
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilizing an exotic dianionic tetrazine bridge in a Ln2 metallocene†
Niki
Mavragani‡
 a,
Alexandros A.
Kitos‡
a,
Akseli
Mansikkamäki
b and
Muralee
Murugesu
*a
a,
Alexandros A.
Kitos‡
a,
Akseli
Mansikkamäki
b and
Muralee
Murugesu
*a
aDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, ON K1N 6N5, Canada. E-mail: m.murugesu@uottawa.ca
bNMR Research Unit, University of Oulu, P.O. Box 8000, FI-90014, Finland
First published on 6th September 2024
Abstract
The unique electronic nature of the 1,2,4,5-tetrazine or s-tetrazine (tz) ring has sparked tremendous scientific interest over the last few years. Tetrazines have found numerous applications, and their ability to coordinate to metal ions has opened the possibility of exploring their chemistry in both molecular systems and extended networks. The rich redox chemistry of s-tetrazines allows them to exchange electrons and switch between their dihydro (H2tz), neutral (tz), and radical (tz˙−) forms. Previous reports in the literature have observed electrochemically that a second electron can potentially be stored in the tetrazinyl ring and form a dianionic species. However, due to its extremely reactive nature, this has not been isolated before. Herein, the combination of strictly anhydrous and inert conditions, strong reducing agents, non-acidic solvents and most importantly blocking the accessibility of the nitrogen atoms by coordinating them to lanthanide ions allowed for the stabilization of a dianionic tetrazine in a lanthanocene complex. Three dinuclear metallocene complexes are reported, [(Cp*2Ln)2(tz˙−)(THF)2](BPh4) (Ln = Y (1-Y); Cp* = pentamethylcyclopentadienyl; THF = tetrahydrofuran) and [(Cp*2Ln)2(tz2−)(THF)2]·2THF (Ln = Gd (2-Gd), or Y (2-Y)), which utilize the unsubstituted tz as the ligand. In 1-Ln, the tz ligand is reduced to the radical anion (tz˙−), while in 2-Ln, the tz ligand is in the −2 charge state. These complexes are the first structurally and physically characterized complexes bearing the dianion radical of an s-tetrazine. Detailed structural analysis, ab initio calculations, and physical characterization support that the tz2− ligand is a closed-shell planar dianion with unique structural features vastly different from those of the tz, tz˙− and H2tz species.
Introduction
Since their first report by Pinner at the end of the 19th century,1–3 tetrazines have been long-known organic heterocycles. They feature a six-membered aromatic ring, where the position of the four nitrogen atoms differentiates them into 1,2,3,4-tetrazines (v-tetrazines), 1,2,3,5-tetrazines (as-tetrazines), and 1,2,4,5-tetrazines (sym-/s-tetrazines; Scheme 1). s-Tetrazines are considered the most stable amongst them,4 and due to their unique physicochemical features, research linked to this family of molecules has skyrocketed in the last four decades.5–8 The presence of four sp2 N-atoms in the six-membered ring leads to a low energy π* type LUMO. As such, all s-tetrazines exhibit a very high electron affinity, which allows them to stabilize radical anions upon one e− reduction at low-to-very-low potentials.9–12 Furthermore, due to an n → π* transition in the visible light region, all s-tetrazines have vivid colours ranging from purple to red to orange and exhibit unique optical properties.11,13 Due to these features, tetrazines hold great promise for a wide range of applications. To name a few, they are widely being used as dienes in inverse Diels–Alder cycloaddition reactions,14–20 in the development of nitrogen-rich energetic materials,21–24 as well as in optoelectronic devices such as organic solar cells and light-emitting diodes or electrofluorochromic windows.22,25–32 Finally, s-tetrazines and their functionally substituted derivatives can act as ditopic or chelating ligands, attracting significant interest in coordination and organometallic chemistry.5,7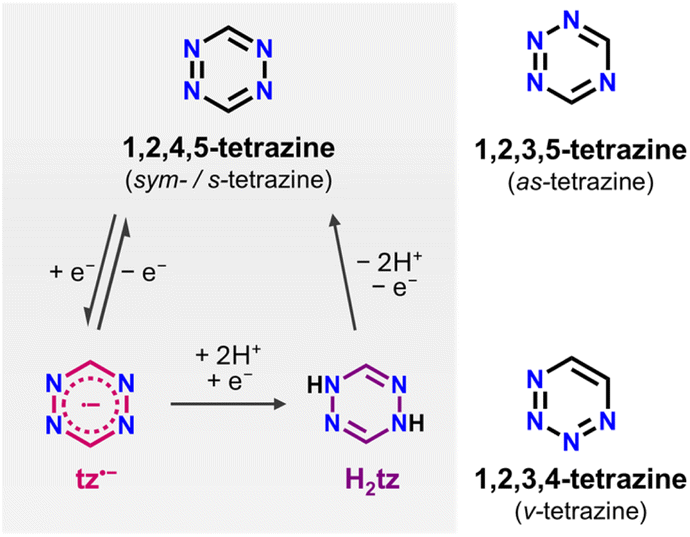 | ||
| Scheme 1 Left: Reduction pathway of s-tetrazine (tz) to dihydrotetrazine (H2tz) via a radical anion. The H2tz can be oxidized back to tz. Right: the structures of as- and v-tetrazine. | ||
The design and development of tetrazine-based metal complexes and metal–organic frameworks have become increasingly sought after due to their numerous applications including, but not limited to, catalysis, sensing, adsorption, energy storage, energetic materials, luminescence, and molecular magnetism.33–46 For the latter, the efficient control of the magnetic interactions between the metal centres is crucial, on the basis of any rational design of polymetallic molecule-based magnets.46,47 Thus, many groups, including ours, have explored s-tetrazines as bridging ligands with both 3d48–61 and 4f metal ions.62–69 Although the core-like nature of the 4f orbitals in lanthanides (LnIII) poses a challenge in the promotion of strong magnetic interactions,70s-tetrazine radical ligands appear to be promising in acting as a magnetic relay between the paramagnetic metal centres.46 This is because the diffuse nature of the spin orbitals of the tetrazinyl radical ring is ideally suited to penetrate the shielded 4f orbitals, promoting strong magnetic coupling. Recently, we presented the first examples of dinuclear [(Cp*2Ln)2(tz˙−)(THF)2]BPh4 (1-Ln; Ln = Gd, Tb or Dy; Cp* = pentamethylcyclopentadienyl, THF = tetrahydrofuran)71 and tetranuclear [(Cp*2Ln)4(tz˙−)4]·3(C6H6) (Ln = Gd, Tb or Dy)72,73 complexes containing LnIII metallocene units bridged by the unsubstituted 1,2,4,5-tetrazine (tz˙−) radical ligand. In both families, the strong magnetic coupling between the Ln centres and the radicals led to the formation of a “giant-spin” model with slow relaxation of the magnetization at zero-field (for the TbIII and DyIII analogues).
As mentioned above, all tetrazines can be reversibly reduced in organic solvents by accepting one electron, resulting in the formation of an anion radical which is very stable in the absence of acids (Scheme 1).74–76 This was first demonstrated in 1963 when Stone and Maki reported the relatively facile (compared to other polyazines) reduction of s-tetrazines.9 Additionally, it has been reported that most tetrazine derivatives can undergo a second electron addition through an electrochemically irreversible process under standard conditions (Scheme 1).77 As proposed by Clavier and Audebert,6 as well as by Neugebauer and co-workers,78 it is likely that the corresponding tetrazine dianion radical has a highly reactive character and immediately reacts with trace amounts of water or protic impurities to form the known dihydrotetrazine (H2tz) or, more likely, its monoanion. Similarly, Fukuzumi et al. reported the reduction of 3,6-diphenyl-s-tetrazine to the dihydrotetrazine by a two-electron/two-proton transfer process.79 Combing through the literature, we found a few reports of metal complexes containing the H2tz ligand or its doubly deprotonated form (H2tz2−), where the tetrazine ring adopts the expected boat conformation. However, to the best of our knowledge, there are no reports to date regarding the isolation of a dianionic tetrazine species. Given the highly basic and reactive character of this dianionic tetrazine ring,80 a potential strategy to avoid its protonation could involve blocking the accessibility of the nitrogen atoms by coordinating them to metal centers. This approach might be key in order to stabilize this exotic and highly reactive tetrazine species.
With this goal in mind, herein we report three new s-tetrazine-bridged dinuclear metallocenes, [(Cp*2Ln)2(tz˙−)(THF)2]BPh4 (Ln = Y (1-Y)) and [(Cp*2Ln)2(tz2−)(THF)2]·2THF (Ln = Gd (2-Gd), or Y (2-Y)), utilizing the unsubstituted 1,2,4,5-tetrazine (tz) as the ligand. In the previously reported cationic complex 1-Ln, the tz ligand is reduced to the radical anion (tz˙−).71 Surprisingly, using a different lanthanide precursor while increasing the amount of the reducing agent used, led to the formation of the neutral complexes 2-Gd and 2-Y. The tz ligand was found to be in the −2 charge state, making these complexes the first structurally and physically characterized complexes bearing the dianion radical of an s-tetrazine. A detailed structural analysis, ab initio calculations, and physical characterization revealed that the tz2− ligand is a closed-shell planar dianion with unique structural features that differ from the neutral tz, tz˙− and H2tz species. Indeed, s-tetrazines can undergo a two-electron reduction to form the dianion radical, which can be isolated under certain conditions: strictly anhydrous and inert conditions, strong reducing agents, and non-acidic solvents (i.e. THF). These results provide a clear mechanistic underpinning for the reduction of s-tetrazines laying out a path for further development of new tetrazine-based magnetic, optical, and energy storage materials.
Results and discussion
Synthesis and structural analysis
The syntheses of all complexes begin with the reduction of tz in THF, where an equimolar mixture of the tz ligand and KC8 in THF is prepared (Fig. 1). Stirring the mixture for 5 h results in the gradual reduction of the tz ligand, which is monitored by the colour change from a vivid red to a dark grey. As previously reported,71 slow addition of this mixture to two equivalents of [Cp*2Ln]BPh4 leads to the isolation of 1-Ln (Ln = Gd or Y). However, addition of the tz˙− mixture to a solution of two equivalents of [Cp*2Ln(C3H5)] and one additional part of KC8 leads to the isolation of 2-Ln (Ln = Gd or Y). Single crystals suitable for single-crystal X-ray diffraction (SCXRD) analysis can be obtained upon slow diffusion of the reaction solutions with Et2O at room temperature for 1-Ln and at −35 °C for 2-Ln, respectively. | ||
| Fig. 1 Top: Synthetic pathways for the isolation of the tz˙−-bridged (1-Ln) and tz2−-bridged (2-Ln) dinuclear complexes. Middle: Molecular structures of 1-Y and 2-Y, where partial labelling and omission of H-atoms, disordered conformers, THF lattice solvents and the BPh4− counter ion for 1-Y have been employed for clarity. Images of the respective single crystals (dark red for 1-Y; violet for 2-Y) under an optical microscope are given for each complex. Bottom: magnification of the Y2-tz core, highlighting the significantly increased tilting of the tz2− ring in 2-Y compared to tz˙− in 1-Y. | ||
SCXRD analysis reveals that 1-Y crystallizes in the monoclinic crystal system, in space group I2/a, and it is isostructural to the previously reported GdIII (1-Gd), TbIII and DyIII congeners71 (Fig. S1†). Complexes 2-Gd and 2-Y crystallize in the monoclinic crystal system, in space group P21/c (Table S1†), and they are also isostructural as revealed by their structural overlay (Fig. S2†). Hence, 1-Y and 2-Y will be used as representatives to describe the salient structural features of each family of complexes. At a first glance, complexes 1-Y and 2-Y appear to be nearly identical where in both cases the centrosymmetric complex consists of two {Cp*2Y(THF)}+ moieties bridged by a tz ligand (Fig. 1). However, complex 1-Y is cationic and thus stabilized by a BPh4− counterion in the crystal lattice. On the other hand, the absence of counterions in complex 2-Y is indicative of its neutral charge state, where only two THF solvent molecules are found in the crystal lattice. This obvious difference stems from the different charges of the tz species that are present in the two complexes.
As previously mentioned, tz is a redox-active ligand and is known to exist in its dihydro, neutral and radical forms as indicated by the respective N–N and C–N bond distances. As seen in Fig. 2, depending on the tetrazine species, the C–N and N–N bond distances vary, adapting to accommodate the respective electronic changes. For instance, upon coordination of the doubly deprotonated dihydrotetrazine (H2tz2−), the average N–N bond distance is 1.417(8) Å, while the six-membered ring is not planar as a result of the presence of both single C–N and double C–N bond distances (on average 1.356(8) Å and 1.301(5) Å, respectively), as seen in the titanocene complex [{Cp2Ti(μ-H2tz2−)}4].81 In the 2D polymeric structure [Ag(tz)(NO3)]n that features the neutral tz, the N–N bond distance is shorter (1.319(2) Å) as a result of the aromatic character of the ligand, while the average C–N bond distance is 1.332(3) Å.36 Upon close inspection of the bond distances and angles of the tetrazine ring in 1-Y, the N–N bond distance of 1.395(2) Å is a clear indication of its radical state, as previously seen in the literature.48,82 However, the respective N–N bond distance increases further in 2-Y, where a bond distance of 1.481(4) Å is observed. Comparing this to the respective N–N bond distance of 1.417(8) Å in the H2tz2−, it is clear that this drastic increase in the N–N bond distances stems from the addition of a second electron in the tetrazine ring, resulting in a formal charge of −2. To the best of our knowledge, although it has been reported in the literature that tetrazines can potentially store a second electron in their six-membered ring,6,78,80 complex 2-Y is the first example where this dianionic tetrazine species is stabilized. The planarity of the six-membered ring and the C–N distances of 1.308(6) and 1.317(7) Å in 2-Y exclude the possibility of the tetrazine being in its doubly deprotonated form (Fig. 2).82,83
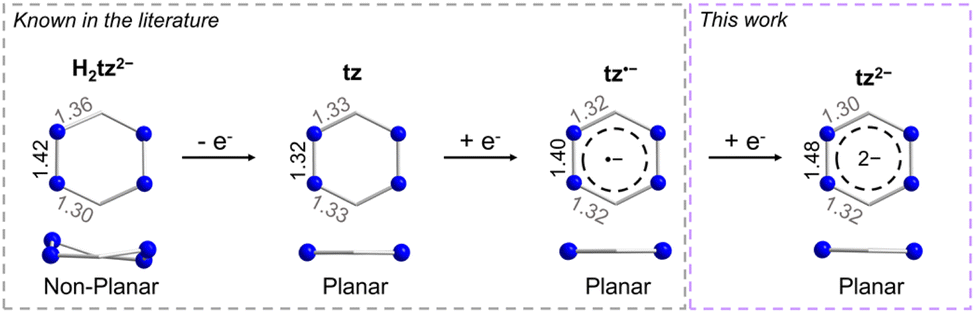 | ||
| Fig. 2 Comparison of the metric parameters of the different tetrazine species. The metric parameters for the doubly deprotonated dihydrotetrazine have been extracted from [{Cp2Ti(μ-H2tz2−)}4],81 while the metric parameters for the neutral tz have been extracted from [Ag(tz)(NO3)]n.36 | ||
The different charge states of the tetrazinyl ring in 1-Y and 2-Y affect the Y–Ntz, Y–OTHF and Y–Cp*cent (cent = centroid of the Cp* ring) bond distances and angles (Table S2†). In 1-Y, the Y–OTHF bond distance of 2.364(1) Å is shorter than the Y–Ntz˙− bond distances of 2.437(2) and 2.462(2) Å. In contrast, the opposite trend is observed for 2-Y, where the Y–OTHF bond distance of 2.439(2) Å is clearly longer than the Y–Ntz2− bond distances of 2.326(3) and 2.320(3) Å, as a result of the increased charge of the tz2− ring compared to its radical anion. The addition of the second electron in the tz2− species also affects the bidentate angle of the tz ring; 37.17(9)° in 2-Y compared to the 33.08(6)° in 1-Y. Additionally, although the ligand remains planar in both complexes, due to the significantly increased tilting of the tz2− ring, the YIII metal centres are found to be out of the tetrazine plane in 2-Y (Y–tz2−cent–Y′ = 165.55°) compared to 1-Y (Y–tz˙−cent–Y′ = 175.67°) where the metal ions are almost coplanar with the tz˙− ligand (Fig. 1). The average Y–Cp*cent bond distance is 2.388(4) Å in 1-Y, while the respective Cp*cent–Y–Cp*cent angle is 136.28(12)°. For 2-Y, although the respective Y–Cp* bond distance is similar to that of 1-Y (av. 2.401(5) Å), a slightly larger Cp*cent–Y–Cp*cent angle of 138.35(12)° is observed.
The formation of 2-Ln can be rationalized by the use of the [Cp*2Y(C3H5)] starting material and the excess of reducing agent that was used during the synthesis. Although attempts were made to synthesize 2-Ln by using [Cp*2Y]BPh4 as a starting material, the doubly reduced tetrazine species was not isolated. It is possible that the presence of the counter ion, or better off the lack of it, plays an intricate role in the isolation of 2-Ln. We hypothesize that upon coordination of the radical tz˙− to the {Cp*2Y(THF)}+ moieties, which are positively charged and, thus, can potentially stabilize the excess negative charge on the nitrogen atoms, the favourable addition of a second electron takes place. Blocking the nitrogen atoms of the tz ring by coordinating them to the metal centres also prevents the protonation of the dianionic tetrazine radical. These act synergistically, forcing the tetrazine ring to stabilize in its doubly reduced dianionic form.
Physicochemical characterization
As already mentioned, the formation of the dianionic tetrazine radical is facilitated by the coordination of the radical tz˙− to the {Cp*2Y(THF)}+ moieties. To validate this hypothesis, cyclic voltammetry (CV) of both the neutral free ligand and 1-Y was performed in DCM solutions with 0.1 M of tetrabutylammonium hexafluorophosphate as a supporting electrolyte. In the case of tz, one quasi-reversible redox feature occurs at −1.195 V vs. Fc/Fc+, which corresponds to the tz/tz˙− redox pair (Fig. S3†). Despite the extension of the voltammogram window to more negative potentials (−2.2 V), no other redox processes were observed (Fig. S3,† inset). However, for 1-Y, two quasi-reversible redox features occur at −1.244 V and +0.199 V vs. Fc/Fc+, which correspond to the tz˙−/tz2− and tz˙−/tz redox pairs, respectively (Fig. 3A). Thus, it is evident that upon complexation, the tz˙− ligand can undergo a second electron addition, which cannot otherwise be observed with the free tz ligand. | ||
| Fig. 3 (A) Cyclic voltammogram of 1-Y measured in DCM at room temperature with [Bu4N][PF6] (0.1 M) as the supporting electrolyte, using a scan rate of 0.1 V s−1. (B) Diffuse reflectance spectra for 1-Y (purple line) and 2-Y (violet line) collected in the 200–800 nm range. (C) Zoomed-in area of the IR spectra of tz (pink), 1-Y (purple) and 2-Y (violet), with the respective wavenumbers. The full spectra are given in Fig. S5.† | ||
The electronic properties of the tz˙− and tz2− rings were investigated by both diffuse reflectance (DR) for 1-Y and 2-Y, as well as UV-vis absorption spectroscopy for 1-Y. For 1-Y, transitions in the UV region can be attributed to absorptions of the Cp* ligands (268, 276, 291 and 341 nm; Fig. S4†). A weak broad absorption band between 450 and 700 nm is ascribed to the tz˙−, which is in accordance with the dark red color of the radical (Fig. S4,† inset). Complementary to this, the DR spectrum for 1-Y reveals broad peaks in the visible range as is expected given the very dark color of tz˙− (Fig. 3B). The DR spectrum for 2-Y also exhibits broad peaks, which, however, are much less pronounced than in 1-Y, suggesting a less electron-rich character for the tz2− in 2-Y compared to the tz˙− in 1-Y (vide infra).
The infrared (IR) spectrum of the neutral tz ligand exhibits strong characteristic bands at 1444 cm−1 (CN-ring stretching), 1197 cm−1 (CH-ring stretching), 1101 cm−1 (NN-ring stretching), and 883 cm−1 (ring bending), consistent with previously published data (Fig. 3C and S5†).84,85 Upon coordination and addition of one or two electrons in 1-Y and 2-Y, respectively, these bands associated with the tz˙− or tz2− ligands were found to be shifted to higher or lower frequencies (Fig. 3C and S5†). More specifically, the CN-ring stretching band was found to be shifted to lower wavenumbers (1434 cm−1) for both 1-Y and 2-Y, in comparison to the neutral tz ligand (1444 cm−1). Similarly, the NN-ring stretching bands and the ring bending band were shifted to lower wavenumbers in 1-Y (1095 and 840 cm−1, respectively) and 2-Y (1066 and 869 cm−1, respectively), in comparison to the neutral tz ligand (1101 and 883 cm−1, respectively). The CH-ring stretching band was found to be shifted to a higher wavenumber in 1-Y (1219 cm−1) and 2-Y (1309 cm−1) compared to the neutral tz ligand (1197 cm−1). Interestingly, a weak broad band at 1589 cm−1 in 1-Y and a strong band at 1581 cm−1 in 2-Y are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching of the tetrazine ligand, which is absent in the neutral tz. This is likely due to the differences in the C–N bond distances where in neutral tz the C–N bond distance is, on average, 1.332(3) Å,36 while in tz˙− and tz2− it is 1.320(3) Å and 1.313(5) Å, respectively. Both spectra of 1-Y (Fig. S6†) and 2-Y (Fig. S7†) exhibit additional bands between 3054 and 2856 cm−1 (C5Me5−), as well as between 1047 and 1022 cm−1 (C–O–C + C–C–H from THF), which are attributed to the Cp* and THF ligands, respectively. Additionally, two strong bands at 704 and 731 cm−1 were found in 1-Y associated with the BPh4− counter ion, which are absent in 2-Y as expected.
N stretching of the tetrazine ligand, which is absent in the neutral tz. This is likely due to the differences in the C–N bond distances where in neutral tz the C–N bond distance is, on average, 1.332(3) Å,36 while in tz˙− and tz2− it is 1.320(3) Å and 1.313(5) Å, respectively. Both spectra of 1-Y (Fig. S6†) and 2-Y (Fig. S7†) exhibit additional bands between 3054 and 2856 cm−1 (C5Me5−), as well as between 1047 and 1022 cm−1 (C–O–C + C–C–H from THF), which are attributed to the Cp* and THF ligands, respectively. Additionally, two strong bands at 704 and 731 cm−1 were found in 1-Y associated with the BPh4− counter ion, which are absent in 2-Y as expected.
It should be noted that efforts to probe the structural or electronic features of the tz2− ring in solution were unsuccessful. It was found that the crystals of complexes 2-Gd or 2-Y were insoluble in all organic solvents except dichloromethane (DCM) and fluorobenzene (F-benzene). However, attempts to dissolve the crystals in these solvents (which are not readily soluble and require stirring to dissolve) resulted in a colour change to bright orange. Attempting to elucidate further the origin of this colour change, crystals of 2-Y were dissolved in F-benzene. Removal of the solvent under vacuum yielded an orange powder for which an IR spectrum was collected and compared to that of 2-Y (Fig. S8†). As evidenced by Fig. S8,† the obtained spectrum is quite complicated. Most importantly, the presence of N–H stretches at 3301 cm−1 is indicative of the presence of amine groups, while the characteristic strong CN band at 1581 cm−1 observed for the crystals of 2-Y is absent. Instead, the strong band at 1708 cm−1 in the IR spectrum of the obtained orange powder represents the CN symmetric stretching.86 Additionally, the bands between 2956 and 2856 cm−1 attributed to the Cp* ligands are relatively shifted for the orange powder in comparison to the respective bands at 2967–2856 cm−1 for the crystals of 2-Y. These suggest that the tz2− most likely dissociates from the metal centres in solution and subsequently reacts further losing its dianionic character. This is in agreement with what is expected given the nature of the tz2−, i.e. that it is a highly reactive species and thus not stable in solution. Consequently, the physical characterization of complexes 2-Gd and 2-Y was based on solid-state measurements.
Thermogravimetric analysis (TGA) was performed to elucidate the thermal stability of 1-Y and 2-Y (Fig. S9†). For complex 1-Y, an initial weight loss of 21% up to 195 °C corresponds to the loss of the two Et2O lattice solvent molecules and the two coordinated THF molecules, which is in excellent agreement with the SCXRD data analysis. Above that temperature, an additional 7% weight loss occurring up to 210 °C corresponds to the loss of the tz˙− ligand. Complex 1-Y undergoes further decomposition through several steps, with an additional weight loss of 44% until 542 °C. For 2-Y, the initial weight loss of 15% up to 84 °C corresponds to the loss of the two THF lattice solvent molecules, followed by an additional 10% weight loss associated with the gradual decomposition of the tz2− ligand up to 205 °C. Above this temperature, an additional decrease of 18% until 328 °C corresponds to the loss of the two coordinated THF solvent molecules. Finally, 2-Y decomposes further with an additional weight loss of 35% until 530 °C. These findings indicate that 2-Y featuring the tz2− ligand is less thermally stable than 1-Y, which features the tz˙− ligand.
EPR spectroscopy and magnetic measurements
The ability of s-tetrazines to store a second electron within their six-membered ring is highly desirable when considering energy storage applications such as organic batteries. Therefore, it is of great importance to elucidate whether this doubly reduced dianionic tetrazine is open (S = 1) or closed-shell (S = 0). To gain further insight into the electronic structure of the tz˙−-bridged complex 1-Y, X-band EPR spectroscopy was employed (Fig. 4A). The presence of a S = 1/2 in 1-Y is supported by the characteristic nine-line pattern of the tz˙− ring, suggesting that the radical interacts strongly with the four 14N nuclei. Simulation of the EPR spectrum using a model based on the hyperfine coupling of four 14N nuclei (Fig. 4A) revealed an isotropic hyperfine coupling constant of aN = 0.584 mT and a g value of 2.0017, which is close to the expected value of a free electron (g = 2.0023). These indicate a negligible interaction with the 89YIII ions and the two 1H for which small hyperfine coupling constants were obtained (Table S3†). Such predominance of the 14N hyperfine coupling in the tz˙− ring has been previously observed for other yttrium complexes with radical tetrazine bridges.69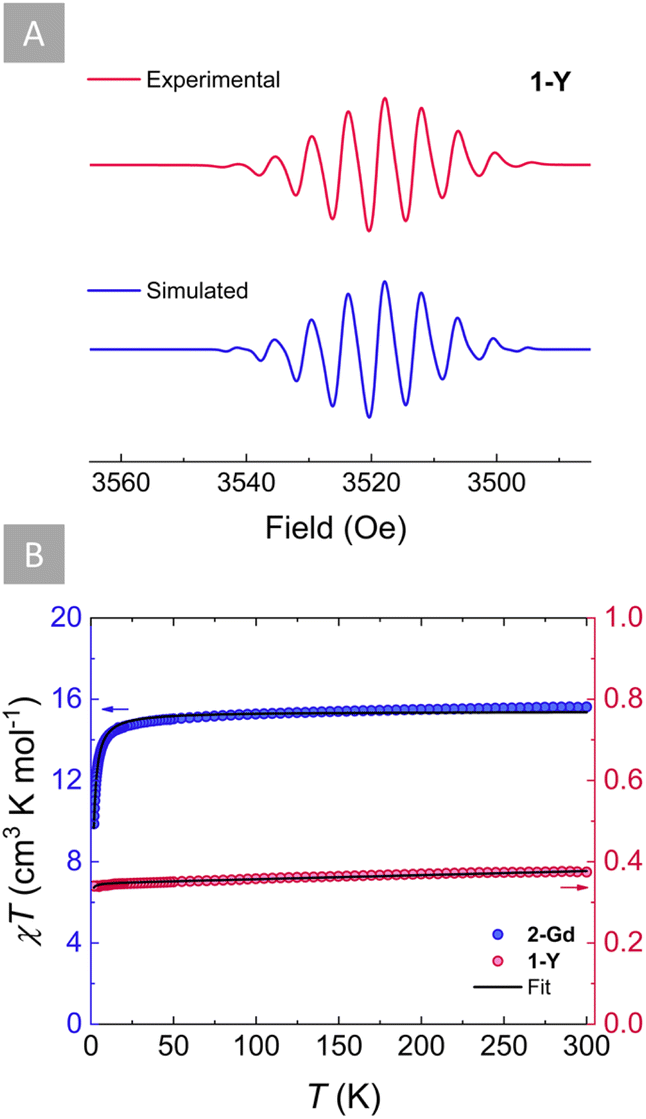 | ||
| Fig. 4 (A) Experimental (magenta) and simulated (blue) EPR spectra for 1-Y at room temperature and 9.86 GHz (in THF) (g = 2.0017; SW = 20 mT; LW = 0.166 mT; aN = 0.584 mT). (B) Variable temperature χT plots of 1-Y (pink circles) and 2-Gd (blue circles) under an applied static field of 1000 Oe. The solid black lines represent the fits for 1-Y and 2-Gd as determined by applying the –2J formalism. | ||
Accordingly, by employing SQUID magnetometry, direct current (dc) magnetic susceptibility measurements were undertaken for 1-Y between 1.8 and 300 K at 1000 Oe (Fig. 4B). The room temperature χT product of 0.38 cm3 K mol−1 is in good agreement with the theoretical value for a S = 1/2 (C = 0.37 cm3 K mol−1). Upon lowering of the temperature, the χT product remains relatively stable, while a very small decrease below 5 K is observed, where the χT product reaches a minimum of 0.34 cm3 K mol−1 at 1.8 K. Fitting of the χT vs. T plot using the PHI software87 (Fig. 4B) revealed small antiferromagnetic intermolecular coupling (zJ′, −0.045 cm−1). Additionally, field-dependent magnetization measurements conducted between 1.9 and 7 K with dc fields ranging from 0 to 70 kOe revealed that the magnetization saturates at 0.83 μB, very close to the calculated value of Ms = 1 μB for an organic radical (Fig. S10†).
Attempts to probe the magnetic behaviour of 2-Y either by EPR spectroscopy or SQUID magnetometry were unsuccessful, since no paramagnetic signal was observed, signifying the diamagnetic nature of the tz2− bridge (S = 0) in 2-Y. To further validate this, dc magnetic susceptibility measurements were undertaken for 2-Gd between 1.8 and 300 K at 1000 Oe (Fig. 4B). At 300 K, the χT product of 15.62 cm3 K mol−1 is in good agreement with the theoretical value of 15.76 cm3 K mol−1 for two non-interacting GdIII ions (S = 7/2, 8S7/2, C = 7.88 cm3 K mol−1), revealing that the tz2− is indeed a closed-shell ligand (S = 0). As the temperature decreases, the χT value remains relatively constant until 200 K. Below that temperature, the χT begins to decrease slightly upon further lowering of the temperature until ∼80 K, where it reaches a value of 15.20 cm3 K mol−1. Beyond this temperature, the χT product decreases rapidly with the temperature drop until it reaches a value of 9.87 cm3 K mol−1 at 1.8 K. This downturn of the χT value at low temperatures can be attributed to the presence of antiferromagnetic intramolecular interactions between the two GdIII ions. To quantify the strength of this magnetic interaction, the χT vs. T plot of 2-Gd was fitted (Fig. 4B) to the spin-only Hamiltonian: Ĥ = –2JŜGdŜGd′ (J represents the intramolecular GdIII–GdIII exchange coupling and Ŝi represents the spin operators for the GdIII ions). The best fit resulted in a small J = −0.048 cm−1 confirming the anticipated weak antiferromagnetic GdIII–GdIII coupling. This small coupling is in line with other Gd2 systems featuring a diamagnetic bridge,88–91 further corroborating the closed-shell nature of tz2− (S = 0). The small antiferromagnetic GdIII–GdIII coupling obtained for 2-Gd (J = −0.048 cm−1) is vastly different from the small ferromagnetic GdIII–GdIII coupling (JGd–Gd = 0.32 cm−1) previously obtained for 1-Gd, which results from the strong antiferromagnetic GdIII-tz˙− coupling (JGd-rad = −7.2 cm−1).71
Electronic structure analysis
In order to provide additional evidence of the −2 charge state of the tetrazine bridge, density functional theory (DFT) calculations were carried out. The structures were optimized at the PBE/TZ2P level.92–94 Calculations were performed on three structures: the hypothetical [Y2]2+, with the tetrazine in a neutral charge state; [Y2]+, with the tetrazine as a radical; and [Y2], with the tetrazine as a dianion. Test calculations were conducted to confirm that both in [Y2]2+ and [Y2], the tetrazine bridge is in a diamagnetic singlet state. The calculated bond distances within the tetrazine bridge, as well as between the tetrazine and the YIII ions in both [Y2]+ and [Y2], are in good agreement with the parameters in the respective crystal structures (Table S4†). The calculated structural parameters of [Y2]2+ deviate considerably from those in the crystal structures of [Y2]+ and [Y2]. Furthermore, the IR absorptions calculated for [Y2]+ and [Y2] (Fig. S11 and S12†) are in reasonably good agreement with their respective experimental IR absorption spectra and are significantly different from the calculated IR absorptions for [Y2]2+ (Fig. S13†).Additionally, the charges of the Y ions and the tetrazine bridge were evaluated at the PBE0/TZ2P level92–96 using the optimized geometries and the quantum theory of atoms in molecules (QTAIM, Table S5†).97,98 In all three structures, the charge of the Y ion is relatively constant: +1.97, +1.98 and +2.01 for [Y2]2+, [Y2]+ and [Y2], respectively. The charge, that is smaller than the formal +3 charge, is indicative of metal–ligand covalency. The minimal variation of the charge in the three structures is strong evidence that the oxidation state of the Y ion remains as +3 in all three complexes, and that the Y ion is not involved in any redox chemistry. The total charge of the tetrazine bridge varies as follows: −0.47, −0.78, and −1.49 for [Y2]2+, [Y2]+ and [Y2], respectively. The values are less negative than the formal charge states, yet they exhibit a clear trend. Most importantly, going from the radical monoanion to the dianion leads to the doubling of the ligand charge in agreement with the −1 to −2 reduction. Thus, the one-electron reduction from [Y2]+ to [Y2] corresponds to the reduction of the monoanionic tetrazine radical to a dianionic tetrazine radical.
In order to probe the aromaticity of the tetrazine ligand in [Y2]2+, [Y2]+ and [Y2], the nuclear-independent chemical shift (NICS)99 was calculated at the ring centre at the PBE0/TZ2P level of theory. The results are listed in Table S7.† The values are 16.4 ppm, 25.0 ppm, and 25.8 ppm for [Y2]2+, [Y2]+ and [Y2], respectively. The values for the freely optimized ligands in the respective charge states are −1.4 ppm, 14.5 ppm, and 9.0 ppm. Thus, aromaticity is only observed in the free neutral tetrazine ligand, and even there in a very small amount. In the complexes, the anti-aromaticity increases with increasing ligand charge. This trend is consistent with the DR spectra where for 1-Y much more pronounced peaks indicate a more electron-rich character of the tz˙− compared to the tz2− in 2-Y (Fig. 3B). For comparison, the values calculated for neutral pyrazine and pyridine at the same level of theory are −5.0 ppm and −6.6 ppm, respectively, indicating that the aromaticity decreases with the increasing number of nitrogen atoms in the ring, as well as with the increasing ligand charge and coordination.
Conclusions
In summary, three new s-tetrazine-bridged dinuclear metallocene complexes have been reported. Although complexes 1-Y and 2-Y are seemingly similar, the tz ring is in its −1 radical charge state in 1-Y, while in 2-Y it has an unprecedented −2 charge state. The combination of strictly anhydrous and inert conditions, strong reducing agents, non-acidic solvents, and most importantly blocking the accessibility of the nitrogen atoms upon coordination to LnIII ions allowed for the stabilization of this dianionic tetrazine in a lanthanocene complex. CV studies on 1-Y reveal that the tz ring can undergo a second electron addition, while DR and IR spectroscopy studies elucidate the electronic differences between the tz˙− and tz2− species. Furthermore, TG analysis reveals that complex 2-Y, featuring the tz2−, exhibits lower thermal stability compared to complex 1-Y, featuring the tz˙− species. EPR and SQUID magnetometry studies reveal that in 2-Ln, the tz2− is a closed-shell bridge, in contrast to the clearly paramagnetic tz˙− species in 1-Ln. The theoretical calculations corroborate the experimental findings, confirming the observed structural differences that suggest the presence of a −2 charge in tz2− and shed light into the anti-aromaticity of this species.Through this work, the first structurally and physically characterized complexes bearing the dianion radical of an s-tetrazine are presented and highlight the extraordinary ability of tetrazines to store more than one e− within their six-membered ring. Although previous literature reports have hypothesized the formation of a highly reactive dianionic tetrazine, the isolation of this species had not been reported. Overcoming the reactivity and stability issues by promoting the coordination of the tz ring with LnIII metal ions opens new possibilities for exploring these fascinating ligands in next-generation energy storage devices and smart materials.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
CRediT: N. M. – conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft. A. A. K. – conceptualization, formal analysis, methodology, writing – original draft. A. M. – data curation, formal analysis, writing original draft. M. M. – funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. M., A. A. K. and M. M. acknowledge the Canada Foundation for Innovation (CFI) and the Natural Sciences and Engineering Research Council of Canada (NSERC) for their financial support. N. M. acknowledges the Stavros Niarchos Foundation for financial support through scholarships. A. M. acknowledges funding provided by the Academy of Finland (grant no. 332294) and the University of Oulu (Kvantum Institute). Computational resources were provided by CSC-IT Center for Science in Finland and the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533). We are grateful to Dr Peter G. Gordon and Kieran G. Lawford for the collection of the TGA data.References
- A. Pinner, Ber. Dtsch. Chem. Ges., 1897, 30, 1871–1890 CrossRef CAS.
- A. Pinner, Adv. Cycloaddit., 1897, 297, 221–271 CAS.
- J. Mondal and A. Sivaramakrishna, Top. Curr. Chem., 2022, 380, 34 CrossRef CAS PubMed.
- A. R. Katritzky, Handbook of heterocyclic chemistry, Elsevier, Oxford, 3rd edn, 2010 Search PubMed.
- W. Kaim, Coord. Chem. Rev., 2002, 230, 127–139 CrossRef CAS.
- G. Clavier and P. Audebert, Chem. Rev., 2010, 110, 3299–3314 CrossRef CAS.
- O. Stetsiuk, A. Abhervé and N. Avarvari, Dalton Trans., 2020, 49, 5759–5777 RSC.
- F. Miomandre and P. Audebert, J. Photochem. Photobiol., C, 2020, 44, 100372 CrossRef CAS.
- E. W. Stone and A. H. Maki, J. Chem. Phys., 1963, 39, 1635–1642 CrossRef CAS.
- E. Kurach, D. Djurado, J. Rimarčik, A. Kornet, M. Wlostowski, V. Lukeš, J. Pécaut, M. Zagorska and A. Pron, Phys. Chem. Chem. Phys., 2011, 13, 2690–2700 RSC.
- Q. Zhou, P. Audebert, G. Clavier, R. Méallet-Renault, F. Miomandre, Z. Shaukat, T.-T. Vu and J. Tang, J. Phys. Chem. C, 2011, 115, 21899–21906 CrossRef CAS.
- B. J. Jordan, M. A. Pollier, L. A. Miller, C. Tiernan, G. Clavier, P. Audebert and V. M. Rotello, Org. Lett., 2007, 9, 2835–2838 CrossRef CAS PubMed.
- E. Jullien-Macchi, V. Alain-Rizzo, C. Allain, C. Dumas-Verdes and P. Audebert, RSC Adv., 2014, 4, 34127–34133 RSC.
- D. L. Boger, Chem. Rev., 1986, 86, 781–793 CrossRef CAS.
- J. Sauer, in Comprehensive Heterocyclic Chemistry II, Elsevier, 1996, pp. 901–955 Search PubMed.
- M. D. Helm, A. Plant and J. P. A. Harrity, Org. Biomol. Chem., 2006, 4, 4278–4280 RSC.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- F. Liu, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 11483–11493 CrossRef CAS.
- O. Roling, A. Mardyukov, S. Lamping, B. Vonhören, S. Rinnen, H. F. Arlinghaus, A. Studer and B. J. Ravoo, Org. Biomol. Chem., 2014, 12, 7828–7835 RSC.
- D. Wang, W. Chen, Y. Zheng, C. Dai, K. Wang, B. Ke and B. Wang, Org. Biomol. Chem., 2014, 12, 3950–3955 RSC.
- D. E. Chavez and M. A. Hiskey, J. Energ. Mater., 1999, 17, 357–377 CrossRef CAS.
- D. E. Chavez, M. A. Hiskey and R. D. Gilardi, Org. Lett., 2004, 6, 2889–2891 CrossRef CAS PubMed.
- H. Gao, R. Wang, B. Twamley, M. A. Hiskey and J. M. Shreeve, Chem. Commun., 2006, 38, 4007–4009 RSC.
- D. Herweyer, A. A. Kitos, P. Richardson, H. Canoe, J. S. Ovens, I. Laroche, B. Jolicoeur, M. Murugesu and J. L. Brusso, Cryst. Growth Des., 2023, 23, 2576–2582 CrossRef CAS.
- M. H. V. Huynh, M. A. Hiskey, D. E. Chavez, D. L. Naud and R. D. Gilardi, J. Am. Chem. Soc., 2005, 127, 12537–12543 CrossRef CAS.
- D. E. Chavez, S. K. Hanson, J. M. Veauthier and D. A. Parrish, Angew. Chem., Int. Ed., 2013, 52, 6876–6879 CrossRef CAS.
- C. J. Snyder, D. E. Chavez, G. H. Imler, E. F. C. Byrd, P. W. Leonard and D. A. Parrish, Chem.–Eur. J., 2017, 23, 16466–16471 CrossRef CAS PubMed.
- J. Ding, N. Song and Z. Li, Chem. Commun., 2010, 46, 8668–8670 RSC.
- W. Cheng, Z. Wu, S. Wen, B. Xu, H. Li, F. Zhu and W. Tian, Org. Electron., 2013, 14, 2124–2131 CrossRef CAS.
- P. Audebert and F. Miomandre, Chem. Sci., 2013, 4, 575–584 RSC.
- Y. Kim, J. Do, E. Kim, G. Clavier, L. Galmiche and P. Audebert, J. Electroanal. Chem., 2009, 632, 201–205 CrossRef CAS.
- S. Seo, C. Allain, J. Na, S. Kim, X. Yang, C. Park, J. Malinge, P. Audebert and E. Kim, Nanoscale, 2013, 5, 7321–7327 RSC.
- M. A. Withersby, A. J. Blake, N. R. Champness, P. A. Cooke, P. Hubberstey, W.-S. Li and M. Schröder, Inorg. Chem., 1999, 38, 2259–2266 CrossRef CAS.
- D. L. Boger, C. W. Boyce, M. A. Labroli, C. A. Sehon and Q. Jin, J. Am. Chem. Soc., 1999, 121, 54–62 CrossRef CAS.
- W.-X. Hu, G.-W. Rao and Y.-Q. Sun, Bioorg. Med. Chem. Lett., 2004, 14, 1177–1181 CrossRef CAS.
- I. A. Gural’skiy, D. Escudero, A. Frontera, P. V. Solntsev, E. B. Rusanov, A. N. Chernega, H. Krautscheid and K. V. Domasevitch, Dalton Trans., 2009, 15, 2856–2864 RSC.
- Y.-H. Gong, F. Miomandre, R. Méallet-Renault, S. Badré, L. Galmiche, J. Tang, P. Audebert and G. Clavier, Eur. J. Org Chem., 2009, 2009, 6121–6128 CrossRef.
- Y. Zhao, Y. Li, Z. Qin, R. Jiang, H. Liu and Y. Li, Dalton Trans., 2012, 41, 13338–13342 RSC.
- J. E. Clements, J. R. Price, S. M. Neville and C. J. Kepert, Angew. Chem., Int. Ed., 2014, 53, 10164–10168 CrossRef CAS PubMed.
- T. Okubo, K. Himoto, K. Tanishima, S. Fukuda, Y. Noda, M. Nakayama, K. Sugimoto, M. Maekawa and T. Kuroda-Sowa, Inorg. Chem., 2018, 57, 2373–2376 CrossRef CAS PubMed.
- E. Parvizi, R. Tayebee, E. Koushki, M. F. Abdizadeh, B. Maleki, P. Audebert and L. Galmiche, RSC Adv., 2019, 9, 23818–23831 RSC.
- G. Beliu, A. J. Kurz, A. C. Kuhlemann, L. Behringer-Pliess, M. Meub, N. Wolf, J. Seibel, Z.-D. Shi, M. Schnermann, J. B. Grimm, L. D. Lavis, S. Doose and M. Sauer, Commun. Biol., 2019, 2, 1–13 CrossRef CAS.
- M. Vinu, K. Sivasankar, S. Prabu, J.-L. Han, C.-H. Lin, C.-C. Yang and J. Demel, Eur. J. Inorg. Chem., 2020, 2020, 461–466 CrossRef CAS.
- N. D. Calvert, A. Kirby, M. Suchý, P. Pallister, A. A. Torrens, D. Burger, G. Melkus, N. Schieda and A. J. Shuhendler, Nat. Commun., 2023, 14, 3965 CrossRef CAS.
- H. Jiang, Q. Gong, R. Zhang and H. Yuan, Coord. Chem. Rev., 2024, 499, 215501 CrossRef CAS.
- N. Mavragani, A. A. Kitos, J. L. Brusso and M. Murugesu, Chem.–Eur. J., 2021, 27, 5091–5106 CrossRef CAS.
- X. Ma, E. A. Suturina, M. Rouzières, M. Platunov, F. Wilhelm, A. Rogalev, R. Clérac and P. Dechambenoit, J. Am. Chem. Soc., 2019, 141, 7721–7725 CrossRef CAS PubMed.
- M. Schwach, H.-D. Hausen and W. Kaim, Inorg. Chem., 1999, 38, 2242–2243 CrossRef CAS.
- S. Ye, W. Kaim, B. Sarkar, B. Schwederski, F. Lissner, T. Schleid, C. Duboc-Toia and J. Fiedler, Inorg. Chem. Commun., 2003, 6, 1196–1200 CrossRef CAS.
- K. C. Gordon, A. K. Burrell, T. J. Simpson, S. E. Page, G. Kelso, M. I. J. Polson and A. Flood, Eur. J. Inorg. Chem., 2002, 2002, 554–563 CrossRef.
- M. Maekawa, H. Konaka, T. Minematsu, T. Kuroda-Sowa, Y. Suenaga and M. Munakata, Inorg. Chim. Acta, 2005, 358, 1317–1321 CrossRef CAS.
- M. Newell, J. D. Ingram, T. L. Easun, S. J. Vickers, H. Adams, M. D. Ward and J. A. Thomas, Inorg. Chem., 2006, 45, 821–827 CrossRef CAS PubMed.
- D. I. Alexandropoulos, B. S. Dolinar, K. R. Vignesh and K. R. Dunbar, J. Am. Chem. Soc., 2017, 139, 11040–11043 CrossRef CAS.
- C. S. Campos-Fernández, B. L. Schottel, H. T. Chifotides, J. K. Bera, J. Bacsa, J. M. Koomen, D. H. Russell and K. R. Dunbar, J. Am. Chem. Soc., 2005, 127, 12909–12923 CrossRef.
- H. T. Chifotides, I. D. Giles and K. R. Dunbar, J. Am. Chem. Soc., 2013, 135, 3039–3055 CrossRef CAS PubMed.
- M. Glöckle, K. Hübler, H.-J. Kümmerer, G. Denninger and W. Kaim, Inorg. Chem., 2001, 40, 2263–2269 CrossRef.
- T. J. Woods, H. D. Stout, B. S. Dolinar, K. R. Vignesh, M. F. Ballesteros-Rivas, C. Achim and K. R. Dunbar, Inorg. Chem., 2017, 56, 12094–12097 CrossRef CAS.
- M. A. Lemes, G. Brunet, A. Pialat, L. Ungur, I. Korobkov and M. Murugesu, Chem. Commun., 2017, 53, 8660–8663 RSC.
- K. Chainok, S. M. Neville, C. M. Forsyth, W. J. Gee, K. S. Murray and S. R. Batten, CrystEngComm, 2012, 14, 3717–3726 RSC.
- D. A. Safin, A. Pialat, A. A. Leitch, N. A. Tumanov, I. Korobkov, Y. Filinchuk, J. L. Brusso and M. Murugesu, Chem. Commun., 2015, 51, 9547–9550 RSC.
- O. Stetsiuk, S. R. Petrusenko, L. Sorace, A. Lupan, A. A. A. Attia, V. N. Kokozay, A. El-Ghayoury and N. Avarvari, Dalton Trans., 2019, 48, 11966–11977 RSC.
- T. Lacelle, G. Brunet, A. Pialat, R. J. Holmberg, Y. Lan, B. Gabidullin, I. Korobkov, W. Wernsdorfer and M. Murugesu, Dalton Trans., 2017, 46, 2471–2478 RSC.
- T. Lacelle, G. Brunet, R. J. Holmberg, B. Gabidullin and M. Murugesu, Cryst. Growth Des., 2017, 17, 5044–5048 CrossRef CAS.
- N. M. Shavaleev, S. J. A. Pope, Z. R. Bell, S. Faulkner and M. D. Ward, Dalton Trans., 2003, 5, 808–814 RSC.
- B. S. Dolinar, S. Gómez-Coca, D. I. Alexandropoulos and K. R. Dunbar, Chem. Commun., 2017, 53, 2283–2286 RSC.
- M. A. Lemes, N. Mavragani, P. Richardson, Y. Zhang, B. Gabidullin, J. L. Brusso, J. O. Moilanen and M. Murugesu, Inorg. Chem. Front., 2020, 7, 2592–2601 RSC.
- G. Brunet, M. Hamwi, M. A. Lemes, B. Gabidullin and M. Murugesu, Commun. Chem., 2018, 1, 1–6 CrossRef CAS.
- J.-T. Chen, H. Yan, T.-T. Wang, T.-D. Zhou and W.-B. Sun, Inorg. Chem., 2022, 61, 19097–19105 CrossRef CAS PubMed.
- N. Mavragani, A. A. Kitos, J. Hrubý, S. Hill, A. Mansikkamäki, J. O. Moilanen and M. Murugesu, Inorg. Chem. Front., 2023, 10, 4197–4208 RSC.
- H.-D. Li, S.-G. Wu and M.-L. Tong, Chem. Commun., 2023, 59, 6159–6170 RSC.
- N. Mavragani, A. A. Kitos, A. Mansikkamäki and M. Murugesu, Inorg. Chem. Front., 2023, 10, 259–266 RSC.
- N. Mavragani, D. Errulat, D. A. Gálico, A. A. Kitos, A. Mansikkamäki and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 24206–24213 CrossRef CAS.
- N. Mavragani, A. A. Kitos, D. A. Gálico, A. Mansikkamäki and M. Murugesu, Chem. Commun., 2023, 59, 13970–13973 RSC.
- T. Troll, Electrochim. Acta, 1982, 27, 1311–1314 CrossRef CAS.
- K. B. Wiberg and T. P. Lewis, J. Am. Chem. Soc., 1970, 92, 7154–7160 CrossRef CAS.
- H. Fischer, T. Müller, I. Umminger, F. A. Neugebauer, H. Chandra and M. C. R. Symons, J. Chem. Soc., Perkin Trans. 2, 1988, 3, 413–421 RSC.
- C. R. Benson, A. K. Hui, K. Parimal, B. J. Cook, C.-H. Chen, R. L. Lord, A. H. Flood and K. G. Caulton, Dalton Trans., 2014, 43, 6513–6524 RSC.
- R. Gleiter, V. Schehlmann, J. Spanget-Larsen, H. Fischer and F. A. Neugebauer, J. Org. Chem., 1988, 53, 5756–5762 CrossRef CAS.
- S. Fukuzumi, J. Yuasa and T. Suenobu, J. Am. Chem. Soc., 2002, 124, 12566–12573 CrossRef CAS PubMed.
- S. Samanta, S. Ray, A. B. Ghosh and P. Biswas, RSC Adv., 2016, 6, 39356–39363 RSC.
- S. Kraft, E. Hanuschek, R. Beckhaus, D. Haase and W. Saak, Chem.–Eur. J., 2005, 11, 969–978 CrossRef CAS PubMed.
- F. A. Neugebauer, C. Kriger, H. Fischer and R. Siegel, Chem. Ber., 1983, 116, 2261–2274 CrossRef CAS.
- W. Kaim, J. Chem. Soc., Perkin Trans. 2, 1985, 10, 1633–1637 RSC.
- W. D. Sigworth and E. L. Pace, Spectrochim. Acta, Part A, 1971, 27, 747–758 CrossRef CAS.
- L. A. Franks, A. J. Merer and K. K. Innes, J. Mol. Spectrosc., 1968, 26, 458–464 CrossRef CAS.
- J. Coates, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS.
- C. Y. Chow, H. Bolvin, V. E. Campbell, R. Guillot, J. W. Kampf, W. Wernsdorfer, F. Gendron, J. Autschbach, V. L. Pecoraro and T. Mallah, Chem. Sci., 2015, 6, 4148–4159 RSC.
- T.-F. Zheng, X.-M. Tian, J. Ji, H. Luo, S.-L. Yao, S.-J. Liu, B.-E. Tang, Y.-J. Zhao, J. Mao, Q. Zhao, K.-H. He and H.-R. Wen, J. Mol. Struct., 2020, 1200, 127094 CrossRef CAS.
- I. Mylonas-Margaritis, J. Mayans, S.-M. Sakellakou, C. P. Raptopoulou, V. Psycharis, A. Escuer and S. P. Perlepes, Magnetochemistry, 2017, 3, 5 CrossRef.
- T. Ekanayaka, T. Jiang, E. Delahaye, O. Perez, J.-P. Sutter, D. Le, A. T. N'Diaye, R. Streubel, T. S. Rahman and P. A. Dowben, Phys. Chem. Chem. Phys., 2023, 25, 6416–6423 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. F. W. Bader, Atoms in molecules: a quantum theory, Clarendon Press, Oxford, 2003, reprinted Search PubMed.
- P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, single-crystal X-ray diffraction data, cyclic voltammograms, additional magnetic, spectroscopic, and computational data. CCDC 2358823, 2358824 and 2358825. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03734k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |