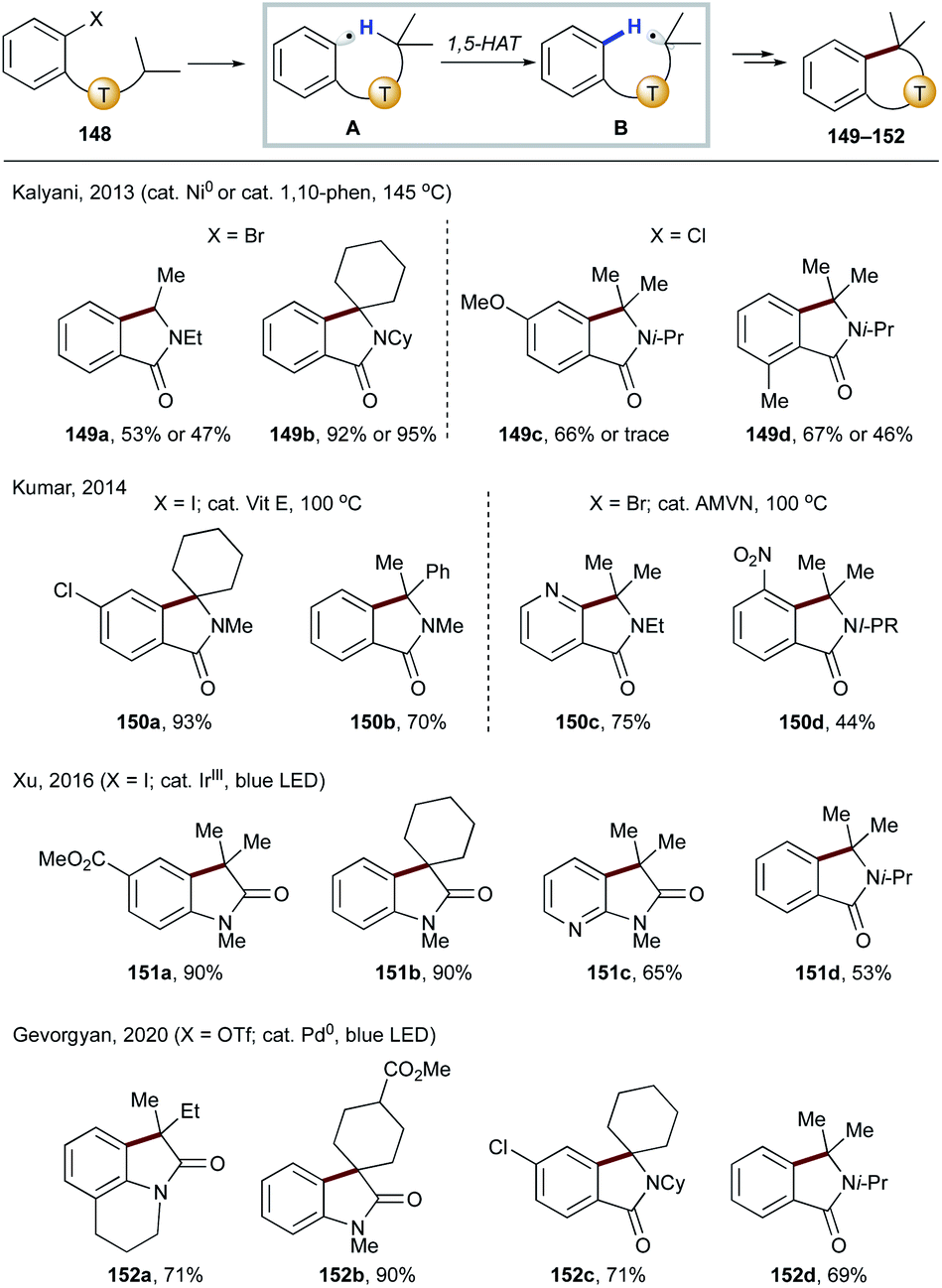 | ||
| Scheme 35 Synthesis of fused systems via 1,5-HAT from the C(sp3)–H to the C(sp2) site followed by radical cyclization at the aromatic ring. | ||
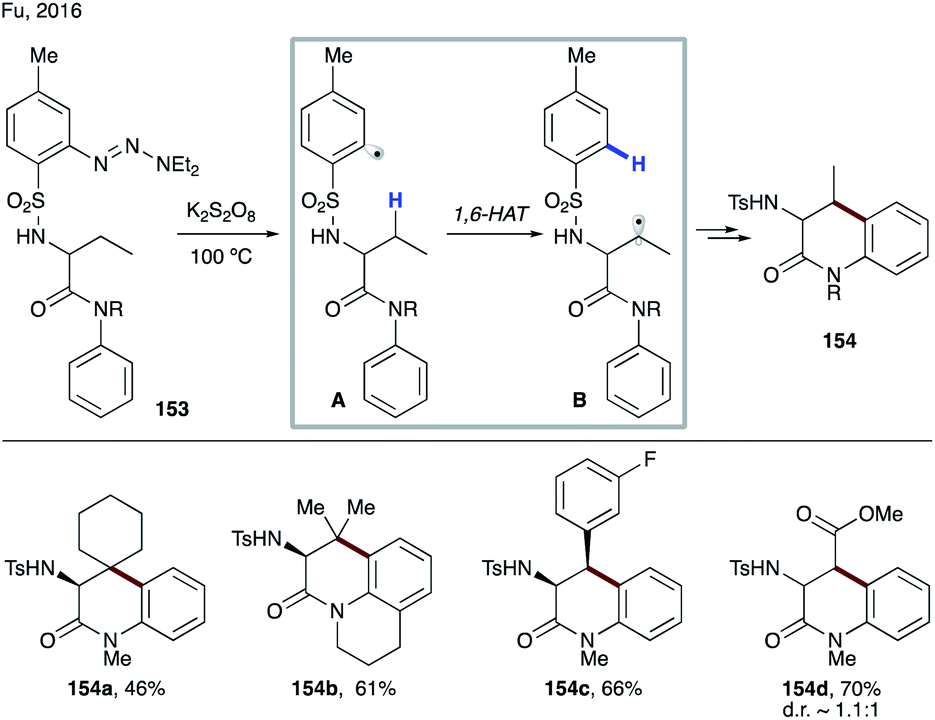 | ||
| Scheme 36 Synthesis of cyclic anilides involving 1,6-HAT from the aliphatic C–H sites. | ||
Recently, Ragains' group developed photocatalytic reduction of ortho-diazoniaphenyl alkyl sulfones for remote C(sp3)–H functionalization (Scheme 37).82 First the photoexcited Ru-catalyst undergoes SET to diazonium salts 155, followed by nitrogen loss leading to aryl radical A. This electrophilic aryl radical then undergoes intramolecular HAT at alkyl or benzylic C–H sites, preferably in a 1,6-fashion (A → B). The formed nucleophilic radical undergoes radical polar crossover via single electron oxidation by the photocatalyst or by the substrate via a chain process into a carbocation. A subsequent trapping of the latter with different nucleophiles affords remote hydroxylation (156a–d), etherification (156e), C–C bond formation (156f), amidation (156g), and desaturation (156h) products. In this protocol, the possibility of 1,5-HAT, as well as 1,7-HAT, was demonstrated, albeit with much lower efficiency.
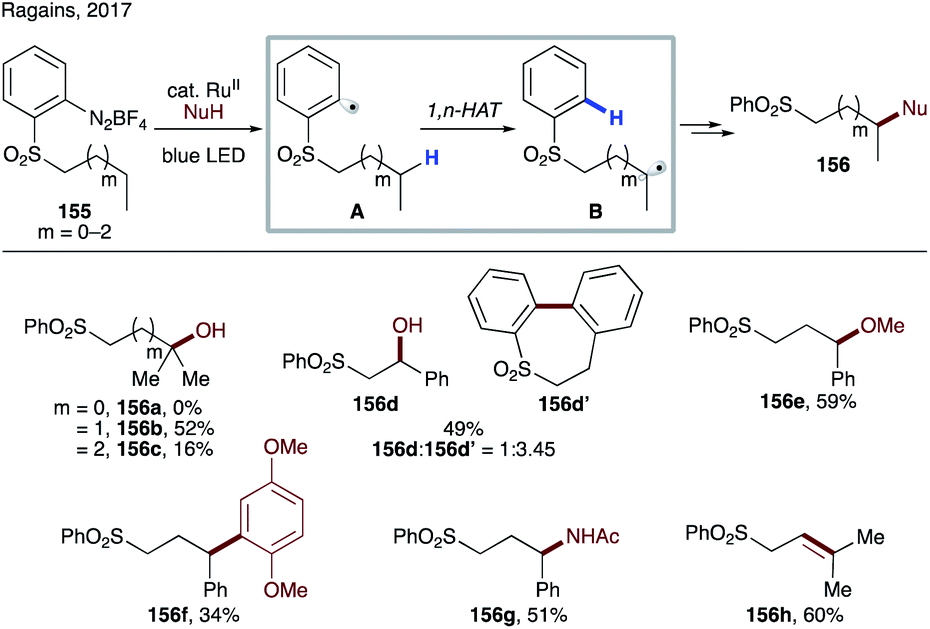 | ||
| Scheme 37 1,6-HAT to the aryl ring of aryl sulfones from aliphatic C(sp3)–H sites. | ||
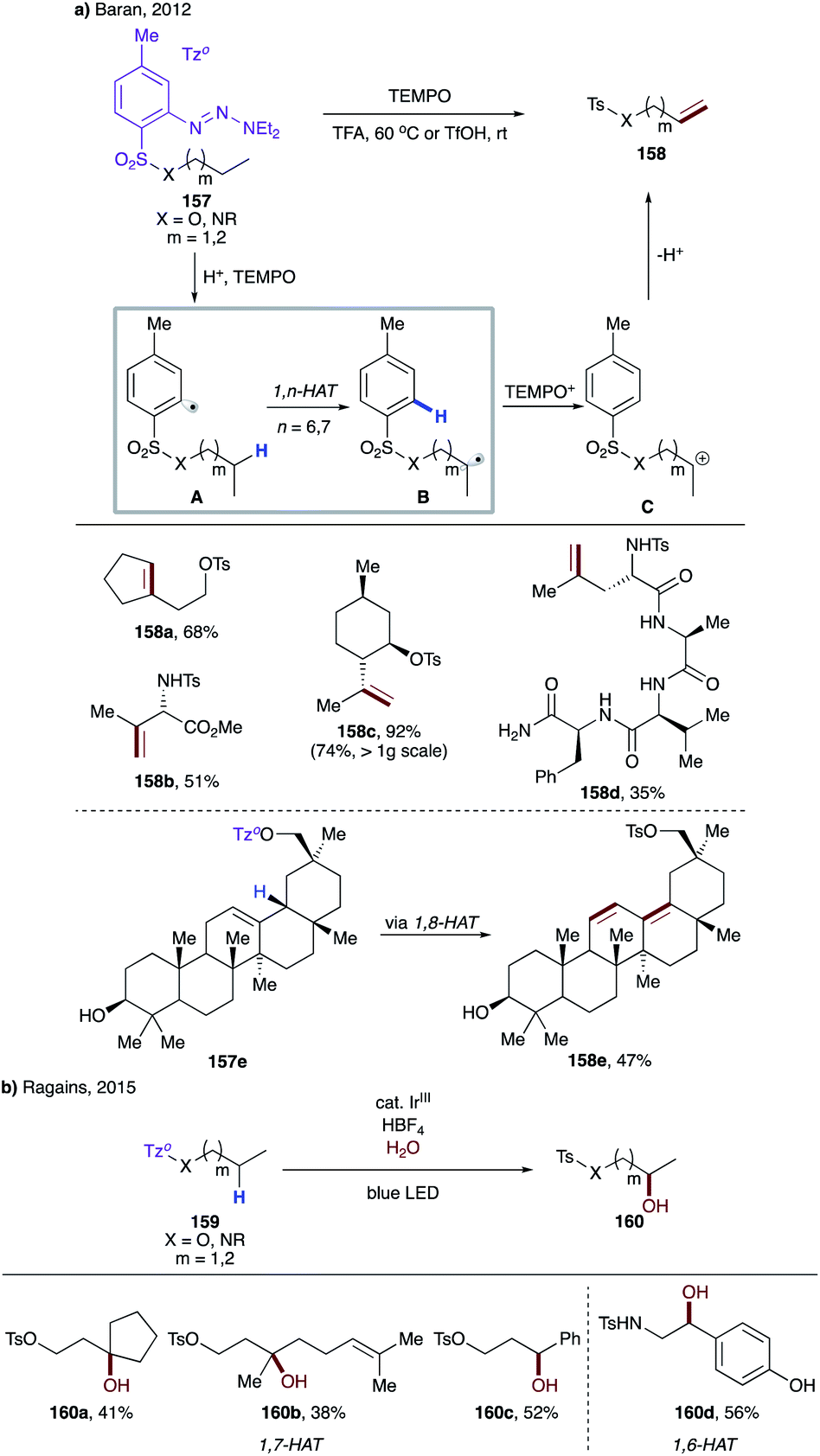 | ||
| Scheme 38 (a) Guided desaturation of aliphatic alcohols and amines via intramolecular HAT. (b) Remote hydroxylation via intramolecular HAT. | ||
In 2015, Ragains and co-workers uncovered remote hydroxylation of alcohols and amines with similar types of aryl sulfonyl tethers under photocatalytic conditions (Scheme 38b).84 In this protocol, instead of TEMPO, a photocatalyst enables the single-electron redox process to afford after solvolysis the hydroxylation products 160a–d. Expectedly, as in the above case, the aryl sulfonyl tether favours 1,7-HAT, unless a weaker benzylic C–H site is present to promote the 1,6-HAT event.
In 2018, Gevorgyan's group developed a mild photo-induced palladium-catalyzed protocol for proximal and remote desaturation of aliphatic amines (Scheme 39).85 This method utilizes readily available 2-iodocarbonyl or 2-iodosulfonyl tethers (162) which can be routinely installed and do not require isolation. Under the reaction conditions, photoexcited palladium undergoes single electron transfer to aryl iodide to form electrophilic aryl radical A, which undergoes 1,n-HAT (n = 5–7) (A → B) to afford the translocated alkyl radical at α-, β-, or γ-C(sp3)–H sites of amines depending upon the type of tether used. The translocated palladium hybrid radical undergoes regioselective β-H loss to afford the desaturation product, either via radical recombination, followed by β-hydride elimination or via direct H-abstraction by Pd(I) species. In this report, the authors utilized the carbonyl tether for 1,5-HAT (163a), while using the aryl sulfonyl tether for 1,7- (163c–d) as well as 1,6-HAT (163b and 163b′).
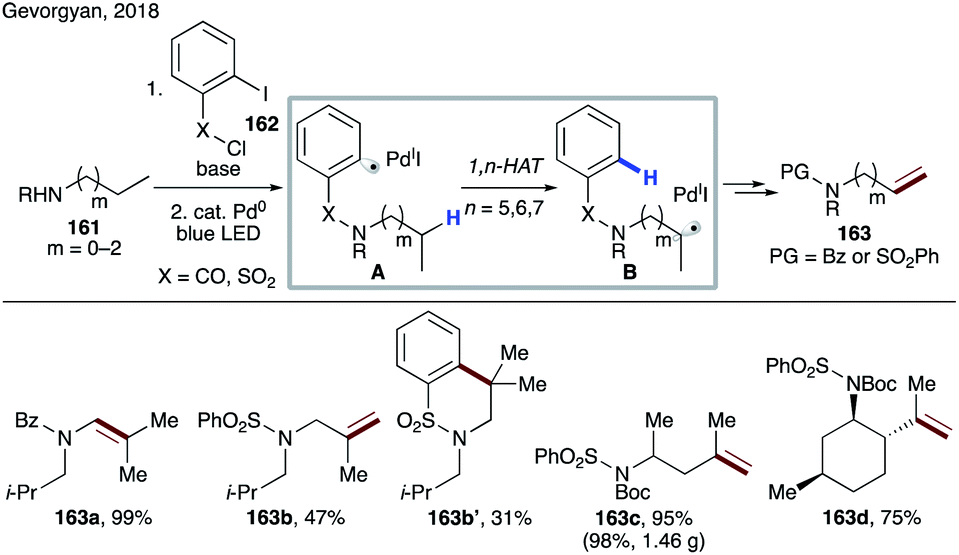 | ||
| Scheme 39 Remote desaturation of aliphatic amines via HAT from the C(sp3)–H site. | ||
In the same year, Studer's group reported translocation of aryl radicals from a sulfonyl group to alkyl C–H sites (Scheme 40).86 In this protocol, they have used thermal conditions and AIBN for radical initiation, which abstracts the weak hydrogen of tris(trimethylsilyl)silane to generate a super silyl radical, which abstracts an iodine atom from substrate 164 to generate aryl radical A, which undergoes 1,7-HAT at unactivated C(sp3)–H sites of alcohols to produce B. The translocated nucleophilic radical undergoes radical Smiles-type rearrangement to afford remote arylated products 165a–c.
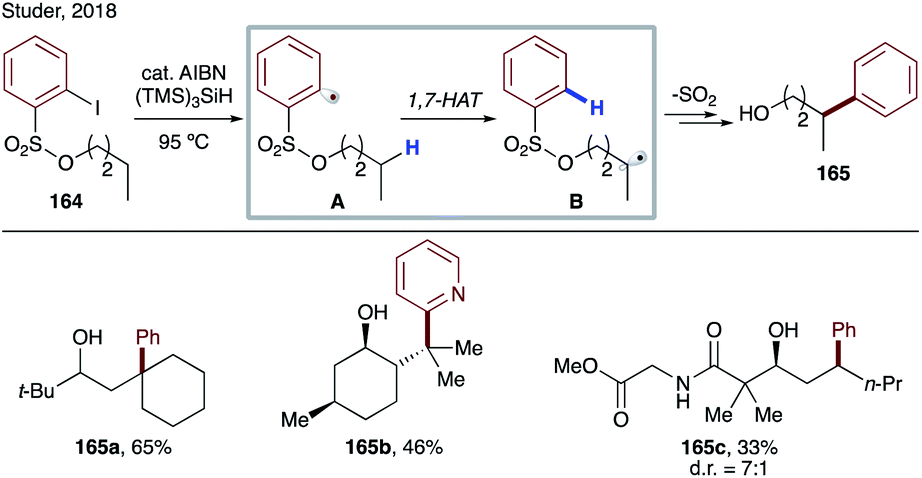 | ||
| Scheme 40 Arylation of alcohols via 1,7-HAT, followed by aryl group migration. | ||
Intermolecular HAT
In contrast to O-centered radicals,7c,87 C-centered radicals are less efficient for intermolecular HAT. Thus, examples of intermolecular HAT by aryl radicals at C(sp3)–H sites are scarce in the literature, mostly limited to activated C–H bonds or use of a C–H source as a solvent. HAT from tetrahydrofuran or toluene by a phenyl radical is a very fast process, with rates of 4.8 × 106 M−1 s−1 and 1.7 × 106 M−1 s−1, respectively.88 Due to the high reactivity of aryl radicals, other side reactions, including addition to an arene and alkene, or halogen abstraction reactions are also capable processes.In 2005, Porta and co-workers reported a systematic study of intermolecular HAT by an aryl radical at C–H sites of tetrahydrofuran and other ethers (Scheme 41).89 They have demonstrated that under aqueous conditions, phenyl radicals, generated from single electron reduction of diazonium salts, are able to intermolecularly abstract a hydrogen atom to form nucleophilic α-alkoxy radicals A. The latter adds to the in situ formed iminium salt B to afford after reduction by Ti(III) species 1,2-aminoalcohol derivatives 168a–c.
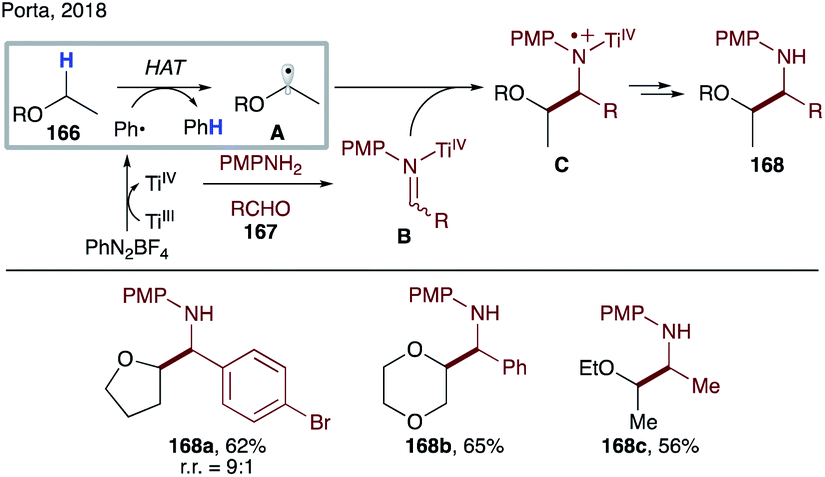 | ||
| Scheme 41 Aminoalkylation of ethers via intermolecular HAT. | ||
Recently, Toste's group reported an interesting light-induced chiral diaryliodonium phosphate-mediated diastereoselective α-acetalization of cyclic ethers (Scheme 42).90 Photochemically active diaryliodonium salt 170 is homolyzed under light irradiation to generate an aryl radical and aryl iodonium radical cation A; the former undergoes a regioselective intermolecular HAT with substrate 169 to produce α-alkoxy radical B, which then recombines with A to give iodonium species C. A subsequent elimination leads to intermediate D or E, which upon chiral diaryliodonium phosphate salt-assisted nucleophilic attack of alcohol 171 delivers the enantioenriched products 172a–b.
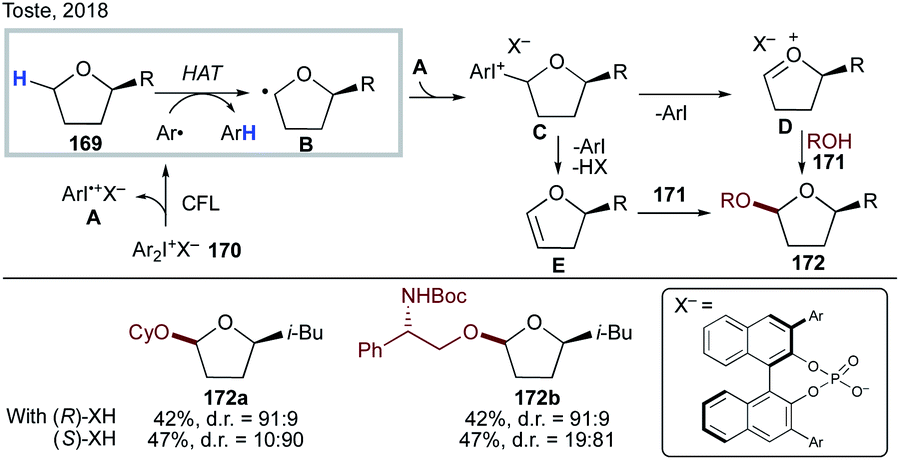 | ||
| Scheme 42 Chiral diaryliodonium phosphate-mediated diastereoselective α-acetalization of cyclic ethers involving intermolecular HAT. | ||
More recently, Li, Zhang, Yang, Walsh and co-workers developed transition metal-free dehydrogenative cross-coupling of N-benzyl amines and saturated heterocycles (Scheme 43).91 They have shown that 2-azaallyl anion A, generated by deprotonation of N-benzyl amines, can undergo SET to an aryl iodide to form an aryl radical and persistent 2-azaallyl radical B. The sterically protected sacrificial aryl radical undergoes intermolecular HAT at activated C(sp3)–H sites to form stabilized nucleophilic radical C, which recombines with persistent 2-azaallyl radical B to deliver the products 174a–d.
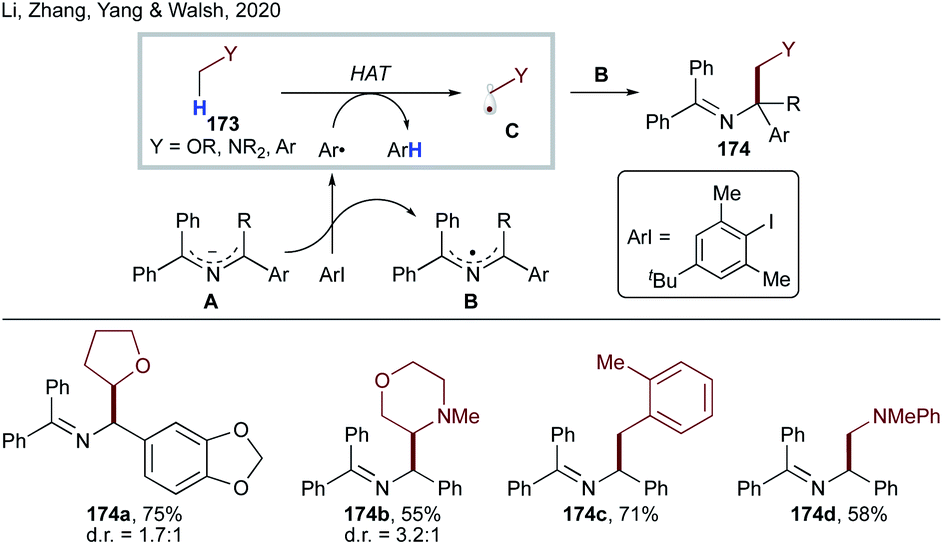 | ||
| Scheme 43 Dehydrogenative cross-coupling via intermolecular HAT. | ||
Conclusions
Organic radical chemistry enjoyed a prosperous period back in the 90's, which resulted in ground-breaking developments of C–H functionalization via the HAT pathway. Recent discoveries of mild methods for generation of radicals, which obviate the use of conventional radical initiators (e.g. AIBN, Et3B) and reagents (e.g. Bu3SnH, (TMS)3SiH), led to a renaissance in the field, as manifested by the booming literature examples. C–H functionalization via HAT often distinguishes itself from transition metal-based two-electron approaches in respect of site selectivity. This complementary feature has opened up numerous opportunities to tackle longstanding problems in organic synthesis, as partially exemplified by the recent advances summarized in this review. With that being said, HAT mediated by a C-centered radical, as compared to the N- and O-centered analogues, is still underdeveloped and mostly confined to intramolecular activation. Thus, a deeper understanding of the nature and guiding parameters of reactivity is needed to harness the full potential of the C-centered radical in more challenging and complex settings, which may in turn lead to the design of novel C-centered radical precursors. Furthermore, enantioselective HAT via N-centered radicals has been achieved very recently,92 thus providing the basis and motivation for the development of enantioselective HAT processes. It is foreseeable that an asymmetric variant of C-centered radical-mediated HAT will mark a major breakthrough in this area. Undoubtedly, the progress of C-centered radical chemistry will further the field of C–H functionalization in the near future.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the National Institute of Health (GM120281), National Science Foundation (CHE-1955663), and Welch Foundation (Chair, AT-0041) for financial support.Notes and references
-
(a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS
; (b) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS
-
(a) R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS
-
(a) H. M. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS
-
(a) J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS
-
(a) A. W. Hofmann, Chem. Ber., 1883, 16, 558–560 CrossRef
-
(a) J. Robertson, J. Pillai and R. K. Lush, Chem. Soc. Rev., 2001, 30, 94–103 RSC
- D. H. Hey and D. G. Turpin, J. Chem. Soc., 1954, 2471–2481 RSC
-
(a) D. P. Curran, D. Kim, H. T. Liu and W. Shen, J. Am. Chem. Soc., 1988, 110, 5900–5902 CrossRef CAS
-
(a) M. D. E. Forbes, J. T. Patton, T. L. Myers, H. D. Maynard, D. W. Smith, G. R. Schulz and K. B. Wagener, J. Am. Chem. Soc., 1992, 114, 10978–10980 CrossRef CAS
- M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS
- W. Shu and C. Nevado, Angew. Chem., Ind. Ed., 2017, 56, 1881–1884 CrossRef CAS
-
(a) K. Héberger and A. Lopata, J. Org. Chem., 1998, 63, 8646–8653 CrossRef
-
(a) J. E. Bennett, B. C. Gilbert, S. Lawrence, A. C. Whitwood and A. J. Holmes, J. Chem. Soc., Perkin Trans. 2, 1996, 1789–1795 RSC
-
(a) S. J. Cole, J. N. Kirwan, B. P. Roberts and C. R. Willis, J. Chem. Soc., Perkin Trans. 1, 1991, 103–112 RSC
-
(a) X. L. Huang and J. J. Dannenberg, J. Org. Chem., 1991, 56, 5421–5424 CrossRef CAS
- D. Kurandina, D. Yadagiri, M. Rivas, A. Kavun, P. Chuentragool, K. Hayama and V. Gevorgyan, J. Am. Chem. Soc., 2019, 141, 8104–8109 CAS
-
(a) D. J. Hart and S. C. Wu, Tetrahedron Lett., 1991, 32, 4099–4102 CrossRef CAS
-
(a) J. Brunckova, D. Crich and Q. W. Yao, Tetrahedron Lett., 1994, 35, 6619–6622 CrossRef CAS
- J. Cassayre and S. Z. Zard, J. Organomet. Chem., 2001, 624, 316–326 CrossRef CAS
- M. Nechab, S. Mondal and M. P. Bertrand, Chem.–Eur. J., 2014, 20, 16034–16059 CrossRef CAS
-
(a) M. Tojino, Y. Uenoyama, T. Fukuyama and I. Ryu, Chem. Commun., 2004, 2482–2483 RSC
- M. S. Wilson, J. C. Woo and G. R. Dake, J. Org. Chem., 2006, 71, 4237–4245 CrossRef CAS
- G. R. Dake, E. E. Fenster and B. O. Patrick, J. Org. Chem., 2008, 73, 6711–6715 CrossRef CAS
- W. Yuan, Y. Wei, M. Shi and Y. Li, Chem.–Eur. J., 2012, 18, 1280–1285 CrossRef CAS
- F. Scully and H. Morrison, J. Chem. Soc., Chem. Commun., 1973, 529–530 RSC
- A. C. Pratt, J. Chem. Soc., Chem. Commun., 1974, 183–184 RSC
-
(a) J. M. Hornback, L. G. Mawhorter, S. E. Carlson and R. A. Bedont, J. Org. Chem., 1979, 44, 3698–3703 CrossRef CAS
- T. Peez, V. Schmalz, K. Harms and U. Koert, Org. Lett., 2019, 21, 4365–4369 CrossRef CAS
-
(a) P. Yu, J. S. Lin, L. Li, S. C. Zheng, Y. P. Xiong, L. J. Zhao, B. Tan and X. Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 11890–11894 CrossRef CAS
-
(a) G. H. Lonca, D. Y. Ong, T. M. H. Tran, C. Tejo, S. Chiba and F. Gagosz, Angew. Chem., Int. Ed., 2017, 56, 11440–11444 CrossRef CAS
-
(a) L. Huang, S. C. Zheng, B. Tan and X. Y. Liu, Chem.–Eur. J., 2015, 21, 6718–6722 CrossRef CAS
-
(a) L. Li, L. Ye, S. F. Ni, Z. L. Li, S. Chen, Y. M. Du, X. H. Li, L. Dang and X. Y. Liu, Org. Chem. Front., 2017, 4, 2139–2146 RSC
-
(a) W. Jin, Y. Zhou, Y. Zhao, Q. Ma, L. Kong and G. Zhu, Org. Lett., 2018, 20, 1435–1438 CrossRef CAS
- K. J. Bian, Y. Li, K. F. Zhang, Y. He, T. R. Wu, C. Y. Wang and X. S. Wang, Chem. Sci., 2020, 11, 10437–10443 RSC
- P. C. Van Dort and P. L. Fuchs, J. Org. Chem., 1997, 62, 7142–7147 CrossRef CAS
- A. Attouche, D. Urban and J. M. Beau, Angew. Chem., Int. Ed., 2013, 52, 9572–9575 CrossRef CAS
- X. Xiao, Z. Xu, Q. D. Zeng, X. B. Chen, W. H. Ji, Y. Han, P. Wu, J. Ren and B. B. Zeng, Chem.–Eur. J., 2015, 21, 8351–8354 CrossRef CAS
- M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857–14860 CrossRef CAS
- For examples on unusual endo-trig radical cyclizations of silyl-tethered radicals due to longer Si–C and Si–O bonds, see:
(a) M. Koreeda and L. G. Hamann, J. Am. Chem. Soc., 1990, 112, 8175–8177 CrossRef CAS
- For selected reviews, see:
(a) D. Kurandina, P. Chuentragool and V. Gevorgyan, Synthesis, 2019, 51, 985–1005 CrossRef CAS
- P. Chuentragool, D. Yadagiri, T. Morita, S. Sarkar, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 1794–1798 CrossRef CAS
- K. Yamada, H. Fujihara, Y. Yamamoto, Y. Miwa, T. Taga and K. Tomioka, Org. Lett., 2002, 4, 3509–3511 CrossRef CAS
- K. Yamada, Y. Yamamoto and K. Tomioka, Org. Lett., 2003, 5, 1797–1799 CrossRef CAS
-
(a) P. Lu, T. Hou, X. Gu and P. Li, Org. Lett., 2015, 17, 1954–1957 CrossRef CAS
- C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS
-
(a)
Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 CrossRef
- B. C. Gilbert and D. J. Parry, J. Chem. Soc., Perkin Trans. 2, 1988, 875–886 RSC
- F. Dénès, F. Beaufils and P. Renaud, Synlett, 2008, 2389–2399 CrossRef
-
(a) U. Wille, Chem. Rev., 2013, 113, 813–853 CrossRef CAS
- M. Gulea, J. M. Lopez-Romero, L. Fensterbank and M. Malacria, Org. Lett., 2000, 2, 2591–2594 CrossRef CAS
- M. M. Logan, T. Toma, R. Thomas-Tran and J. Du Bois, Science, 2016, 354, 865–869 CrossRef CAS
- D. P. Curran and W. Shen, J. Am. Chem. Soc., 1993, 115, 6051–6059 CrossRef CAS
- M. Ratushnyy, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2018, 57, 2712–2715 CrossRef CAS
- E.-A. I. Heiba and R. M. Dessau, J. Am. Chem. Soc., 1967, 89, 3772–3777 CrossRef CAS
- E. Bosch and M. D. Bachi, J. Org. Chem., 1993, 58, 5581–5582 CrossRef CAS
- S. Bogen, M. Gulea, L. Fensterbank and M. Malacria, J. Org. Chem., 1999, 64, 4920–4925 CrossRef CAS
- R. Bénéteau, C. F. Despiau, J. C. Rouaud, A. Boussonnière, V. Silvestre, J. Lebreton and F. Dénés, Chem.–Eur. J., 2015, 21, 11378–11386 CrossRef
-
(a) S. Wu, X. Wu, D. Wang and C. Zhu, Angew. Chem., Int. Ed., 2019, 58, 1499–1503 CrossRef CAS
-
(a) T. Shang, J. Zhang, Y. Zhang, F. Zhang, X. S. Li and G. Zhu, Org. Lett., 2020, 22, 3667–3672 CrossRef CAS
- H. Li, L. Guo, X. L. Feng, L. P. Huo, S. Q. Zhu and L. L. Chu, Chem. Sci., 2020, 11, 4904–4910 RSC
- P. Devin, L. Fensterbank and M. Malacria, J. Org. Chem., 1998, 63, 6764–6765 CrossRef CAS
- G. M. Allan, A. F. Parsons and J. F. Pons, Synlett, 2002, 1431–1434 CAS
- H. Lin, A. Schall and O. Reiser, Synlett, 2005, 2603–2606 CAS
- S. K. Pagire and O. Reiser, Green Chem., 2017, 19, 1721–1725 RSC
-
(a) J. K. Vellucci and C. M. Beaudry, Org. Lett., 2015, 17, 4558–4560 CrossRef CAS
- Y. Yoshimura, Y. Yamazaki, K. Wachi, S. Satoh and H. Takahata, Synlett, 2007, 111–114 CrossRef CAS
-
(a) A. H. Lewin, A. H. Dinwoodie and T. Cohen, Tetrahedron, 1966, 22, 1527–1537 CrossRef CAS
- S. H. Pines, R. M. Purick, R. A. Reamer and G. Gal, J. Org. Chem., 1978, 43, 1337–1342 CrossRef CAS
- D. P. Curran and J. Xu, J. Am. Chem. Soc., 1996, 118, 3142–3147 CrossRef CAS
- D. P. Curran and H. S. Yu, Synthesis, 1992, 123–127 CrossRef CAS
-
(a) V. Snieckus, J. C. Cuevas, C. P. Sloan, H. T. Liu and D. P. Curran, J. Am. Chem. Soc., 1990, 112, 896–898 CrossRef CAS
- M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef CAS
- G. H. Han, M. C. Mcintosh and S. M. Weinreb, Tetrahedron Lett., 1994, 35, 5813–5816 CrossRef CAS
- F.-Q. Huang, X. Dong, L.-W. Qi and B. Zhang, Tetrahedron Lett., 2016, 57, 1600–1604 CrossRef CAS
- P. Liu, J. Tang and X. Zeng, Org. Lett., 2016, 18, 5536–5539 CrossRef CAS
-
(a) W. C. Wertjes, L. C. Wolfe, P. J. Waller and D. Kalyani, Org. Lett., 2013, 15, 5986–5989 CrossRef CAS
- B. S. Bhakuni, A. Yadav, S. Kumar, S. Patel, S. Sharma and S. Kumar, J. Org. Chem., 2014, 79, 2944–2954 CrossRef CAS
- J. Q. Chen, Y. L. Wei, G. Q. Xu, Y. M. Liang and P. F. Xu, Chem. Commun., 2016, 52, 6455–6458 RSC
- M. Ratushnyy, N. Kvasovs, S. Sarkar and V. Gevorgyan, Angew. Chem., Int. Ed., 2020, 59, 10316–10320 CrossRef CAS
- H. Tian, H. Yang, C. Zhu and H. Fu, Sci. Rep., 2016, 6, 19931 CrossRef
- S. Du, E. A. Kimball and J. R. Ragains, Org. Lett., 2017, 19, 5553–5556 CrossRef CAS
- A. F. Voica, A. Mendoza, W. R. Gutekunst, J. O. Fraga and P. S. Baran, Nat. Chem., 2012, 4, 629–635 CrossRef CAS
- K. A. Hollister, E. S. Conner, M. L. Spell, K. Deveaux, L. Maneval, M. W. Beal and J. R. Ragains, Angew. Chem., Int. Ed., 2015, 54, 7837–7841 CrossRef CAS
- P. Chuentragool, M. Parasram, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2018, 140, 2465–2468 CrossRef CAS
- F. W. Friese, C. Muck-Lichtenfeld and A. Studer, Nat. Commun., 2018, 9, 2808 CrossRef
-
(a) K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213–4217 CrossRef CAS
- J. C. Scaiano and L. C. Stewart, J. Am. Chem. Soc., 1983, 105, 3609–3614 CrossRef CAS
- A. Clerici, R. Cannella, W. Panzeri, N. Pastori, E. Regolini and O. Porta, Tetrahedron Lett., 2005, 46, 8351–8354 CrossRef CAS
- B. Ye, J. Zhao, K. Zhao, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2018, 140, 8350–8356 CrossRef CAS
- Z. Liu, M. Li, G. Deng, W. Wei, P. Feng, Q. Zi, T. Li, H. Zhang, X. Yang and P. J. Walsh, Chem. Sci., 2020, 11, 7619–7625 RSC
- K. M. Nakafuku, Z. Zhang, E. A. Wappes, L. M. Stateman, A. D. Chen and D. A. Nagib, Nat. Chem., 2020, 12, 697–704 CrossRef
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |