
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyperstable alkenes: are they remarkably unreactive?†
Matthew D.
Summersgill
 a,
Lawrence R.
Gahan
a,
Sharon
Chow
a,
Gregory K.
Pierens
b,
Paul V.
Bernhardt
a,
Elizabeth H.
Krenske
a and
Craig M.
Williams
*a
a,
Lawrence R.
Gahan
a,
Sharon
Chow
a,
Gregory K.
Pierens
b,
Paul V.
Bernhardt
a,
Elizabeth H.
Krenske
a and
Craig M.
Williams
*a
aSchool of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, 4072, Queensland, Australia. E-mail: c.williams3@uq.edu.au
bCentre for Advanced Imaging, University of Queensland, Brisbane, 4072, Queensland, Australia
First published on 5th November 2024
Abstract
In 1981, Maier and Schleyer first identified a select number of cage bicyclic olefins (alkenes) as “hyperstable”, and predicted them to be “remarkably unreactive”, based solely on theoretical methods. Since that time only three ad hoc systems meeting the criteria of a hyperstable alkene have been reported in the literature. A one-pot, telescoped synthesis, of four hyperstable alkenes is reported herein, which has uncovered unexpected reactivity towards oxidation. Although, this work represents a new benchmark in hyperstable alkenes, it concomitantly emphasised the need to clarify the definition based on a long-held computational prediction.
Introduction
Hyperstable olefins (alkenes) were first postulated by Maier and Schleyer in the early 1980s in the course of developing predictive theoretical methods to classify the stability of cage bicyclic bridgehead alkenes1,2i.e., in an effort to better define Bredt's Rule.3Utilising Allinger's MM1 empirical force field program, a vast array of cage bicyclic systems were evaluated, and subsequently ranked using olefin strain energy (OSE) values. This molecular mechanics-based approach led to the posit that “Hyperstable olefins should be remarkably unreactive”.1a Given, for example, that the anti-cancer agent, taxol, contains a cage bridgehead alkene, this research endeavor was also of particular importance to rationalise bridgehead double bond stability4 and that of an increasing number of related natural products being reported in the modern era.3
Five theoretical bicyclo[m.n.o] (1) hyperstable systems were initially identified in 1981 (e.g., 2–5, Fig. 1), which led to a further prediction that bicyclo[4.4.4]tetradec-1-ene (5) “may even resist hydrogenation”.1a In a later study by McEwen and Schleyer, the list of examples was expanded to include additional cases that compared E-(e.g., 6) and Z-alkene (e.g., 7) configurations, and pyramidalisation through homeomorphic isomerisation (e.g., in-8) (Fig. 1).6 A decade later Kim performed a more systematic investigation of over 70 bicyclo[m.n.o] bridgehead alkenes, and ranked 15 of the top hyperstable predictions, which ranged from bicyclo[4.4.3]tridec-11-ene (9) through to bicyclo[4.4.4]tetradec-1-ene (5) (Fig. 1).5,7 Although larger ring systems were identified through the studies undertaken by Kim (e.g., bicyclo[7.4.4] and [8.3.3]), both Schleyer and Kim recognised that a hyperstability optimal zone existed, which preferred medium-sized rings (e.g., the bicyclo[4.4.4] (5), bicyclo[4.4.3] (6), and bicyclo[5.3.3] (10) systems). In fact, the major contribution to polycyclic alkene stability is suggested to arise from sp2 flattening at the bridgehead carbon, which provides significant reductions in angle and torsional strain and in non-bonded interactions (e.g., van der Waals forces), all controlled by changes in ring size.7,8 That is, combinations of much larger rings (or bicyclic bridges) do not provide additional stabilisation, while smaller ring sizes give rise to unstable bridgehead double bonds and/or anti-Bredt systems.3,4
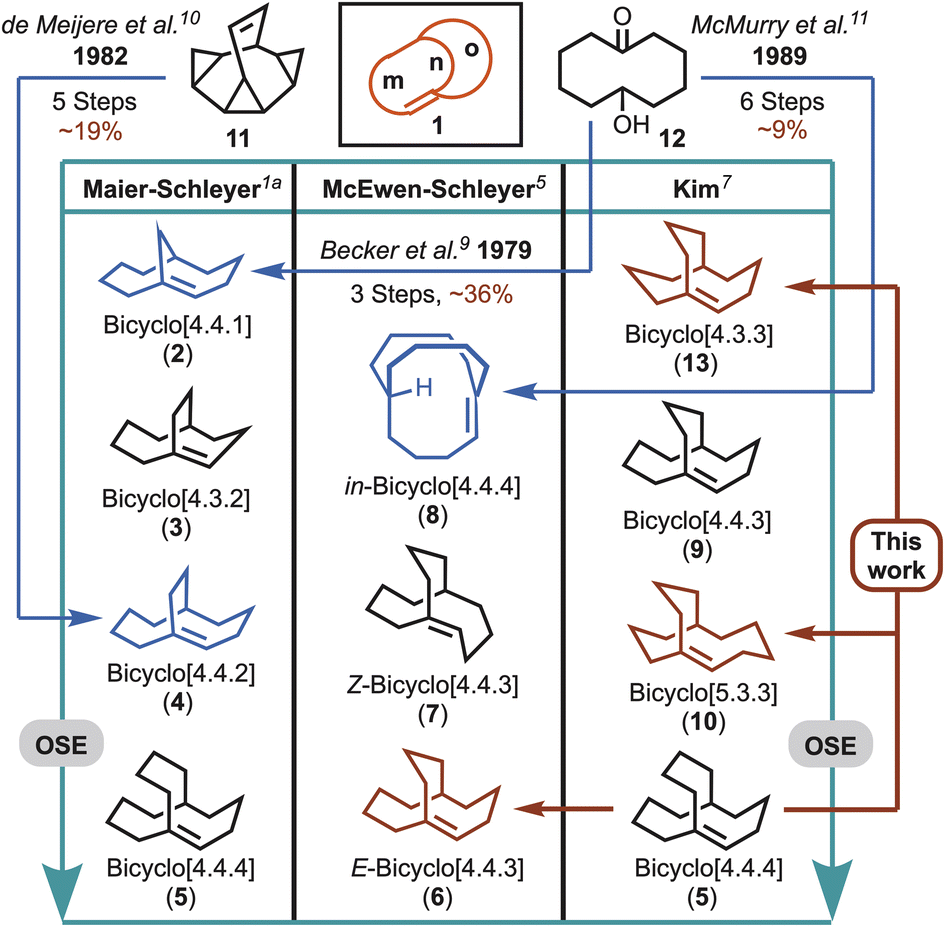 | ||
| Fig. 1 Hyperstable bicyclo[m.n.o] bridgehead alkenes ranked according to olefin strain energy (OSE). Note: alkenes listed in order of increasing stability as predicted in three separate bodies of work using different theoretical methods to determine OSE values.5 The systems highlighted in dark red are the focus of this work, and those in blue have previously been synthesised ad hoc. | ||
Despite putative hyperstable systems being identified through Maier–Schleyer–Kim in silico studies, a substantial limitation preventing the interrogation of alkene hyperstability has been an inability to readily synthesise the computationally predicted targets. To highlight this point only three examples that lie in the hyperstability optimal zone have been synthesised previously (i.e., 2, 4 and 8). In 1979, Becker et al.9 reported the synthesis of bicyclo[4.4.1]undec-1-ene (2), but it predated the hyperstability hypothesis by two years, and was detected as a serendipitous side product in pursuit of anti-Bredt systems. Similarly, a few years later de Meijere et al.10 reported having unexpectedly obtained bicyclo[4.4.2]dodeca-1-ene (4), while working with exo,exo-bishomobullvalene 11, but subsequently demonstrated 4 was a hyperstable alkene system. Lastly, in the course of pursuing stable three-center, two-electron C–H–C bonds McMurry et al. reported the synthesis of hyperstable bridgehead alkene, in-bicyclo[4.4.4]tetradec-1-ene (8), in 6 steps commencing from 6-hydroxy-cyclodecan-1-one (12).11,12 Interestingly, in the case of 2, 4 and 8, all could be hydrogenated, but conditions ranged from mild to forcing.9,10,11b Unfortunately, however, in the intervening years there have been no attempts to access the systems with even greater predicted stability,13i.e., 5, 6 and 10 (Fig. 1).
Disclosed herein are efficient methods to access these cage bicyclic systems, which has enabled refinement of the “unreactive” definition, and determination of whether the term hyperstable alkene is more broadly applicable.
Results and discussion
Alkene synthesis
Brown et al. demonstrated that boracyclanes (e.g., B-methoxy-9-borabicylo[3.3.1]nonane, 14) (Scheme 1) undergo facile ring enlargement.14 Expanding upon the methodology of Matteson,15 treatment with (dichloromethyl)lithium (i.e., LiCHCl2) was shown to give the corresponding (α-haloalkyl)borinic ester homologue (15), via boronate 16. Subsequently, Brown et al. developed a procedure utilising (chloromethyl)lithium (i.e., LiCH2Cl),16 which enabled sequential formation of the unsubstituted B-methoxy-9-borabicyclo[3.3.2]decane (17), via boronate 19, or B-methoxy-2-borabicyclo[3.3.3]undecane (18) on addition of a second equivalent (Scheme 1). Only in the case of 17 was the boracyclane used synthetically, whereas the other borabicycles have remained unexplored. Inspired by the boracyclane chemistry, synthesis of the proposed hyperstable alkenes bicyclo[5.3.3] tridec-1-ene (10) and bicyclo[4.3.3]dodec-1-ene (13) (Fig. 1), were attempted using this synthetic methodological approach.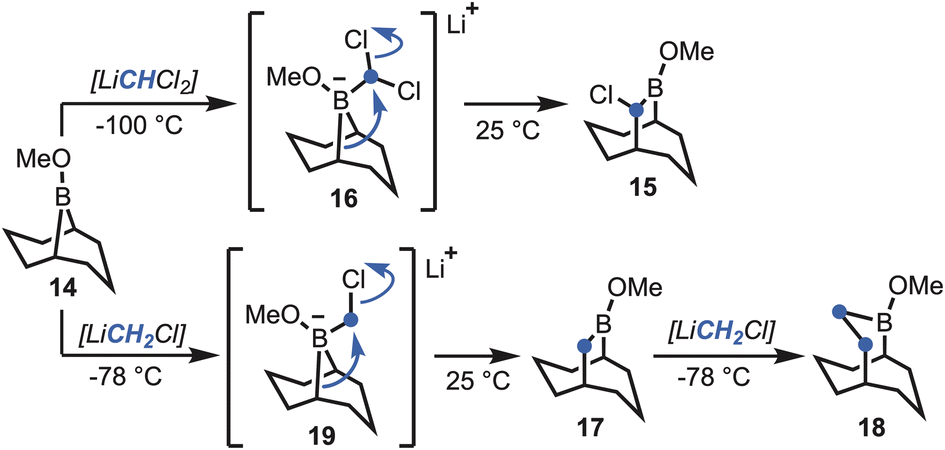 | ||
| Scheme 1 The Matteson homologation-based boracyclane homologation methodology developed by Brown et al.16 | ||
In the course of preparing fresh 9-BBN (20), via cyclooctadiene (21) (Scheme 2),17 to investigate the first homologation, it soon became apparent that the entire process to access the desired carbocycles might be feasible through a one-pot sequence.18 That is, synthesis of 9-BBN provided a clean precipitate that could be used directly to afford the borinic ester 14 (δB 57.2 ppm). In pursuit of the first ring expansion, (bromomethyl)lithium was selected as the α-halomethyllithium nucleophile of choice, leveraging the widespread availability of dibromomethane and straightforward lithium–halogen exchange.19 Homologation was achieved through addition of n-butyllithium to a solution of dibromomethane and 14 at −78 °C, with formation of the ring-expanded borinic ester 17 visible by 11B NMR (δB 55.3 ppm) upon warming. Essential to maintaining the one-pot procedure was adjustment for the growing volume of the solution (i.e., all depending on reaction scale). However, the hexanes introduced alongside the n-butyllithium could be carefully removed under reduced pressure, in addition to a small quantity of tetrahydrofuran (THF), whereupon the solution was diluted to approximately 1 M by the addition of anhydrous THF.
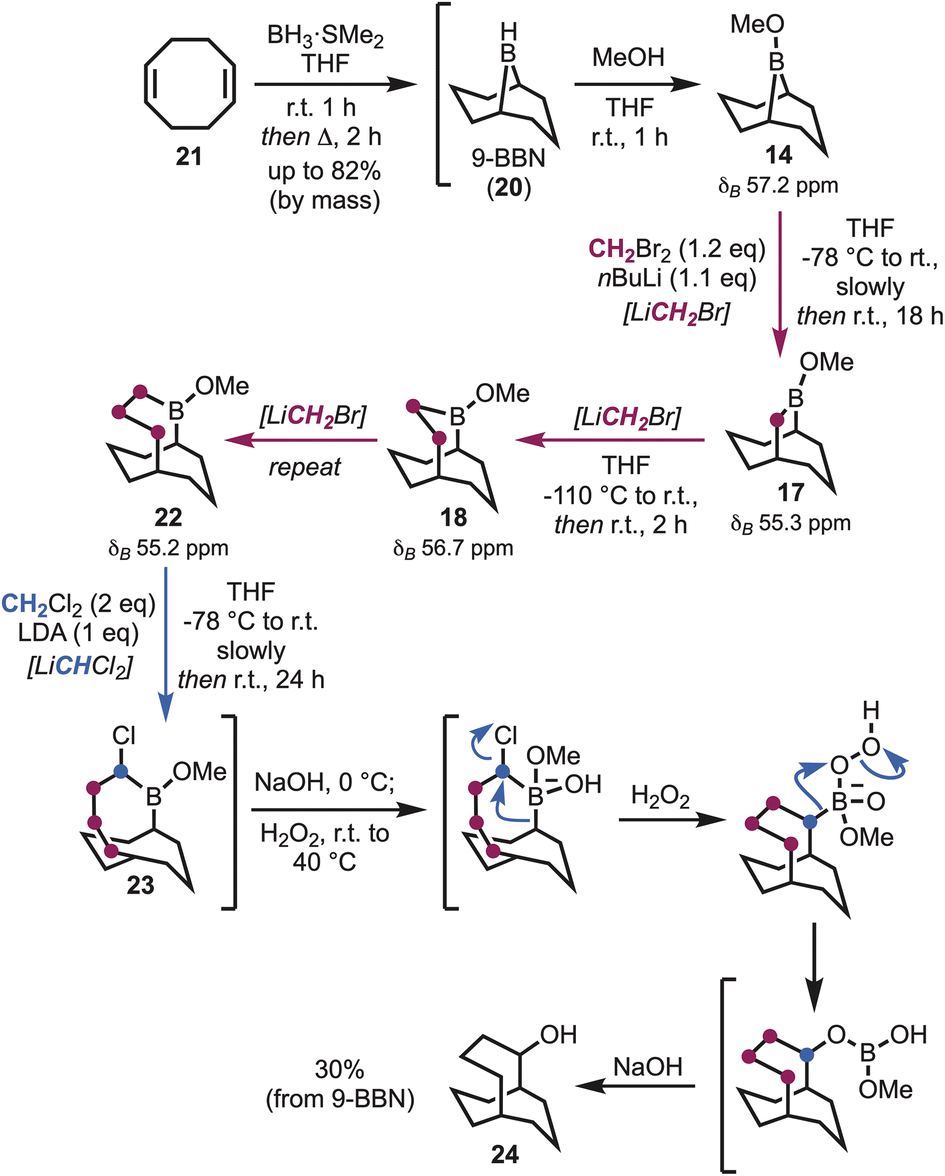 | ||
| Scheme 2 One-pot synthesis of bicyclo[4.3.3]docecan-2-ol (24). | ||
Following the formation of 17, further homologation could be achieved utilising (bromomethyl)lithium, but this transformation required significantly reduced temperatures to prevent over-homologation, suggesting that the 1,2-migration responsible for ring expansion occurs at temperatures as low as −78 °C within this system (Scheme 2). Formation of (bromomethyl)lithium at −110 °C saw significantly improved selectivity during the second homologation, with formation of the ring-expanded borinic ester 18 observed within 2 hours of stirring at room temperature (δB 56.7 ppm). Formation of the tri-homologated B-methoxy-2-borabicyclo[4.3.3]dodecane (22) was achieved on repetition of this procedure (δB 55.2 ppm), however formation of the all-carbon framework necessitated the use of LiCHCl2, in order to facilitate migration of the boron atom out of the ring. Lithium N,N-diisopropylamide (LDA) was the optimum base in this regard and was added to a solution of 22 and dichloromethane (DCM) at −78 °C [Note: typically prompting a slow colour change on, or when, approaching the completion of addition]. Workup of the α-chloroborane (23), on sequential treatment with aqueous sodium hydroxide and hydrogen peroxide, furnished the secondary alcohol bicyclo[4.3.3]dodecan-2-ol (24) in 30% overall yield (∼80% yield for each step following methanolysis) (Scheme 2).
With alcohol 24 in hand, elimination reactions could be explored in an attempt to access the first target, i.e., bicyclo[4.3.3]dodec-1-ene (13). Simple treatment of the alcohol with mesyl chloride (MsCl) in the presence of base at room temperature gave bridgehead alkene 13, and traces of 25. However, while dissolved in deuterated chloroform 13 fully converted into the bridgehead alkene, bicyclo[4.3.3]dodec-6-ene (25), within 24 hours, suggesting a more stable system. This unexpected transformation likely occurs via carbocation formation (i.e., 26), mediated by the presence of traces of acid in the CDCl3 (Scheme 3).
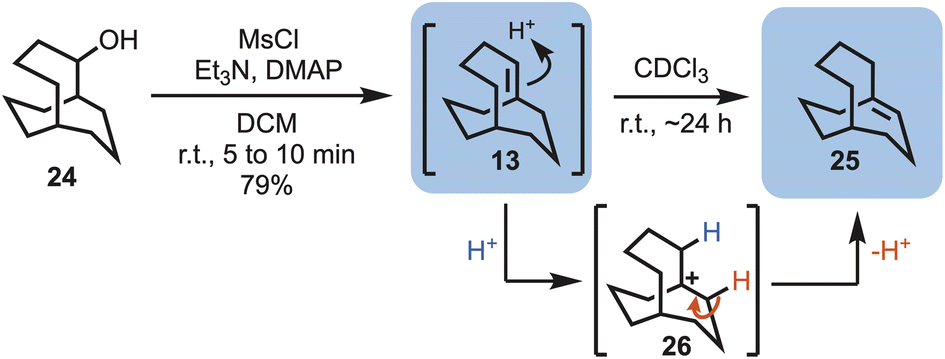 | ||
| Scheme 3 Elimination of bicyclo[4.3.3]dodecan-2-ol (24) to give bicyclo[4.3.3]dodec-1-ene (13) and bicyclo[4.3.3]dodec-6-ene (25). | ||
With an iterative homologation strategy in place, further ring expansion was explored in an effort to access the larger bicyclo[5.3.3]tridecane system, and to determine the upper limit of the methodology. Therefore, the same homologation sequence was utilised to access the previously generated boracyclane 22 [i.e., starting from cyclooctadiene (21)], whereupon further treatment with (bromomethyl)lithium was expected to give the ring enlarged boracyclane (27). Interestingly, on subsequent treatment with (dichloromethyl)lithium, followed by oxidative workup, not only was the desired bicyclo[5.3.3]tridecan-2-ol (28) obtained, but also bicyclo[4.3.3]dodecan-2-ol (24) and cyclooctanol derivative 29 in an inseparable 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 ratio, respectively (Scheme 4). This observation suggested that controlling the formation of boracyclane 27 is more difficult compared with the preceding boracyclanes 17, 18 and 22, especially given the developing excess of (bromomethyl)lithium through each iterative homologation. This was particularly evident through the isolation of the ring-opened cyclooctanol 29, which is a result of undesired over-homologation of boracyclane 27 giving boracyclane 30 (Path A, Scheme 4). However, in this case the preference for the carbon–boron bond migration switched to the bridgehead carbon of boronate complex 31, which then underwent dehydroboration20 to afford 32, and subsequently 29 on oxidation. Brown has suggested that increasing ring size within medium-ring boracyclanes can encroach the strain limits of the labile carbon–boron bond.16a This notion is supported by conformational analysis of 31 whereby conformer 31a, required for the desired homologation via a staggered conformation, shows that the bromine atom approaches the bicyclic ring hydrogens as the ring expands. To avoid the ensuing steric clash the alternate anti-periplanar conformer 31b is adopted, and this change in conformation facilitates the observed bridgehead carbon-boron migration to give boracyclane 30 (Scheme 4). The formation of undesired bicyclo[4.3.3]dodecan-2-ol (24) arose from under homologation of boracyclane 22 (Path B, Scheme 4), whereas desired product bicyclo[5.3.3]tridecan-2-ol (28) was obtained via boracyclane 27 (Path C, Scheme 4).
:1:3 ratio, respectively (Scheme 4). This observation suggested that controlling the formation of boracyclane 27 is more difficult compared with the preceding boracyclanes 17, 18 and 22, especially given the developing excess of (bromomethyl)lithium through each iterative homologation. This was particularly evident through the isolation of the ring-opened cyclooctanol 29, which is a result of undesired over-homologation of boracyclane 27 giving boracyclane 30 (Path A, Scheme 4). However, in this case the preference for the carbon–boron bond migration switched to the bridgehead carbon of boronate complex 31, which then underwent dehydroboration20 to afford 32, and subsequently 29 on oxidation. Brown has suggested that increasing ring size within medium-ring boracyclanes can encroach the strain limits of the labile carbon–boron bond.16a This notion is supported by conformational analysis of 31 whereby conformer 31a, required for the desired homologation via a staggered conformation, shows that the bromine atom approaches the bicyclic ring hydrogens as the ring expands. To avoid the ensuing steric clash the alternate anti-periplanar conformer 31b is adopted, and this change in conformation facilitates the observed bridgehead carbon-boron migration to give boracyclane 30 (Scheme 4). The formation of undesired bicyclo[4.3.3]dodecan-2-ol (24) arose from under homologation of boracyclane 22 (Path B, Scheme 4), whereas desired product bicyclo[5.3.3]tridecan-2-ol (28) was obtained via boracyclane 27 (Path C, Scheme 4).
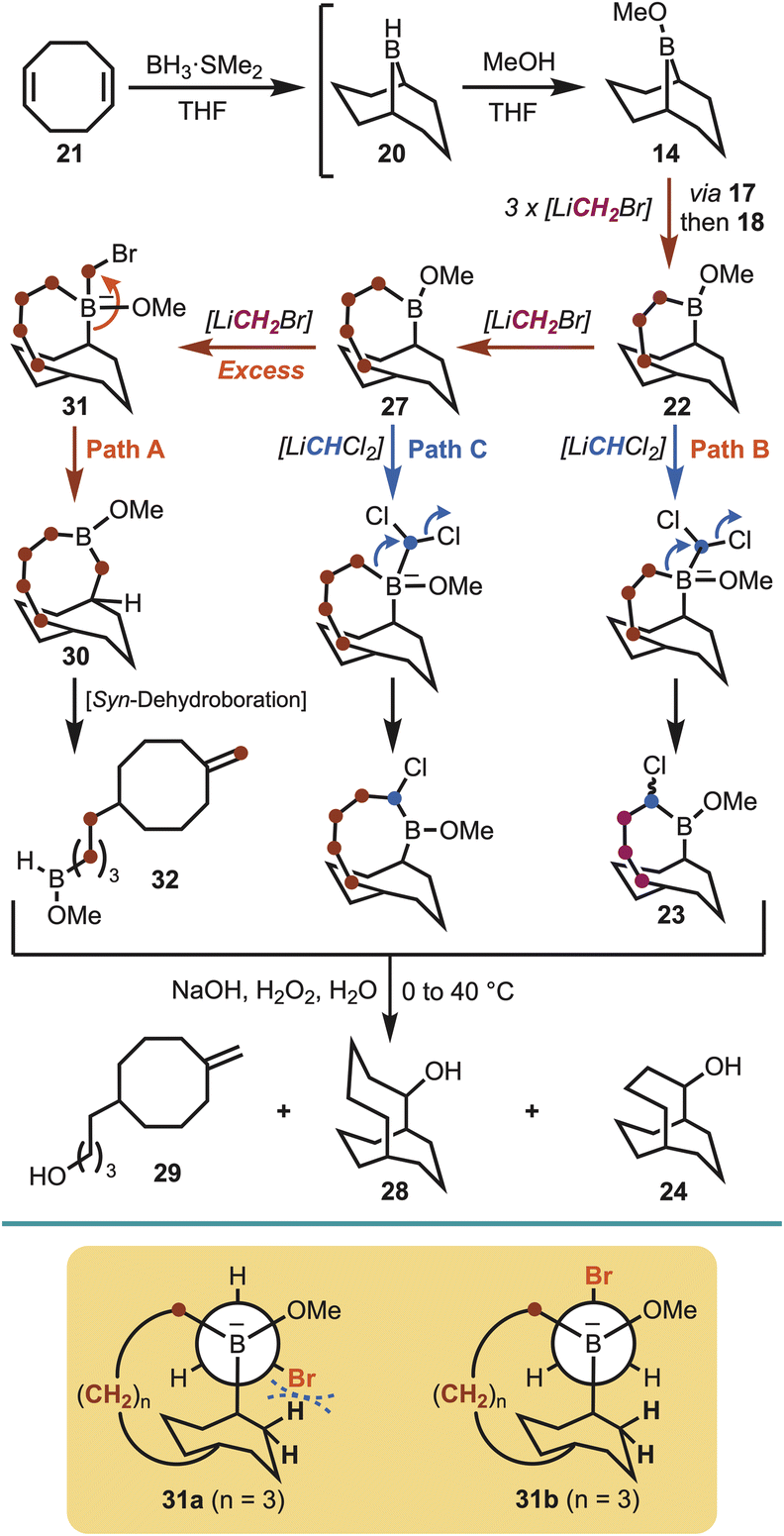 | ||
| Scheme 4 One-pot synthesis of bicyclo[5.3.3]tridecan-2-ol (28). | ||
Treating the mixture of 24, 28 and 29 with t-butyldimethylsilyl chloride (TBSCl) enabled removal of the corresponding bicyclo[4.3.3]dodecan-2-yl (not shown) and cyclooctanyl silyl ethers (33), to deliver the desired TBS-protected bicyclo[5.3.3]tridecane 34 (Scheme 5). Exposure of 34 to a catalytic amount of methanesulfonic acid (MsOH) afforded a mixture of the bicyclo[5.3.3] alkene (10), and unexpectedly the rearranged bicyclo[4.4.3] alkene 6 as a minor product (Scheme 5). The ratio of alkene isomers could be improved from a ratio ∼4:1 to ∼9:1 in favour of 10 by lowering the temperature of the reaction from room temperature to −78 °C. The competing elimination and rearrangement pathways can be envisaged as arising from carbocation 35 (Scheme 5). The major product is derived from elimination of a proton to afford the Kim system, i.e., bicyclo[5.3.3]tridec-1-ene (10). Carbocation 35 also undergoes a Wagner–Meerwein rearrangement to 36. This is followed by a conformational change to give 37, that relieves steric clashing, and then loss of a proton to afford the McEwen–Schleyer system i.e., E-bicyclo[4.4.3]tridec-1-ene (6) (Scheme 5).
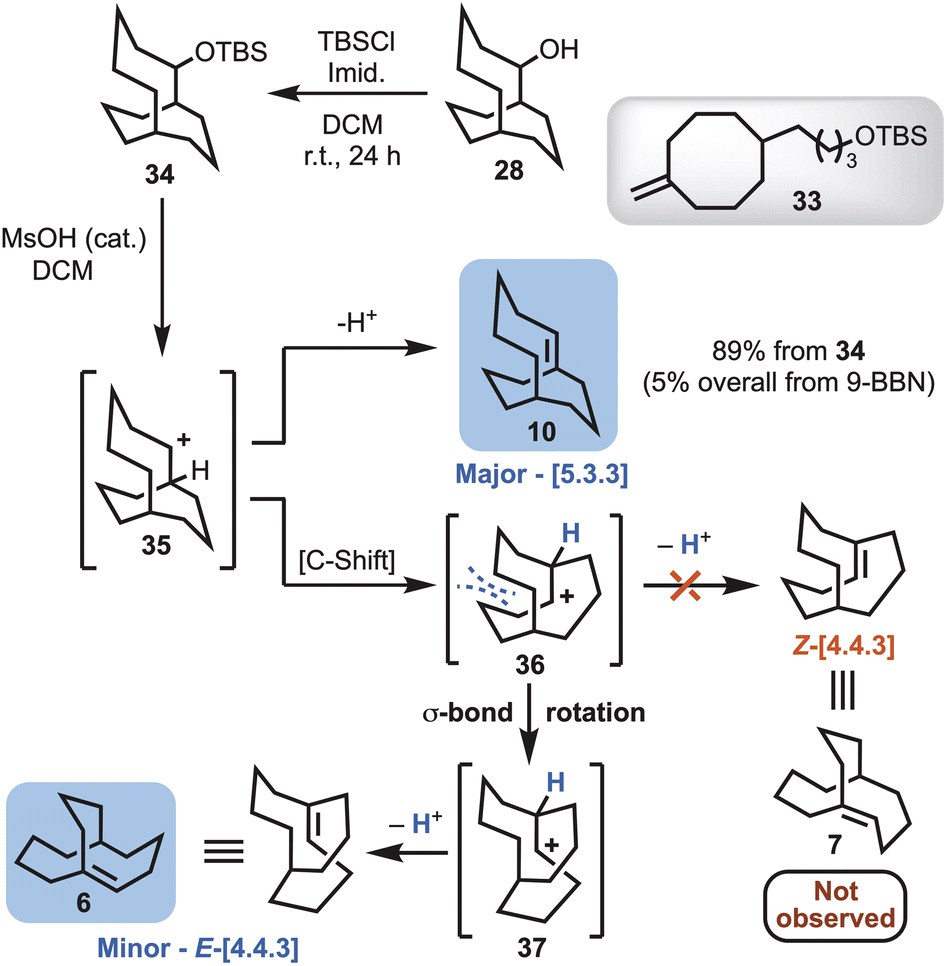 | ||
| Scheme 5 Synthetic route affording bicyclo[5.3.3]tridec-1-ene (10) and E-bicyclo[4.4.3]tridec-1-ene (6) and proposed mechanistic pathway of formation. | ||
Hydrogenation
The sole experimental criteria put in place by Maier and Schleyer to determine alkene hyperstability status (i.e., “unreactive”) resides on whether the alkene resists catalytic hydrogenation. However, Maier and Schleyer did not detail specific hydrogenation conditions. Simply, the bicyclo[4.4.4] system 5 (Fig. 1) was described as thus: “Therefore, [5] should be an unusually unreactive olefin and may even resist hydrogenation under normal conditions”.1a Although, it would not be unreasonable to consider normal conditions to consist of palladium suspended on carbon under one atmosphere of hydrogen gas overnight, a variety of conditions could be imagined. With that in mind, previous work by Becker et al. demonstrated that bicyclo[4.4.1]undec-1-ene (2) could be successfully hydrogenated using palladium on carbon under a hydrogen atmosphere (i.e., say normal conditions) to give 38,9 whereas the bicyclo[4.4.2]dodeca-1-ene (4) system reported by de Meijere et al. required Adam's catalyst (platinum oxide) over a three day duration (i.e., slightly harsher conditions) to afford 39,10 and lastly McMurry et al.11 showed that in,out-bicyclo[4.4.4]tetradec-1-ene (8) required more forcing conditions (platinum oxide and 50 psi of pressure) to access 40 (Scheme 6).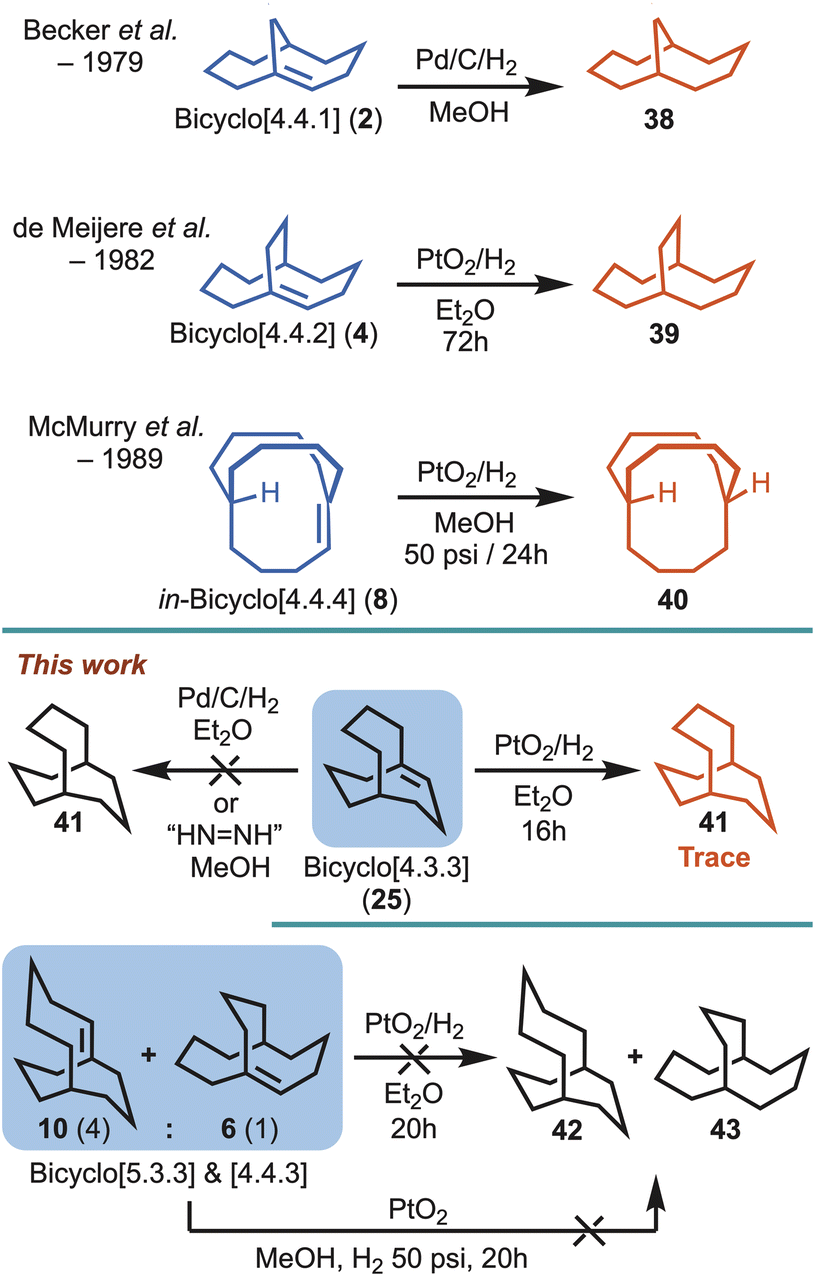 | ||
| Scheme 6 Hydrogenation outcomes for known and new bridgehead alkenes. | ||
Concerning the alkenes investigated herein, the bicyclo[4.3.3] alkene 25 did not undergo hydrogenation using Pd/C/H2, and only provided trace amounts of the fully saturated alkane 41 when applying slightly harsher conditions (PtO2/H2). For the major and minor mixture of bicyclo[5.3.3] (10) and bicyclo[4.4.3] (6) alkenes (∼4:1), both of these completely resisted hydrogenation using platinum oxide at atmospheric pressure, but also on hydrogenation under more forcing conditions (PtO2/H2 at 50 psi), with no detection of the corresponding alkanes 42 and 43 by 1H NMR or GC/MS (Scheme 6). Furthermore, when alkene 25 was exposed to in situ generated diimide (HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)21 no reduction was observed.
NH)21 no reduction was observed.
Computational investigations
Density functional theory22 calculations with M06-2X/def2-TZVPP23 were performed to investigate the stabilities and reactivities of the four new alkenes, 6, 10, 13, and 25 (Fig. 2). [Note: calculated heats of hydrogenation are used in lieu of experimentally determined values, because the rate of hyperstable alkene hydrogenation is too slow to measure, as reported by Roth.24] In the case of the 13/25 pair, alkene 25 was found to be 4.3 kcal mol−1 lower in energy than 13 (Fig. 2A). The result is consistent with the observed formation of 25 from 13 upon standing in CDCl3.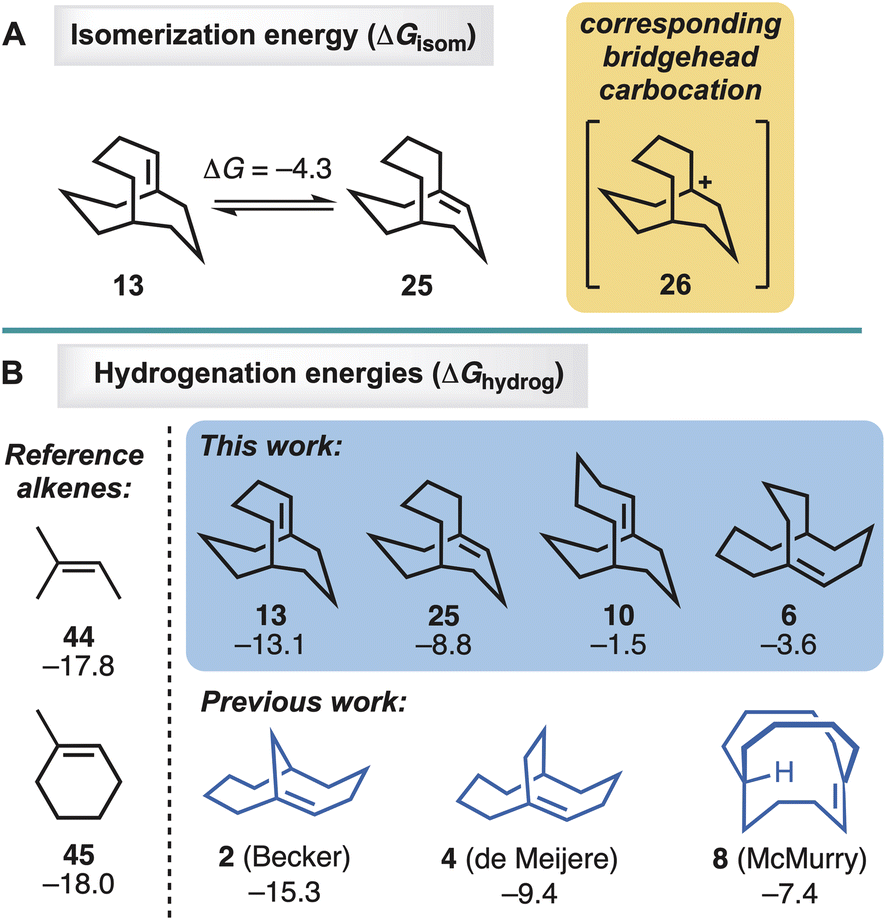 | ||
| Fig. 2 Density functional theory computations of (A) alkene isomerisations and (B) alkene hydrogenations. ΔG in kcal mol−1 (M06-2X/def2-TZVPP). | ||
The original hyperstability predictions proposed by Maier, Schleyer, and Kim, were established on the basis of olefin strain energies (OSE) as calculated with molecular mechanics. In the present study, however, DFT-computed free energies of hydrogenation (ΔGhydrog, Fig. 2B) were used as a direct measure of the propensities of the alkenes to undergo hydrogenation. For comparison, OSEs were also calculated, using Rablen's recently reported quantum mechanical group increment method25 (see ESI†). The ΔGhydrog and OSE values were found to be linearly correlated (R2 = 0.98). A linear correlation was also detected between OSE and the enthalpy of hydrogenation, ΔHhydrog, a quantity considered by Maier and Schleyer in their original study1a (see ESI†).
The two simple alkenes 44 and 45 represent relatively strain-free trisubstituted systems lacking a bicyclic ring system. Their hydrogenation energies (both −18 kcal mol−1) provide reference values against which the new and previously synthesised bridgehead alkenes can be compared (Fig. 2). In general, a hyperstable alkene would be expected to release less energy upon hydrogenation than a strain-free reference alkene, and its ΔGhydrog would therefore be expected to be less negative than those of 44 and 45. This was observed to be the case with all three of the previously synthesised hyperstable alkenes, for which the hydrogenation energies range from −15 kcal mol−1 (2) to −9 kcal mol−1 (4) to −7 kcal mol−1 (8). This trend in energies mirrors the trend in reactivity toward hydrogenation observed experimentally by others, viz. 2 > 4 > 8 (Scheme 6).
For the series of alkenes 6, 10, 13, and 25, the hydrogenation energies range from −2 kcal mol−1 to −13 kcal mol−1 (Fig. 2). These values follow the same trend as observed experimentally: the theoretically least hyperstable alkene that was studied in hydrogenation experiments, 25 (ΔGhydrog = −9 kcal mol−1), gave trace amounts of hydrogenated product under harsher conditions, while the theoretically more hyperstable alkenes in the series, 6 and 10 (ΔGhydrog = −4 and −2 kcal mol−1, respectively), failed to undergo hydrogenation even under forcing conditions. There are some variations between the series worth noting. Firstly, theory predicts that alkene 25 has a smaller driving force for hydrogenation than the Becker et al.9 alkene 2, consistent with the observation that experimentally 25 required harsher conditions than reported for 2, and only afforded trace amounts of product (i.e.,41) (Scheme 6). Furthermore, the hydrogenation energies do not provide information about the barrier heights for the hydrogenation processes, nor do they take into account any differences between the mechanisms of the hydrogenations catalysed by different heterogeneous catalysts in the different solvents used experimentally. Secondly, the McMurry et al.11in-bicyclo[4.4.4]tetradec-1-ene (8) system has a 7 kcal mol−1 driving force for hydrogenation according to theory, but unlike alkenes 6 and 10, it does not survive hydrogenation conditions involving platinum oxide at a pressure of 50 psi. Beyond the potential limitations of utilising thermodynamic hydrogenation energies, however, the McMurry et al. case is considerably different due to the in-bridgehead hydrogen atom (i.e., all other systems are out-bridgehead hydrogen atoms). Such systems can stabilise any developing δ+ charge through contact with the catalyst and the double bond (i.e., lowering the energy barrier of hydrogenation).
Osmylation and X-ray crystallography
In an effort to gain an understanding of whether bridgehead alkenes 6, 10, and 25, could be considered hyperstable alkenes more broadly (i.e., in the context of resisting other alkene reaction conditions), they were subjected to osmylation. Donohoe et al.26 have shown that the combination of osmium tetroxide (OsO4), and N,N,N′,N′-tetramethylethylenediamine (TMEDA), is a very reactive and efficient oxidant of a wide range of alkenes. Furthermore, Rychnovsky et al.,27 have demonstrated that the method can be utilised for structure determination by X-ray crystallography, as the TMEDA–osmium(VI) complex of the vicinal diol product is typically crystalline.Therefore, alkene 25 was treated with OsO4, and TMEDA at −78 °C, which afforded 46. The X-ray crystal structure of 46 was determined (Fig. 3A), and although disordered, the C-atom positions and connectivity were all clearly resolved (see ESI†). This analysis also confirmed the structure of the parent [4.3.3] alkene 25 and revealed that the bridgehead alkene was susceptible to attack by OsO4 in the usual manner. In light of these results the isomeric mixture of 10 and 6 was similarly reacted with OsO4/TMEDA, which afforded osmium(VI) complexes 47 and 48. Recrystallisation gave a co-crystal comprising both isomers 47 and 48 (in a ratio of 62:38 respectively) where the Os, TMEDA and all O-donor atoms occupy identical positions within the structure, but the C-atoms of the bicyclic cages are disordered between positions corresponding to their [5.3.3] or [4.4.3] parent (see ESI† for a more detailed discussion). The Os(VI)-coordinated [5.3.3] bicycle 47 is shown in Fig. 3B (derived from 10) and the [4.4.3] isomer 48 (derived from 6) is shown in Fig. 3C.
 | ||
| Fig. 3 X-ray crystal structures of the Os(VI) complexes 46 (A), 47 (B) and 48 (C). | ||
Epoxidation
Given that the OsO4/TMEDA oxidant system is reported to be 10000 times more reactive than OsO4,24a it was deemed instructive to explore whether a less reactive oxidant would oxidise the bridgehead alkenes 6, 10, and 25. Although, a number of oxidants could be envisaged, meta-chloroperbenzoic acid (mCPBA) was first chosen, and was found to afford the corresponding epoxides 49, 50 and 51. However, 49 was found to be quite unstable to moisture, and could not be sufficiently purified, although it could be obtained in high purity using the oxidant dimethyldioxirane (DMDO) (Scheme 7).
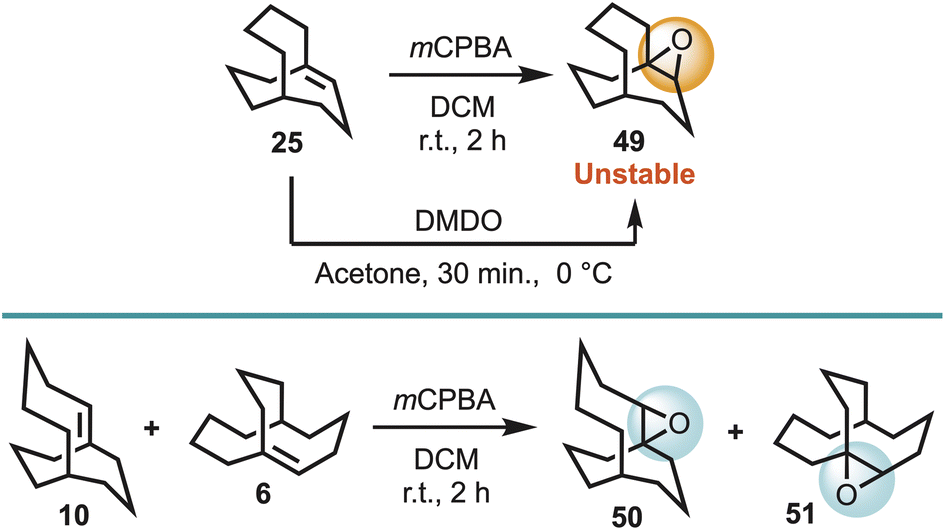 | ||
| Scheme 7 Reaction of bridgehead alkenes 6, 10, and 25 with the common oxidants mCPBA and DMDO. | ||
Interestingly, the structure of cerorubenic acid-I, which contains a bicyclo[4.4.1] hyperstable alkene skeleton (see 2 in Fig. 1), slowly oxidises in air to give the corresponding epoxide28 (i.e., a result of the oxidant triplet oxygen29). Therefore, as a class, hyperstable alkenes are seemingly not resistant to oxidation. These observations are not too surprising given that oxidants (oxidation reagents) are highly reactive species, that operate via a redox mechanism, which transfers an electronegative oxygen atom(s).30 This process is in stark contrast to hydrogenation.
Conclusions
In summary, while developing predictive theoretical methods to classify the stability of cage bicyclic bridgehead alkenes, Schleyer, Maier and McEwen introduced the term “hyperstable alkene”,1a,6 followed by Kim some years later.7 Together, they predicted over 60 bicyclo[m.n.o] alkenes to be hyperstable, using as their criteria olefin strain energy calculations, in addition to predicting resistance of these alkenes to hydrogenation under normal conditions. Of these 60 theoretical examples the syntheses of only three alkenes (i.e.,2, 4 and 8) that resided in this theoretical optimal hyperstability zone had been reported, albeit independently; 2 being reported before the term hyperstable alkene was even developed. A major limitation to testing this theory more extensively was the inability to access bridgehead double bond containing systems which matched the Schleyer–Maier–McEwen–Kim predicted hyperstable alkenes.Herein described are further examples of hyperstable systems (i.e.,6, 10, 13, and 25) obtained via an optimised Brown–Matteson homologation sequence. Three of these were found to be isolable (i.e.,6, 10, and 25) and resistant to hydrogenation under a variety of conditions, consistent with the previously reported Schleyer and Maier definition of an “unreactive” hyperstable alkene i.e.,6 and 10 represent the most stable hyperstable systems reported to-date. However, the bridgehead alkenes 6, 10, and 25 were observed to undergo reaction with both strong and mild oxidants to afford osmate esters and epoxides, respectively. As a result of these studies, it is apparent that the computationally derived term “hyperstable alkene” only applies to resistance of hydrogenation, whether it be normal transition metal catalysed hydrogenation or non-metal based conditions. Lastly, it is important to recognise that alkene hyperstability is not a blanket term for all reaction conditions, and that bridgehead alkenes continue to be reported in the natural product literature that can likely attribute their stability to unique cage bicyclic structure i.e., “Such olefins should be very unreactive-not due to steric hindrance or to enhanced π-bond strength but due to special stability afforded by the cage structure of the olefin and to the greater strain of the parent polycycloalkane”.1a
Data availability
All experimental data are provided in the ESI.†Author contributions
M. D. S. and S. C. performed chemical synthesis. L. R. G. and E. H. K. carried out in silico calculations. G. K. P. performed high field NMR measurements of target alkenes and associated osmium complexes. P. V. B. acquired the X-ray crystallographic data and performed the analysis. C. M. W. conceived the project, coordinated the study, assisted in analysing and interpreting the data, and wrote the manuscript. All authors read and agreed on the content of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Queensland (UQ) and the Australian Research Council for funding to C. M. W. (FT110100851, DP180103004 and LE190100106) and E. H. K (FT120100632 and 180103047). M. D. S. is grateful for Research Training Program Scholarship from UQ (UQ RTP). The Australian National Computational Infrastructure and the UQ Research Computing Centre are gratefully acknowledged for computational resources. Dr Michael Gallen (QAEHS, UQ) is gratefully acknowledged for performing GC/MS accurate mass measurements.Notes and references
- (a) W. F. Maier and P. v. R. Schleyer, Evaluation and prediction of the stability of bridgehead olefins, J. Am. Chem. Soc., 1981, 103, 1891–1900 CrossRef CAS; (b) See also, P. M. Warner and S. Peacock, Strain energies of some bridgehead olefins as calculated with the MM2 force field, J. Comput. Chem., 1982, 3, 417–420 CrossRef CAS.
- For a recent discussion of the meaning of “stability” see: R. Hoffmann, P. v. R. Schleyer and H. F. Schaefer III, Predicting Molecules—More Realism, Please!, Angew. Chem., Int. Ed., 2008, 47, 7164–7167 CrossRef PubMed.
- (a) J. Y. W. Mak, R. H. Pouwer and C. M. Williams, Natural Products with Anti-Bredt and Bridgehead Double Bonds, Angew. Chem., Int. Ed., 2014, 53, 13664–13688 CrossRef CAS; (b) E. H. Krenske and C. M. Williams, Do Anti-Bredt Natural Products Exist? Olefin Strain Energy as a Predictor of Isolability, Angew. Chem., Int. Ed., 2015, 54, 10608–10612 CrossRef CAS; (c) A. G. Kutateladze, E. H. Krenske and C. M. Williams, Reassignments and Corroborations of Oxo-Bridged Natural Products Directed by OSE and DU8+ NMR Computation, Angew. Chem., Int. Ed., 2019, 58, 7107–7112 CrossRef CAS PubMed; (d) J. Liu, X. Liu, J. Wu and C.-C. Li, Chem, 2020, 6, 579–615 CrossRef CAS; (e) L. McDermott, Z. G. Walters, S. A. French, A. M. Clark, J. Ding, A. V. Kelleghan, K. N. Houk and N. K. Garg, A solution to the anti-Bredt olefin synthesis problem, Science, 2024, 386, eadq3519 CrossRef PubMed.
- C. S. Swindell, T. F. Isaacs and K. J. Kanes, Bicyclo[5.3.1]Undec-1(10)-Ene Bridgehead Olefin Stability and the Taxane Bridgehead Olefin, Tetrahedron Lett., 1985, 26, 289–292 CrossRef CAS.
- OSE discrepancies between the work of Schleyer and Kim for the bicyclo[4.4.3]tridec-1(11)-ene systems were noted by Kim, but Kim seemed not to have evaluated E- and Z-isomers (see ref. 6). Therefore, although 6 and 9 are drawn the same they are numbered differently for this reason.
- A. B. McEwen and P. v. R. Schleyer, Hyperstable olefins: further calculational explorations and predictions, J. Am. Chem. Soc., 1986, 108, 3951–3960 CrossRef CAS.
- J.-S. Kim, The Molecular Mechanics Evaluation of the Stability of Bridgehead Olefins Containing Medium Rings, Bull. Korean Chem. Soc., 1997, 18, 488–495 CAS.
- W. Parker, R. L. Tranter, C. I. F. Watt, L. W. K. Chang and P. v. R. Schleyer, Enhancement of reactivity at bridgehead positions, J. Am. Chem. Soc., 1974, 96, 7121–7122 CrossRef CAS.
- K. B. Becker and J. L. Chappuis, Synthesis and Dimerization of Bicyclo [4.4.1]undec-1(11)-ene, a Bridged trans-Cycloheptene, Helv. Chim. Acta, 1979, 62, 34–43 CrossRef CAS.
- H. Kukuk, E. Proksch and A. de Meijere, Bicyclo[4.4.2]dodeca-1-ene – The First “Hyperstable” Bridgehead Olefin, Angew. Chem., Int. Ed., 1982, 21, 306 CrossRef.
- (a) J. E. McMurry and C. N. Hodge, Synthesis of in,out-Bicyclo[4.4.4]tetradecane. Generation of a Stable μ-Hydrido-Bridged Carbocation, J. Am. Chem. Soc., 1984, 106, 6450–6451 CrossRef CAS; (b) J. E. McMurry, T. Lectka and C. N. Hodge, The in-bicyclo[4.4.4]-1-tetradecyl cation: a stable substance with a three-center, two-electron carbon-hydrogen-carbon bond, J. Am. Chem. Soc., 1989, 111, 8867–8872 CrossRef CAS.
- Larger in,out-systems were later reported, see J. E. McMurry and T. Lectka, Carbocations with bent three-center, two-electron carbon-hydrogen-carbon bonds, J. Am. Chem. Soc., 1993, 115, 10167–10173 CrossRef CAS.
- Bicyclo[n.2.2] hyperstable systems have been prepared although these lie outside the computed optimal zone, see, (a) S. Yasuo, T. Shingo, O. Masaru, M. Masaharu, T. Yoshito and O. Yoshinobu, Synthesis and Properties of Bridgehead-substituted Bicyclo[n.2.2] Bridgehead Alkenes, Bull. Chem. Soc. Jpn., 1981, 54, 1474–1480 CrossRef; (b) K.-L. Noble, H. Hopf and L. Ernst, Cyclophane XXI, Über das chemische Verhalten von [8]Paracyclophan: Reaktionen des Benzolrings, Chem. Ber., 1984, 117, 455–473 CrossRef CAS; (c) W. Grimme, A. Bertsch, H. Flock, T. Noack and S. Krauthäuser, Bridged Medium Rings with Hyperstable Double Bonds - Syntheses and Reactions, Synlett, 1998, 1175–1181 CrossRef CAS.
- H. C. Brown, T. Imai, P. T. Perumal and B. Singaram, Organoboranes. 41. Reaction of organoboranes with (dichloromethyl)lithium. Scope and limitations. Synthesis of homologated primary and secondary alcohols, J. Org. Chem., 1985, 50, 4032–4036 CrossRef.
- (a) D. S. Matteson and R. Ray, Directed chiral synthesis with pinanediol boronic esters, J. Am. Chem. Soc., 1980, 102, 7590–7591 CrossRef CAS; (b) D. S. Matteson, Boronic Esters in Asymmetric Synthesis, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS.
- (a) H. C. Brown, A. S. Phadke and M. V. Rangaishenvi, Ring enlargement of boracyclanes via sequential one carbon homologation. The first synthesis of boracyclanes in the strained medium ring range, J. Am. Chem. Soc., 1988, 110, 6263–6264 CrossRef CAS PubMed; (b) H. C. Brown and M. V. Rangaishenvi, Some recent applications of hydroboration/organoborane chemistry to heterocycles, J. Heterocycl. Chem., 1990, 27, 13–24 CrossRef CAS; (c) H. C. Brown and S. Jayaraman, Organoboranes. 57. Relative efficacy of representative lithium dialkylamide bases for the in situ metalation of allyl chloride to produce (α-chloroallyl)lithium. Application of the reagent to B-alkoxy-9-borabicyclononanes producing new routes to interesting cyclooctane derivatives, J. Org. Chem., 1993, 58, 6791–6794 CrossRef CAS.
- H. C. Brown, A. K. Mandal and S. U. Kulkarni, Hydroboration. 45. New, convenient preparations of representative borane reagents utilizing borane-methyl sulfide, J. Org. Chem., 1977, 42, 1392–1398 CrossRef CAS.
- Matteson homologations have previously been used in a one pot sequence to construct acyclic (linear) systems using boronic esters, see for example, M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Assembly-line synthesis of organic molecules with tailored shapes, Nature, 2014, 513, 183–188 CrossRef CAS PubMed.
- T. J. Michnick and D. S. Matteson, (Bromomethyl) lithium: Efficient in situ Reactions, Synlett, 1991, 31, 631–632 CrossRef.
- N. M. Weliange, D. S. McGuinness and J. Patel, Thermal Dehydroboration: Experimental and Theoretical Studies of Olefin Elimination from Trialkylboranes and Its Relationship to Alkylborane Isomerization and Transalkylation, Organometallics, 2014, 33, 4251–4259 CrossRef CAS.
- N. J. Cusack, C. B. Reese, A. C. Risius and B. Roozpeikar, 2,4,6-Tri-isopropylbenzenesulphonyl Hydrazide: A Convenient Source of Di-imide, Tetrahedron, 1976, 32, 2157–2162 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) W. R. Roth, Hydrierwärmen, Nachr. Chem., Tech. Lab., 1983, 31, 964–968 CrossRef CAS; (b) See also, Z.-H. Li and M. Jones Jr, Hydrogenation of [7]Paracyclophane. Formation of a Hyperstable Bridgehead Olefin, Tetrahedron Lett., 1987, 28, 753–754 CrossRef CAS.
- P. R. Rablen, A Procedure for Computing Hydrocarbon Strain Energies Using Computational Group Equivalents, with Application to 66 Molecules, Chemistry, 2020, 2, 347–360 CrossRef.
- (a) T. J. Donohoe, K. Blades, P. R. Moore, M. J. Waring, J. J. G. Winter, M. Helliwell, N. J. Newcombe and G. Stemp, Directed Dihydroxylation of Cyclic Allylic Alcohols and Trichloroacetamides Using OsO4/TMEDA, J. Org. Chem., 2002, 67, 7946–7956 CrossRef CAS PubMed; (b) T. J. Donohoe, L. Mitchell, M. J. Waring, M. Helliwell, A. Bell and N. J. Newcombe, Scope of the directed dihydroxylation: application to cyclic homoallylic alcohols and trihaloacetamides, Org. Biomol. Chem., 2003, 1, 2173–2186 RSC.
- A. S. Burns, C. Dooley III, P. R. Carlson, J. W. Ziller and S. D. Rychnovsky, Relative and Absolute Structure Assignments of Alkenes Using Crystalline Osmate Derivatives for X-ray Analysis, Org. Lett., 2019, 21, 10125–10129 CrossRef CAS.
- M. S. Tempesta, T. Iwashita, F. Miyamoto, K. Yoshihara and Y. Naya, A New Class of Sesterterpenoids from the Secretion of Ceropiastes rubens (Coccidae), J. Chem. Soc. Chem. Commun., 1983, 1182–1183 RSC.
- P. D. Bartlett and R. Banavali, Spontaneous Oxygenation of Cyclic Olefins: Effects of Strain, J. Org. Chem., 1991, 56, 6043–6050 CrossRef CAS.
- Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2nd edn, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2378473 and 2378474. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06697a |
| This journal is © The Royal Society of Chemistry 2024 |