
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIs aromaticity loss necessary for transition-metal promoted arene–alkene cycloadditions?†
João V.
Schober
 *,
Croix J.
Laconsay
and
Judy I.
Wu
*
*,
Croix J.
Laconsay
and
Judy I.
Wu
*
Department of Chemistry, University of Houston, Houston, TX 77204, USA. E-mail: jiwu@central.uh.edu; jvoliveisoares@uh.edu
First published on 8th October 2024
Abstract
Computed gas-phase reaction profiles for Diels–Alder reactions, nucleus-independent chemical shifts, and bonding analyses for substituted and metal complexed, η2-, η4-, η6-coordinated arenes reveal that dearomatization is necessary for arenes to exhibit ene- and diene-like reactivity. Substituted arenes and η6-arenes exhibit high free energy barriers towards cycloaddition reactions, but strongly dearomatized η2- and η4-arenes can display low free energy barriers (∼20 kcal mol−1 or less) for [4 + 2] cycloaddition reactions.
Introduction
Arenes are rarely envisioned as dienes or dienophiles for Diels–Alder reactions because of the energetic penalty for disrupting aromaticity. For this reason, known examples of arene Diels–Alder reactions typically involve harsh conditions (i.e., high temperature and pressure),1,2 photoreactions,3–6 strained scaffolds,7,8 or the use of highly potent dienes or dienophiles.9 Transition metal coordination can profoundly alter the reactivity of aromatic systems,10–14 and in some cases, instill diene- or ene-like reactivity in much milder conditions.15–19 It was suggested that increased diene- and ene-like reactivity is linked to a loss of aromaticity in the metal coordinated arene, but such relationships have only been speculated. In this computational investigation, we show that diene- and ene-like reactivity in transition metal coordinated arenes can only be expected when the metal complexed arene displays a notable degree of aromaticity loss.Even though examples of dihapto (η2) and tetrahapto (η4) coordinated arenes showing diene- and ene-like reactivity are known, the role of aromaticity loss of the coordinated arene in driving such reactivity has never been quantified. In a series of papers, Harman et al. reported that η2-arenes, heteroarenes, and fused arenes could mimic the cycloaddition reactivity of dienes and undergo Diels–Alder reactions (Fig. 1a and b)11,16–18,20 Cooper et al. found that η4-Mn(CO)3-benzene could undergo two sequential electrophilic addition steps followed by ring closure to yield cyclohexadienyl complexes via a [2 + 2 + 2] cycloaddition (Fig. 1c).15 Fe(CO)3 complexes of tropone show enone-like reactivity.21,22 Yet, not all metal-complexed arenes show diene- or ene-like reactivity. η6-Cr(CO)3, η6-Mn(CO)3+, η6-FeCp+, and η6-RuCp+ readily undergo nucleophilic substitution and addition reactions to form substituted benzenes and cyclohexadienes, but do not show diene- or ene-like reactivity.10 Likewise, substituents can effectively promote electrophilic and nucleophilic reactions in arenes,23 but do not show diene- or ene-like reactivity either. What are the necessary criteria for making arenes react like dienes and dienophiles?
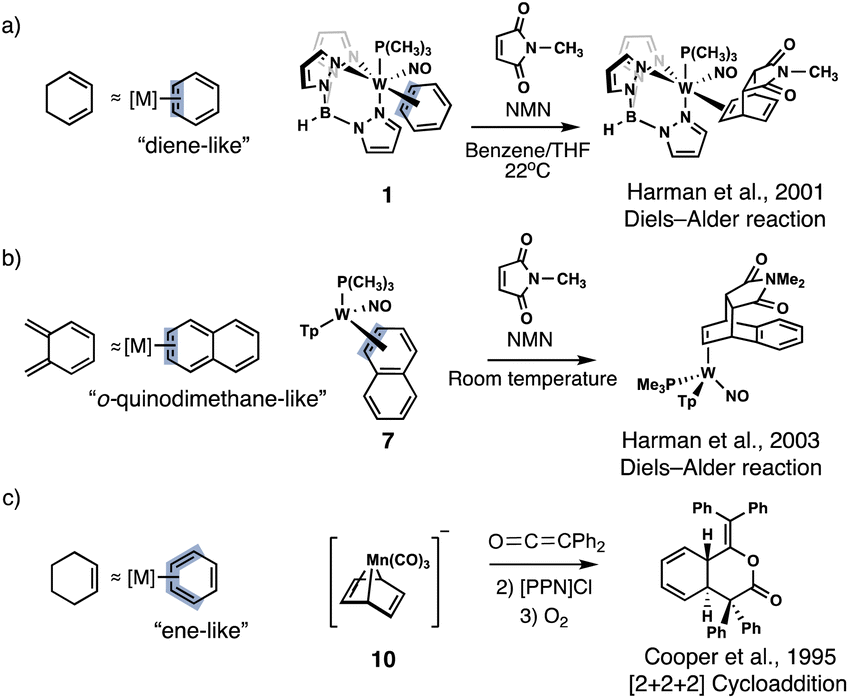 | ||
| Fig. 1 Examples of η2- and η4- arenes and fused arenes showing diene- and ene-like reactivity. | ||
Results and discussion
We selected three aromatic systems (benzene, naphthalene, indole) and first examined the dearomatization effects of η2 (1–8), η4 (9–14), and η6 (15–17) coordination, as quantified by dissected nucleus-independent chemical shifts,24 NICS(1)zz, a magnetic index of aromaticity that measures the diatropicity and paratropicity of a system (Fig. 2). Negative NICS(1)zz values indicate aromaticity (diatropicity), and positive NICS(1)zz values indicate antiaromaticity (paratropicity). Noting that a nearby transition-metal could cause deshielding effects on the computed NICS values,25 NICS probes were placed 1 Å above the π-ring opposite to the transition-metal coordinating side. The complexes shown in Fig. 2 are organized based on increasing effective nuclear charge of the metal (left to right across a Row and up to down across a Group). Large negative NICS(1)zz values indicate that all of the uncomplexed parent compounds are strongly aromatic (Fig. 2). Computed NICS(1)zz values for selected substituted benzenes: aniline (−25.7 ppm), nitrobenzene (−28.8 ppm), and p-nitrobenzene (−22.5 ppm) are only modestly less negative compared to benzene and exemplify the robustness of aromaticity towards ring substitution.26 A small decrease in diatropicity occurs only when push–pull groups are placed strategically around the arene; and even then, there is only modest aromaticity loss. | ||
| Fig. 2 NICS(1)zz values for selected arenes and their η2-, η4-, and η6-complexes. | ||
The η2-benzenes (1–6) exhibit a range of NICS(1)zz values. Going across a row, the series: 1 (−10.7 ppm), 2 (−16.3 ppm), 3 (−24.9 ppm) (W → Re → Os), and the series: 4 (+5.3 ppm), 5 (−11.2 ppm), 6 (−26.5 ppm) (Cr → Mn → Fe), both show increasing diatropicity at the arene as the metal identity shifts from an early to late transition metal. Sums of Wiberg Bond Index (WBI)27 values for the two metal-carbon contacts in each of the complexes decrease in the order: 1 (sum of WBI = 1.23, W) > 2 (1.17, Re) > 3 (0.82, Os) (Row 6), and 4 (1.03, Cr) > 5 (0.92, Mn) > 6 (0.54, Fe) (Row 4), suggesting that metals with a lower effective nuclear charge have stronger back-bonding interactions with the arene (i.e., metal dπ orbital to arene pπ*). These findings are consistent with prior studies showing that 1 and 2 readily undergo Diels–Alder reactions with N-methylmaleimide (NMM) at room temperature, while 3 is unreactive.16,17
Fused arenes also can be activated for electrophilic addition by η2-coordination. Harman et al. reported a Diels–Alder reactions for 7 with NMM.18 The authors speculated that the thermodynamically favored 1,2-η2-coordinated species, 7, first ring walks to a 2,3-η2-binding mode over a ∼19 kcal mol−1 barrier, and then NMM adds to the complexed ring (ring A, see 7′ in Fig. 2 and S2†).18 Experiments carried out at elevated temperatures produced the cycloadduct with shorter reaction times.18 Computed NICS(1)zz values for the 2,3-η2-binding mode of 7 (ring A: −4.9 ppm, ring B: −27.2 ppm) and 7′ (ring A: −18.9 ppm, ring B: −9.3 ppm) suggest that ring walking from a 1,2-η2 to a 2,3-η2 binding mode is a relevant step. In 7, adding NMM to ring A disrupts aromaticity in ring B (ring B in adduct: −1.3 ppm). But in 7′, adding NMM to ring A restores aromaticity in ring B (ring B in adduct: −30.9 ppm). No Diels–Alder reactions were observed for 8 with NMM.18,28 Computed NICS(1)zz values for 8′ (ring A: −25.2 ppm, ring B: −13.4 ppm) reveal a strongly aromatic ring A, which may explain the absence of a Diels–Alder reaction.
The η4-benzenes (9–14) all show largely reduced diatropicities as the coordinated ring distorts from planarity. To estimate the NICS(1)zz values, an “out-of-plane” tensor component (zz) was defined by placing carbons C1, C2, C4, and C5 in the xy plane (see ring carbon numbering for 9, Fig. 2). Going across Row 4: 9 (−3.5 ppm), 10 (−7.4 ppm), 11 (−7.8 ppm) (Cr → Mn → Fe), and down Group 9: 12 (−5.6 ppm), 13 (−9.6 ppm), 14 (−13.2 ppm) (Co → Rh → Ir), increased effective nuclear charge is accompanied by a more negative NICS(1)zz values. Since all of the η4-arenes are nonplanar, we realize that the trends of the estimated NICS(1)zz values do not necessarily reflect changes in aromaticity. Nevertheless, it is reasonable to expect that all of the η4-arenes are largely dearomatized.
The η6-arenes (15-17) show minimal dearomatization (Fig. 2). Complexation to the electron-withdrawing Cr(CO)3 group has been shown to activate arenes towards nucleophilic addition.29 Yet, known reactions typically involve the formation of an η5-coordinated complex and the η6-binding mode is not the reacting species. Though, notably, it has been pointed out that Pauli repulsion lowering contributes to the interaction energies in different Ru moieties η6-coordinated to corannulene.30 In agreement with prior studies,3115 (NICS(1)zz = −26.0 ppm) is nearly as aromatic as benzene (−30.3 ppm). The computed NICS(1)zz values for 16 (ring A: −24.8 ppm, ring B: −27.3 ppm) are comparable to those of naphthalene (−29.5 ppm), and those for 17 (ring A: −25.6 ppm, ring B: −28.2 ppm) are comparable to indole (ring A: −30.3 ppm, ring B: −29.6 ppm). All three η6-complexes show only a slight decrease in negative NICS(1)zz values for the complexed arene (ring A); in 16 and 17, ring B retains aromaticity and thus is unlikely to show “diene-like” reactivity. Hydrogenation and substitution reactions for 16 and 17 have been observed,32,33 but their dearomatized analogs are even more reactive. This is consistent with experiments showing that tricarbonyl(naphthoquinone)chromium complexes could add to cyclic and acyclic dienes giving endo Diels–Alder products in good yield.33
We further note that the computed NICS(1)zz values for complexes 1-8, 7′, 8′, 15, 16, benzene, naphthalene, and the substituted benzenes (aniline, nitrobenzene, and p-nitroaniline) correlate satisfactorily with geometric indices of aromaticity, based on computed harmonic oscillator measure of aromaticity (HOMA)34 values (r2 = 0.771) (see data in the ESI†).
We next computed gas-phase reaction profiles for Diels–Alder reactions for benzene, p-nitroaniline, and selected η2-, η4- and η6-arenes. Activation free energy barriers (ΔG‡) were calculated based on the energy of the transition state structure relative to the σ-complex of the reactants. Since some of the reactions studied can proceed in an endo or exo fashion, we show the lower energy reaction pathway in Fig. 3a–d and include data for the alternative pathway in the ESI.† The reaction profile for a Diels–Alder reaction for benzene and cyclopentadiene is endothermic and reveals a high barrier (ΔG‡ = 46.9 kcal mol−1, Fig. 3a). This is comparable to the computed Diels–Alder reaction barrier for p-nitroaniline and cyclopentadiene, which also shows a high barrier (ΔG‡ = 41.0 kcal mol−1, endo, Fig. 3a), as anticipated by the largely intact aromatic character of the substituted arene. Computed HOMO and LUMO energies for benzene and p-nitroaniline reveal a lower HOMO–LUMO energy gap for p-nitroaniline, consistent with its higher reactivity (Table 1).
 | ||
| Fig. 3 Computed reaction profiles for Diels–Alder reactions at ωB97X-D/def2-TZVPP//ωB97X-D/def2-SVP. Single-point energies at DLPNO-CCSD(T)/def2-TZVPP//ωB97X-D/def2-SVP are in parenthesis. Cf.Fig. 4 shows computed transition state structures. All reaction barriers are computed relative to a σ-complex of the reactants. | ||
| Compound | HOMO | LUMO | ΔEH–L |
|---|---|---|---|
| Benzene | −8.49 | −3.00 | 5.49 |
| p-Nitroaniline | −7.84 | −3.37 | 4.47 |
| 1 | −5.54 | −2.08 | 3.46 |
| 2 | −5.47 | −1.37 | 4.10 |
| 3 | −13.80 | −10.4 | 3.37 |
| 14 | −6.40 | −2.96 | 3.44 |
| Naphthalene | −7.43 | −2.82 | 4.61 |
| 7′ | −5.07 | −2.64 | 2.43 |
| 16 | −7.04 | −4.19 | 2.85 |
Computed energy profiles of Diels–Alder reactions for NMM with benzene and the η2-benzenes (1–3) show decreasing ΔG‡ values for benzene (38.3 kcal mol−1) > 3 (32.8 kcal mol−1, endo) > 2 (23.3 kcal mol−1, exo) > 1 (22.9 kcal mol−1, exo) (Fig. 3b), which is consistent with decreasing aromatic character of the arene ring: benzene > 3 > 2 > 1 (cf.Fig. 2b). Dearomatization prompts an earlier transition state, as suggested by the longer incipient C–C bonds of the transition state structures of 1 > 2 > 3 (Fig. 4). These findings are consistent with the observed “diene” reactivity of 1 and 2versus the unreactive 3.16,17 Notably, the computed HOMO–LUMO energy gaps for 1 (3.46 eV), 2 (4.10 eV), and 3 (3.37 eV) are all lower than that of benzene (5.46 eV) (Table 1). Yet, there is no clear relationship between the HOMO–LUMO energy gaps of 1–3 and their computed Diels–Alder reaction barriers; 3 exhibits a lower HOMO–LUMO energy gap than 1 and 2 (Table 1), but a higher reaction barrier (Fig. 3b). In this regard, computed NICS(1)zz values, which quantify the extent of aromaticity loss, appear to be a more effective predictor for “diene-like” reactivity for η2-coordinated species—less aromatic species are more reactive and exhibit lower Diels–Alder reaction barriers.
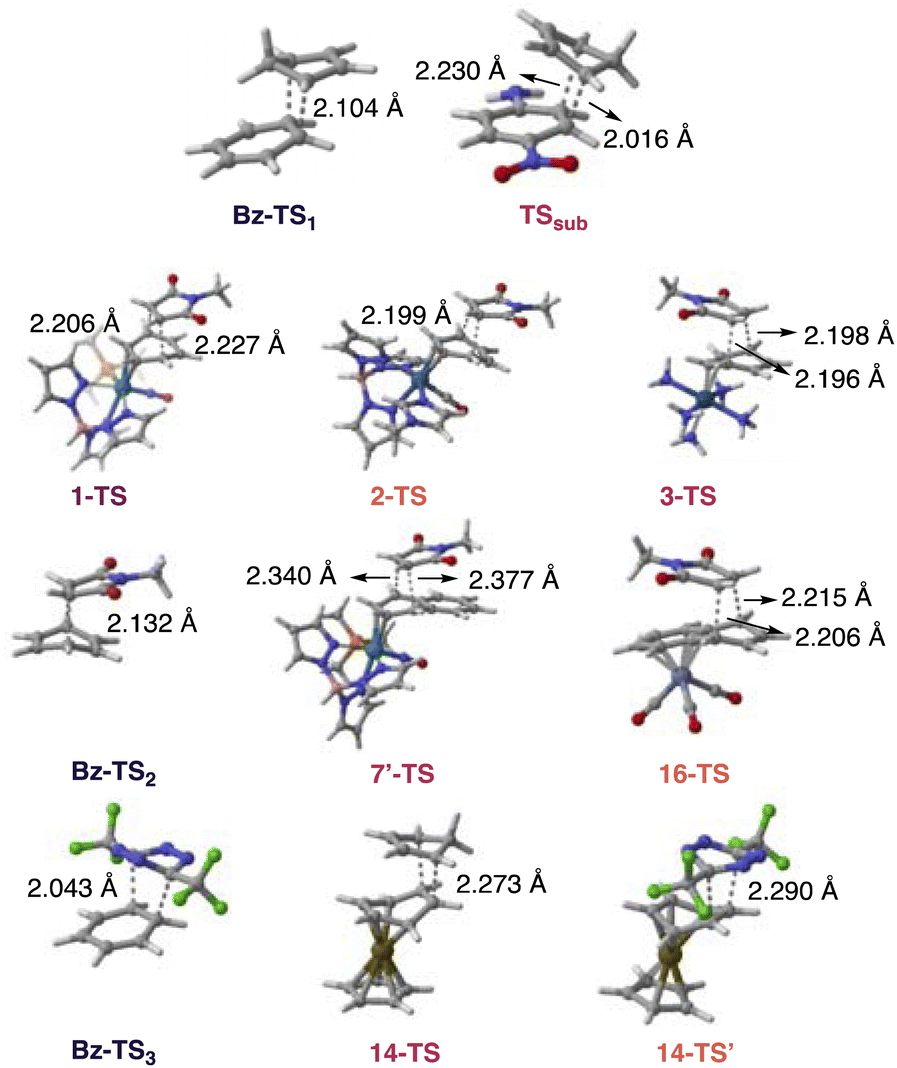 | ||
| Fig. 4 Computed transition state structures for the Diels–Alder reactions shown in Fig. 3 at ωB97X-D/def2-SVP. Transition structures including only one bond length denotes that both forming bond lengths are identical. | ||
We examined Harman et al.’s proposal that a 2,3-η2-coordinated naphthalene could mimic the cycloaddition reactivity of an o-quinodimethane. Cycloaddition adducts were reported for a hypothesized intermediate 7′ reacting with NMM. Indeed, the computed Diels–Alder reaction barrier for 7′ with NMM at the metal complexed ring (ring A) (ΔG‡ = 10.6 kcal mol−1, exo, Fig. 3c) is reasonably low. For comparison, the computed barrier for cycloaddition to the uncomplexed ring (ring B) (29.7 kcal mol−1, exo) is much higher. These results are consistent with experimental observations of cycloaddition occurring at the complexed ring (ring A). In sharp contrast, computed Diels–Alder reaction barriers for an η6-naphthalene complex, 16, and NMM (ΔG‡ = 31.0 kcal mol−1, endo, Fig. 3c) is comparable to that of naphthalene with NMM (ΔG‡ = 32.5 kcal mol−1, endo). This suggests that the uncomplexed “diene” fragment of 16 does not display diene reactivity (note that ring B remains largely aromatic, see Fig. 2, η6-set). Both 7′ (2.43 eV) and 16 (2.85 eV), exhibit narrower HOMO–LUMO energy gaps than naphthalene (4.61 eV) (Table 1), which further exemplifies that aromaticity loss is obligatory for complexed arenes to show “diene-like” reactivity. Even though Cr(CO)3 coordination activates 16 for nucleophilic addition,29 this has little effect on promoting “diene-like” reactivity as the uncomplexed ring remains largely aromatic.
The η4-benzenes have a preference for inverse electron-demand Diels–Alder reactions (Fig. 3d) as the uncomplexed “ene” fragment is strained (i.e., like that of 7-norbornadiene) and electron-rich.19 Computed energy profiles of Diels–Alder reactions for 14 with cyclopentadiene (ΔG‡ = 29.2 kcal mol−1, endo) versus with bis(trifluoromethyl)tetrazine (ΔG‡ = 14.7 kcal mol−1, inverse electron-demand) reveal a lower barrier for the latter (Fig. 3d). An inverse electron-demand also is observed when the complexing metal fragment is replaced by Fe(CO)3 (reaction profiles for 11 with bis(trifluoromethyl)tetrazine, with cyclopentadiene, and with an electron-rich Danishefsky diene are included to Fig. S3 of the ESI†). The electron richness of the uncomplexed “ene” fragment in 14 also is evident from a significantly raised HOMO energy compared to benzene (note the relatively unchanged LUMO energy of 14 relative to benzene).
Conclusions
Metal-coordinated arenes containing “planar” aromatic fragments can exhibit aromaticity loss and reactivity gain depending on the nature of the complexed metal fragment. It is obvious, for the η4-arenes, that metal complexation will distort the arene from planarity, and activate the leftover ene fragment. But the effect of aromaticity loss on reactivity is less intuitive for the η2- and η6-arenes, which remain largely planar upon metal coordination. We show here that aromaticity loss, but not distortion from planarity, is necessary for activating “ene-like” and “diene-like” reactivity in metal-complexed arenes. Our computational investigation demonstrates a protocol for how experimentalists might approach predicting ene-like and diene-like reactivity for transition metal coordinated arenes. NICS values can be convenient probes for predicting “ene-like” and “diene-like” reactivity in metal complexed arenes.Computational methods
Geometry optimizations and frequency calculations were performed at ωB97X-D/def2-TZVPP//ωB97X-D/def2-SVP, employing Gaussian 16, Revision C.01.35 The def2-SVP and def2-TZVPP basis sets include effective core potentials (ECPs) for atoms with Z > 36. Vibrational frequency analysis and intrinsic reaction coordinate (IRC) calculations verified the nature of the minima and transition state structures. Wiberg Bond Indices (WBIs) were computed using Natural Bond Orbital (NBO) Version 7.0.36 HOMO–LUMO gaps (ΔEH–L) were obtained from TD-DFT calculations at CAM-B3LYP/def2-TZVPP//ωB97X-D/def2-SVP. LUMO energies were derived by the sum of the computed HOMO energy and the ΔEH–L energies. Nucleus-independent chemical shifts, NICS(1)zz, were computed at ωB97X-D/def2-TZVPP. Bq points were placed at 1 Å above the ring centers opposite to the metal complexed face. NICS(1)zz values for the η4-complexes were estimated by defining an xy plane with C1, C2, C4, and C5 (see ring carbon numbering for 9, in Fig. 2).Single-point energies for stationary points in the energetic profiles shown in Fig. 3 were computed at DLPNO-CCSD(T)/def2-TZVPP//ωB97X-D/def2-SVP using ORCA 5.0.37 DFT functionals (ωB97X-D, PW6B95, M06, M06-L, MN15, PBE0-D3 all with def2-TZVPP) known to accurately simulate organometallic systems based on literature precedent38 were tested against each other and benchmarked against data at the DLPNO-CCSD(T)/def2-TZVPP level. We found that ωB97X-D outperformed the other functionals in estimating activation barriers and reactions energies, and provided a reasonable cost-to-insight level that tracks well with what is reported by others.39 Specifically, ωB97X-D/def2-TZVPP//ωB97X-D/def2-SVP yielded a free energy activation barrier (ΔG‡) that is within 3 kcal mol−1 and a free energy of reaction that is within 1 kcal mol−1 of the DLPNO-CCSD(T)/def2-TZVPP//ωB97X-D/def2-SVP results. Images of three-dimensional (3D) structures were produced using the CYLView20 software.40
Data availability
The data supporting this article have been included as part of the ESI.† Details of computational methods, Cartesian coordinates, and computed reaction barriers for alternative Diels–Alder reaction pathways are included to the ESI.†Author contributions
J. V. S. conceptualized the project. J. V. S. and C. L. performed data analysis. Writing, editing, and investigation was performed by all authors. J. I. W. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. I. W. thanks the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) (R35GM133548) and the Alfred P. Sloan Research Foundation (FG-2020-12811) for financial support. We acknowledge the use of the Carya cluster and support from the Research Computing Data Core at the University of Houston. We also thank Professor Thomas A. Albright and Dr Lucas Karas for friendly discussions.Notes and references
- W. Jarre, D. Bieniek and F. Korte, Benzene as Dienophile in the Diels-Alder Reaction, Angew. Chem., Int. Ed., 1975, 14, 181–182 CrossRef.
- G. Seitz, R. H. Hoferichter and R. Mohr, Benzene and Linearly Annelated Arenes as Dienophiles in Diels–Alder Reactions with Inverse Electron Demand, Angew. Chem., Int. Ed., 1987, 26, 332–334 CrossRef.
- C. Baralotto, M. Chanon and M. Julliard, Total Synthesis of the Tricyclic Sesquiterpene (±)-Ceratopicanol. An Illustration of the Holosynthon Concept, J. Org. Chem., 1996, 61, 3576–3577 CrossRef CAS PubMed.
- T. Gaich and J. Mulzer, From Silphinenes to Penifulvins: A Biomimetic Approach to Penifulvins B and C, Org. Lett., 2010, 12, 272–275 CrossRef CAS PubMed.
- R. Remy and C. G. Bochet, Arene–Alkene Cycloaddition, Chem. Rev., 2016, 116, 9816–9849 CrossRef CAS PubMed.
- J. J. Serrano-Pérez, F. de Vleeschouwer, F. De Proft, D. Mendive-Tapia, M. J. Bearpark and M. A. Robb, How the Conical Intersection Seam Controls Chemical Selectivity in the Photocycloaddition of Ethylene and Benzene, J. Org. Chem., 2013, 78, 1874–1886 CrossRef PubMed.
- A. A. Aly, S. Ehrhardt, H. Hopf, I. Dix and P. G. Jones, Cycloadditions to Alkenyl[2.2]Paracyclophanes, Eur. J. Org Chem., 2006, 335–350 CrossRef CAS.
- A. F. Murad, J. Kleinschroth and H. Hopf, Cyclophanes as Diene Components in Diels-Alder Reactions, Angew. Chem., Int. Ed., 1980, 19, 389–390 CrossRef.
- Y. Inagaki, M. Nakamoto and A. Sekiguchi, A Diels-Alder Super Diene Breaking Benzene into C2H2 and C4H4 Units, Nat. Commun., 2014, 5, 3018 CrossRef PubMed.
- E. P. Kündig, Transition Metal Arene π-Complexes in Organic Synthesis and Catalysis, Springer, Berlin and Heidelberg, Germany, 2004 Search PubMed.
- B. K. Liebova and W. D. Harman, Group 6 Dihapto-Coordinate Dearomatization Agents for Organic Synthesis, Chem. Rev., 2017, 117, 13721–13755 CrossRef PubMed.
- L. J. Williams, Y. Bhonoah, L. A. Wilkinson and J. W. Walton, As Nice as π: Aromatic Reactions Activated by π-Coordination to Transition Metals, Chem. –Eur. J., 2021, 27, 3650–3660 CrossRef CAS PubMed.
- M. Jakoobi, Y. Tian, R. Boulatov and A. G. Sergeev, Reversible Insertion of Ir into Arene Ring C-C Bonds with Improved Regioselectivity at a Higher Reaction Temperature, J. Am. Chem. Soc., 2019, 141, 6048–6053 CrossRef CAS PubMed.
- A. P. Y. Chan, M. Jakoobi, C. Wang, R. T. O'Neill, G. S. S. Aydin, N. Halcovitch, R. Boulatov and A. G. Sergeev, Selective Ortho-C–H Activation in Arenes without Functional Groups, J. Am. Chem. Soc., 2022, 144, 11564–11568 CrossRef CAS PubMed.
- S. Lee, S. J. Geib and N. J. Cooper, [2 + 2 + 2] Addition of Diphenylketene to the Reductively Activated Benzene in the Transition Metal Complex [Mn(.eta.4-C6H6)(CO)3]- To Give a Dihydroisochroman-3-One, J. Am. Chem. Soc., 1995, 117, 9572–9573 CrossRef CAS.
- M. D. Chordia, P. L. Smith, S. H. Meiere, M. Sabat and W. D. Harman, A Facile Diels–Alder Reaction with Benzene: Synthesis of the Bicyclo[2.2.2]Octene Skeleton Promoted by Rhenium, J. Am. Chem. Soc., 2001, 123, 10756–10757 CrossRef CAS PubMed.
- P. M. Graham, S. H. Meiere, M. Sabat and W. D Harman, Dearomatization of Benzene, Deamidization of N,N-Dimethylformamide, and a Versatile New Tungsten π Base, Organometallics, 2003, 22, 4364–4366 CrossRef CAS.
- L. Strausberg, M. Li, D. P. Harrison, W. H. Myers, M. Sabat and W. D. Harman, Exploiting the O- Quinodimethane Nature of Naphthalene: Cycloaddition Reactions with η2-Coordinated Tungsten-Naphthalene Complexes, Organometallics, 2013, 32, 915–925 CrossRef CAS.
- Q. H. Luu, T. Fiedler and J. A. Gladysz, Gigging Benzene, Angew. Chem., Int. Ed., 2017, 129, 5756–5758 CrossRef.
- J. M. Keane and W. D Harman, A New Generation of π-Basic Dearomatization Agents, Organometallics, 2005, 24, 1786–1798 CrossRef CAS.
- M. Franck-Neumann and D. Martina, Cycloadditions de la Tropone Avec le Cyclopentadiene: Synthese d’un Intermediaire Potentiel Par Utilisation de Complexe Metallique, Tetrahedron Lett., 1977, 31, 2293–2296 CrossRef.
- J. H. Rigby and C. O. Ogbu, Tricarbonyl(Tropone)Iron as a Useful Functionalized Enone Equivalent, Tetrahedron Lett., 1990, 31, 3385–3388 CrossRef CAS.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, Oxford, 2nd edn, 2012 Search PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- Q. Zhu, S. Chen, D. Chen, L. Lin, K. Xiao, L. Zhao, M. Solà and J. Zhu, The Application of Aromaticity and Antiaromaticity to Reaction Mechanisms, Fundam. Res., 2023, 3, 926–993 CrossRef CAS PubMed.
- T. M. Krygowski, K. Ejsmont, B. T. Stepień, M. K. Cyrański, J. Poater and M. Solà, Relation between the Substituent Effect and Aromaticity, J. Org. Chem., 2004, 69, 6634–6640 CrossRef CAS PubMed.
- K. B. Wiberg, Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- F. Ding, M. T. Valahovic, J. M. Keane, M. R. Anstey, M. Sabat, C. O. Trindle and W. D. Harman, Diastereo- and Enantioselective Dearomatization of Rhenium-Bound Naphthalenes, J. Org. Chem., 2004, 69, 2257–2267 CrossRef CAS PubMed.
- C. A. Merlic, M. M. Miller, B. N. Hietbrink and K. N. Houk, Reactivity of (η6-Arene)Tricarbonylchromium Complexes toward Additions of Anions, Cations, and Radicals, J. Am. Chem. Soc., 2001, 123, 4904–4918 CrossRef CAS PubMed.
- A. Lebcir, A. Boukhari, M. Solà and Y. García-Rodeja, [4+2]-Cycloaddition Reactions to Corannulene Accelerated by η6 Coordination of Ruthenium Complexes, Eur. J. Org Chem., 2024, e202400697 CrossRef.
- P. v. R. Schleyer, B. Kiran, D. V. Simin and T. S. Sorensen, Does Cr(CO)3 Complexation Reduce the Aromaticity of Benzene?, J. Am. Chem. Soc., 2000, 122, 510–513 CrossRef CAS.
- M. F. Semmelhack, W. Wulff and J. L. Garcia, New Substitution Eeactions on Indole Promoted by Cr(CO)3 Unit, J. Organomet. Chem., 1982, 240, C5–C10 CrossRef CAS.
- D. Möhring, M. Nieger, B. Lewall and K. H. Dötz, Tricarbonyl(naphthoquinone)chromium: Synthesis and Application in [4+2] Cycloaddition Reactions, Eur. J. Org Chem., 2005, 2620–2628 CrossRef.
- T. M. Krygowski, Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of .pi.-electron systems, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohman, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018, https://nbo7.chem.wisc.edu/ Search PubMed.
- F. Neese, Software Update: The ORCA program System, Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 4–9 Search PubMed.
- T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Computational Studies of Synthetically Relevant Homogeneous Organometallic Catalysis Involving Ni, Pd, Ir, and Rh: An Overview of Commonly Employed DFT Methods and Mechanistic Insights, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Best-Practice DFT Protocols for Basic Molecular Computational Chemistry, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05337k |
| This journal is © The Royal Society of Chemistry 2024 |