
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bridge editing of spin-flip emitters gives insight into excited state energies and dynamics†
Florian
Reichenauer
a,
Robert
Naumann
 a,
Christoph
Förster
a,
Winald R.
Kitzmann
a,
Antti-Pekka M.
Reponen
b,
Sascha
Feldmann
bc and
Katja
Heinze
*a
a,
Christoph
Förster
a,
Winald R.
Kitzmann
a,
Antti-Pekka M.
Reponen
b,
Sascha
Feldmann
bc and
Katja
Heinze
*a
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: Katja.Heinze@uni-mainz.de
bRowland Institute, Harvard University, 100 Edwin H. Land Boulevard, Cambridge, MA 02142, USA
cInstitute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland
First published on 11th November 2024
Abstract
Six-coordinate chromium(III) complexes with high spin-flip (SF) photoluminescence quantum yields and lifetimes (molecular rubies) have attracted huge interest in the past years due to their applicability in sensing, photocatalysis or circularly polarised emission. However, clearcut design rules for high quantum yields and lifetimes are still lacking due to the multidimensional problem of the non-radiative decay of the SF states. Based on an isostructural series of complexes differing in the ligand backbone, we disentangle decisive structural and electronic features for SF excited state energies and non-radiative decays promoted by spin–orbit coupling, Jahn–Teller distortions and (thermally activated) multiphonon relaxation. This analysis goes beyond the classical increasing of the ligand field strength or the metal–ligand covalency to reduce non-radiative decay or to tune the SF energy. The results underscore the utility of the combination of near-infrared absorption, variable temperature emission and fs-transient absorption spectroscopy as well as photolysis and high-level quantum chemical calculations to obtain a comprehensive picture of the excited dynamics on ultrafast and long timescales.
1 Introduction
Understanding and ultimately rationally modulating energies of metal-centered (MC) and charge-transfer (CT) excited states as well as their dynamics after light absorption are at the heart of contemporary design of transition metal complex photosensitisers and photocatalysts based on abundant elements.1–14 Yet, design rules for MC states typically rely on qualitative arguments based on the ligand field splitting Δo, i.e. the energy gap between t2g and eg orbitals at the ground state geometry. This splitting is defined by the metal ion, the ligands and the coordination geometry according to well-known rules.15–17 For CT excited states, ligand modification can shift the CT states to higher or lower energies based on clear-cut substituent effects, although this chemical modification might also indirectly affect the d orbital energies.2–4 The dynamics of excited states18,19 are in general governed by their relative energies, their different geometries and electronic couplings, yet in most cases a comprehensive picture for transition metal complexes is unavailable or still subject of debate.20–23Tuning of excited state energies and dynamics of metal complexes with nested spin-flip (SF)1,4,8,9 states as lowest energy excited states is even less understood. Yet, several high-performing 3d3-chromium(III) based complexes possessing high phosphorescence quantum yields and long excited state lifetimes have been developed in recent years.24–30 Their useful excited state reactivities have been exploited in various applications, from sensing and photoredox catalysis to circularly polarised emission.31–42
The energies of the intraconfigurational excited states 2E/2T1 with t32g electron configuration in (pseudo-octahedral) transition metal complexes are essentially independent of Δo.8,43,44 Their energies rather depend on the electron–electron repulsion as described by the Racah parameters B and C (with C being often approximated as multiple of B) in ligand field theory.8,15,16 Qualitatively, these parameters can be related to the nephelauxetic (cloud-expanding) effect which describes the covalent vs. ionic character of the bonds between the metal and the ligands.5,8 The expansion of the orbitals onto the ligands reduces the repulsion between individual d electrons and hence gives in principle a synthetic handle to modify SF state energies, although straightforward design rules are lacking.45–52
With pyridine and amine based ligands, chromium(III) complexes typically emit between ca. 680–780 nm (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700–12820 cm−1).1 π-Electron donating amido (carbazolato or isoindolinato) donors46,50 and cyclometalating ligands45,47 shift the ruby-like luminescence above 900 nm (Scheme 1).1 Apart from changing the donors from amines/pyridines to anionic donors, even the bridging unit between coordinating pyridines affects the SF energies. In particular, the highly emissive polypyridyl chromium(III) complexes [Cr(ddpd)2]3+ ([1NMe]3+) and [Cr(bpmp)2]3+ ([1CH2]3+) (molecular rubies) with NMe and CH2 bridges emit at 775/738 and 709 nm, respectively, although both complexes share a very similar six-fold pyridine coordination environment (Scheme 1).24,29 While the correct energy level ordering of the two complexes could be predicted by high-level quantum chemical calculations, the underlying origin of the 1200 cm−1 emission energy difference remained unclear.29 Under hydrostatic pressure, the emission energy of [1NMe]3+ shifts by 13.9 ± 0.9 cm−1 kbar−1 to lower energy slightly depending on the environment.33 Compared to the pressure sensitivity of the 2E emission of Al2O3:Cr with 0.7 cm−1 kbar−1, this sensitivity is much more pronounced.53 A compression of the Cr–N bonds and a co-planarisation of the central pyridine rings (Pyc) of the tridentate ligands leads to better overlap with ligand orbitals and thus to “cloud expansion”.54 The lowest doublet state is of 2T1 parentage with two paired electrons in a t2g-derived orbital showing the larger shift under pressure as compared to the classical lowest doublet states of 2E parentage with singly occupied t2g orbitals.1,8,9
700–12820 cm−1).1 π-Electron donating amido (carbazolato or isoindolinato) donors46,50 and cyclometalating ligands45,47 shift the ruby-like luminescence above 900 nm (Scheme 1).1 Apart from changing the donors from amines/pyridines to anionic donors, even the bridging unit between coordinating pyridines affects the SF energies. In particular, the highly emissive polypyridyl chromium(III) complexes [Cr(ddpd)2]3+ ([1NMe]3+) and [Cr(bpmp)2]3+ ([1CH2]3+) (molecular rubies) with NMe and CH2 bridges emit at 775/738 and 709 nm, respectively, although both complexes share a very similar six-fold pyridine coordination environment (Scheme 1).24,29 While the correct energy level ordering of the two complexes could be predicted by high-level quantum chemical calculations, the underlying origin of the 1200 cm−1 emission energy difference remained unclear.29 Under hydrostatic pressure, the emission energy of [1NMe]3+ shifts by 13.9 ± 0.9 cm−1 kbar−1 to lower energy slightly depending on the environment.33 Compared to the pressure sensitivity of the 2E emission of Al2O3:Cr with 0.7 cm−1 kbar−1, this sensitivity is much more pronounced.53 A compression of the Cr–N bonds and a co-planarisation of the central pyridine rings (Pyc) of the tridentate ligands leads to better overlap with ligand orbitals and thus to “cloud expansion”.54 The lowest doublet state is of 2T1 parentage with two paired electrons in a t2g-derived orbital showing the larger shift under pressure as compared to the classical lowest doublet states of 2E parentage with singly occupied t2g orbitals.1,8,9
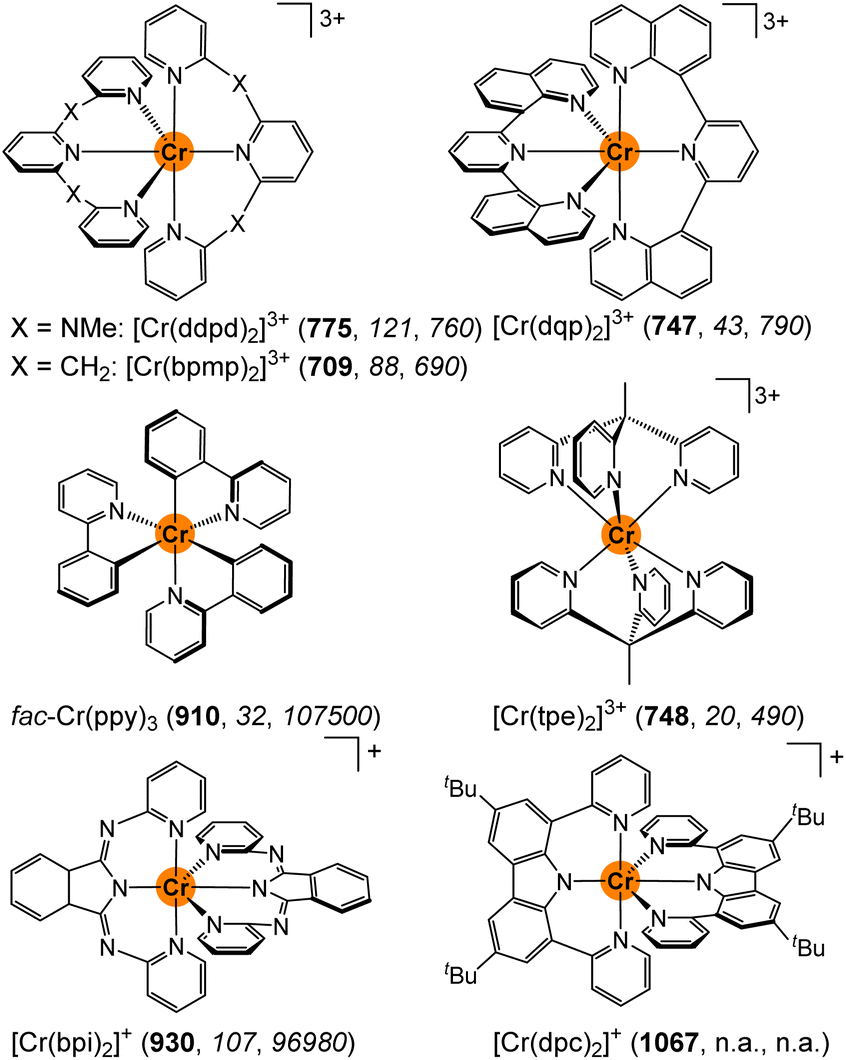 | ||
| Scheme 1 Molecular structures of [Cr(ddpd)2]3+ (ddpd = N,N′-dimethyl-N,N′-dipyridine-2-yl-2,6-diamine),24,25 [Cr(bpmp)2]3+ (bpmp = 2,6-bis(2-pyridylmethyl)pyridine),29 [Cr(dqp)2]3+ (dqp = 2,6-di(quinoline-8-yl)pyridine),28fac-Cr(ppy)3 (Hppy = 2-phenylpyridine),47 [Cr(tpe)2]3+ (tpe = 1,1,1-tris(pyrid-2-yl)ethane),27 [Cr(bpi)2]+ (Hbpi = 1,3-bis(2′-pyridylimino)-isoindoline),50 and [Cr(dpc)2]+ (Hdpc = 3,6-di-tert-butyl-1,8-di(pyridine-2-yl)-carbazole),46 along with their emission wavelengths (λem/nm) in bold and the radiative and non-radiative rate constants (kr/s−1, knr/s−1) in italics in solution at room temperature (n.a. = not available). | ||
Beyond the energies of the emissive SF states, the excited state dynamics are of particular importance for the overall performance of a SF luminescent complex. This includes the efficiency of the population of the doublet states via intersystem crossing (ISC),55–57 as well as the radiative and non-radiative decay to the ground state (kr and knr). The radiative rate constant kr of the Laporte-forbidden58 emission depends on the symmetry of the complex, with centrosymmetric complexes such as [Cr(tpe)2]3+ (Scheme 1) or [Cr(CN)6]3– exhibiting very small radiative rate constants down to 20 and 0.4 s−1, respectively.1,27
Several processes contribute to the non-radiative rate knr. One reason for the small knr is the comparably large Δo at the Franck-Condon geometry which likely mitigates back-ISC from the SF states to the non-emissive 4T2 states with t22ge1g electron configuration. However, the relaxed 4T2 states are strongly Jahn–Teller distorted and experience a huge stabilisation upon distortion.59–61 Hence, for the excited state decay via back-ISC, distortional coordinates play major roles in addition to Δo at the ground state geometry. These back-ISC dynamics could be severely affected by subtle changes in the complex geometry and ligand properties, yet this has not been fully appreciated in the literature of molecular chromium(III) emitters. For ISC in general and the 2E/2T1 → 4T2 back-ISC processes in particular, spin–orbit coupling (SOC) and/or spin-vibronic coupling is required.55,56
A further non-radiative decay path of low-energy emitters is provided through overtones of nearby CH groups as energy acceptors.62,63 This multiphonon relaxation depends on the distance of the CH oscillators to the metal confined excited state wave function and the spectral overlap integral of the luminescence bands with absorption bands of CH overtones.62,63 For most chromium(III)-based SF emitters with pyridine ligands, the 4th aromatic CH overtone of a pyridine ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) CH4 = 14065 cm−1 is in the region of the emission energy providing a non-radiative decay path.25
CH4 = 14065 cm−1 is in the region of the emission energy providing a non-radiative decay path.25
In this study, we shed light on the specific factors that (i) determine the SF state energies and (ii) the population and decay (knr) of the SF states of molecular rubies. To this end, we expand the complex series [1NMe]3+ and [1CH2]3+ by two isolobal complexes with chalcogen bridges [1O]3+ and [1S]3+. Elucidation of the ground state geometries by X-ray diffraction (XRD) analyses and excited state properties by UV/Vis/NIR absorption and emission spectroscopy is combined with high level quantum chemical modelling ((time-dependent) density functional theory, (TD)DFT and multi-reference, CASSCF-SC-NEVPT2). The excited state dynamics are probed by fs-transient absorption (TA) spectroscopy, variable-temperature (VT) emission spectroscopy, photolysis experiments and quantum chemical modelling of excited states. The combined information draw a consistent picture of excited state energies and dynamics of molecular rubies and informs about future design strategies to tune the thermodynamics and kinetics after light excitation.
2 Results and discussion
2.1 Syntheses, structures and ground state properties
The known tridentate pyridyl thioether ligand bptp (2,6-bis(pyridine-2-ylthio)pyridine)64,65 was prepared from 2-mercaptopyridine in 79% yield. Crystals suitable for single crystal XRD were obtained by crystallisation at low temperature. Expectedly, the C–S–C angle is with 102.7° much smaller than the angles observed at CH2, NMe and O bridged pyridines with 111.7°, 122.9° and 120.3°, respectively66–68 (ESI, Fig. S1†). The analogous synthetic route for the pyridyl ether ligand 2,6-bis(pyridine-2-yloxy)pyridine (bpop) starting from 2-hydroxypyridine (2-pyridone), however, yields mainly 2,6-bis(2-oxopyridin-1(2H)-yl)pyridine69 due to the lower nucleophilicity of oxygen compared to sulphur and the higher resonance contribution of the keto form of the deprotonated 2-pyridone compared to its enolate form.70,71 After extensive optimisation using various bases and solvents at different temperatures, the oxygen bridged ligand bpop could be obtained in low yields via substitution of 2,6-dihydroxypyridine with 2-bromopyridine besides 2,6-bis(2-oxopyridin-1(2H)-yl)pyridine as the major product. Both tripyridine pincer ligands bpop and bptp are fully characterised by elemental analyses, ESI+ mass spectrometry (ESI, Fig. S2–S4†), as well as 1H/13C NMR (ESI, Fig. S5–S15†), IR (ESI, Fig. S16–S18†) and optical spectroscopy (ESI, Fig. S19–S21†).The orange complex salts [Cr(bpop)2][OTf]3[1O][OTf]3 and [Cr(bptp)2][OTf]3[1S][OTf]3 were prepared in 24% and 31% isolated yields, respectively, by heating an acetonitrile solution of anhydrous chromium(III) triflate (see ESI† for a convenient route and characterisation, Fig. S22–S24†)72 and the corresponding ligand (Scheme 1). The purity and composition of the complexes are confirmed by elemental analyses, ESI+ mass spectrometry (ESI, Fig. S25 and S26†), IR spectroscopy (ESI, Fig. S27 and S28†), optical spectroscopy (ESI, Fig. S29–S32†) and electrochemistry (ESI, Fig. S33–S36†).
Electrochemical experiments reveal irreversible reduction waves at Ep = −0.48, −1.91 V and −0.40, −1.52 V vs. ferrocene for [1O]3+ and [1S]3+, respectively (ESI, Fig. S33–S36†), while [1NMe]3+ and [1CH2]3+ are reversibly reduced at −1.11 V and −0.81 V vs. ferrocene.24,29 DFT calculations on the dications [1O]2+ and [1S]2+ and monocations [1O]+ and [1S]+ suggest a metal centered first reduction event forming chromium(II) species followed by a second reduction with significant ligand localisation. As chromium(II) complexes are typically very labile,73,74 ligand loss or partial ligand dissociation leading to solvent coordination similar to analogous ruthenium(II) complexes might account for the irreversibility of the reduction processes.65
Single crystals of the complex salts [1O][OTf]3 and [1S][OTf]3 suitable for XRD were obtained from acetonitrile solutions at 5 °C confirming the constitution and meridional configuration (Fig. 1, ESI, Fig. S37, S38 and Tables S1, S2†). The [CrN6] core of the complexes is highly octahedral with N–Cr–N bond angles close to 90°/180° and almost uniform Cr–N bond lengths analogous to [1NMe][BF4]3 (CCDC 1059802†) and [1CH2][OTf]3 (CCDC 1989537†).24,29 Distinct differences are apparent in the four complex cations [1X]3+, which arise from the different bridging units X. The sp3-atom bridging units X = NMe, O, CH2, S with their averaged C–X–C angles decreasing from 122°, 121°, 115° to 104° are responsible for the chelate ring conformations and the pyridine orientations.
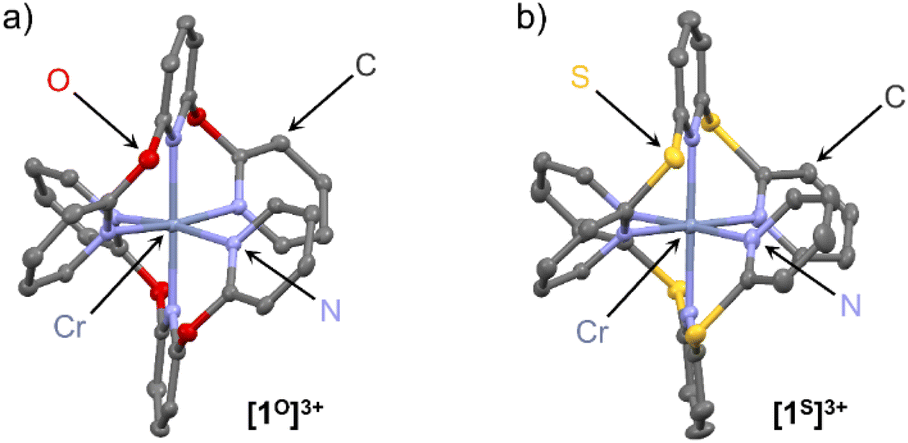 | ||
| Fig. 1 Molecular structures of the trications of the chromium complexes (a) [1O][OTf]3 and (b) [1S][OTf]3 determined by XRD. Hydrogen atoms, counter ions and co-crystallised solvent molecules omitted. Thermal ellipsoids at 50% probability level. | ||
For a detailed structure description, we denote the atoms of two tridentate ligands LX of [1X]3+ with (1) and (2) shown in red and blue, respectively (Scheme 2). The atoms of the central and terminal pyridines LX are denoted with subscripts c and t, respectively (Scheme 2). The averaged Cr–Nt distances dCrNt (Table S1†) increase from 2.042(4), 2.047(6) to 2.075(3)/2.076(3) Å for X = NMe, O, S, CH2. The averaged Cr–Nc distances dCrNc increase from 2.038(3), 2.038(4), 2.069(2) to 2.094(2) Å for X = NMe, O, CH2, S. The overall size of the [CrN6] coordination polyhedron thus increases in the series [1X]3+ with X = NMe, O, CH2 and S. The Nt atoms of the tridentate ligands in [1O]3+ and [1NMe]3+ form Nt(1)–Cr–Nt(1) angles α (Table S1†) below 180° (α = 172.6(2) and 171.9(1)°), while α of [1S]3+ and [1CH2]3+ is closer to 180° (α = 177.8(1) and 177.6(1)°). The smallest Nt(1)–Cr–Nt(2) angle β between the Nt atoms of the two different ligands (Table S1†) is close to 90° for [1O]3+ and [1NMe]3+ (β = 89.7(2) and 89.8(1)°), but below 90° for [1S]3+ and [1CH2]3+ (β = 85.3(1) and 84.9(1)°). The angles between the planes of the terminal pyridines within a ligand are denoted by the torsion angles δ1/δ2 Calpha–Nt(1)–Nt(1)–Calpha and Calpha–Nt(2)–Nt(2)–Calpha, respectively (Table S2†). These angles δ1 and δ2 increase from 73.5/69.8°, 72.3/71.2°, 79.1/80.6° to 81.7/86.3° for X = O, CH2, NMe and S approaching a more orthogonal orientation. The angles between the planes of the central pyridines of the two ligands ϕ, described by the torsion angle Calpha–Nc(1)–Nc(2)–Calpha (Table S2†), decrease from 29.5°, 21.8°, 18.5° to 18.3° for X = CH2, O, S, NMe. The structural parameters dCrN, α and β of the coordination polyhedron and the orientation of the pyridines δ1,2 and ϕ serve to identify the most relevant structural aspects for the SF state energies in the quantum chemical modeling. The optical properties and the excited state dynamics of the complexes will be discussed next.
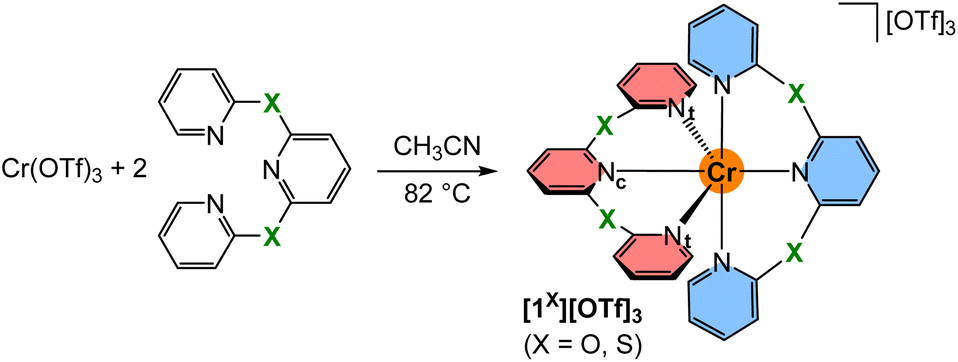 | ||
| Scheme 2 Synthesis of chromium(III) complexes with tripyridine ligands with chalcogen atoms X = O, S in the six-membered chelate rings. Coordinating terminal and central pyridine nitrogen atoms are denoted Nt and Nc, respectively. The two tridentate ligands (1) and (2) are highlighted in red and blue, respectively. | ||
2.2 Excited state energies
To understand the optical properties of the complexes, we first describe the properties of the chalcogen bridged pyridine ligands bpop and bptp (ESI, Fig. S19–S21†). The S0 → S1 absorption bands at λmax = 267/308 nm and the corresponding broad S1 → S0 fluorescence bands at λemis = 307/349 nm show large Stokes shifts of 4880 and 3810 cm−1, respectively. This agrees with the X → pyridine CT character of the S1 state assigned by TDDFT calculations (ESI, Fig. S19 and S20†) and the higher-energy sulphur lone pairs as compared to the oxygen lone pairs.In the absorption spectra of the chromium(III) complexes [1O]3+ and [1S]3+, high intensity bands of 4LMCT/4(π–π*) character, weak spin-allowed, Laporte-forbidden 4A2 → 4T2 bands and very weak spin- and Laporte-forbidden 4A2 → 2T1/2E bands are present in the UV, visible and NIR spectral regions, respectively (Fig. 2; ESI, Fig. S29 and S30†). The 4A2 → 4T2 bands of [1O]3+ appear isolated according to TDDFT calculations (ESI, Table S3†) at 463 nm with ε = 100 M−1 cm−1 (Fig. 2a), similar to 4A2 → 4T2 bands of [1CH2]3+ (465 nm; ε = 70 M−1 cm−1; ESI, Fig. S39†).29 In contrast, the analogous dd band of [1S]3+ is hidden by low energy S → Cr LMCT transitions according to TDDFT calculations (ESI, Table S4†), which increases the intensity of this band at 452 nm (Fig. 2b; ε = 1880 M−1 cm−1). This is analogous to the NMe → Cr LMCT transition in [1NMe]3+ at around 435 nm (ε = 4100 M−1 cm−1) overlapping the 4A2 → 4T2 dd transitions (ESI, Fig. S40†).9,24
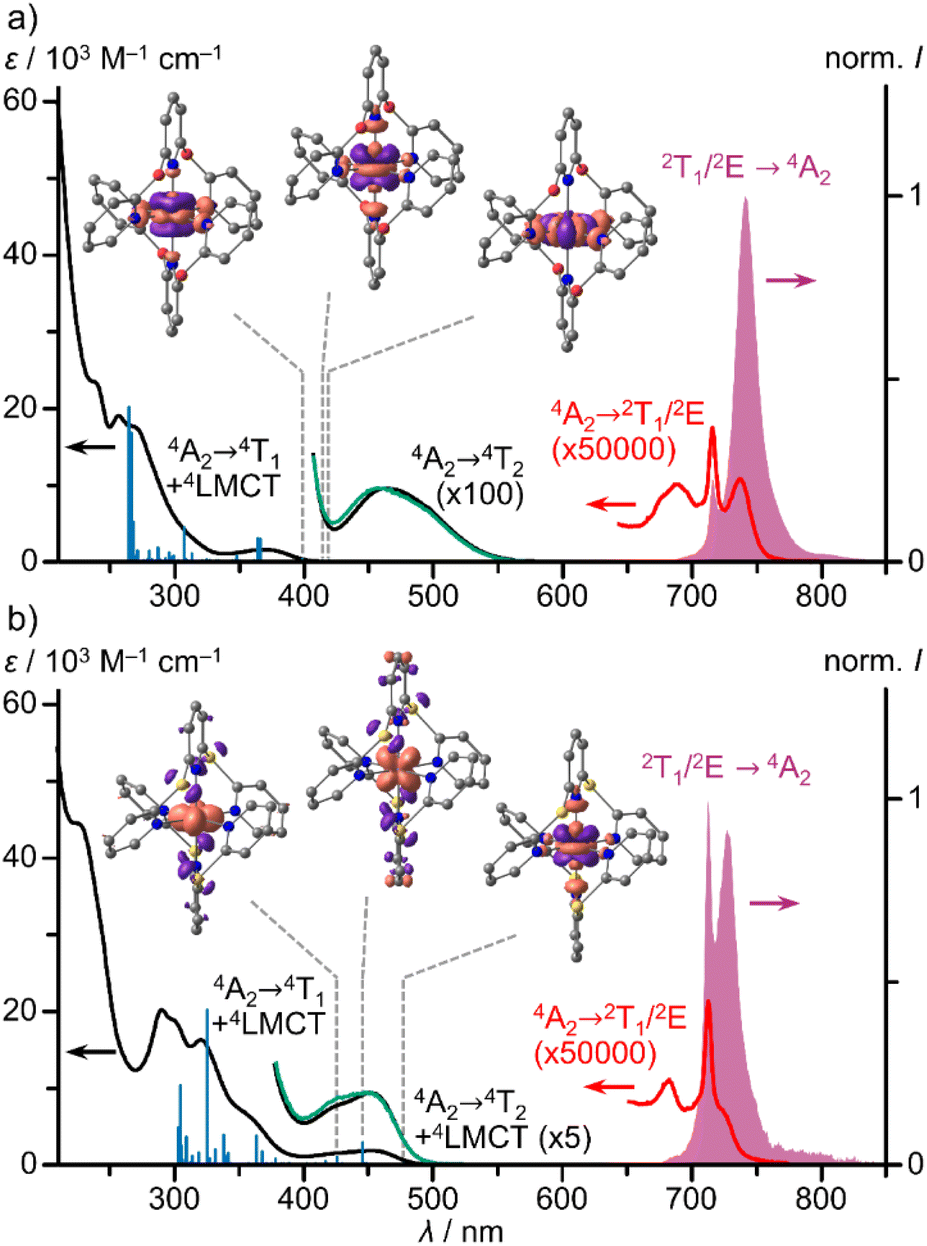 | ||
| Fig. 2 UV/Vis/NIR absorption spectra (black, red), excitation spectra (λem = 714 and 742 nm, green) and emission spectra (λexc = 450 nm, purple) of (a) [10]3+ and (b) [1S]3+ in deaerated acetonitrile at room temperature, TDDFT calculated oscillator strengths (blue sticks) and difference electron densities of three low-energy transitions of 4LMCT and 4MC character. The regions of the spin-forbidden absorption bands, the lowest energy spin allowed/LMCT absorption band and the excitation spectrum are scaled with the indicated factors. | ||
Due to the superimposed LMCT bands and the symmetry deviating from octahedral, the ligand field splitting Δo can only roughly be estimated from the dd bands as 21510, 21600, 22120 and 22990 cm−1 for [1X]3+ with X = CH2, O, S and NMe, respectively. For a more reliable ordering of the complexes in a spectrochemical series we resorted to quantum chemical calculations. TDDFT calculated lowest energy 4A2 → 4T2 transitions at the optimised geometries, which can be assigned to the ligand field splitting Δo, increase from 20960, 23120, 23790 to 23870 cm−1 for [1X]3+ with X = S, CH2, NMe and O, respectively. CASSCF(7,12)-SC-NEVPT2 calculations at the same geometries deliver 20290, 21740, 22660 and 23220 cm−1 for [1X]3+ with X = S, CH2, O and NMe, respectively (ESI, Table S5, Fig. S41†). Hence, we suggest a weaker ligand field in the [1S]3+ derivative, a medium field in [1CH2]3+ and stronger fields in the [1O]3+/[1NMe]3+ complexes. Yet, all ligand field strengths can be classified as very strong.
The NIR spectral region shows a characteristic absorption band pattern consisting of three weak bands with discernible maxima at 689, 716, 737 nm and at 683, 713, 724 (sh) nm for [1O]3+ and [1S]3+, respectively. Similar absorption band patterns of 697, 736, 771 nm and 674, 699, 706 nm were found for [1NMe]3+ and [1CH2]3+, respectively (Fig. 3a, ESI, Fig. S39 and S40†).24,29
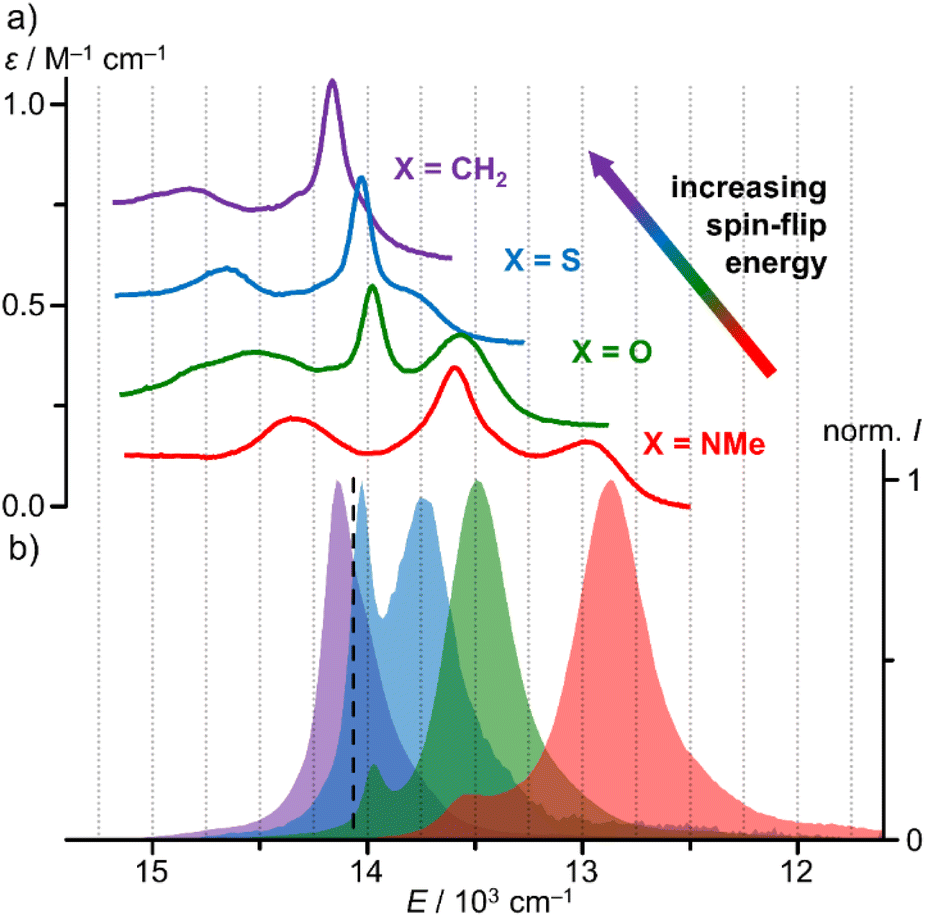 | ||
| Fig. 3 (a) NIR absorption spectra and (b) normalised emission spectra of [1CH2]3+ (purple, λexc = 460 nm), [1S]3+ (blue, λexc = 450 nm), [1O]3+ (green, λexc = 450 nm) and [1NMe]3+ (red, λexc = 435 nm) in CH3CN at room temperature. For clarity, the absorption spectra of [1X]3+ with X = O, S, CH2 were shifted vertically by +0.2, +0.4 and +0.6 M−1 cm−1. The dashed black line marks the energy of the 4th pyridine CH overtone CH4.25 | ||
As the NIR absorption bands correspond to five SF transitions (2E, 2T1 in octahedral symmetry), we fitted the spectral patterns with five Voigt functions each (after baseline correction as described in the ESI; Fig. S42–S45†). Indeed, these five Voigt functions excellently reproduce the experimental band patterns. The data is compiled in Table 1.
| No. | #5 | #4 | #3 | #2 | #1 | ΔE(#5–#1) | ΔE(#2–#1) |
|---|---|---|---|---|---|---|---|
| a This is the band with a smaller experimental full width at half maximum (FWHM). | |||||||
| X = CH 2 | |||||||
| Exp. | 15060 |
14780 |
14320 |
14160 |
14050 |
1010 | 110 |
| Calcd |
14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 660 660
|
14640
|
14430
|
14140
|
13970
|
690 | 170 |
| Term | 2T1(3) | 2E(2) | 2T1(2) | 2E(1)a | 2T1(1) | ||
|
|||||||
| X = S | |||||||
| Exp. | 14770 |
14630 |
14210 |
14030 |
13800 |
970 | 230 |
| Calcd |
14750
|
14720
|
14500
|
14200
|
14000
|
750 | 200 |
| Term | 2T1(3) | 2E(2) | 2T1(2) | 2E(1)a | 2T1(1) | ||
|
|||||||
| X = O | |||||||
| Exp. | 14780 |
14520 |
14360 |
13980 |
13560 |
1220 | 420 |
| Calcd |
14510
|
14420
|
14160
|
14000
|
13590
|
920 | 410 |
| Term | 2E(2) | 2T1(3) | 2T1(2) | 2E(1)a | 2T1(1) | ||
|
|||||||
| X = NMe | |||||||
| Exp. | 14430 |
14300 |
13600 |
13530 |
12960 |
1470 | 570 |
| Calcd |
14400
|
14250
|
13880
|
14180
|
13400
|
1000 | 480 |
| Term | 2E(2) | 2T1(3) | 2E(1)a | 2T1(2) | 2T1(1) | ||
From the CASSCF(7,12)-SC-NEVPT2 calculations of all [1X]3+ complexes at their respective DFT-optimised ground state geometries we obtained the five lowest doublet state energies (ESI, Table S5, Fig. S41†). With a common scaling factor of 0.89 these energies excellently fit to the experimentally derived energies allowing a detailed assignment of the individual states (Table 1). Importantly, the calculations reproduce the experimental nephelauxetic series of the complexes [1X]3+ with X = CH2, S, O and NMe derived from the SF energies, the increased lifting of the degeneracies ΔE(#5–#1) in the series CH2/S < O < NMe and the energy difference of the two lowest energy SF absorptions ΔE(#2–#1) (Table 1). The very sharp central band (doublet state #3 for X = NMe, #2 for the other complexes) reflects a miniscule geometric distortion of an excited state and can most likely be assigned to a (0,0) transition of a nested, i.e. undistorted, microstate of 2E character.1,8 The tentative assignment of the SF state #2 as 2E(1) is confirmed by the (7,12)-SC-NEVPT2 calculations (Table 1). Based on these agreements, we attest a high fidelity to the quantum chemical calculations and assignments. In all cases, the lowest doublet states #1 and #2 are of 2T1 and 2E character, respectively.
Irradiation of a solution of [1O]3+ and [1S]3+ at room temperature in acetonitrile with λexc = 450 nm gives rise to two sharp emission bands peaking at 716/741 nm and 713/727 nm with FWHM of 140/350 and 150/420 cm−1, respectively (Fig. 3b, ESI, Fig. S31†). The energies of the two phosphorescence bands of all complexes [1X]3+ match the energies of the two lowest-energy absorption bands (Fig. 3; ESI, Fig. S39 and S40†).9,24,29 Small Stokes shifts of 80, 60, 70 and 70 cm−1 are observed for the lowest energy emission band (2T1(1) → 4A2), while even smaller Stokes shifts <40 cm−1 are determined for the second lowest energy emission band (2E(1) → 4A2) in agreement with slightly distorted 2T1(1) states and almost perfectly nested 2E(1) states (Fig. 3).
Doublet states of 2E and 2T1 parentage could be localised by excited state geometry optimization via TDDFT. The Cr–N distances dCrN slightly decrease from dCrNc/dCrNt = 2.051/2.071 Å (4A2 ground state) to 2.044/2.064 Å (2E) and 2.042/2.057 Å (2T1) for X = O and from dCrNc/dCrNt = 2.140/2.118 Å (4A2 ground state) to 2.103/2.087 Å (2E) and 2.098/2.079 Å (2T1) for X = S. In both cases, the compression is larger in the 2T1(1) excited states than in the 2E(1) states. This further confirms that the broader low-energy bands with a larger Stokes shifts arise from the slightly more distorted 2T1(1) states.
In order to correlate structural effects of the hexapyridine chromium(III) complexes with their doublet state energies, we first disentangle primary electronic from secondary structural effects (dCrN, α, β, δ1,2 and ϕ) of the bridging unit X. In a first series of calculations, the bridging unit was substituted by a different bridge while retaining the geometry (ESI, Fig. S46, Table S6†). For example, the NMe unit of [1NMe]3+ was replaced by X = O and CH2, while the original geometry of [1NMe]3+ was retained. According to the CASSCF(7,12)-SC-NEVPT2 calculations, NMe → O replacement increases the SF energy, while NMe → CH2 replacement decreases it (ESI, Fig. S46, Table S6†). The latter is at odds with the experiment, suggesting that electronic substituent effects (at the ortho positions) alone do not correctly describe the SF energy shifts. Furthermore, the energy variation at fixed geometries but with different X bridges is smaller than the energy variation due to geometric distortions (ESI, Fig. S46†). Hence, the indirect effects of structural modification (dCrN, α, β, δ1,2 and ϕ) exerted by the bridge X are more relevant for the SF energies than primary electronic effects.
To explore the effects of the geometry on the SF energies, we employed a model system [Cr(py)6]3+ where contributions from the bridging atoms are excluded (Scheme 3). The parameters dCrN, α, β, δ and ϕ were varied within the value ranges as determined from XRD analyses and DFT optimisations and also extrapolated to smaller and larger values (ESI, Fig. S47–S57†). Within this set of parameters, decreasing the Cr–N distances dCrNc = 2.140 → 2.046 Å and dCrNt = 2.118 → 2.058 Å shifts the SF energies to lower energy by 225 and 228 cm−1, respectively, at fixed α, β, δ and ϕ angles in the calculations (Fig. S58†). Hence, symmetric compression of the [CrN6] coordination polyhedron in local D4h symmetry results in bathochromic shifts of the SF energies. This bathochromic shift is also observed for ruby53 and molecular rubies under pressure (ESI, Fig. S48–S51†).35,54 Shorter Cr–N bonds enhance the covalency of the bonds, thereby increasing the electron delocalization and hence the nephelauxetic effect resulting in lower SF energies. Expectedly the energetic order of the five individual SF microstates is essentially unaffected by the symmetrical Cr–N modes in the observed distance range. Decreasing the angle α in the model [Cr(py)6]3+ (ESI, Fig. S52†) from 179° (as found in [1S]3+) to 173° (as found in [1NMe]3+) at fixed Cr–N distances and ϕ angles (β and δ had to be adjusted in order to avoid close H⋯H contacts) lowers the SF energy by 75 cm−1 (ESI, Fig. S58†). This unsymmetrical distortion also changes the 2T1/2E contributions to the individual microstates. Expansion of β from 84° (as found in [1CH2]3+) to 90° (as found in [1O]3+) lowers the SF energy by 28 cm−1 (ESI, Fig. S58†). The torsional modes δ (ESI, Fig. S54 and S55†) and ϕ (ESI, Fig. S56 and S57†) exert weaker effects. Creating a less orthogonal orientation of terminal pyridines, i.e. adjusting δ = 83 → 68° ([1S]3+ → [1O]3+) and more co-planar orientations of the central pyridines, i.e. adjusting ϕ = 26 → 17° ([1CH2]3+ → [1S]3+) leads to very small bathochromic shifts of 9 and 5 cm−1, respectively (ESI, Fig. S58†).
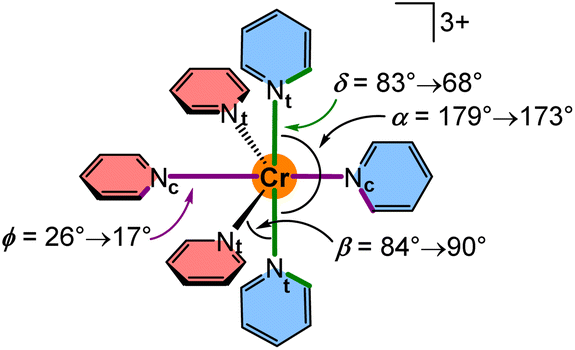 | ||
| Scheme 3 Structure of the model system [Cr(py)6]3+ and the definition and relevant ranges of the bond angles α/β and the dihedral angles δ/ϕ. | ||
Consequently within the present complex series [1X]3+, the bridging unit X indirectly affects both the average energy of the 2T1/2E manifold and the relative energies of the five doublet microstates with 2T1 and 2E parentage. Bond compression exerts the largest bathochromic effect, followed by lowering the Nt–Cr–Nt bite angle α, although the angular modifications are not independent from each other in the chelate complexes. Having assigned the character of the lowest energy excited states and the structural origins of the relative energies, we now turn to the excited state dynamics.
2.3 Excited state dynamics
Excitation spectra of [1O]3+ and [1S]3+ (λem = 742 and 714 nm) closely follow the respective absorption spectra in the region of the lowest spin-allowed 4A2 → 4T2 and 4LMCT transitions (ca. 380–600 nm, Fig. 3). This substantiates that both types of quartet states efficiently populate the SF states via ISC without branching to other decay channels.Femtosecond TA spectroscopy on [1O]3+ and [1S]3+ confirms that the vibrationally cold doublet states are populated within τ1,τ2 = 1.0, 47 ps and τ1,τ2 = 1.2, 90 ps (ESI, Fig. S59–S64†) after excitation at 343 nm ([1O]3+) and 515 nm ([1S]3+), respectively. In both complexes [1O]3+ and [1S]3+, ISC appears to occur on an ultrafast time scale. This rapid ISC likely arises from the high density of doublet states both for 4LMCT ([1O]3+) and 4MC/4LMCT ([1S]3+) excitation. For example the 2T2 derived states are close in energy to the 4T2 states according to CASSCF calculations (ESI, Table S6, Fig. S41†). No loss channels are apparent on the fast time scale and hence the decisive non-radiative and radiative decay occurs from the lowest energy SF states, similar to [1NMe]3+ and [1CH2]3+.24,29
The photoluminescence quantum yield Φ = 11.5% and SF excited state lifetime τP = 836 μs of [1O]3+ in deaerated CH3CN are in very high ranges, similar to the record values of [1NMe]3+ and [1CH2]3+.24,29 The observed excited state lifetime corresponds to the common lifetime of the equilibrating emissive lowest energy doublet states 2T1(1) and 2E(1).31 The radiative and non-radiative rate constants kr = 138 s−1 and knr = 1059 s−1 are similar to the values of [1NMe]3+ and [1CH2]3+ (Scheme 1). On the other hand, the values of the sulphur derivative [1S]3+ are lower by orders of magnitude with Φ = 0.01% and τP = 1.65 μs. This gives a somewhat smaller kr = 61 s−1, while the non-radiative rate constant knr = 606000 s−1 increased by orders of magnitude.
As this huge difference of knr is completely unexpected in light of the similar ground state geometries and excited state energies, we investigated the excited decay of [1O]3+ and [1S]3+ by variable-temperature (time-resolved) emission spectroscopy in order to identify thermally accessible loss channels. Emission spectra and lifetimes of [1O]3+ and [1S]3+ were determined between 293 and 77 K in ethanol/methanol (3:2 v/v) solution, which freezes around 130 K (Fig. 4).
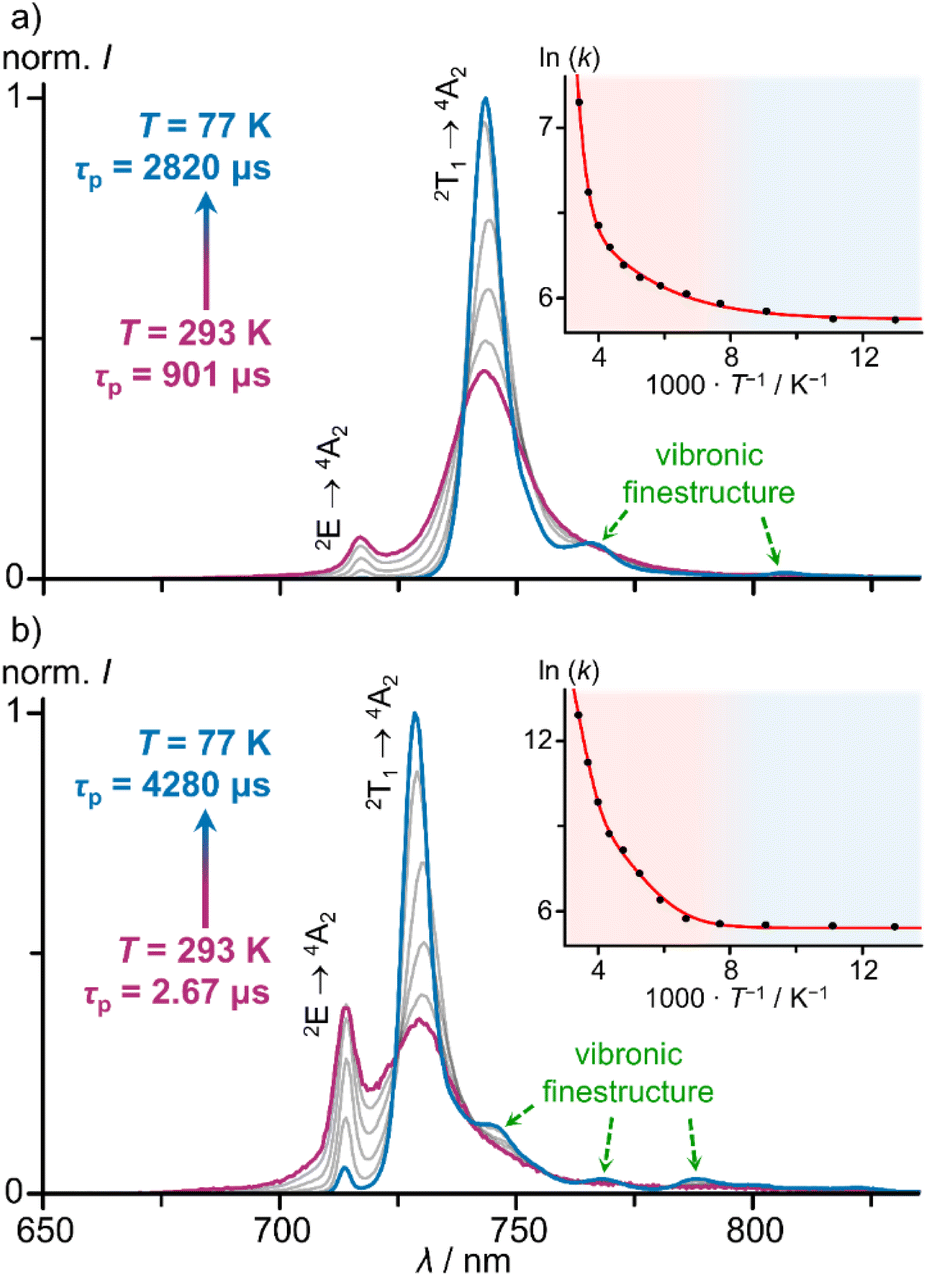 | ||
| Fig. 4 Integration area normalised low temperature emission spectra (λexc = 450 nm) of (a) [10]3+ and (b) [1S]3+ in a mixture of ethanol/methanol (3:2 v/v) between 77 and 293 K under deaerated conditions. Insets show corresponding Arrhenius plots of the photoluminescence rate constants k(T) with fit and coloured backgrounds for temperature ranges of liquid (red) and frozen solvent mixture (blue). | ||
Temperature-dependent emission spectra clearly reveal thermal population of the two lowest energy SF states for all complexes under study and even population of the next higher energy SF state #3 2T1(2) to a small degree (ESI, Fig. S65–S68†). At lower temperatures, the lowest SF state 2T1(1) is increasingly populated at the expense of the other SF states. The 2T1(1) → 4A2 emission bands develop vibrational fine structure at lower temperature with maxima centred at 13050, 12390, 12210, 11830 cm−1 and 13440, 13020, 12690, 12480, 12150 cm−1 for [1O]3+ and [1S]3+, respectively (ESI, Fig. S65 and S66†). Similar vibrational progressions have been reported for [1NMe]3+ and [1CH2]3+ (ESI, Fig. S67 and S68†).24,29 The energy differences are compatible with Cr–N vibrational modes.75
Cooling the solution of [1O]3+ from 293 K to 77 K sharpens the low-energy 2T1(1) → 4A2 band (FWHM = 350 → 150 cm−1) and increases the lifetime τP = 901 → 2820 μs (Fig. 4a; ESI, Fig. S69†). At 250 K, the quantum yield reaches approximately Φ = 22%. Similarly, cooling a solution of [1S]3+ from 293 K to 77 K sharpens the low-energy 2T1(1) → 4A2 band (FWHM = 420–140 cm−1) and drastically increases the lifetime τP = 2.67 → 4280 μs (Fig. 4b, ESI, Fig. S70†). Concomitantly, the integrated luminescence intensity increases 900-fold, i.e. approaching approximate values of Φ = 9% at 77 K (assuming a temperature independent absorbance at the excitation energy).
The thermally activated decay of [1O]3+ and [1S]3+ was successfully modelled with an Arrhenius-like behaviour according to eqn (1) (Fig. 4). The photoluminescence rate constants k(T) = 1/τp(T) of the complexes (ESI, Fig. S69 and S70†) were fitted as a sum of a T-independent rate constant k0, describing non-thermally activated radiative and non-radiative processes, and two T-dependent rate constants k1(T) and k2(T) (eqn (1)).
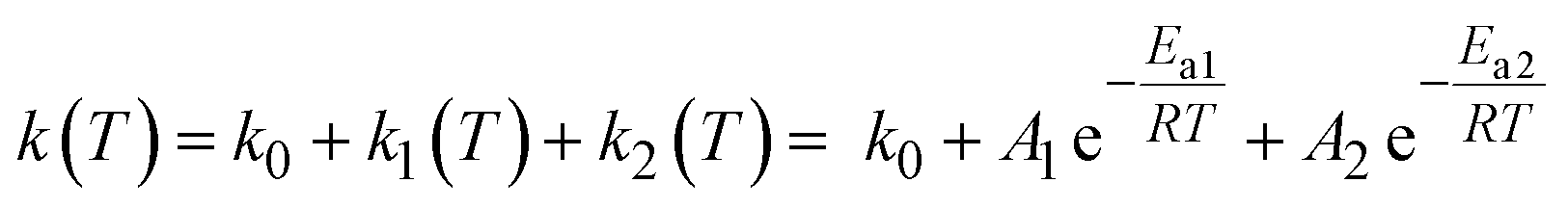 | (1) |
Two thermally activated processes (A1, Ea1; A2, Ea2) were required to satisfactorily fit the experimental data (Table 2). k1 (A1, Ea1) and k2 (A2, Ea2) dominate the high and low temperature regions, respectively.
| [1O]3+ | [1S]3+ | |
|---|---|---|
| k 0/s−1 | 356 | 225 |
| E a1/cm−1 (eV) | 3360 (0.417) | 4320 (0.536) |
| E a2/cm−1 (eV) | 370 (0.046) | 1090 (0.135) |
| A 1/s−1 | 9.18 × 109 | 6.27 × 1014 |
| A 2/s−1 | 1.80 × 103 | 4.65 × 106 |
Based on the two experimentally determined barriers Ea1 and Ea2 for thermally activated decay pathways with high and small frequency factors A1 and A2 (Table 2), we now propose a kinetic model with physical assignments to the processes. The larger barriers Ea1 of 3360 and 4320 cm−1 for [1O]3+ and [1S]3+ are likely associated with the thermally activated back-ISC from the 2T1(1)/2E(1) levels to the quartet levels, possibly along several conceivable Jahn–Teller modes associated with the 4T2 or 4LMCT states similar to 3MC states of d6 metal complexes such as [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine).76–79
Excited state geometry optimisations of the complex cations by TDDFT (ESI, Table S7†) delivered quartet excited states with 4T2 character and one or two elongated Cr–Nc distances. The symmetric Nc–Cr–Nc breathing elongation amounts to 9% and 6–8% for [1O]3+ and [1S]3+, while the more asymmetric Cr–Nc elongation reaches 22% and 13% for [1O]3+ and [1S]3+, respectively. Asymmetric 4T2 states were also localised for X = [1CH2]3+ and [1NMe]3+ where a single Cr–Nc bond is elongated by 20% and 13%, respectively. In addition to the distorted metal-centered 4T2 states of [1S]3+, two distorted 4LMCT states are located at similar energies. One optimised 4LMCT coordination polyhedron is elongated along the symmetric Nc–Cr–Nc breathing mode (10–15% elongation) and the other optimised state exhibits a pincer-like deformation with a 14% increase of two Cr–Nt bond lengths of a single bptp ligand. In essence, all complexes [1X]3+ possess low-energy distorted metal-centered quartet states, while [1S]3+ additionally possesses distorted low-energy 4LMCT states with significant S → Cr CT character.
The experimental barrier Ea1 of the oxygen derivative [1O]3+ is smaller than that of the sulphur derivative [1S]3+. This suggests favoured thermally activated non-radiative decay for [1O]3+, which is at odds with the experimentally observed lower knr of [1O]3+ at first sight. However, the frequency factor A1 determined for [1S]3+ is higher than that for [1O]3+ by orders of magnitude, which overcompensates the somewhat higher barrier of [1S]3+ (Table 2). In a picture of semi-classical Marcus theory, larger frequency factors are associated with a stronger electronic coupling of the involved states.80 In other words, although [1O]3+ has smaller barriers, its electronic coupling is much weaker leading to a smaller ISC transition probability. For [1S]3+, the large frequency factor A1 enables rapid excited state decay. The larger A1 might arise from a higher density of accessible distorted quartet states in [1S]3+ (Table S7†) and/or higher SOC, i.e. higher electronic coupling. We hypothesise that the heavier sulphur atoms invoke larger SOCs16,81 as compared to the lighter bridging carbon, nitrogen or oxygen atoms of [1CH2]3+, [1NMe]3+ and [1O]3+ and hence promote the 2T1(1)/2E(1) → 4T2/4LMCT back-ISC process via the heavy atom effect.82–85 The sulphur orbital contributions to the 4LMCT state could further assist the 2T1(1)/2E(1) → 4LMCT back-ISC process via SOC.55,56
Following this simple argument, the lower Φ of [1S]3+ at room temperature is associated with a heavy-atom promoted thermally activated back-ISC to 4T2/4LMCT levels. As these distorted 4T2/4LMCT states can cross the 4A2 ground state potential surface or can lead to ligand dissociation (see below), this back-ISC process can be irreversible.
The second barriers Ea2 of a few hundred wavenumbers might be associated with the thermal population of the five lowest doublet states (Table 1 and Fig. 4). This reversible equilibration of doublet states, however, should not promote non-radiative decay per se as these doublet states are essentially nested states. Yet, multiphonon relaxation, which requires a resonant energy transfer to a vibrational overtone, might become relevant upon populating higher energy SF states. The 4th aromatic CH overtone of a pyridine of CH4 = 14065 cm−1 is in the region of the SF states for both complexes (Table 1 and Fig. 3b).25 Accidently, the 2E(1) state of [1S]3+ is even almost resonant with CH4. Hence, a diminished 2E(1) population of [1S]3+ at lower temperature will mitigate this pathway significantly. Inspection of Fig. 4b shows that the 2E(1) → 4A2 emission band of [1S]3+ dramatically diminishes in intensity at 77 K relative to the 2T1(1) → 4A2 emission band. This effect is much less pronounced for [10]3+ due to its larger E(2E(1)–2T1(1)) energy difference (Table 1). In addition, its 2E(1) emission band has a much smaller spectral overlap integral with the CH overtone anyway thanks to the larger energy gap E(2E(1)–CH4). Consequently, we propose that multiphonon relaxation via CH overtones is more relevant for [1S]3+ and that this pathway is T-dependent due to thermally activated population of its (accidently resonant) 2E(1) state.
The extremely efficient non-radiative decay of [1S]3+ with knr(293 K) = 606000 s−1via the 4T2/4LMCT and 2E(1) levels is strongly diminished at 77 K with knr(77 K) = 213 s−1. The first decay path is associated with the presence of a sulphur atom with larger SOC for the 2T1(1)/2E(1) → 4T2/4LMCT ISC (internal heavy atom effect) and the second decay path is associated with the thermally populated 2E(1) level that is accidently resonant with a CH overtone (thermally activated multiphonon relaxation).
2.4 Excited state reactivity
The photostability of the complexes [1X]3+ was probed by irradiating them in deaerated CH3CN (X = CH2, S, O, NMe). High light intensities are required for significant photochemical responses which is why we used a 460 nm UHP-LED with an output power of 1.1 W for these experiments. Under these conditions, which also might include a rise in temperature, the absorption spectra change and the luminescence intensities decrease in all cases (Fig. S71–S74†). The resulting spectra are compatible with (partial) ligand dissociation with the dissociated tripyridine ligands possibly undergoing follow-up reactions such as protonation (from the environment) or oxidation by excited chromium complexes.67The ligand photodissociation is not very efficient for [1O]3+, [1NMe]3+ and [1CH2]3+, but appears more efficient for [1S]3+, although the rates are not entirely comparable due to the different absorption changes during the photolysis. As discussed above, the heavy-atom effect of sulphur82–85 and the higher density of states might again be made responsible for the more efficient 2E/2T1 → 4T2(1)/4LMCT back-ISC of [1S]3+ and hence the more efficient unimolecular ligand dissociation. A potentially dissociative state could be the 4LMCT state with a pincer-like deformation of one bptp ligand (ESI, Table S7†).
For all complexes, the excited state lifetimes are sufficiently high (>1.5 μs) at room temperature to allow for bimolecular quenching. The doublet states of all sensitisers [1X]3+ are quenched by Dexter energy transfer to triplet oxygen with rate constants kq = 0.17 × 107 M−1 s−1, 0.29 × 107 M−1 s−1,29 1.77 × 107 M−1 s−1,36 8.55 × 107 M−1 s−1 for X = O, CH2, NMe, S, respectively (Fig. S75 and S76†). This series might be associated with the different accessibility of the Cr centre by O2 due to the different bridges X. Singlet oxygen quantum yields were already reported for [1NMe]3+ and [1CH2]3+ as 61% (DMF) and 55% (DMF/HClO4), respectively.29,32
Based on the high excited state reduction potentials of [1O]3+ and [1S]3+ of E(*[1X]3+/[1X]2+) = 1.25 and 1.34 V (derived from the 2E(1) energies at 293 K), these sensitisers should react with suitable redox-active quenchers. Anthracene (E1/2 = 0.69 V vs. ferrocene, ET = 1.85 eV)86,87 and trans-stilbene (E1/2 = 1.03 V vs. ferrocene, ET = 2.14 eV)86,87 should quench excited [1O]3+ and [1S]3+ exclusively by electron transfer, but not by energy transfer due to their high triplet energies ET. The larger driving force for the oxidation of anthracene compared to trans-stilbene gives quenching rate constants kq(anthracene) = 7.21 × 108/44 × 108 M−1 s−1 larger by two orders of magnitude than for trans-stilbene with kq(trans-stilbene) = 2.06 × 106/27.3 × 106 M−1 s−1 for [1O]3+ and [1S]3+, respectively (Fig. S77–S80†). Both quenchers react faster with the sulphur derivative [1S]3+ than with [1O]3+ due to the higher excited state potential of [1S]3+ in agreement with Marcus theory.
4 Conclusions
Two new molecular rubies [1X]3+ with X = O, S bridging atoms in the tripyridine ligands were structurally and spectroscopically characterised complementing the analogous known spin-flip emitters with X = NMe, CH2. This series allows an unprecedented insight into excited state energies and dynamics. The energies of all five lowest doublet levels derived from 2E/2T1 terms in octahedral symmetry of all complexes were experimentally identified by absorption spectroscopy for the first time. High-level quantum chemical calculations reproduced these doublet state energies very well. Calculations on model systems revealed the underlying structure–property relationship with the dominant parameter for the emission energy being the Cr–N distances: shorter distances lead to bathochromic shifts, which can be explained on the basis of the nephelauxetic effect.All four molecular rubies [1X]3+ undergo ultrafast ISC to the doublet manifold. The excited state dynamics of [1X]3+ with X = O, CH2, NMe on longer times scales are similar and high quantum yields and lifetimes are obtained. The sulphur derivative [1S]3+, however, experiences extremely rapid non-radiative decay via two pathways at room temperature: The first decay path is likely associated with stronger spin–orbit coupling caused by the sulphur atoms leading to faster doublet-quartet back-intersystem crossing (internal heavy atom effect). The second decay pathway is opened by thermal population of the 2E(1) level that is accidently resonant with a CH overtone enabling multiphonon relaxation. The efficient population of the distorted quartet states in [1S]3+ furthermore promotes more facile ligand dissociation. All sensitisers [1X]3+ engage in photoinduced energy and electron transfer reactions from their doublet levels thanks to their microsecond excited state lifetime.
Key results from this study are as follows: (i) the Cr–L distances appear as the most important parameter to tune the luminescence energies of spin-flip emitters (nephelauxetic effect); (ii) with a large ligand field splitting, the density of doublet states at the Franck-Condon geometry is high, which enables efficient ISC without requiring SOC by further heavy atoms; (iii) heavy atoms should indeed be avoided as they can promote thermally activated back-ISC to the quartet levels along Jahn–Teller modes, increasing knr (heavy atom effect); (iv) Jahn–Teller distortions in 4T2/4LMCT states are responsible for photodissociation and (v) CH overtones of the ligands (in particular CH oscillators those close to the metal centre) can define spectral regions of the luminescence with higher and lower non-radiative decay via (thermally activated) multiphonon relaxation. These key aspects will aid in future design concepts of improved molecular rubies.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
F. R. performed the syntheses and spectroscopic characterization as well as the quantum chemical calculations. R. N. and F. R. measured and interpreted the emission data. C. F. solved and refined the single crystal structures and assisted with the quantum chemical calculations. W. R. K., A. M. R. and S. F. conducted TA measurements. K. H. conceived and designed the project. F. R. and K. H. wrote the manuscript with contributions of all authors. K. H. supervised and C. F. co-supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been financially supported by the Deutsche Forschungsgemeinschaft (DFG) under grants INST 247/1018-1, HE2778/10-2 and HE2778/15-2. W. R. K. is thankful for the support from the German Academic Scholarship Foundation and financial support from the Max Planck Graduate Center (MPGC) with the Johannes Gutenberg University of Mainz. Parts of this research were conducted using the supercomputer Elwetritsch and advisory services offered by the RPTU Kaiserslautern-Landau (https://elwe.rhrk.uni-kl.de), which is a member of the AHRP. We thank Dr Dieter Schollmeyer and Dr Luca M. Carrella for collecting the XRD data.Notes and references
- C. Förster and K. Heinze, The Photophysics and Applications of Molecular Rubies, Adv. Inorg. Chem., 2024, 83, 111–159 CrossRef
.
- A. M. May and J. L. Dempsey, A new era of LMCT: leveraging ligand-to-metal charge transfer excited states for photochemical reactions, Chem. Sci., 2024, 15, 6661–6678 RSC
- N. Sinha and O. S. Wenger, Photoactive Metal-to-Ligand Charge Transfer Excited States in 3d6 Complexes with Cr0, MnI, FeII, and CoIII, J. Am. Chem. Soc., 2023, 145, 4903–4920 CrossRef CAS PubMed
- W. R. Kitzmann and K. Heinze, Charge-Transfer and Spin-Flip States: Thriving as Complements, Angew. Chem., Int. Ed., 2023, 62, e202213207 CrossRef CAS PubMed
- N. Sinha, P. Yaltseva and O. S. Wenger, The Nephelauxetic Effect Becomes an Important Design Factor for Photoactive First-Row Transition Metal Complexes, Angew. Chem., Int. Ed., 2023, 62, e202303864 CrossRef CAS PubMed
- C. Förster and K. Heinze, Bimolecular Reactivity of 3d Metal-Centered Excited States (Cr, Mn, Fe, Co), Chem. Phys. Rev., 2022, 3, 041302 CrossRef
- P. Dierks, Y. Vukadinovic and M. Bauer, Photoactive iron complexes: more sustainable, but still a challenge, Inorg. Chem. Front., 2022, 9, 206–220 RSC
- W. R. Kitzmann, J. Moll and K. Heinze, Spin-Flip Luminescence, Photochem. Photobiol. Sci., 2022, 21, 1309–1331 CrossRef CAS PubMed
- W. R. Kitzmann, C. Ramanan, R. Naumann and K. Heinze, Molecular Ruby: Exploring the Excited State Landscape, Dalton Trans., 2022, 51, 6519–6525 RSC
- C. Wegeberg and O. S. Wenger, Luminescent First-Row Transition Metal Complexes, JACS Au, 2021, 1, 1860–1876 CrossRef CAS
- C. Förster and K. Heinze, Photophysics and photochemistry with Earth-abundant metals – fundamentals and concepts, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC
- O. S. Wenger, Photoactive Complexes with Earth-Abundant Metals, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed
- L. A. Büldt and O. S. Wenger, Chromium complexes for luminescence, solar cells, photoredox catalysis, upconversion, and phototriggered NO release, Chem. Sci., 2017, 8, 7359–7367 RSC
- Y. Liu, P. Persson, V. Sundström and K. Wärnmark, Fe N-Heterocyclic Carbene Complexes as Promising Photosensitizers, Acc. Chem. Res., 2016, 49, 1477–1485 CrossRef CAS PubMed
-
B. N. Figgis and M. A. Hitchman, Ligand field theory and its applications, Wiley, Chichester, 2000 Search PubMed
-
A. B. P. Lever and E. I. Solomon, Ligand Field Theory and the Properties of Transition Metal Complexes in Inorganic Electronic Structure and Spectroscopy, ed. E. I. Solomon and A. B. P. Lever, John Wiley & Sons, Inc, 1999, vol. I, pp. 1–91 Search PubMed
- J. K. McCusker, Electronic structure in the transition metal block and its implications for light harvesting, Science, 2019, 363, 484–488 CrossRef CAS PubMed
- P. M. Kraus, M. Zürch, S. K. Cushing, D. M. Neumark and S. R. Leone, The ultrafast X-ray spectroscopic revolution in chemical dynamics, Nat. Rev. Chem, 2018, 2, 82–94 CrossRef CAS
- K. J. Gaffney, Capturing photochemical and photophysical transformations in iron complexes with ultrafast X-ray spectroscopy and scattering, Chem. Sci., 2021, 12, 8010–8025 RSC
- L. Lindh, T. Pascher, S. Persson, Y. Goriya, K. Wärnmark, J. Uhlig, P. Chábera, P. Persson and A. Yartsev, Multifaceted Deactivation Dynamics of Fe(II) N-Heterocyclic Carbene Photosensitizers, J. Phys. Chem. A, 2023, 127, 10210–10222 CrossRef CAS PubMed
- M. E. Reinhard, B. K. Sidhu, I. B. Lozada, N. Powers-Riggs, R. J. Ortiz, H. Lim, R. Nickel, J. van Lierop, R. Alonso-Mori, M. Chollet, L. B. Gee, P. L. Kramer, T. Kroll, S. L. Raj, T. B. van Driel, A. A. Cordones, D. Sokaras, D. E. Herbert and K. J. Gaffney, Time-Resolved X-ray Emission Spectroscopy and Synthetic High-Spin Model Complexes Resolve Ambiguities in Excited-State Assignments of Transition-Metal Chromophores: A Case Study of Fe-Amido Complexes, J. Am. Chem. Soc., 2024, 146, 17908–17916 CrossRef CAS
- J. N. Schrauben, K. L. Dillman, W. F. Beck and J. K. McCusker, Vibrational coherence in the excited state dynamics of Cr(acac)3: probing the reaction coordinate for ultrafast intersystem crossing, Chem. Sci., 2010, 1, 405–410 RSC
- B. C. Paulus and J. K. McCusker, On the use of vibronic coherence to identify reaction coordinates for ultrafast excited-state dynamics of transition metal-based chromophores, Faraday Discuss., 2022, 237, 274–299 RSC
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, [Cr(ddpd)2]3+: a molecular, water-soluble, highly NIR-emissive ruby analogue, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed
- C. Wang, S. Otto, M. Dorn, E. Kreidt, J. Lebon, L. Sršan, P. Di Martino-Fumo, M. Gerhards, U. Resch-Genger, M. Seitz and K. Heinze, Deuterated Molecular Ruby with Record Luminescence Quantum Yield, Angew. Chem., Int. Ed., 2018, 57, 1112–1116 CrossRef CAS PubMed
- S. Otto, C. Förster, C. Wang, U. Resch-Genger and K. Heinze, A strongly luminescent chromium(III) complex acid, Chem.–Eur. J., 2018, 24, 12555–12563 CrossRef CAS
- S. Treiling, C. Wang, C. Förster, F. Reichenauer, J. Kalmbach, P. Boden, J. P. Harris, L. Carrella, E. Rentschler, U. Resch-Genger, C. Reber, M. Seitz, M. Gerhards and K. Heinze, Luminescence and Light-driven Energy and Electron Transfer from an Exceptionally Long-lived Excited State of a Non-innocent Chromium(III) Complex, Angew. Chem., Int. Ed., 2019, 58, 18075–18085 CrossRef CAS
- J.-R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Chiral Molecular Ruby [Cr(dqp)2]3+ with Long-Lived Circularly Polarized Luminescence, J. Am. Chem. Soc., 2019, 141, 13244 CrossRef
- F. Reichenauer, C. Wang, C. Förster, P. Boden, N. Ugur, R. Báez-Cruz, J. Kalmbach, L. M. Carrella, E. Rentschler, C. Ramanan, G. Niedner-Schatteburg, M. Gerhards, M. Seitz, U. Resch-Genger and K. Heinze, Strongly Red-Emissive Molecular Ruby [Cr(bpmp)2]3+ surpasses [Ru(bpy)3]2+, J. Am. Chem. Soc., 2021, 143, 11843–11855 CrossRef CAS PubMed
- J.-R. Jiménez, M. Poncet, S. Míguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Bright Long-Lived Circularly Polarized Luminescence in Chiral Chromium(III) Complexes, Angew. Chem., Int. Ed., 2021, 60, 10095–10102 CrossRef PubMed
- S. Otto, N. Scholz, T. Behnke, U. Resch-Genger and K. Heinze, Thermo-Chromium: A Contactless Optical Molecular Thermometer, Chem.–Eur. J., 2017, 23, 12131–12135 CrossRef CAS
- S. Otto, A. M. Nauth, E. Ermilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, Photo-Chromium: Sensitizer for Visible Light-Induced Oxidative C-H Bond Functionalization – Electron or Energy Transfer?, ChemPhotoChem, 2017, 1, 344–349 CrossRef CAS
- S. Otto, J. Harris, K. Heinze and C. Reber, Molecular ruby under pressure, Angew. Chem., Int. Ed., 2018, 57, 11069–11073 CrossRef CAS PubMed
- C. Wang, S. Otto, M. Dorn, K. Heinze and U. Resch-Genger, Luminescent TOP Nanosensors for Simultaneously Measuring Temperature, Oxygen, and pH at a Single Excitation Wavelength, Anal. Chem., 2019, 91, 2337–2344 CrossRef CAS PubMed
- C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. Di Bari and M. Seitz, Strong Circularly Polarized Luminescence of an Octahedral Chromium(III) Complex, Chem. Commun., 2019, 55, 13078–13081 RSC
- L. Stein, C. Wang, C. Förster, U. Resch-Genger and K. Heinze, Bulky Ligands protect Molecular Ruby from Oxygen Quenching, Dalton Trans., 2022, 51, 17664–17670 RSC
- C. Wang, W. R. Kitzmann, F. Weigert, C. Förster, X. Wang, K. Heinze and U. Resch-Genger, Matrix Effects on Photoluminescence and Oxygen Sensitivity of a Molecular Ruby, ChemPhotoChem, 2022, 6, e202100296 CrossRef CAS
- C. Wang, F. Reichenauer, W. R. Kitzmann, C. Kerzig, K. Heinze and U. Resch-Genger, Efficient Triplet-Triplet Annihilation Upconversion Sensitized by a Chromium(III) Complex via an Underexplored Energy Transfer Mechanism, Angew. Chem., Int. Ed., 2022, 61, e202202238 CrossRef CAS
- T. H. Bürgin, F. Glaser and O. S. Wenger, Shedding Light on the Oxidizing Properties of Spin-Flip Excited States in a CrIII Polypyridine Complex and Their Use in Photoredox Catalysis, J. Am. Chem. Soc., 2022, 144, 14181–14194 CrossRef
- S. Sittel, R. Naumann and K. Heinze, Molecular Rubies in Photoredox Catalysis, Front. Chem., 2022, 10, 887439 CrossRef CAS
- S. Sittel, A. C. Sell, K. Hofman, C. Wiedemann, J. P. Nau, C. Kerzig, G. Manolikakes and K. Heinze, Visible-Light Induced Fixation of SO2 into Organic Molecules with Polypyridine Chromium(III) Complexes, ChemCatChem, 2023, 15, e202201562 CrossRef CAS
- C. Wang, H. Li, T. H. Bürgin and O. S. Wenger, Cage escape governs photoredox reaction rates and quantum yields, Nat. Chem., 2024, 16, 1151–1159 CrossRef CAS PubMed
- Y. Tanabe and S. Sugano, On the Absorption Spectra of Complex Ions. I, J. Phys. Soc. Jpn., 1954, 9, 753–766 CrossRef CAS
- Y. Tanabe and S. Sugano, On the Absorption Spectra of Complex Ions. II, J. Phys. Soc. Jpn., 1954, 9, 766–779 CrossRef CAS
- J.-R. Jiménez, M. Poncet, B. Doistau, C. Besnard and C. Piguet, Luminescent polypyridyl heteroleptic CrIII complexes with high quantum yields and long excited state lifetimes, Dalton Trans., 2020, 49, 13528–13532 RSC
- N. Sinha, J.-R. Jiménez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, A Near-Infrared-II Emissive Chromium(III) Complex, Angew. Chem., Int. Ed., 2021, 60, 23722–23728 CrossRef PubMed
- L. Stein, P. Boden, R. Naumann, C. Förster, G. Niedner-Schatteburg and K. Heinze, The overlooked NIR luminescence of Cr(ppy)3, Chem. Commun., 2022, 58, 3701–3704 RSC
- N. Sawicka, C. J. Craze, P. N. Horton, S. J. Coles, E. Richards and S. J. A. Pope, Long-lived, near-IR emission from Cr(III) under ambient conditions, Chem. Commun., 2022, 58, 5733–5736 RSC
- J. Chong, C. Besnard, C. M. Cruz, C. Piguet and J.-R. Jiménez, Heteroleptic mer-[Cr(N∩N∩N)(CN)3] complexes: synthetic challenge, structural characterization and photophysical properties, Dalton Trans., 2022, 51, 4297–4309 RSC
- Y. Cheng, Q. Yang, J. He, W. Zou, K. Liao, X. Chang, C. Zou and W. Lu, The energy gap law for NIR-phosphorescent Cr(III) complexes, Dalton Trans., 2023, 52, 2561–2565 RSC
- L. Stein, C. Förster and K. Heinze, Luminescent Cyclometalated Chromium(III) Complexes, Organometallics, 2024, 43, 1766–1774 CrossRef CAS
- R. W. Jones, R. A. Cowin, I. I. Ivalo, D. Chekulaev, T. M. Roseveare, C. R. Rice, J. A. Weinstein, P. I. P. Elliott and P. A. Scattergood, A Near-Infrared Luminescent Cr(III) N-Heterocyclic Carbene Complex, Inorg. Chem., 2024, 63, 8526–8530 CrossRef CAS
- R. A. Forman, G. J. Piermarini, J. D. Barnett and S. Block, Pressure Measurement Made by the Utilization of Ruby Sharp-Line Luminescence, Science, 1972, 176, 284–285 CrossRef CAS
- C. Förster, H. Osthues, D. Schwab, N. L. Doltsinis and K. Heinze, Quantum Chemical Study of the Pressure-dependent Phosphorescence of [Cr(ddpd)2]3+ in the Solid State, ChemPhysChem, 2023, 24, e202300165 CrossRef
- C. M. Marian, Understanding and Controlling Intersystem Crossing in Molecules, Annu. Rev. Phys. Chem., 2021, 72, 617–640 CrossRef CAS
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Spin-Vibronic Mechanism for Intersystem Crossing, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed
- R. W. Jones, A. J. Auty, G. Wu, P. Persson, M. V. Appleby, D. Chekulaev, C. R. Rice, J. A. Weinstein, P. I. P. Elliott and P. A. Scattergood, Direct Determination of the Rate of Intersystem Crossing in a Near-IR Luminescent Cr(III) Triazolyl Complex, J. Am. Chem. Soc., 2023, 145, 12081–12092 CrossRef CAS PubMed
- O. Laporte and W. F. Meggers, Some rules of spectral structure, J. Opt. Soc. Am., 1925, 11, 459–463 CrossRef CAS
- H. U. Guedel and T. R. Snellgrove, Jahn-Teller effect in the 4T2g state of chromium(III) in dicesium sodium indium(III) hexachloride, Inorg. Chem., 1978, 17, 1617–1620 CrossRef CAS
- R. B. Wilson and E. I. Solomon, Spectroscopic Studies of the Photoactive 4T2g Excited State of Hexaamminechromium(lII), Inorg. Chem., 1978, 17, 1729–1736 CrossRef CAS
- I. B. Bersuker, Pseudo-Jahn–Teller Effect—A Two-State Paradigm in Formation, Deformation, and Transformation of Molecular Systems and Solids, Chem. Rev., 2013, 113, 1351–1390 CrossRef CAS PubMed
- V. L. Ermolaev and E. B. Sveshnikova, The application of luminescence-kinetic methods in the study of the formation of lanthanide ion complexes in solution, Russ. Chem. Rev., 1994, 63, 905–922 CrossRef
-
E. Kreidt, C. Kruck and M. Seitz, Nonradiative Deactivation of Lanthanoid Luminescence by Multiphonon Relaxation in Molecular Complexes, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 2018, vol. 53, pp. 35–79 Search PubMed
- S. Balamurugan, S. Ganesan, S. Kamaraj, V. Mathew, J. Kim, N. Arumugam and A. I. Almansour, Effect of poly (ethylene glycol) gel polymer electrolyte consist of novel heteroleptic cobalt redox shuttle and pyridine based organic additive on performance of dye sensitized solar cells, Opt. Mater., 2022, 125, 112082 CrossRef CAS
- C. M. Brown, N. E. Arsenault, T. N. K. Cross, D. Hean, Z. Xu and M. O. Wolf, Structural, electrochemical and photophysical behavior of Ru(II) complexes with large bite angle sulfur-bridged terpyridyl ligands, Inorg. Chem. Front., 2020, 7, 117–127 RSC
- M. Nieger, E. Niecke and C. Volkholz, CCDC 258237: Experimental Crystal Structure Determination, 2005, DOI:10.5517/cc8nq7v.
- C. Förster, M. Dorn, T. Reuter, S. Otto, G. Davarci, T. Reich, L. Carrella, E. Rentschler and K. Heinze, Ddpd as expanded terpyridine: dramatic effects of symmetry and electronic properties in first row transition metal complexes, Inorganics, 2018, 6, 86 CrossRef
- S. J. Dunne, E. I. von Nagy-Felsobuki and M. F. Mackay, 2,2’-Oxybispyridine and 2,2’-Selenobispyridine, Acta Cryst., 1995, C51, 1454–1457 CAS
- P.-S. Wang, C.-K. Liang and M.-k. Leung, An improved Ullmann–Ukita–Buchwald–Li conditions for CuI-catalyzed coupling reaction of 2-pyridones with aryl halides, Tetrahedron, 2005, 61, 2931–2939 CrossRef CAS
- M. J. Cook, A. R. Katritzky, P. Linda and R. D. Tack, Aromaticity and Tautomerism. Part 1. The Aromatic Resonance Energy of 2-Pyridone and the Related Thione, Methide, and lmine, J. Chem. Soc., Perkin Trans. 2, 1972, 1295–1301 RSC
- S. Millefiori and A. Millefiori, On Tautomerism and Self-Association of 2-Pyridinol/2-Pyridone System, Bull. Chem. Soc. Jpn., 1990, 63, 2981–2984 CrossRef CAS
- R. D. Köhn, A. G. N. Coxon, S. Chunawat, C. Heron, S. Mihan, C. L. Lyall, S. B. Reeksting and G. Kociok-Köhn, Triazacyclohexane chromium triflate complexes as precursors for the catalytic selective olefin trimerisation and its investigation by mass spectrometry, Polyhedron, 2020, 185, 114572 CrossRef
- P. M. Becker, C. Förster, L. M. Carrella, P. Boden, D. Hunger, J. van Slageren, M. Gerhards, E. Rentschler and K. Heinze, Spin Crossover and Long-lived Excited States in a Reduced Molecular Ruby, Chem.–Eur. J., 2020, 26, 7199–7204 CrossRef CAS
- M. Kranz, A. Witkowska and J. T. Yoke, Preparation of Labile Compounds Under Protective Conditions. Chromium(II) Salts, Inorg. Synth., 2007, 6, 144–146 CrossRef
- R. Acevedo, G. Díaz and C. D. Flint, Normal coordinate analysis of the 25-atom system Cr(NH3)63+ octahedral symmetry, Spectrochim. Acta, 1985, 41, 1397–1403 CrossRef
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Recent progress in ligand photorelease reaction mechanisms: Theoretical insights focusing on Ru(II) 3MC states, Coord. Chem. Rev., 2020, 408, 213184 CrossRef CAS
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Theoretical Study of the Full Photosolvolysis Mechanism of [Ru(bpy)3]2+: Providing a General Mechanistic Roadmap for the Photochemistry of [Ru(N^N)3]2+-Type Complexes toward Both Cis and Trans Photoproducts, Inorg. Chem., 2020, 59, 14679–14695 CrossRef CAS PubMed
- K. Eastham, P. A. Scattergood, D. Chu, R. Z. Boota, A. Soupart, F. Alary, I. M. Dixon, C. R. Rice, S. J. O. Hardman and P. I. P. Elliott, Not All 3MC States Are the Same: The Role of 3MCcis States in the Photochemical N∧N Ligand Release from [Ru(bpy)2(N∧N)]2+ Complexes, Inorg. Chem., 2022, 61, 19907–19924 CrossRef CAS PubMed
- D. Hernández-Castillo, R. E. P. Nau, M.-A. Schmid, S. Tschierlei, S. Rau and L. González, Multiple Triplet Metal-Centered Jahn-Teller Isomers Determine Temperature-Dependent Luminescence Lifetimes in [Ru(bpy)3]2+, Angew. Chem., Int. Ed., 2023, 62, e202308803 CrossRef
- M. C. Carey, S. L. Adelman and J. K. McCusker, Insights into the excited state dynamics of Fe(II) polypyridyl complexes from variable-temperature ultrafast spectroscopy, Chem. Sci., 2019, 10, 134–144 RSC
- M. Blume and R. E. Watson, Theory of spin-orbit coupling in atoms II. Comparison of theory with experiment, Proc. R. Soc. London, A, 1963, 271, 565–578 CAS
- M. Mitra, O. Mrózek, M. Putscher, J. Guhl, B. Hupp, A. Belyaev, C. M. Marian and A. Steffen, Structural Control of Highly Efficient Thermally Activated Delayed Fluorescence in Carbene Zinc(II) Dithiolates, Angew. Chem., Int. Ed., 2024, 63, e202316300 CrossRef CAS
- S. D. Ezquerra Riega, M. E. Gutierrez Suburu, H. B. Rodríguez, B. Lantaño, M. Kleinschmidt, C. M. Marian and C. A. Strassert, A Case-Study on the Photophysics of Chalcogen-Substituted Zinc(II) Phthalocyanines, Chem.–Eur. J., 2024, 30, e202304083 CrossRef CAS
- W. Sang-aroon, M. E. Alberto, M. Toscano and N. Russo, Chalcogen atom effect on the intersystem crossing kinetic constant of oxygen- and sulfur disubstituted heteroporphyrins, J. Comput. Chem., 2024, 45, 1322–1328 CrossRef CAS
- M. Mońka, D. Grzywacz, E. Hoffman, V. Ievtukhov, K. Kozakiewicz, R. Rogowski, A. Kubickia, B. Liberek, P. Bojarski and I. E. Serdiuk, Decisive role of heavy-atom orientation for efficient enhancement of spin–orbit coupling in organic thermally activated delayed fluorescence emitters, J. Mater. Chem. C, 2022, 10, 11719–11729 RSC
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Energy transfer catalysis mediated by visible light: principles, applications, directions, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC
-
M. Montalti, A. Credi, L. Prodi, M. T. Gandolfi, J. Michl and V. Balzani, Handbook of photochemistry, CRC/Taylor & Francis, Boca Raton, 2020 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic details, quantum chemical results. CCDC 2380706–2380708. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05860g |
| This journal is © The Royal Society of Chemistry 2024 |