
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvation structure dependent ion transport and desolvation mechanism for fast-charging Li-ion batteries†
Zhenyu
Fan
a,
Jingwei
Zhang
a,
Lanqing
Wu
a,
Huaqing
Yu
a,
Jia
Li
a,
Kun
Li
a and
Qing
Zhao
 *ab
*ab
aFrontiers Science Center for New Organic Matter, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), State Key Laboratory of Advanced Chemical Power Sources, College of Chemistry, Nankai University, Tianjin, 300071, China. E-mail: zhaoq@nankai.edu.cn
bHaihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192, China
First published on 20th September 2024
Abstract
The solvation structures of Li+ in electrolytes play prominent roles in determining the fast-charging capabilities of lithium-ion batteries (LIBs), which are in urgent demand for smart electronic devices and electric vehicles. Nevertheless, a comprehensive understanding of how solvation structures affect ion transport through the electrolyte bulk and interfacial charge transfer reactions remains elusive. We report that the charge transfer reaction involving the desolvation process is the rate-determining step of the fast charging when ion conductivity reaches a certain value as determined by investigating electrolytes with eight conventional solvents (linear/cyclic carbonate/ether). The physicochemical characteristics of solvent molecules can result in strong ion–ion, moderate ion–dipole, strong ion–dipole, and weak ion–dipole/ion–ion interactions, respectively, in which the speed of the charge transfer reaction follows the above order of interactions. Among all solvents, dioxolane (DOL) is found to enable strong ion–ion interactions in electrolytes and thus exhibits exceptional fast-charging performance and it can still retain 60% of the initial capacity at 20C (1C = 170 mA g−1) with a polarization of merely 0.35 V. Further experimental characterization and theoretical calculation reveal that the aggregates in DOL electrolytes contribute to hopping assisted ion transport and facilitate the desolvation process of Li+. Our results deepen the fundamental understanding of the behavior of Li+ solvation and provide an effective guiding principle for electrolyte design for fast-charging batteries.
Introduction
Since the commercialization in the 1990s, lithium-ion batteries (LIBs) have boosted the development of mobile devices and electric vehicles, with ever-growing calendar life and energy density.1–3 Nevertheless, compared with the low refuelling time of traditional internal combustion engine vehicles, battery electric vehicles have a clear disadvantage in terms of charging time, stemming from the various kinetic limitations in the process of Li+/electron transport during battery operation.4,5 Without considering the formation of an electrode–electrolyte interphase, the charging process of LIBs is generally as follows (Scheme 1a): (1) Li+ diffuses into the cathode material; (2) Li+ enters the electrolyte with a simultaneous solvation process; (3) Li+ transport in the bulk electrolyte; (4) Li+ desolvation at the anode surface with charge transfer; (5) Li+ diffuses into the anode material. Therefore, the migration rate or activation energy of each process through the electrolyte and electrode affects the fast-charging performance of LIBs. Multiple previous studies have stated that ion diffusion through electrode particles is the rate-limiting step in the charging process. According to eqn (1), in order to reduce the minimum time (τ*) required for charging, plenty of efforts have been made by either reducing the length (L*) of Li+ diffusion such as electrode structure engineering,6–8 or increasing the diffusion coefficient (DLi+δ) of Li+ such as chemically modifying electrode materials through element doping.9–11 Simultaneously, the geometry and charging mode determining the constant (α) also influence τ*. Thanks to these strategies, the fast-charging performance of electrode materials has been greatly improved over the past few years. | (1) |
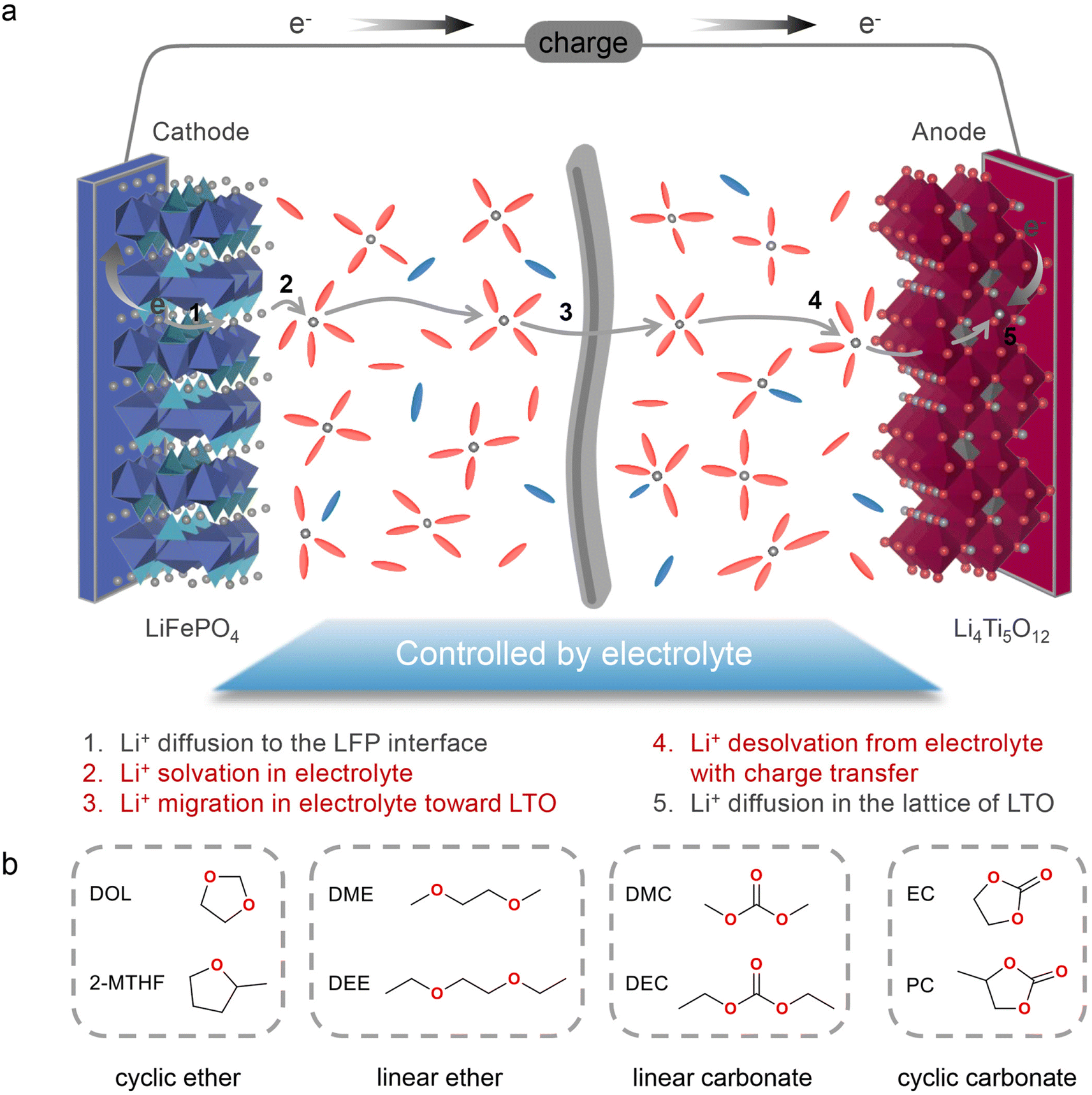 | ||
| Scheme 1 (a) Schematic description of Li+ migration during the charging process. (b) Molecular structures of eight solvents investigated in this work. | ||
In spite of this intriguing progress, the electrochemical properties of the battery are determined by “Cask Effect Theory”, meaning that the electrolyte is also crucial for fast-charging performance. Electrolytes can not only regulate the ion transport and desolvation process, but also influence the formation of the solid electrolyte interphase (SEI), a passivated film at the electrode–electrolyte interface that prevents continuous parasitic reactions between the electrode and electrolyte. Disregarding the chemical/electrochemical instability at the interface, the ion-transport and desolvation process are mainly controlled by the interactions between electrolyte components, including dipole–dipole interaction between solvents (eqn (2)), ion–dipole interaction between the solvent and cation/anion (eqn (3)), ion–ion interaction between the cation and anion (eqn (4)).12 In detail, the strength of interaction is determined by the distance between two components (r), the charge of the ion (ze), the solvent molecule angle (θ), the dielectric constant (ε) and the dipole moment of solvent (μ). For electrolytes with high ε solvents such as ethylene carbonate-EC,13 the Li+ and anion can be well dissociated, but the strong dipole–dipole interactions may hinder the transport of Li+ especially at low temperatures.14–16 For electrolytes with solvent that is endowed with high ion-chelating capability and low viscosity such as 1,2-dimethoxyethane (DME), the ionic conductivity is outstanding, while the desolvation process may be sluggish.17,18 For electrolytes with a highly concentrated Li salt,19–21 the reduction of the solvent ratio contributes to the enhancement of ion–ion interaction, which may facilitate the ion transport in a hopping manner, but largely increase the viscosity and cost. Therefore, the complexity of electrolyte solvation structures increases the challenge of evaluating and designing electrolytes for fast charging batteries.
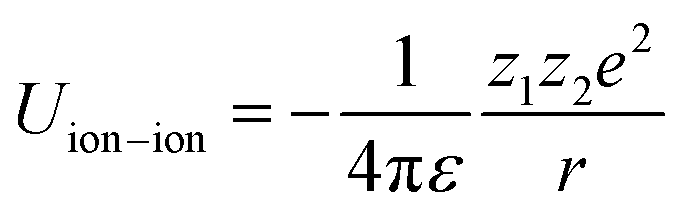 | (2) |
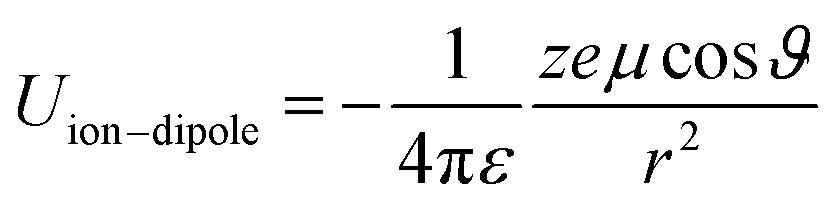 | (3) |
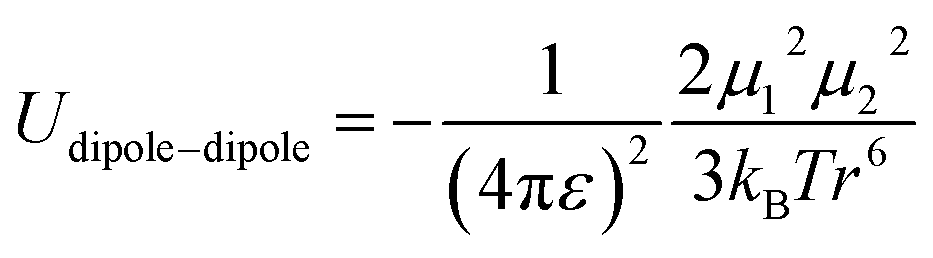 | (4) |
To date, there are ongoing debates on whether ion transport22–24 or ion desolvation25–29 in electrolytes is the limiting factor of fast charging performance. For the former, strategies such as adding acetonitrile (AN) to the electrolyte to realize Li+ jump transport30 and utilizing low molecular weight solvents to improve the conductivity and the diffusion coefficient of electrolyte31 have been adopted. For the latter, quite a few studies have suggested that the process of desolvation and charge transfer32,33 is the rate-determining step of fast charging. Approaches including utilizing highly concentrated electrolytes34,35 to modulate the solvation structure to reduce the desolvation energy and adding additives (e.g. LiNO3)27,36,37 to reduce the coordination of Li+-solvent have been proposed. These inconsistent conclusions on the one hand indicate that both ion transport and desolvation are crucial for fast charging, while on the other hand they may come from the battery system used for studies. Most research focuses on LIBs using high-voltage cathodes38,39 (e.g. LiNi0.8Co0.1Mn0.1O2) and low-voltage graphite anodes, in which the formation of a cathode–electrolyte interphase (CEI) and SEI complicates the distinction between the interfacial transport (RSEI) and desolvation process (RCT). Therefore, to better clarify the effect of ion transport and ion desolvation processes, the electrodes should be carefully selected to minimize the impact of the interphase. In this case, LiFePO4 (LFP) and Li4Ti5O12 (LTO) should be a suitable cathode and anode, respectively, benefiting from their excellent cycling performance and negligible interphase formation.40 In addition, possessing low interfacial energy (0.343 eV) of the metastable intermediates at the two-phase boundary,41 LTO is a good electrode material for fast charging LIBs.
Herein in this work, with a LFP‖LTO full cell as the model system, we systematically study how solvation structures affect the ion transport and desolvation process of electrolytes and further demonstrate their effect on the fast-charging performance of batteries. Four types of solvents (Scheme 1b), including cyclic ethers (1,3-dioxolane (DOL) and 2-methyltetrahydrofuran (2-MTHF)), linear ethers (DME and ethylene glycol diethyl ether (DEE)), linear carbonates (dimethyl carbonate (DMC) and diethyl carbonate (DEC)), and cyclic carbonates (EC and propylene carbonate (PC)), are used to prepare electrolytes with a concentration of 1 M lithium bis(trifluoromethanesulphonyl)imide (LiTFSI). Based on ion transport characteristics and solvation structures, we propose various hopping or vehicle ion transport behaviors depending on the solvent of electrolytes and reveal that the hopping mechanism with high ionic conductivity is most favorable for fast charging performance. Among all electrolytes, the dominated Li-ion aggregation structure with low-viscosity DOL contributes to decent ionic conductivity, extremely low activity energy and a high Li+ transference number. The aggregation of ions in DOL electrolytes also contributes to a hopping assisted desolvation process, thus exhibiting the lowest charge transfer resistance. Therefore, the DOL-based electrolyte shows the highest specific capacity of 82.6 mA h g−1 and the lowest polarization of only 0.35 V at 20C (1C = 170 mA g−1).
Results and discussion
Fast charging electrochemical performance
To obviate the influence of ion-transport in the electrode on fast charging performance, relatively thin electrodes (LFP: ∼2.5 mg cm−2; LTO: ∼2.9 mg cm−2) are used in this study. The electrolyte concentration is set at 1.0 M due to the notable solubility, ionic conductivity, and good stability of LiTFSI in the electrochemical window of battery operation. For the sake of simplicity, all electrolytes are denoted as “solvent Ele.”. The LFP‖LTO full cells using these eight electrolytes were charged/discharged at 1C, 2C, 5C, 8C, 10C, 15C, and 20C, respectively (Fig. S1†). As demonstrated in Fig. 1a and b, there is no obvious difference in the kinetic properties of the eight electrolytes at low current density, with similar discharge specific capacity and polarization potential (the polarization in this work is calculated from the difference in median voltage between charging and discharging). However, as current density increases, both capacity and polarization begin to show evident differences. At 20C, the potential of polarization ranks as DOL Ele.< DEE Ele.< EC Ele.< 2-MTHF Ele.< DMC Ele.< PC Ele.< DME Ele.< DEC Ele.< DEC Ele.< DEC Ele. Among the studied electrolytes, the fast-charging performance of DOL Ele., DEE Ele. and EC Ele. is better than that of the others with stable charging/discharging platforms and lower polarization under 0.45 V at 20C (Fig. 1c–i), among which DOL Ele. delivers the lowest overpotential (0.35 V) with the highest specific capacity of 82.6 mA h g−1. The low polarization of DOL Ele. can be well maintained in the subsequent cycles (Fig. S2†). Moreover, it is worth noting that the polarization can be further reduced to ∼0.23 V when the battery is discharged at a normal rate (20C charge/1C discharge). (Fig. S3†). In contrast, the electrochemical performance of DEC is the worst at all rates, especially at 20C, where the specific capacity decays to 49.9 mA h g−1 and the polarization reaches 1.17 V.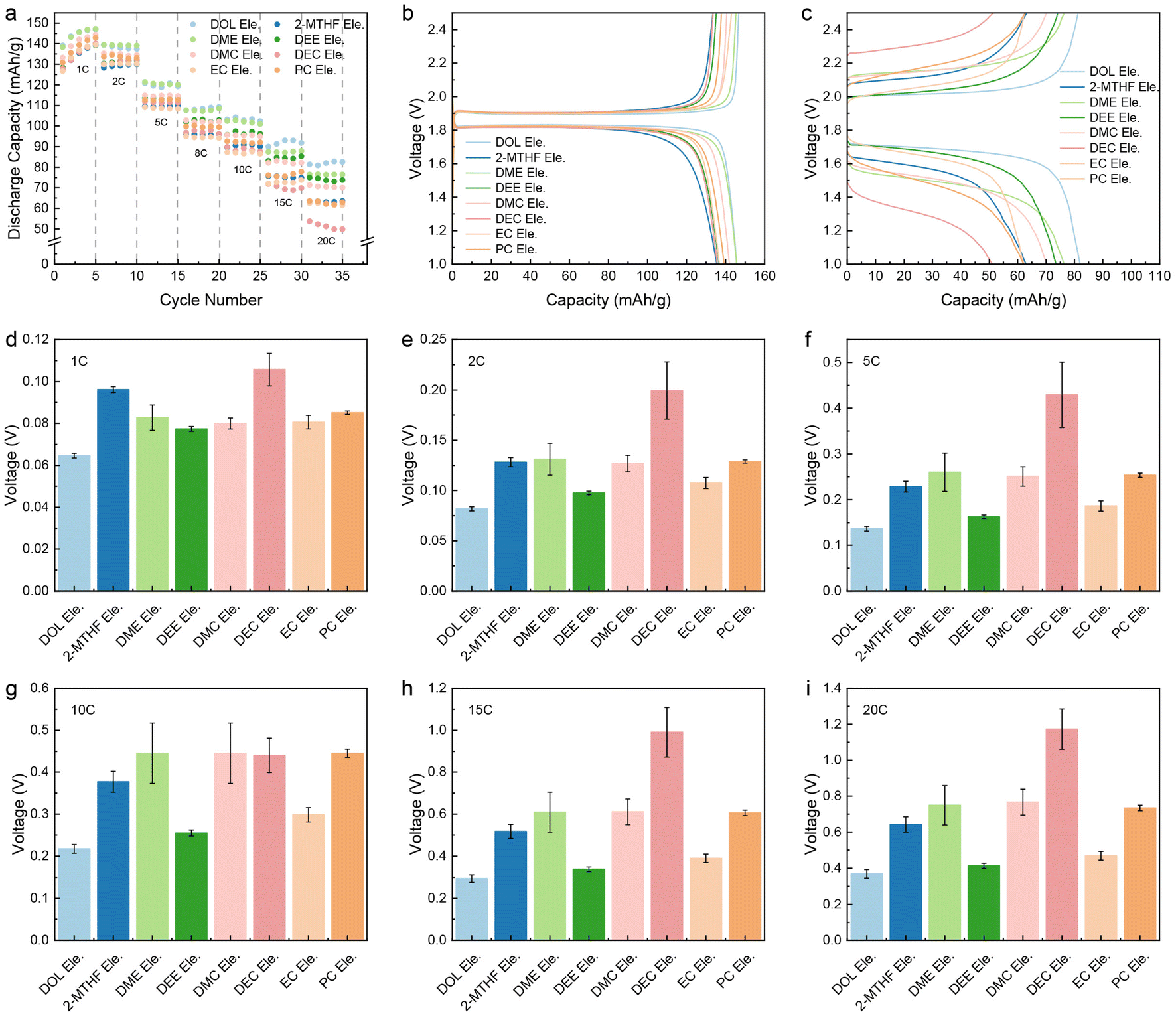 | ||
| Fig. 1 Rate performance of LTO‖LFP full cells with the electrolytes of interest. (a) The comparison of discharge capacity at 1C, 2C, 5C, 8C, 10C, 15C and 20C during cycling. Corresponding charge and discharge voltage curves at (b) 1C and (c) 20C. Corresponding polarization potential at (d) 1C, (e) 2C, (f) 5C, (g) 10C, (h) 15C and (i) 20C. The error bars for polarization potential are calculated using parallel cells of each electrolyte. | ||
In order to unveil the factors that result in the distinct capacity and polarization at fast charging, we first tested the conductivity of the electrolytes, which are usually positively correlated with the number of ions in the electrolyte and the speed of ion movement. Fig. 2a shows the ionic conductivity of electrolytes at different temperatures. At room temperature (20 °C), except for 2-MTHF Ele. and DEC Ele., the ionic conductivity of all electrolytes is higher than 3.5 mS cm−1, which indicates that the ion transport in bulk electrolyte is not a decisive factor for these electrolytes. In comparison, the lower ionic conductivity of 2-MTHF Ele. (1.70 mS cm−1) and DEC Ele. (2.08 mS cm−1) may be one of the main factors limiting the fast-charging performance.
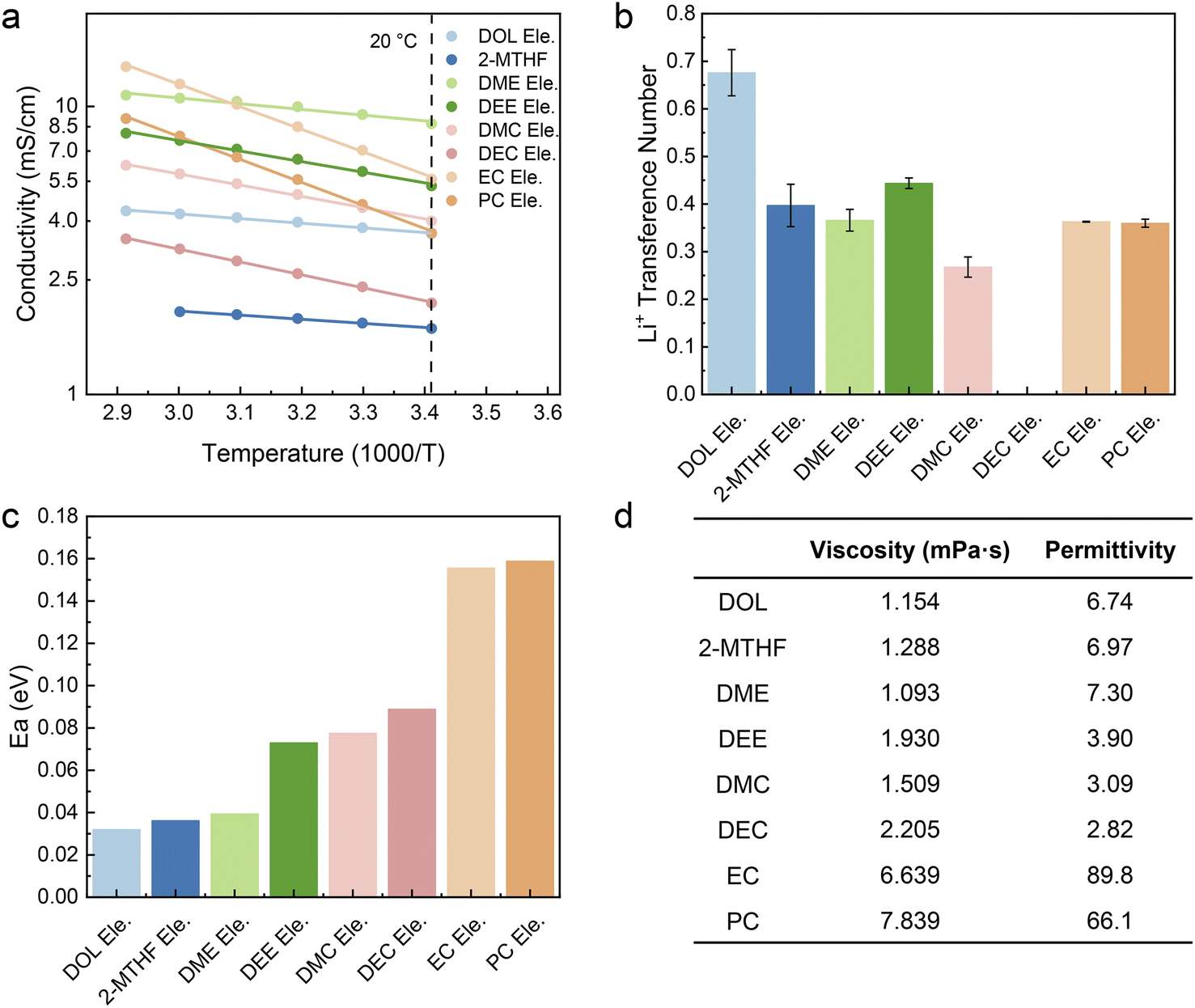 | ||
| Fig. 2 Ion motion kinetics of various electrolytes. (a) Ionic conductivity versus temperature and (b) Li+ transference number (tLi+) of the selected electrolytes. The error bars represent the maximum and minimum values of tLi+. (c) Activation energy (Ea) obtained by fitting the ionic conductivity versus temperature through the Arrhenius equation. (d) Measured viscosity of selected electrolytes with the permittivity of the corresponding solvents.42,43 | ||
The ionic conductivity of all the electrolytes increases as the temperature rises, which can be attributed to the faster thermal movement, especially for solvent molecules at high temperatures. Through fitting the ionic conductivity using the Arrhenius equation, we can illustrate the migration barriers of different electrolytes using activation energy (Ea). As shown in Fig. 2c, the activation energy of the ethers is lower than those of the esters, which may come from the characteristics that ethers are generally endowed with lower viscosity or weaker ion–dipole interaction compared to ester solvents (Fig. 2d). Among all the studied electrolytes, DOL Ele. has the lowest barrier of ion transport with an activation energy of 0.032 eV. It is worth mentioning that EC Ele. and PC Ele. have the highest activation energies (Ea > 0.15 eV), which is mainly due to the high viscosity (Fig. 2d). The high viscosity indicates strong dipole–dipole interactions, which retard the motion of ions in solvent vehicle mode. Therefore, Ea may not accurately reflect the transport behavior of Li+ due to its complex influencing factors.
In addition to ionic conductivity, the transference number of Li+ (t+) that reflects the ratio of cation movement under an electric field also plays a crucial role in determining whether enough ions can be provided at the electrode interface under fast charging conditions. Sand's time (tSand) usually indicates the time of Li+ depletion near the electrode of LIBs. In accordance with eqn (5), higher t+ results in longer Sand's time, indicating higher Li-ion transport capacity in the bulk phase.
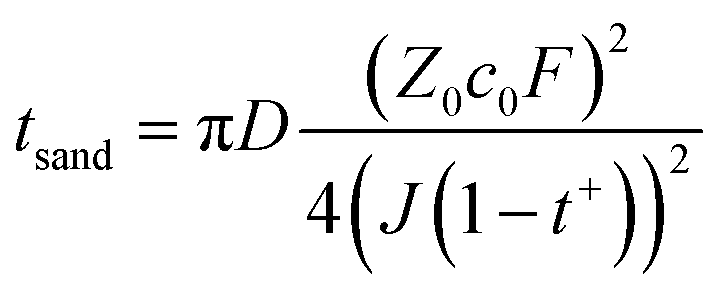 | (5) |
D, c0, t+, Z0, F, and J correspond to the diffusion coefficient, concentration, transference number of Li+, charge of cations, Faraday's constant, and current density, respectively. As depicted in Fig. 2b and S4–S10,† the t+ of most electrolytes is around 0.35 and DMC Ele. has the lowest transference number (0.26). In contrast, DOL Ele. and DEE Ele. have higher transference numbers than other electrolytes (tDOL Ele.+ = 0.68; tDEE Ele.+ = 0.45), which also corresponds to their superior fast charging performance. It should be noted that due to the instability towards Li metal, the transference number of DEC could not be accurately obtained.
To sum up, the ionic conductivity is not considered to play a decisive role in fast charging performance when it exceeds a certain value (for instance, >3.5 mS cm−1). The Ea for ion transport is also not directly correlated with fast charging behavior, but it may reflect the ion transport mechanism that will be illustrated in subsequent research. Although high t+ is beneficial for fast charging performance, it is similar for most electrolytes, except for DOL Ele. In addition, DOL Ele. also reveals the lowest Ea. We speculate that DOL Ele. is endowed with abundant aggregates (AGGs), where Li+ is transported through the hopping mechanism,1,44–47 resulting in a lower Li+ migration barrier and high t+, and further making DOL Ele. optimal for fast charging batteries.
Solvation structures of electrolytes
To further clarify the principles behind the discrepancy of fast charging performance, we then studied the solvation structures of electrolytes, which usually result from the competition between different interactions.48–52 The solvation structure can be generally classified into three types according to the coordination number of Li+ with TFSI−, namely AGGs (>1 Li+), CIPs (contact ion pairs = 1 Li+), and SSIPs (solvent-separated ion pairs, no Li+ and free TFSI−). Symmetric S–N–S stretching vibration in TFSI−, located at around 750 cm−1 in Raman and Fourier Transform Infrared (FTIR) spectra, is sensitive to the coordination of Li+. As shown in Fig. 3a and b, S11 and Table S1,† the S–N–S peaks will gradually move to higher Raman shifts (Raman spectra) or wavenumbers (FTIR spectra) due to the transition from SSIPs and CIPs to AGGs.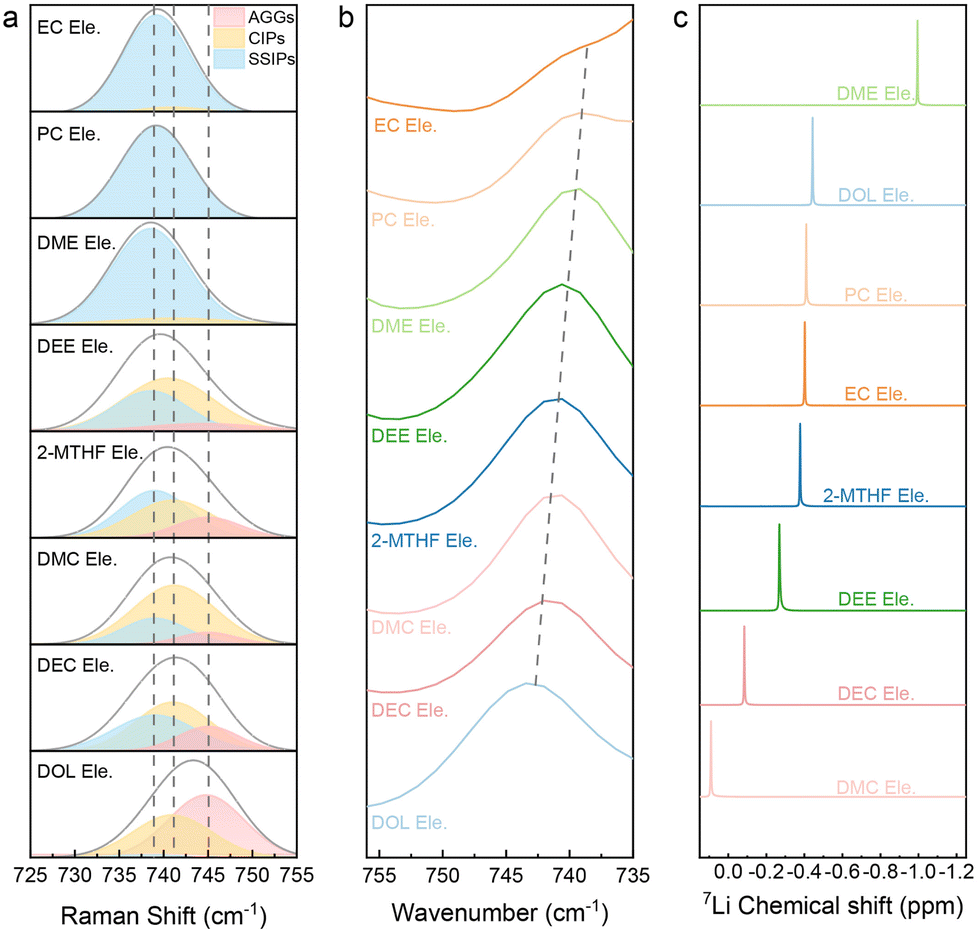 | ||
| Fig. 3 Spectra analysis of various electrolytes. (a) Raman and (b) FTIR spectra focused on the range of S–N–S stretch vibration. The peaks in Raman spectra are fitted with AGGs, CIPs and SSIPs, respectively. (c) 7Li NMR spectra of selected electrolytes. | ||
Among them, the S–N–S Raman peak of DOL Ele. appears at a high value of 742.8 cm−1, which proves more AGGs, implying a strong ion–ion interaction. This behavior can be caused by the weak solvation capability of DOL. The Raman peaks of DMC Ele., DEC Ele., 2-MTHF Ele. and DEE Ele. appear in the middle position, indicating that the ion–ion interactions are weaker than those of DOL Ele. The Raman peak of DME Ele. (737.5 cm−1) appears at the lowest value, indicating the weakest Li+–TFSI− interaction. As the two oxygen atoms in the DME molecule are favorable to form a chelating coordination structure with Li+, the ion–dipole interactions are extremely strong, making it difficult for Li+ to coordinate with the anion. The S–N–S peak position of EC Ele. and PC Ele. is also located at a low value and slightly higher than that of DME Ele. This phenomenon can be ascribed to the high dielectric constant of the solvent, which can well dissociate Li+ and TFSI−. The results of FTIR (Fig. 3b) are consistent with those of Raman; however it should be noted that the S–N–S vibrations in EC Ele. and PC Ele. are partially overlapped with solvent peaks.
Although the S–N–S variation can partially manifest the Li+ solvation structure, the strength of Li+ with the solvent or anion is still ambiguous. Therefore, 7Li NMR (Fig. 3c) analysis was further used to study all the electrolytes. In general, an upfield shift (more negative) of 7Li indicates increased electron density around Li+, which can be caused by either stronger anion binding or stronger solvent binding. DME Ele. shows the obviously most negative 7Li peak, unveiling the strong coordination of Li+ with the DME molecule rather than the anion, as Raman spectra already confirm the weak Li+ – anion intercalation. The next one is DOL Ele., which should be ascribed to the strong coordination with the anion, which is consistent with Raman spectra. The electrolyte with 2-MTHF that also has a ring structure is considered similar to DOL Ele., but the total interactions are weaker. Although DEE is also a linear ether like DME, the steric effect of DEE was previously reported to reveal an anion rich solvation structure.53 Therefore, we classify DEE Ele. in the same category as 2-MTHF Ele. For EC Ele. and PC Ele., in spite of their similar SSIP dominated solvation structure in Raman spectra, the 7Li in these electrolytes show an apparent downshift compared to DME Ele., indicating a lower electron density. The results suggest that although solvents with a high dielectric constant (EC and PC) can well dissociate salt, the coordination strength is not as strong as that of the DME molecule. Finally, the peaks of Li+ in DMC Ele. and DEC Ele. have the lowest electron cloud density. Based on Raman spectra, we propose that both the ion–ion and ion–dipole interactions are relatively weak in these electrolytes.
To provide a quantitative solvation structure in the electrolyte, molecule dynamic (MD) simulations were carried out (Fig. 4, S12–S20 and Table S2–S5†). In general, there are abundant AGGs in DOL Ele., 2-MTHF Ele., and DEE Ele. (Fig. 4a, e and f), indicating strong ion–ion interactions, in which the anion coordination number is 2.65, 1.58 and 1.79, respectively. Particularly in DOL Ele., a significant proportion of AGGs (61.9%) was formed. In comparison, Fig. 4b, d and h, and Table S3† show that DME Ele., EC Ele., and PC Ele. have abundant charges and predominantly SSIP structures, with negligible anion coordination (0.256, 0.042 and 0.049, respectively). For EC and PC, the phenomenon is caused by the high dielectric constant of solvent, LiTFSI can be fully dissociated, resulting in a higher coordination number of Li+-solvent (EC: 4.4; PC: 4.3). For DME Ele., the phenomenon is caused by the high solvating power of DME that strongly coordinates with Li to prevent anions from entering the primary solvation sheath of Li+. Therefore, the coordination number of Li+-DME is the highest, reaching 5.7, which further verifies the result of the highest electron cloud density of Li in DME Ele. Finally, DMC Ele. and DEC Ele. also have few AGGs but are more abundant in CIP structures (DMC: 39.2%; DEC: 47.9%) (Fig. 4c and g), in which the anion coordination number is 0.75 and 0.71, respectively. We suppose that on one hand the low dielectric constants of DMC and DEC lead to insufficient dissociation of LiTFSI, and on the other hand the higher electron density of carbonyl than that of ether oxygen atoms in cyclic ethers weakens the ion–ion interactions and leads to less aggregation. Therefore, Li in the above electrolytes has the lowest electron cloud density due to weak interactions with both the solvent and the anion.
 | ||
| Fig. 4 Solvation structures obtained from MD simulations. The radial distribution function (RDF) and coordination number of anions and solvents extracted from MD of (a) DOL Ele., (b) DME Ele., (c) DMC Ele., (d) EC Ele., (e) 2-MTHF Ele., (f) DEE Ele., (g) DEC Ele. and (h) PC Ele. The inset shows the proportion of three solvation structures in different electrolytes. | ||
In short, we can classify four types of solvents according to the spectral analysis and MD simulations, strong ion–ion interactions (DOL, 2-MTHF and DEE Ele.), strong ion–dipole and weak ion–ion interactions (DME Ele.), moderate ion–dipole and weak ion–ion interactions (EC and PC Ele.), and weak ion–dipole and ion–ion interactions (DMC and DEC Ele.). These interaction characteristics not only effect the ionic transport, but also influence the desolvation process, which will be discussed in detail in the following text.
Charge transfer reactions influenced by various electrolytes
The above results illustrate that the solvation structures are quite distinct depending on the properties of solvents. As mentioned before, the rate performance has little correlation with the ion conductivity under the circumstance that the conductivity reaches a certain value. Therefore, we consider that the resistance of the charge transfer reaction (Rct) that reflects the desolvation process is the main factor responsible for the fast charging. Electrochemical impedance spectroscopy (EIS) was then performed to verify our hypothesis (Fig. S21†). The LFP‖LTO full cells were charged and discharged at a rate of 1C and EIS was performed respectively in the 1st, 3rd, and 5th cycles during the charging platform. As shown in Fig. 5a–c, although the SEI is also observed in all the electrolytes due to their decomposition, the resistance of the SEI (RSEI) in most electrolytes is between 10 and 20 Ω, much lower than Rct (Table S6†). Therefore, the desolvation process plays a leading role compared with RSEI at the interface. In general, both RSEI and Rct tended to be stable after three cycles (Fig. S21†). In the fifth cycle, the Rct values of the eight electrolytes follow the order of DOL Ele. EC Ele.< DEE Ele., 2-MTHF Ele.< PC Ele.< DME Ele.< DMC Ele., DEC Ele. (Fig. 5d), matching well with their electrochemical performance (Fig. 1i). In addition, the order of bulk resistance (Rb) is generally consistent with the ionic conductivity, confirming the accuracy of EIS measurement. Among them, the Rct of DOL Ele. is only 12 Ω, while those of DMC Ele. and DEC Ele. reach 100 Ω. Therefore, EIS demonstrates that Rct is the dominant factor contributing to the fast-charging performance of LIBs. Rct can be affected by various factors, such as the electrode structure, charge number in the electrolyte, solvation structure, etc. As the electrode structures of all electrolytes are considered identical, the charge number and solvation structure may be the decisive factors for charge transfer reactions. On the basis of the spectroscopic and electrochemical results, we divide the eight electrolytes into four categories (Scheme 2), according to the solvation structures and the interactions between the components. In addition, in situ Raman spectra were further recorded to monitor the desolvation process near the LTO anode under fast-charging conditions of LTO‖LFP cells (Fig. 6), which was found to be well consistent with our hypothesis (Scheme 2). | ||
| Fig. 5 EIS analysis of LTO‖LFP batteries with different electrolytes. (a) 1st, (b) 3rd and (c) 5th cycles. (d) Resistance of bulk (Rb), resistance of the SEI (RSEI) and resistance of the charge transfer reaction (Rct) calculated by fitting the data with the inserted equivalent circuit model. The batteries were charged and discharged at 1C, and the EIS were obtained during the charging platform of the batteries. | ||
 | ||
| Scheme 2 Schematic diagram of the Li+ transport mechanism and charge transfer process in various electrolytes. | ||
 | ||
| Fig. 6 In situ Raman measurement of the electrolytes near the surface of the LTO anode in LTO‖LFP batteries during the fast charging process. Galvanostatic charging voltage curves of LTO‖LFP full cells at 10C in (a) DOL Ele., (c) EC Ele., (e) DME Ele., and (g) DEC Ele. Corresponding variations of S–N–S stretching vibrations in (b) DOL Ele., (d) EC Ele., (f) DME Ele., and (h) DEC Ele. | ||
For electrolytes with AGG-rich solvation structures such as DOL Ele., 2-MTHF Ele., and DEE Ele., the migration is possibly carried out in hopping-assisted ionic transport mode. Compared with the traditional vehicle mechanism in which Li+ moves together with solvation sheaves, Li+ jumps between adjacent positions of AGGs, which greatly reduces the transport barrier of the electrolyte bulk. Therefore, the activation energies of DOL Ele. and 2-MTHF Ele. are only 0.032 eV and 0.036 eV, which have the fastest bulk dynamics. Meanwhile, the relative higher activation energy of DEE Ele. (0.073 eV) may be caused by the higher viscosity. Furthermore, as less solvent is involved, when AGGs move to the vicinity of the electrode–electrolyte interface, the desolvation process is more feasible without the accumulation of ions near the electrode–electrolyte interface or even the necessity of the desolvation process. In the in situ Raman spectra of DOL Ele. (Fig. 6a and b), we found that the intensity of TFSI− is basically unchanged after entering the charging platform, which confirms this hopping-assisted mechanism. As a result, the barriers of the charge transfer reaction are largely reduced, resulting in both low Rct and excellent fast charging performance of DOL Ele. and DEE Ele. In addition, the relative high polarization of 2-MTHF Ele. is mainly caused by the low ionic conductivity as mentioned before.
In comparison, EC Ele., PC Ele., and DME Ele., which mainly consist of SSIPs with abundant charges, are transported through the vehicle mechanism. Among them, EC and PC dissociate Li+ and anions by shielding the Coulomb force due to their high dielectric constant, which exhibit weaker interaction with Li+ than the DME molecule that dissociates Li+ by strong chelation intercalation. The coordination number and 7Li NMR have well confirmed this hypothesis. In in situ Raman spectra (Fig. 6c and d), the concentration of anions near the LTO electrode in EC Ele. gradually decreases with the progress of the charging process, indicating the continuous movement of anions under the electric field. Nevertheless, the concentration of anions in DME Ele. first decreases and then increases in the later part of the charging process (Fig. 6e and f). We ascribe this occurrence to the increased difficulty of Li+ desolvation in DME Ele., leading to the accumulation of Li+ near the anode, where anions are also attracted under the influence of Coulomb forces. Therefore, EC Ele. and PC Ele. exhibit faster desolvation kinetics at the interface than DME Ele., although all of them are composed of SSIPs with plentiful charges, which is also consistent with their electrochemical behavior in the fast charging process.
Finally, although DMC Ele. and DEC Ele. are also transported through the vehicle mechanism, the salts in these solvents are not fully dissociated with the CIP-rich solvation structure due to both weak ion–dipole and ion–ion interactions. The net charge of CIPs is neutral, indicating that they move more slowly than AGGs and SSIPs under an electric field, which is further supported by the in situ Raman spectra of DEC Ele. (Fig. 6g and h). The general trend of anions in DEC Ele. is similar to that of EC Ele. However, due to few charges and slow motion of solvation structures, a fast decrease of anions is found near the interface of the anode during the charging process. Therefore, the charge transfer reactions in DMC Ele. and DEC Ele. are most sluggish with the worst fast charging performance.
As a result, we can conclude that the electrolyte with weak ion–dipole/dipole–dipole and strong ion–ion interactions is the most favorable for fast charging performance. The strong ion–ion interactions facilitate ion transport/desolvation in hopping assisted mode and increase the Li+ transference number. Meanwhile, small molecules with low viscosity (such as DOL) are favorable to reduce the dipole–dipole interaction, which also contributes to high ionic conductivity of electrolytes.
Conclusions
In summary, through systematically investigating the influence of eight solvents on the fast-charging performance of LTO‖LFP full cells, we found that the charge transfer reaction is the rate-limiting step when ionic conductivity is high enough (for example > 3.5 mS cm−1). Furthermore, the underlying reason for the charge transfer reaction speed is ascribed to the different solvation structures formed by the competition of ion–ion, ion–dipole, and dipole–dipole interactions. Strong ion–ion interactions, strong ion–dipole interactions, moderate ion–dipole interactions, and weak ion–dipole/ion–ion interactions result in AGG, SSIP, SSIP, and CIP dominated solvation structures, respectively, in which the kinetic speeds are in the order of AGGs > SSIPs with moderate ion–dipole interactions > SSIPs with strong ion–dipole interactions > CIPs. Owing to the hopping assisted mechanism, AGG-rich electrolytes (DOL Ele., etc.) can realize rapid ion transport and a low interfacial desolvation barrier enabling them to reach 60% of their specific capacity in less than 3 mins with a polarization of only 0.35 V. Following this principle, we anticipate that solvents with weak solvation ability and low viscosity are promising candidates for fast charging electrolytes. Through molecule design, the high oxidation and long-term cycle stability can be further improved for practical applications.Data availability
Additional data, including the preparation of electrolyte and electrode, battery assembly, potentiostatic polarization test, Raman spectra, IR spectra, 7Li nuclear magnetic resonance spectroscopy, MD simulations and parameters of MD calculation, EIS analysis and additional information of LTO‖LFP full cell tests are available in the ESI.†Author contributions
Zhenyu Fan: conceptualization, methodology, investigation, formal analysis, validation and writing – original draft; Jingwei Zhang: software and formal analysis; Lanqing Wu: investigation, methodology and formal analysis; Huaqing Yu: investigation and formal analysis; Kun Li: investigation and formal analysis; Jia Li: investigation and formal analysis; Qing Zhao: conceptualization, writing – review and editing, supervision, resources, and funding acquisition.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22372083 and 52201259), the National Key R&D Program of China (2021YFB2500300), the Natural Science Foundation of Tianjin (No. 22JCZDJC00380), and the Young Elite Scientist Sponsorship Program by CAST.Notes and references
- B. Dunn, H. Kamath and J.-M. Tarascon, Electrical Energy Storage for the Grid: A Battery of Choices, Science, 2011, 334, 928–935 CrossRef CAS.
- D. Lu, R. H. Li, M. M. Rahman, P. Y. Yu, L. Lv, S. Yang, Y. Q. Huang, C. C. Sun, S. Q. Zhang, H. K. Zhang, J. B. Zhang, X. Z. Xiao, T. Deng, L. W. Fan, L. X. Chen, J. P. Wang, E. Y. Hu, C. S. Wang and X. L. Fan, Ligand-channel-enabled ultrafast Li-ion conduction, Nature, 2024, 627, 101–107 CrossRef CAS.
- C. Y. Wang, T. Liu, X. G. Yang, S. H. Ge, N. V. Stanley, E. S. Rountree, Y. J. Leng and B. D. McCarthy, Fast charging of energy-dense lithium-ion batteries, Nature, 2022, 611, 485–490 CrossRef CAS.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X.-Q. Yang, O. Borodin and C. Wang, Electrolyte design for Li-ion batteries under extreme operating conditions, Nature, 2023, 614, 694–700 CrossRef CAS.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Fast Charging of Lithium-Ion Batteries: A Review of Materials Aspects, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- C. B. Zhu, R. E. Usiskin, Y. Yu and J. Maier, The nanoscale circuitry of battery electrodes, Science, 2017, 358, eaao2808 CrossRef.
- S. N. Lauro, J. N. Burrow and C. B. Mullins, Restructuring the lithium-ion battery: A perspective on electrode architectures, eScience, 2023, 3, 100152 CrossRef.
- Z. M. Konz, B. M. Wirtz, A. Verma, T. Y. Huang, H. K. Bergstrom, M. J. Crafton, D. E. Brown, E. J. McShane, A. M. Colclasure and B. D. McCloskey, High-throughput Li plating quantification for fast-charging battery design, Nat. Energy, 2023, 8, 450–461 CrossRef CAS.
- K. H. Chen, V. Goel, M. J. Namkoong, M. Wied, S. Müller, V. Wood, J. Sakamoto, K. Thornton and N. P. Dasgupta, Enabling 6C Fast Charging of Li-Ion Batteries with Graphite/Hard Carbon Hybrid Anodes, Adv. Energy Mater., 2020, 11, 2003336 CrossRef.
- C. Wang, Y. Xie, Y. Huang, S. Zhou, H. Xie, H. Jin and H. Ji, Li3PO4-Enriched SEI on Graphite Anode Boosts Li+ De-Solvation Enabling Fast-Charging and Low-Temperature Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2024, 63, e202402301 CrossRef CAS PubMed.
- U. H. Kim, S. B. Lee, N. Y. Park, S. J. Kim, C. S. Yoon and Y. K. Sun, High-Energy-Density Li-Ion Battery Reaching Full Charge in 12 min, ACS Energy Lett., 2022, 7, 3880–3888 CrossRef CAS.
- X. Chen and Q. Zhang, Atomic Insights into the Fundamental Interactions in Lithium Battery Electrolytes, Acc. Chem. Res., 2020, 53, 1992–2002 CrossRef CAS PubMed.
- N. Yao, L. G. Yu, Z. H. Fu, X. Shen, T. Z. Hou, X. Y. Liu, Y. C. Gao, R. Zhang, C. Z. Zhao, X. Chen and Q. Zhang, Probing the Origin of Viscosity of Liquid Electrolytes for Lithium Batteries, Angew. Chem., Int. Ed., 2023, 62, e202305331 CrossRef CAS PubMed.
- R. Hou, S. Guo and H. Zhou, Atomic Insights into Advances and Issues in Low-Temperature Electrolytes, Adv. Energy Mater., 2023, 13, 2300053 CrossRef CAS.
- X. P. Li, M. H. Li, Y. Liu, Y. L. Jie, W. X. Li, Y. W. Chen, F. Y. Huang, Y. C. Zhang, T. M. Sohail, S. Y. Wang, X. B. Zhu, T. Cheng, M. D. Gu, S. H. Jiao and R. G. Cao, Fast Interfacial Defluorination Kinetics Enables Stable Cycling of Low-Temperature Lithium Metal Batteries, J. Am. Chem. Soc., 2024, 146, 17023–17031 CrossRef CAS PubMed.
- R. D. Guo, Y. X. Che, G. Y. Lan, J. L. Lan, J. H. Li, L. D. Xing, K. Xu, W. Z. Fan, L. Yu and W. S. Li, Tailoring Low-Temperature Performance of a Lithium-Ion Battery via Rational Designing Interphase on an Anode, ACS Appl. Mater. Interfaces, 2019, 11, 38285–38293 CrossRef CAS PubMed.
- D. W. Xia, E. P. Kamphaus, A. Y. Hu, S. Hwang, L. Tao, S. Sainio, D. Nordlund, Y. B. Fu, H. B. Huang, L. Cheng and F. Lin, Design Criteria of Dilute Ether Electrolytes toward Reversible and Fast Intercalation Chemistry of Graphite Anode in Li-Ion Batteries, ACS Energy Lett., 2023, 8, 1379–1389 CrossRef CAS.
- H. R. Cheng, Q. J. Sun, L. L. Li, Y. G. Zou, Y. Q. Wang, T. Cai, F. Zhao, G. Liu, Z. Ma, W. Wahyudi, Q. Li and J. Ming, Emerging Era of Electrolyte Solvation Structure and Interfacial Model in Batteries, ACS Energy Lett., 2022, 7, 490–513 CrossRef CAS.
- Z. C. Wu, R. H. Li, S. Q. Zhang, L. Lv, T. Deng, H. Zhang, R. X. Zhang, J. J. Liu, S. H. Ding, L. W. Fan, L. X. Chen and X. L. Fan, Deciphering and modulating energetics of solvation structure enables aggressive high-voltage chemistry of Li metal batteries, Chem, 2023, 9, 650–664 CAS.
- X. Cao, P. Gao, X. Ren, L. Zou, M. H. Engelhard, B. E. Matthews, J. Hu, C. Niu, D. Liu, B. W. Arey, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Effects of fluorinated solvents on electrolyte solvation structures and electrode/electrolyte interphases for lithium metal batteries, Proc. Natl. Acad. Sci. U.S.A., 2021, 118, e2020357118 CrossRef CAS PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Advances and issues in developing salt-concentrated battery electrolytes, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, Oxidative-Stability Enhancement and Charge Transport Mechanism in Glyme-Lithium Salt Equimolar Complexes, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- M. Fang, X. Yue, Y. Dong, Y. Chen and Z. Liang, A temperature-dependent solvating electrolyte for wide-temperature and fast-charging lithium metal batteries, Joule, 2024, 8, 91–103 CrossRef CAS.
- C. Y. Yang, J. Chen, T. T. Qing, X. L. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. S. Wang and K. Xu, 4.0 V Aqueous Li-Ion Batteries, Joule, 2017, 1, 122–132 Search PubMed.
- C. Sun, X. Ji, S. Weng, R. Li, X. Huang, C. Zhu, X. Xiao, T. Deng, L. Fan, L. Chen, X. Wang, C. Wang and X. Fan, 50C Fast-Charge Li-Ion Batteries using a Graphite Anode, Adv. Mater., 2022, 34, 2206020 Search PubMed.
- J. Xu, V. Koverga, A. Phan, A. min Li, N. Zhang, M. Baek, C. Jayawardana, B. L. Lucht, A. T. Ngo and C. Wang, Revealing the Anion-Solvent Interaction for Ultralow Temperature Lithium Metal Batteries, Adv. Mater., 2023, 36, 2306462 Search PubMed.
- S. Yuan, S. Cao, X. Chen, J. Wei, Z. Lv, H. Xia, J. Li, H. Zhang, L. Liu, C. Tian, L. Chen, W. Zhang, Z. Xing, H. Li, S. Li, Q. Zhu, X. Feng and X. Chen, Deshielding Anions Enable Solvation Chemistry Control of LiPF6-Based Electrolyte toward Low-Temperature Lithium-Ion Batteries, Adv. Mater., 2024, 36, 2311327 CrossRef CAS PubMed.
- L. Q. Wu, Z. Li, Z. Y. Fan, K. Li, J. Li, D. B. Huang, A. J. Li, Y. Yang, W. W. Xie and Q. Zhao, Unveiling the Role of Fluorination in Hexacyclic Coordinated Ether Electrolytes for High-Voltage Lithium Metal Batteries, J. Am. Chem. Soc., 2024, 146, 5964–5976 CrossRef CAS PubMed.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, Unusual Stability of Acetonitrile-Based Superconcentrated Electrolytes for Fast-Charging Lithium-Ion Batteries, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- X. Huang, R. Li, C. Sun, H. Zhang, S. Zhang, L. Lv, Y. Huang, L. Fan, L. Chen, M. Noked and X. Fan, Solvent-Assisted Hopping Mechanism Enables Ultrafast Charging of Lithium-Ion Batteries, ACS Energy Lett., 2022, 7, 3947–3957 CrossRef CAS.
- N. Gao, S. Kim, P. Chinnam, E. J. Dufek, A. M. Colclasure, A. Jansen, S.-B. Son, I. Bloom, A. Dunlop, S. Trask and K. L. Gering, Methodologies for Design, Characterization and Testing of Electrolytes that Enable Extreme Fast Charging of Lithium-ion Cells, Energy Storage Mater., 2022, 44, 296–312 CrossRef.
- J. H. He, J. K. Meng and Y. H. Huang, Challenges and recent progress in fast-charging lithium-ion battery materials, J. Power Sources, 2023, 570, 232965 CrossRef CAS.
- Y. X. Yao, X. Chen, N. Yao, J. H. Gao, G. Xu, J. F. Ding, C. L. Song, W. L. Cai, C. Yan and Q. Zhang, Unlocking Charge Transfer Limitations for Extreme Fast Charging of Li-Ion Batteries, Angew. Chem., Int. Ed., 2022, 62, e202214828 CrossRef PubMed.
- X. T. Yang, C. Han, Y. M. Xie, R. Fang, S. Zheng, J. H. Tian, X. M. Lin, H. Zhang, B. W. Mao, Y. Gu, Y. H. Wang and J. F. Li, Highly Stable Lithium Metal Batteries Enabled by Tuning the Molecular Polarity of Diluents in Localized High-Concentration Electrolytes, Small, 2024, 20, 2311393 CrossRef CAS PubMed.
- L. L. Jiang, C. Yan, Y. X. Yao, W. Cai, J. Q. Huang and Q. Zhang, Inhibiting Solvent Co-Intercalation in a Graphite Anode by a Localized High-Concentration Electrolyte in Fast-Charging Batteries, Angew. Chem., Int. Ed., 2020, 60, 3402–3406 CrossRef PubMed.
- X. Liu, J. Zhang, X. Yun, J. Li, H. Yu, L. Peng, Z. Xi, R. Wang, L. Yang, W. Xie, J. Chen and Q. Zhao, Anchored Weakly-Solvated Electrolytes for High-Voltage and Low-Temperature Lithium-ion Batteries, Angew. Chem., Int. Ed., 2024, 63, e202406596 CrossRef CAS PubMed.
- J. Chen, Y. Zhang, H. Lu, J. Ding, X. Wang, Y. Huang, H. Ma and J. Wang, Electrolyte solvation chemistry to construct an anion-tuned interphase for stable high-temperature lithium metal batteries, eScience, 2023, 3, 100135 CrossRef.
- Y. Zhao, T. H. Zhou, T. Ashirov, M. El Kazzi, C. Cancellieri, L. P. H. Jeurgens, J. W. Choi and A. Coskun, Fluorinated ether electrolyte with controlled solvation structure for high voltage lithium metal batteries, Nat. Commun., 2022, 13, 2575 CrossRef CAS PubMed.
- B. Roy, P. Cherepanov, C. Nguyen, C. Forsyth, U. Pal, T. C. Mendes, P. Howlett, M. Forsyth, D. MacFarlane and M. Kar, Lithium Borate Ester Salts for Electrolyte Application in Next-Generation High Voltage Lithium Batteries, Adv. Energy Mater., 2021, 11, 2101422 CrossRef CAS.
- T. R. Jow, S. A. Delp, J. L. Allen, J. P. Jones and M. C. Smart, Factors Limiting Li+ Charge Transfer Kinetics in Li-Ion Batteries, J. Electrochem. Soc., 2018, 165, A361–A367 CrossRef CAS.
- W. Zhang, D. H. Seo, T. Chen, L. J. Wu, M. Topsakal, Y. M. Zhu, D. Y. Lu, G. Ceder and F. Wang, Kinetic pathways of ionic transport in fast-charging lithium titanate, Science, 2020, 367, 1030–1034 CrossRef CAS PubMed.
- Y. Zhang, Y. Lu, J. Jin, M. Wu, H. Yuan, S. Zhang, K. Davey, Z. Guo and Z. Wen, Electrolyte Design for Lithium-Ion Batteries for Extreme Temperature Applications, Adv. Mater., 2023, 36, 2308484 CrossRef PubMed.
- Z. H. Piao, R. H. Gao, Y. Q. Liu, G. M. Zhou and H. M. Cheng, A Review on Regulating Li+ Solvation Structures in Carbonate Electrolytes for Lithium Metal Batteries, Adv. Mater., 2023, 35, 2206009 CrossRef CAS PubMed.
- S. Kondou, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Ionic transport in highly concentrated lithium bis(fluorosulfonyl)amide electrolytes with keto ester solvents: structural implications for ion hopping conduction in liquid electrolytes, Phys. Chem. Chem. Phys., 2019, 21, 5097–5105 RSC.
- Y. Ugata, S. Sasagawa, R. Tatara, K. Ueno, M. Watanabe and K. Dokko, Structural Effects of Solvents on Li-Ion-Hopping Conduction in Highly Concentrated LiBF4/Sulfone Solutions, J. Phys. Chem. B, 2021, 125, 6600–6608 CrossRef CAS PubMed.
- S. Mukherji, N. V. S. Avula, R. Kumar and S. Balasubramanian, Hopping in High Concentration Electrolytes – Long Time Bulk and Single-Particle Signatures, Free Energy Barriers, and Structural Insights, J. Phys. Chem. Lett., 2020, 11, 9613–9620 CrossRef CAS PubMed.
- Y. Ugata, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Li-ion hopping conduction in highly concentrated lithium bis(fluorosulfonyl)amide/dinitrile liquid electrolytes, Phys. Chem. Chem. Phys., 2019, 21, 9759–9768 RSC.
- Q. Sun, Z. Cao, Z. Ma, J. Zhang, H. Cheng, X. Guo, G.-T. Park, Q. Li, E. Xie, L. Cavallo, Y.-K. Sun and J. Ming, Dipole–Dipole Interaction Induced Electrolyte Interfacial Model to Stabilize Antimony Anode for High-Safety Lithium-Ion Batteries, ACS Energy Lett., 2022, 7, 3545–3556 CrossRef CAS.
- K. Chen, X. Shen, L. Luo, H. Chen, R. Cao, X. Feng, W. Chen, Y. Fang and Y. Cao, Correlating the Solvating Power of Solvents with the Strength of Ion-Dipole Interaction in Electrolytes of Lithium-ion Batteries, Angew. Chem., Int. Ed., 2023, 62, e202312373 CrossRef CAS PubMed.
- Z. S. Wang, H. P. Wang, S. A. Qi, D. X. Wu, J. D. Huang, X. Li, C. Y. Wang and J. M. Ma, Structural regulation chemistry of lithium ion solvation for lithium batteries, Ecomat, 2022, 4, e12200 CrossRef CAS.
- T. Hou, K. Fong, J. Wang and K. Persson, The solvation structure, transport properties and reduction behavior of carbonate-based electrolytes of lithium-ion batteries, Chem. Sci., 2021, 12, 14740–14751 RSC.
- J. Wang, J. Luo, H. Wu, X. Yu, X. Wu, Z. Li, H. Luo, H. Zhang, Y. Hong, Y. Zou, S. Cao, Y. Qiao and S. G. Sun, Visualizing and Regulating Dynamic Evolution of Interfacial Electrolyte Configuration during De-solvation Process on Lithium-Metal Anode, Angew. Chem., Int. Ed., 2024, 63, e202400254 CrossRef CAS PubMed.
- Y. L. Chen, Z. Yu, P. Rudnicki, H. X. Gong, Z. J. Huang, S. C. Kim, J. C. Lai, X. Kong, J. Qin, Y. Cui and Z. N. Bao, Steric Effect Tuned Ion Solvation Enabling Stable Cycling of High-Voltage Lithium Metal Battery, J. Am. Chem. Soc., 2021, 143, 18703–18713 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05464d |
| This journal is © The Royal Society of Chemistry 2024 |