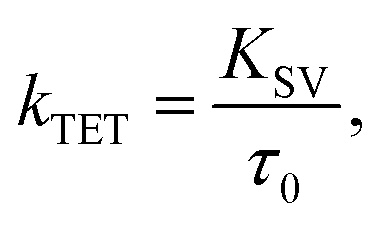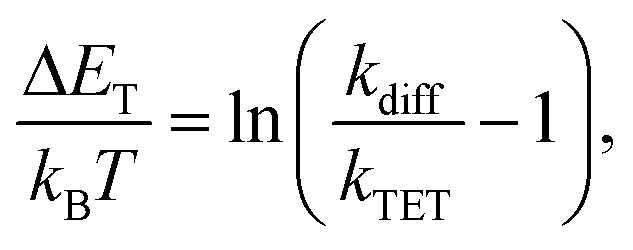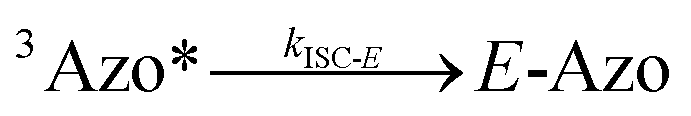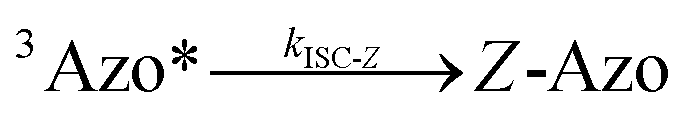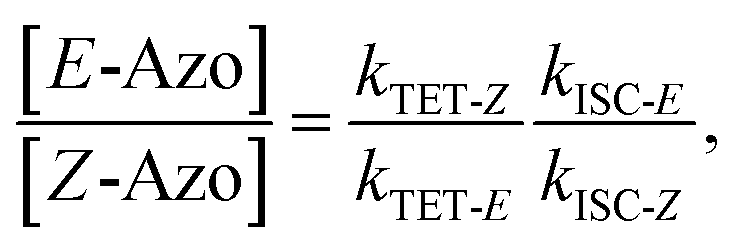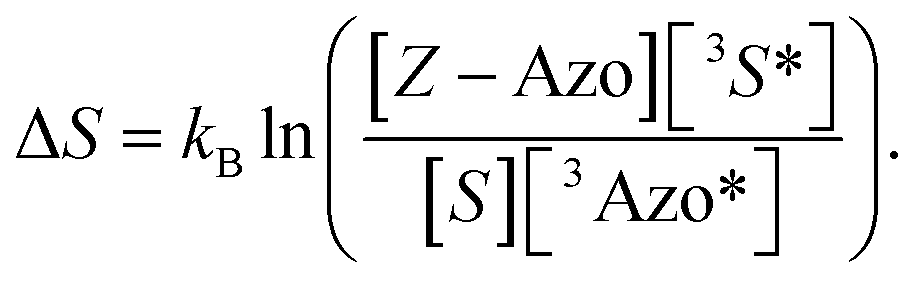DOI:
10.1039/D1SC01717A
(Edge Article)
Chem. Sci., 2021,
12, 7504-7509
Expanding excitation wavelengths for azobenzene photoswitching into the near-infrared range via endothermic triplet energy transfer†
Received
26th March 2021
, Accepted 25th April 2021
First published on 27th April 2021
Abstract
Developing azobenzene photoswitches capable of selective and efficient photoisomerization by long-wavelength excitation is an enduring challenge. Herein, rapid isomerization from the Z- to E-state of two ortho-functionalized bistable azobenzenes with near-unity photoconversion efficiency was driven by triplet energy transfer upon red and near-infrared (up to 770 nm) excitation of porphyrin photosensitizers in catalytic micromolar concentrations. We show that the process of triplet-sensitized isomerization is efficient even when the sensitizer triplet energy is substantially lower (>200 meV) than that of the azobenzene used. This makes the approach applicable for a wide variety of sensitizer-azobenzene combinations and enables the expansion of excitation wavelengths into the near-infrared spectral range. Therefore, indirect excitation via endothermic triplet energy transfer provides efficient and precise means for photoswitching upon 770 nm near-infared light illumination with no chemical modification of the azobenzene chromophore, a desirable feature in photocontrollable biomaterials.
Introduction
Light is a versatile, non-invasive and efficient stimulus with high spatial and temporal resolution and facile modulation. Photoswitches, of which azobenzenes are arguably the most common class, enable the control of functional materials with light.1,2 The photoswitching of azobenzenes is based on their reversible trans(E)-to-cis(Z) isomerization. Depending on their structure, azobenzenes have varying Z-isomer thermal lifetimes and varying degrees of spectral separation between the absorption bands of the isomers. Conventional azobenzenes absorb ultraviolet or visible light with relatively high energy, which limits their applicability for emerging fields such as solar energy harvesting,3 3D printing,4 photosensors,5 photoactuation6 and photocatalysis.7 When compared to longer wavelengths, UV excitation is hampered by, for example, its limited solar availability, non-specific absorption, and harmfulness to organic materials and live cells.8 Moreover, due to the optical properties of biological tissue, photoswitching systems capable of operating under red or even near-infrared (NIR) light are of particular interest for biomedicine.9–14
Developing azobenzenes suitable for these aforementioned fields in terms of properties such as red-shifted excitation wavelengths, spectral resolution of E- and Z-isomers and thermal stability of the Z-isomer, requires structural modification;15 a complex approach that generally involves extensive computational studies and synthesis. For example, ortho-functionalization of azobenzenes is an established approach to create photoswitches with a long Z-lifetime,16,17 which is required for efficient photoswitching in both directions and precise control of the isomer composition. Despite recent advances, creating bistable red-absorbing azobenzenes remains a challenge, since red-shifting typically leads to drastically decreased thermal stability of the Z-isomer. Furthermore, there are no reports on bistable NIR-absorbing azobenzenes.18,19
Indirect excitation, i.e., excitation that does not rely on the thermal relaxation or absorption bands of the E- and Z-isomers, but instead makes use of a proxy molecule to absorb light-activation signals, may overcome the foregoing issues of azobenzenes while retaining their beneficial intrinsic properties such as bistability and robustness. Intriguingly, indirect excitation allows tailoring and expansion of the photoswitching properties of the system without structural modification, even beyond the capabilities of the azobenzene itself. Isomerization by indirect excitation can be effected in various ways, such as photoinduced electron transfer,20–22 two-photon absorption induced energy transfer,23 photon upconversion24,25 and triplet sensitization.26–30
Triplet-sensitized Z-to-E isomerization of azobenzenes is an especially viable approach for indirect excitation due to its reversibility, non-destructivity and near-unity conversion efficiency.26,29,31 Triplet sensitization has also been utilized to isomerize other types of photoswitches, such as overcrowded alkenes,32 diarylethenes,33 stilbenes34 and indigos.35 However, in previous reports triplet sensitizers have been excited in the UV-to-yellow (<580 nm) range, while the capabilities of potent red and NIR-absorbing triplet sensitizers, such as porphyrins, have not been utilized in azobenzene photoswitching. Thus, we set out to study this orthogonal excitation pathway for bistable azobenzenes and explore the wavelength limits of triplet-sensitized photoisomerization and how it improves the efficiency and control over photoswitching.
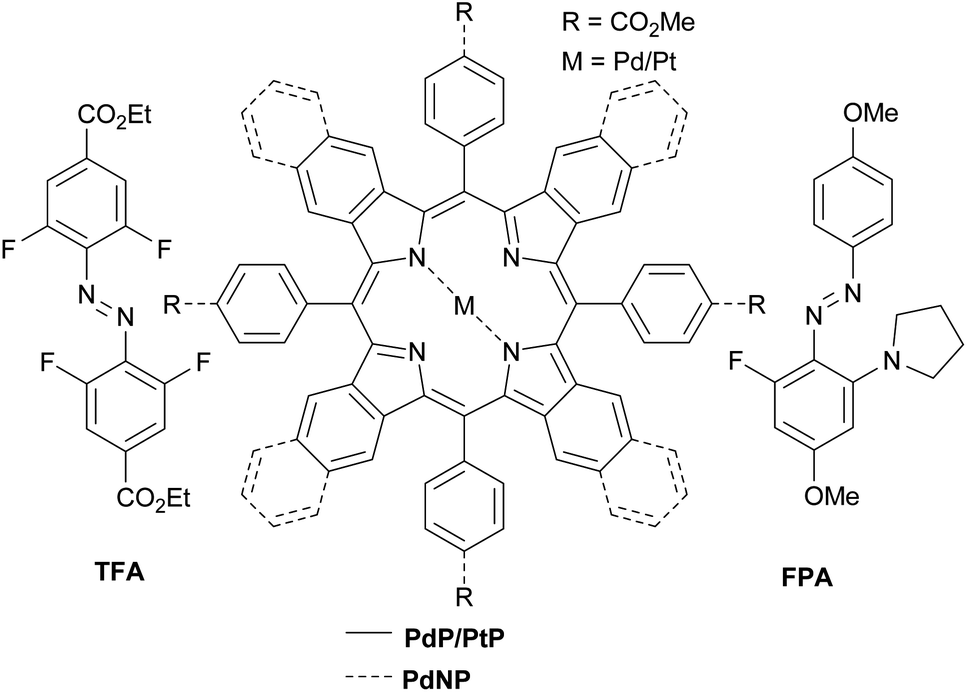
Here we demonstrate triplet-sensitized Z-to-E isomerization of azobenzenes under red and NIR excitation and establish its efficiency via kinetics studies. We have employed a NIR-absorbing porphyrin, PdNP (Pd(II)meso-tetraphenyltetranaphthoporhyrin36–38) and two commercially available red-absorbing porphyrins, PdP and PtP (Pd(II) and Pt(II)meso-tetraphenyltetrabenzoporphyrin, see Chart 1), as triplet photosensitizers. The sensitizers were used in combination with tetrafluoroazobenzene (TFA39,40) and fluoropyrrolidineazobenzene (FPA,41 see Chart 1). Both ortho-functionalized azobenzenes exhibit efficient photoisomerization (>80% conversion to both Z- and E-isomers) under visible light and remarkably long thermal half-lives (days). TFA requires blue light (410 nm) for Z-to-E photoisomerization. In contrast, Z-FPA absorbs red light, albeit weakly, and thus it was used to compare photoisomerization under direct and triplet-sensitized excitation.
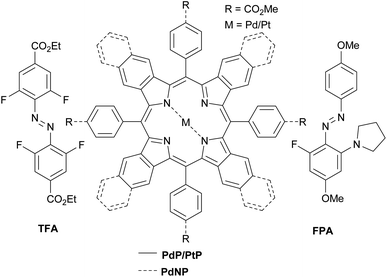 |
| | Chart 1 Sensitizers and azobenzenes used in this study. | |
Results and discussion
Triplet energy transfer studies
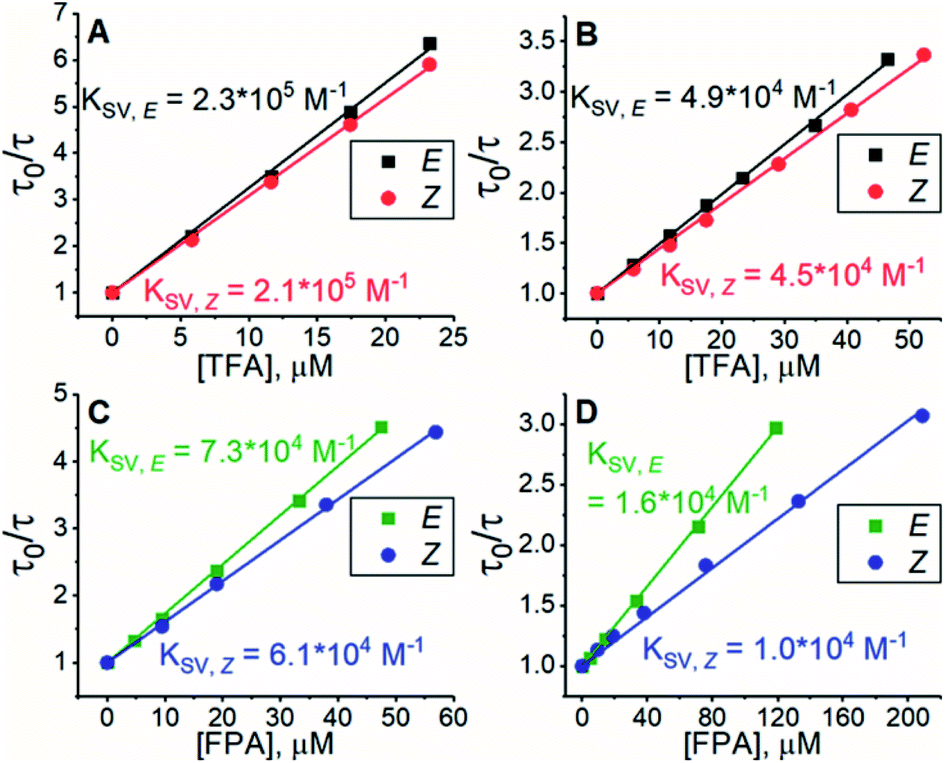
All experiments were conducted in dimethyl sulfoxide (DMSO). Since triplet sensitizers generate reactive oxygen species under illumination, bis(methylthio)methane was added as an oxygen scavenger.42 To study the triplet energy transfer (TET) from PdP and PtP to the azobenzenes, we performed quenching studies by measuring the phosphorescence lifetimes of both sensitizers in the presence of E- or Z-isomers of both azobenzenes. The resulting Stern–Volmer plots of the quenching studies are shown in Fig. 1.
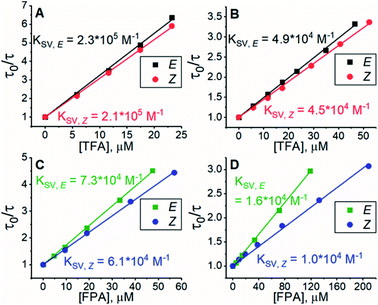 |
| | Fig. 1 Results of phosphorescence quenching. Stern–Volmer plots and the corresponding KSV values of (A) TFA and PdP, (B) TFA and PtP, (C) FPA and PdP and (D) FPA and PtP. | |
The Stern–Volmer rate constant (KSV) yielded by the linear fit on the quenching data was then used to evaluate the rate constant of triplet energy transfer:
| | 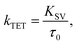 | (1) |
where
τ0 is the unquenched phosphorescence lifetime of the sensitizer (260 μs and 46 μs for PdP and PtP, respectively). The quenching experiments are discussed in more detail in the ESI.
†kTET values were then used to calculate the triplet energy gap (Δ
ET) between the sensitizer and azobenzene triplet states:
43| | 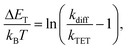 | (2) |
where
kB is the Boltzmann constant,
T is the temperature and
kdiff (= 1.1 × 10
9 M
−1 s
−1) is the diffusion rate constant of the system (see the ESI
†). Finally, the obtained Δ
ET values were used together with the sensitizer triplet energies (1.55 and 1.61 eV for PdP and PtP, respectively, see Fig. S3
†) to evaluate the triplet energies (
ET) of both
E- and
Z-isomers of the azobenzenes. To our knowledge, these are the first reported triplet state energies of
ortho-substituted azobenzenes. The
kTET, Δ
ET and
ET values of each pair are given in
Table 1.
Table 1 Rate constant of triplet energy transfer (kTET), the triplet energy gap (ΔET) between the sensitizer and azobenzene, and the triplet energy (ET) of the azobenzene derived from the quenching results
| Pair |
k
TET (M−1 s−1) |
ΔET (kBT) |
ΔET (meV) |
E
T (eV) |
|
E-TFA/PdP |
8.7 × 108 |
−1.3 |
−34 |
1.49–1.52 |
|
E-TFA/PtP |
1.1 × 109 |
−4.7 |
−120 |
|
Z-TFA/PdP |
8.1 × 108 |
−1.0 |
−26 |
1.52–1.56 |
|
Z-TFA/PtP |
9.7 × 108 |
−2.0 |
−52 |
|
E-FPA/PdP |
2.8 × 108 |
1.1 |
29 |
1.58–1.63 |
|
E-FPA/PtP |
3.6 × 108 |
0.7 |
18 |
|
Z-FPA/PdP |
2.4 × 108 |
1.3 |
34 |
1.58–1.65 |
|
Z-FPA/PtP |
2.2 × 108 |
1.4 |
36 |
Interestingly, the Z-isomer of TFA has higher ET than the E-isomer, which appears contrary to the unsubstituted azobenzene (ET of Z-isomer 29 kcal mol−1i.e. 1.29 eV), whereas the ET values of E-TFA and E-FPA are comparable to the previously reported values for unsubstituted and para-substituted azobenzenes (33–35 kcal mol−1i.e. 1.43–1.52 eV).44,45 The kinetics studies results also reveal the thermodynamics between the sensitizer and azobenzene: triplet energy transfer from both PdP and PtP appears exothermic for TFA (negative ΔET), while the lower values of kTET between the sensitizers and FPA indicate endothermic energy transfer (positive ΔET).
Triplet-sensitized isomerization with red light
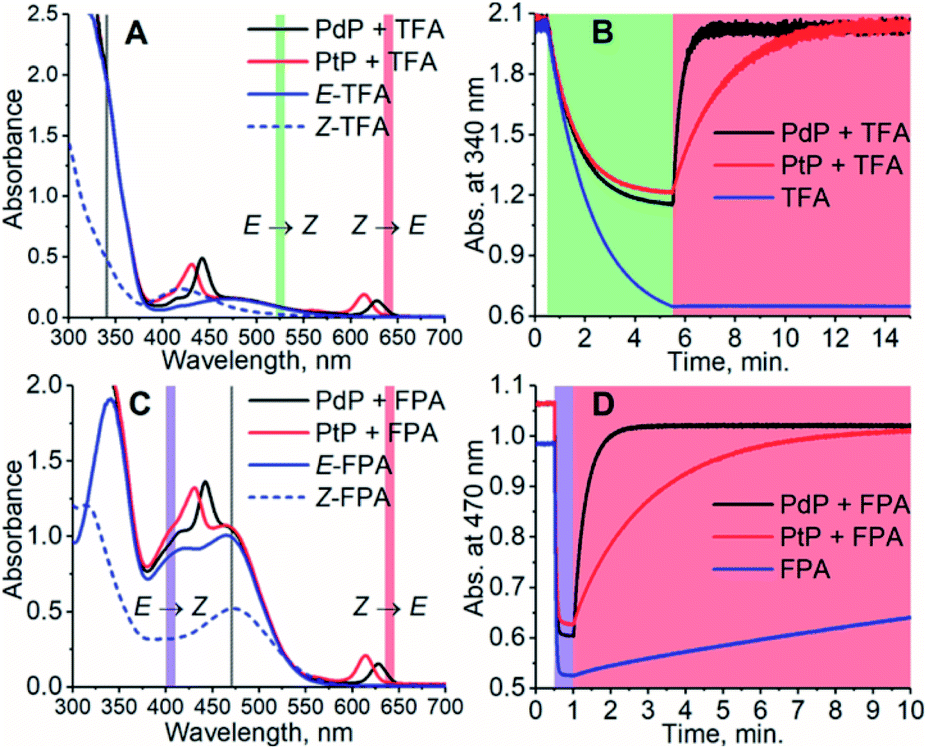
To study the effect of the triplet energies on photoisomerization, we employed both PdP and PtP for photoswitching of TFA and FPA. In agreement with previous works,26,28,29E-to-Z isomerization was negligible on excitation of the sensitizer. This was observed although kTET of the E-isomer is larger than for the Z-isomer for both TFA and FPA. However, after isomerizing the azobenzenes to their Z-configuration by direct excitation of the E-isomer (525 nm for TFA and 405 nm for FPA), rapid and nearly complete Z-to-E isomerization could be readily induced under 640 nm excitation using catalytic amounts (1.8 μM i.e. 1.2 mol% with respect to the azobenzene) of each sensitizer (see Fig. 2). An exponential function was fitted to the isomerization data to determine the rates of isomerization (rZ→E). The resulting fits are shown in Fig. S12–S16.† The rates of Z-to-E isomerization are clearly increased by several orders of magnitude by indirect excitation via the triplet energy transfer route. For example, the photoisomerization rate for FPA by triplet sensitization was over 100 times faster than that under 640 nm direct excitation (3.10 min−1versus 0.028 min−1). As expected, no isomerization of TFA without sensitizers was observed under 640 nm excitation. Notably, the sensitized photoswitching of FPA, for which TET is endothermic and thus slower, occurs as rapidly as photoswitching of TFA.
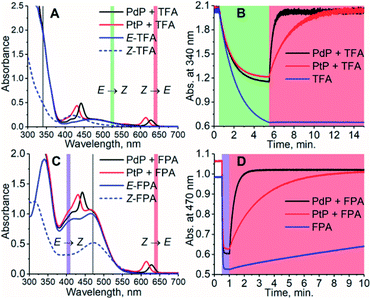 |
| | Fig. 2 The absorption spectra of the photoswitching systems consisting of PdP and PtP and TFA (A) and FPA (C) and their photoisomerization curves (B) and (D). The concentration of the azobenzene was 150 μM and the concentration of the sensitizer was 1.2 mol% of the azobenzene, i.e. 1.8 μM. The colored sections indicate the wavelength and time ranges used for photoswitching. The excitation light is on during the times indicated by the representative colors. Gray lines in (A) and (C) indicate the wavelength used for monitoring the isomerization. The observed partial E-to-Z isomerization of the azobenzenes, especially in case of TFA due to its small molar extinction coefficient at 525 nm, is caused by the competitive absorption of the azobenzene and the sensitizer. | |
The conversion efficiency of sensitized Z-to-E isomerization (ΦZ→E) was determined from the curves as the ratio of initial (dark) and final (achieved upon sensitization) absorbances. All values were close to unity, and only a minor difference in the conversion efficiency was observed between TFA and FPA. rZ→E and ΦZ→E values of each sensitizer/azobenzene pair are shown in Table 2.
Table 2 Rates (rZ→E) and efficiency (ΦZ→E) of the photoisomerization under 640 nm excitation
| Pair |
TFA/PdP |
TFA/PtP |
FPA/PdP |
FPA/PtP |
|
The smaller ΦZ→E yielded by PtP results mainly from a smaller spectral overlap between the absorption and the excitation (see Fig. 2).
|
|
r
Z→E (min−1) |
3.10 |
0.50a |
3.10 |
0.49a |
|
Φ
Z→E (%) |
99 |
99 |
96 |
96 |
Surprisingly, the sensitizers appear to catalyze the Z-to-E isomerization of FPA even in the dark (see Fig. S18†). This increase in the rate of the thermal isomerization is especially pronounced with PtP as the contribution of the dark reaction to the overall observed rate is 31% (0.15 min−1). In case of FPA/PdP, the contribution is only ca. 2% (0.06 min−1). Also, the free base of the porphyrin (H2P) catalyzes the isomerization of FPA in the dark (0.09 min−1). This catalytic effect is perhaps a result of ground state coordination46–48 between Z-FPA and the porphyrin. FPA has three π donors in ortho- and para-positions and only one electron-withdrawing fluorine substituent, increasing the affinity of the azo bridge to an electron-deficient site such as a metal cation. This effect is not observed with the drastically electron-poorer TFA, which supports this explanation.
Mechanism of triplet-sensitized isomerization
To better understand the observed results of triplet-sensitized photoswitching and the considerably increased isomerization rates, we conducted an analysis of the underlying reaction mechanisms that can be described with the following set of equations:26,29| |  | (3) |
| |  | (4) |
| | 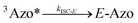 | (5) |
| | 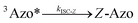 | (6) |
where S is the sensitizer, superscript 3 is the triplet state, ISC is the intersystem crossing between the singlet and triplet states and k denotes the rate constant of each process. kTET-E and kTET-Z are determined experimentally and given in Table 1. It is also important to notice, that 3Azo* can be considered as a common state between the isomers, since there is no energy barrier associated with the CNNC twist in the triplet manifold.49,50 While there are no direct observations reported on the triplet lifetime of azobenzenes, the commendable computational work49 by Cembran et al. estimates kISC-E as 1011 s−1. The ultrafast ISC results from the degeneracy between the minimum of the triplet state and the intersection between the triplet state and ground state of E-Azo. Since the photostationary composition yielded by sensitized isomerization can be derived from the equations above as26,29| | 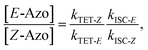 | (7) |
we can use ΦZ→E and the TET results to estimate relative kISC-Z for TFA (109 s−1) and FPA (3 × 109 s−1), corresponding roughly to 1–3% of kISC-E. This also explains why triplet sensitization of azobenzenes leads almost exclusively to the formation of the E-isomer.26,29,49
Based on the isomerization kinetics, even endothermic TET (as is the case with FPA) is apparently capable of driving isomerization efficiently. This indicates that the whole process of triplet-sensitized isomerization is largely entropy-driven,51,52 since the rate of the isomerization is fairly decoupled from the change in enthalpy involved in TET. The change in entropy (ΔS) of TET is51
| | 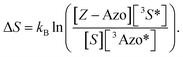 | (8) |
The ultrafast crossing between the triplet state and the ground state of E-Azo (eqn (5)) leads to [3Azo*] ≪ [Z-Azo], [3S*], [S] and ensures a large entropy component in the triplet-sensitized isomerization. This also effectively eliminates the azobenzene-to-sensitizer reverse TET.52 Therefore, even photosensitizers with considerably lower triplet energies are still capable of sensitizing the Z-to-E isomerization of azobenzenes, which enables the expansion of excitation wavelengths to the deeper red and even into NIR regions.
Triplet-sensitized isomerization with near-infrared light
Prompted by this finding, we combined TFA with PdNP, a sensitizer with a triplet energy of 1.30 eV and strong absorption in the NIR region (see Fig. S2 and S4†). The triplet energy gap of this pair is thus ≥220 meV, i.e., 8.5 kBT, and as a result, TET is highly endothermic. Nonetheless, photoisomerization under 740 nm excitation was, to our delight, efficient despite this remarkably high endothermic energy gap (see Fig. 3). PdNP was capable of sensitizing isomerization of TFA even under 770 nm excitation (rZ→E = 0.93 min−1, see Fig. S14†).
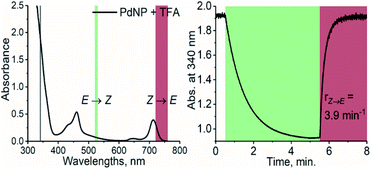 |
| | Fig. 3 Absorption spectrum of PdNP and TFA (left) and the photoisomerization curve (right) under 525 nm (green color) and 740 nm (dark red) excitation with the resulting rates of isomerization. | |
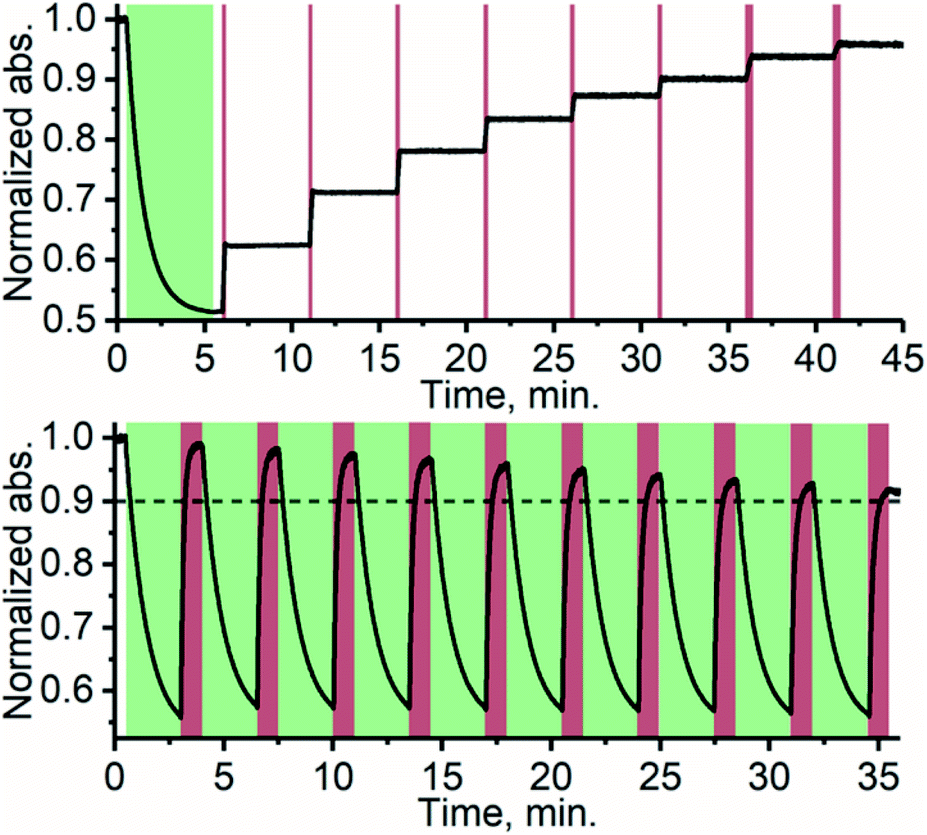
Thanks to the bistable nature and rapid triplet-sensitized isomerization of TFA under NIR excitation, the isomer composition can be precisely controlled by modulating the excitation dose (duration and/or intensity). This stepwise photoisomerization by dosed excitation is shown in Fig. 4. Repeatable cyclic switching between isomers is also typically desired for applications of photoswitching. This can be achieved in triplet-sensitized photoswitching systems by alternating excitation of the azobenzene and sensitizer as shown in Fig. 4 No discernible change in the rate of sensitized isomerization and only a slight decrease in efficiency (less than 10%, likely due to photobleaching) was observed in 10 cycles. Photoswitching is achievable even in the presence of oxygen (Fig. S19†), when the effects of singlet oxygen generated by the photosensitizer are mitigated by employing oxygen-scavengers.
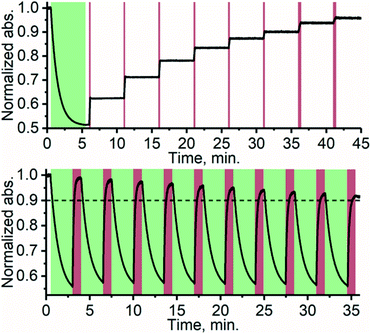 |
| | Fig. 4 Stepwise photoisomerization (above) of TFA by exciting PdNP with 10 s (last two were 20 s) doses of 740 nm excitation (dark red bars) in 5 min intervals. Cyclic photoswitching (below) of TFA/PdNP with 10 cycles of alternating 525 nm (direct excitation of E-TFA, green sections) and 740 nm (excitation of PdNP, dark red sections) excitation wavelengths. | |
Conclusions
We have shown that photoisomerization of bistable ortho-functionalized azobenzenes via triplet energy transfer is rapid with near-unity efficiency under red and near-infrared (up to 770 nm) excitation. Detailed studies of the kinetics indicate that triplet-sensitized isomerization is largely entropy-driven. Thus, even sensitizers with triplet energies considerably lower than those of the azobenzenes used are still capable of effectively sensitizing the isomerization. This was confirmed by using an azobenzene/sensitizer pair with an endothermic triplet energy gap of over 200 meV between them. The major entropy-factor involved in the process also projects that this excitation pathway is efficient for any azobenzene. This expands the properties of photoswitching systems without chemically modifying the photoswitch itself. Combined with the desirable use of red/NIR excitation, precise control of isomer composition, and repeatable cyclic isomerization, we envision that this approach will emerge as a potent tool for low-energy photoswitching in light-responsive materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge financial support from the Academy of Finland (Grant No. 316893) and the European Research Council (ERC Starting Grant Project PHOTOTUNE, decision number 679646). K. Kuntze is grateful for financial support from the Tampere University graduate school. This work was conducted as part of the Academy of Finland Flagship Programme, Photonics Research and Innovation (PREIN), Decision Number 321065.
Notes and references
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310–433 CrossRef CAS.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- D. Ahn, L. M. Stevens, K. Zhou and Z. A. Page, ACS Cent. Sci., 2020, 6, 1555–1563 CrossRef CAS.
- X. Zhou, T. Zifer, B. M. Wong, K. L. Krafcik, F. Léonard and A. L. Vance, Nano Lett., 2009, 9, 1028–1033 CrossRef PubMed.
- J. Boelke and S. Hecht, Adv. Opt. Mater., 2019, 7, 1900404 CrossRef.
- B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS.
- M. Zayat, P. Garcia-Parejo and D. Levy, Chem. Soc. Rev., 2007, 36, 1270–1281 RSC.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436–11447 CrossRef CAS PubMed.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymański, Chem. Sci., 2020, 11, 11672–11691 RSC.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef.
- M. J. Hansen, M. M. Lerch, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 13514–13518 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, C. V Collins, K. Jarrah, O. Sadovski, Q. Dai and G. A. Woolley, J. Am. Chem. Soc., 2017, 139, 13483–13486 CrossRef CAS PubMed.
- D. B. Konrad, G. Savasci, L. Allmendinger, D. Trauner, C. Ochsenfeld and A. M. Ali, J. Am. Chem. Soc., 2020, 142, 6538–6547 CrossRef CAS PubMed.
- A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS PubMed.
- A. Goulet-Hanssens, C. Rietze, E. Titov, L. Abdullahu, L. Grubert, P. Saalfrank and S. Hecht, Chem, 2018, 4, 1740–1755 CAS.
- G. L. Hallett-Tapley, C. D'Alfonso, N. L. Pacioni, C. D. McTiernan, M. González-Béjar, O. Lanzalunga, E. I. Alarcon and J. C. Scaiano, Chem. Commun., 2013, 49, 10073–10075 RSC.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1544–1547 CrossRef CAS PubMed.
- Z. Jiang, M. Xu, F. Li and Y. Yu, J. Am. Chem. Soc., 2013, 135, 16446–16453 CrossRef CAS PubMed.
- L. Wang, H. Dong, Y. Li, C. Xue, L.-D. Sun, C.-H. Yan and Q. Li, J. Am. Chem. Soc., 2014, 136, 4480–4483 CrossRef CAS PubMed.
- L. B. Jones and G. S. Hammond, J. Am. Chem. Soc., 1965, 87, 4219–4220 CrossRef CAS.
- J. Ronayette, R. Arnaud, P. Lebourgeois and J. Lemaire, Can. J. Chem., 1974, 52, 1848–1857 CrossRef CAS.
- J. Ronayette, R. Arnaud and J. Lemaire, Can. J. Chem., 1974, 52, 1858–1867 CrossRef CAS.
- P. Bortolus and S. Monti, J. Phys. Chem., 1979, 83, 648–652 CrossRef CAS.
- A. Goulet-Hanssens, C. Rietze, E. Titov, L. Abdullahu, L. Grubert, P. Saalfrank and S. Hecht, Chem, 2018, 4, 1740–1755 CAS.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS PubMed.
- A. Cnossen, L. Hou, M. M. Pollard, P. V Wesenhagen, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2012, 134, 17613–17619 CrossRef CAS PubMed.
- S. Fredrich, T. Morack, M. Sliwa and S. Hecht, Chem.–Eur. J., 2020, 26, 7672–7677 CrossRef CAS PubMed.
- J. A. Mercer-Smith and D. G. Whitten, J. Am. Chem. Soc., 1978, 100, 2620–2625 CrossRef CAS.
- G. M. Wyman, B. M. Zarnegar and D. G. Whitten, J. Phys. Chem., 1973, 77, 2584–2586 CrossRef CAS.
- J. E. Rogers, K. A. Nguyen, D. C. Hufnagle, D. G. McLean, W. Su, K. M. Gossett, A. R. Burke, S. A. Vinogradov, R. Pachter and P. A. Fleitz, J. Phys. Chem. A, 2003, 107, 11331–11339 CrossRef CAS.
- V. V Rozhkov, M. Khajehpour and S. A. Vinogradov, Inorg. Chem., 2003, 42, 4253–4255 CrossRef PubMed.
- O. S. Finikova, S. E. Aleshchenkov, R. P. Briñas, A. V Cheprakov, P. J. Carroll and S. A. Vinogradov, J. Org. Chem., 2005, 70, 4617–4628 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem.–Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed.
- Z. Ahmed, A. Siiskonen, M. Virkki and A. Priimagi, Chem. Commun., 2017, 53, 12520–12523 RSC.
- D. Dzebo, K. Moth-Poulsen and B. Albinsson, Photochem. Photobiol. Sci., 2017, 16, 1327–1334 CrossRef CAS PubMed.
- K. Sandros, Acta Chem. Scand., 1964, 18, 2355–2374 CrossRef.
- S. Monti, E. Gardini, P. Bortolus and E. Amouyal, Chem. Phys. Lett., 1981, 77, 115–119 CrossRef CAS.
- S. Monti, S. Dellonte and P. Bortolus, J. Photochem., 1983, 23, 249–256 CrossRef CAS.
- A. Nakamura, K. Doi, K. Tatsumi and S. Otsuka, J. Mol. Catal., 1976, 1, 417–429 CrossRef CAS.
- S. Ciccone and J. Halpern, Can. J. Chem., 1959, 37, 1903–1910 CrossRef CAS.
- Z. Wang, R. Losantos, D. Sampedro, M. Morikawa, K. Börjesson, N. Kimizuka and K. Moth-Poulsen, J. Mater. Chem. A, 2019, 7, 15042–15047 RSC.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS PubMed.
- L. Gagliardi, G. Orlandi, F. Bernardi, A. Cembran and M. Garavelli, Theor. Chem. Acc., 2004, 111, 363–372 Search PubMed.
- Y. Y. Cheng, B. Fückel, T. Khoury, R. G. C. R. Clady, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. A, 2011, 115, 1047–1053 CrossRef CAS PubMed.
- J. Isokuortti, S. R. Allu, A. Efimov, E. Vuorimaa-Laukkanen, N. V. Tkachenko, S. A. Vinogradov, T. Laaksonen and N. A. Durandin, J. Phys. Chem. Lett., 2020, 11, 318–324 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01717a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Elina
Vuorimaa-Laukkanen
a,
Mikhail A.
Filatov
b,
Andrey
Turshatov
a,
Elina
Vuorimaa-Laukkanen
a,
Mikhail A.
Filatov
b,
Andrey
Turshatov