
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unraveling the mechanism of the CO2-assisted oxidative dehydrogenation of propane over VOx/CeO2: an operando spectroscopic study†
Leon
Schumacher
,
Marius
Funke
and
Christian
Hess
 *
*
Department of Chemistry, Technical University of Darmstadt, Eduard-Zintl-Institut für Anorganische und Physikalische Chemie, Peter-Grünberg-Str. 8, 64287 Darmstadt, Germany. E-mail: christian.hess@tu-darmstadt.de
First published on 7th November 2024
Abstract
The CO2-assisted oxidative dehydrogenation (ODH) of propane is of great interest for the usage of CO2 in chemical industry. Vanadia-based catalysts are a promising material class, which can replace highly toxic CrOx, the current state-of-the-art catalyst. Ceria is a commonly used support material in CO2 activation but has not yet been used as a vanadia support for CO2-assisted propane ODH. In this study, we address the interplay between vanadia and ceria as well as the nuclearity-dependent reaction behavior of VOx/CeO2 catalysts using XRD, multi-wavelength Raman, UV-Vis, and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). While the vanadia loading increases the selectivity, the catalysts exhibit a significant amount of side reactions, including most prominently the total oxidation over bare ceria on surface oxygen sites due to their high reducibility and propane dry reforming (PDR) over catalysts with high vanadia loading. Mechanistic analysis reveals that dimers can transfer hydrogen from propane to the ceria lattice, forming Ce–H or to a monodentate carbonate, facilitating the reverse water–gas shift reaction (RWGSR), whereas a transfer to bridged Ce–OH surface species leads to total oxidation due to the high reactivity of the formed surface species. Oligomers facilitate PDR due to their high reducibility and the active oxygen site shifts from ceria to vanadia. The catalyst can be regenerated via carbonates, which are highly stable and can subsequently deactivate the catalyst surface. Our results highlight the benefit of applying multiple operando spectroscopies to enhance the mechanistic understanding of materials relevant for CO2 activation and further the knowledge-based optimization of catalytic performance.
Sustainability spotlightThe CO2-assisted oxidative dehydrogenation (ODH) of propane is of great technical importance and enables the use of CO2 in a value-adding process. Supported vanadium oxide (VOx) catalysts are a promising alternative to more active but toxic chromium oxide catalysts. Ceria is a of great interest in oxidation catalysis showing synergistic effects with VOx and we have therefore used it as a support material for VOx in the CO2-ODH of propane. In this study, we disentangle the complex network, and elucidate the mechanism and deactivation behavior for this reaction, based on a multiple in situ/operando approach (DRIFTS, multi-wavelength Raman, UV-Vis, XRD). This combination allows to unravel the vanadia–ceria interaction essential for CO2 activation, as well as the nuclearity-dependent reactivity behavior of the supported VOx species. Because of the great significance of vanadia/ceria systems for both future and existing oxidation catalysts, the environmental perspective of using CO2 as soft-oxidant, as well as the general applicability of our in situ/operando spectroscopic approach for mechanistic analysis, we believe our results to be of immediate importance to a broad readership. |
1. Introduction
Propylene is an important basis chemical that is used e.g. in the fabrication of polypropylene.1,2 Different propylene production methods such as steam cracking, fluid catalytic cracking etc. are employed, which are however not sufficient to satisfy the high industry demand, resulting in the ‘propylene gap’.3,4 Therefore, additional methods to produce propylene are required, of which propane ODH is of great interest, as the introduction of oxygen into the gas feed leads to lower reaction temperatures and less catalyst leaching and the exothermicity of the reaction allows for heat integration of the process. However, to stop the reaction at the selective oxidation product, preventing its overoxidation to COx, is challenging.1,2,5 One approach to solve this issue is to use CO2 as a soft oxidant instead of O2, which increases the selectivity towards propylene. Additional benefits include a more homogeneous thermal distribution in the reactor due to the high heat capacity of CO2, a lower flammability, less catalyst deactivation by carbon formation due to coal gasification, and the valorization of CO2 as an unwanted green-house gas.6,7 The latter is especially important in the industrial context to mitigate the influence of CO2 on global climate change.8–10 However, due to the high thermodynamic stability of CO2, a suitable catalyst is required to make the reaction viable.11–13The current state of the art catalyst used for the CO2-assisted ODH of propane is supported CrOx due to its high activity.7,14–17 In this catalyst, chromium was described to change its oxidation state under reactions conditions, that is, Cr6+ species are reduced to Cr2+/3+, which are then involved in non-oxidative pathways towards propylene, namely direct dehydrogenation (DDH) followed by subsequent RWGSR. Alternatively, the direct ODH pathway can occur.16 However, due to the high toxicity and potential damage to the environment when using chromium, its replacement with other active catalytic phases is highly desirable.18 To that end, vanadia has been of interest due to its established commercial uses in oxidation reactions and high catalytic activity in propane ODH using O2 as the oxidizing agent.19–24 However, the activity of vanadia strongly depends on its support, which can be categorized as inactive or active.5,25,26 Inactive supports are not actively participating in the reaction and include SiO2 (ref. 20 and 23) or Al2O3,19 while active supports can actively participate in the reaction by e.g. supplying lattice oxygen and include CeO2, TiO2 (ref. 24) or In2O3.21,22 Recently, the use of TiO2 as vanadia support during CO2-assisted propane ODH was studied and showed promising activities that were even comparable to those of CrOx.24 However, the use of ceria as a support, which is commonly used in reactions that require the activation of CO2, has not yet been reported to the literature despite its high potential.
Ceria is an established material for the conversion of C1 substrates, including CO oxidation,27 CO2 hydrogenation to methanol28 and RWGSR.29 So far, its potential for CO2-assisted propane ODH is undisputed. Ceria in combination with vanadia has been used for ODH reactions using O2 as the oxidizing agent, including alcohol ODH (methanol and ethanol),30,31 and short alkane ODH (ethane, propane, butane).32–34 In the context of propane ODH, it was shown that the complex interplay between vanadia and ceria is highly relevant to understand the reactivity behavior of the catalyst. On the ceria surface, vanadia is present in different nuclearities,35 which perform different functions.36 While monomeric vanadia is able to modulate the ceria oxygen mobility by interacting with surface vacancies,33 dimeric and oligomeric nuclearities catalyze the initial C–H bond breakage and facilitate the rapid transfer of hydrogen from propane to ceria lattice oxygen, while the trimeric nuclearity is very stable and does not participate.36 In addition, ceria was shown to keep vanadia in oxidation state V5+, while being reduced itself.33,34,36–40 However, when using CO2, it was shown that especially monomeric vanadia cannot be fully reoxidized after being reduced.41 Further recent studies showed that the ceria lattice can be re-oxidized by CO2 after regenerating vanadia with nuclearities >1 via carbonate intermediates that are reduced to CO leading to a pristine lattice again.42 Understanding how these properties translate to the CO2-assisted propane ODH will be an important step in understanding the reaction mechanism over VOx/CeO2.
In addition to elucidating the catalyst's behavior, understanding the reaction network is of high importance to obtain a full mechanistic picture and assign the identified active sites to the individual reactions. Besides the ODH reaction, multiple side reactions can occur, which include PDR, coking, cracking, and coal gasification of the formed carbon.13 In addition, the dehydrogenation pathway was shown by experimental and theoretical work of Ascoop et al.20 to occur via two routes, that is, a direct ODH pathway and an indirect two-step pathway. The latter is based on direct dehydrogenation, resulting in hydrogen and propylene, and subsequent reaction of hydrogen with CO2 yielding CO and water via RWGSR. The presence of these two pathways was further confirmed experimentally by Rogg et al.23 for VOx/SiO2 using operando UV-Raman spectroscopy, highlighting the importance of understanding this highly complex reaction network and the mode of operation of the vanadia catalyst under reaction conditions.
In this study, we investigate the CO2-assisted propane ODH over VOx/CeO2 with the goal of elucidating the detailed reaction mechanism using multiple operando and in situ spectroscopies. These include in situ XRD, operando multi-wavelength Raman, operando UV-Vis, and quasi in situ DRIFT spectroscopy. In addition, the reaction network will be analyzed in detail to understand the reactivity behavior. Using the above methods the question of the interaction between ceria and vanadia, the identification of the active site, and the nuclearity-dependent reactivity behavior will be addressed to obtain a full mechanistic picture. Furthermore, we will address the active sites involved in potential side reactions and the common problem of catalyst deactivation during CO2-assisted propane ODH. Our results demonstrate the potential of combining multiple methods to obtain detailed mechanistic insights into a catalytic reaction with potential industrial applicability. The approach can be readily transferred to other reactions and materials relevant to CO2 activation and valorization.
2. Experimental section
2.1. Catalyst preparation
The ceria support was prepared by two-fold calcination of cerium(III) nitrate hexahydrate (≥99.99%, Sigma-Aldrich) staying at 600 °C for 12 h after heating using a rate of 1.5 °C min−1, as described previously.43 Ceria was loaded with vanadia by incipient wetness impregnation. Three different loadings were prepared by mixing 1 g of ceria with 0.5 mL of different-concentration precursor solutions (1.07 mol L−1, 0.51 mol L−1, and 0.21 mol L−1) containing vanadium(V) oxytriisopropoxide (≥97%, Sigma-Aldrich) and 2-propanol (99.5%, Sigma-Aldrich). The samples were then heated to 600 °C at a heating rate of 1.5 °C min−1 and calcined for 12 h. The specific surface area of bare ceria was determined to be 61.4 m2 g−1 by nitrogen physisorption experiments and the use of the Brunauer–Emmett–Teller (BET) method, yielding vanadium loadings of 2.83 V nm−2 (2.32 wt% V2O5), 1.36 V nm−2 (1.11 wt% V2O5), and 0.57 V nm−2 (0.47 wt% V2O5), respectively. Higher vanadium loadings were not considered since vanadia crystallites were shown to be present at loadings >2.9 V nm−2.44 The resulting catalyst powders were subsequently pressed at a pressure of 2000 kg m−2 for 20 s, ground and then sieved using a combination of sieves to obtain 200–300 μm particles.2.2. Catalytic testing
Catalytic testing was performed in a CCR 1000 reaction cell (Linkam Scientific) equipped with a membrane pump to allow for operation in a fluidized-bed mode, using 70 mg of catalyst. The samples were first dehydrated in 12.5% O2/He for 1 h at 550 °C, subsequently cooled to 50 °C, exposed to 12.5% CO2/12.5% C3H8/He with a total flow rate of 40 mln min−1, and then heated in 45 °C steps up to 550 °C, staying at each temperature for 1 h. The direct dehydrogenation of propane (DDH) was tested in the same way but no CO2 was added to the gas feed. The gas-phase composition was analyzed continuously using a gas chromatograph (GC, Agilent Technologies 7890B) equipped with a PoraPlot Q and a Molsieve column. The separated gases were analyzed by a thermal conductivity detector (TCD) and a flame ionization detector (FID) in series. The set-up is connected through a twelve-way valve. One chromatogram is measured every 29 min, resulting in two chromatograms for each temperature, which were averaged. The pressure before and after the GC was monitored to correct the detected areas for pressure fluctuations. To analyze the reaction network, the temporal evolution of the reactivity behavior was analyzed at 550 °C. The samples were first dehydrated in 12.5% O2/He for 1 h, then treated consecutively in 7.5% H2/Ar, 12.5% CO2/He, 12.5% CO2/12.5% C3H8/He, and finally regenerated in 12.5% CO2/He (for 2 h for each gas phase), respectively. The reaction products were analyzed and ratios between different products were calculated to determine the relative contributions of the different reaction pathways to the overall product distribution.2.3. X-ray diffraction
X-ray diffraction patterns were recorded on an Empyrean system (Malvern Panalytical) in Bragg–Brentano geometry using CuKα radiation and a PIXcel1D detector. For in situ analysis, 90 mg of catalyst was placed in an XRK 900 reaction chamber (Anton Paar) equipped with a NiCr–NiAl thermocouple to measure the temperature directly next to the sample surface. The samples were first dehydrated in 12.5% O2/N2, cooled to room temperature, and subsequently treated in 12.5% CO2/12.5% C3H8/He. The samples were then heated in 100 °C steps up to 750 °C, with an equilibration time of 30 minutes at each temperature, and measured for 10 minutes in a 2θ range from 20 to 70° with a step-width of 0.025° for each step. A Rietveld analysis was performed for the diffraction pattern recorded at 750 °C using the TOPAS software together with reference structures from the ICSD database. The background was corrected by using a Chebyshev function with ten polynomials.2.4. UV-Raman spectroscopy
UV-Raman spectroscopy was performed at an excitation wavelength of 385 nm generated by a laser system based on a Ti:Sa solid-state laser pumped by a frequency-doubled Nd:YAG laser (Coherent, Indigo). The fundamental wavelength was frequency doubled to 385 nm using a LiB3O5 crystal. The light was focused onto the sample, and the scattered light was collected by a confocal mirror setup and focused into a triple-stage spectrometer (Princeton Instruments, TriVista 555).44 Finally, the Raman contribution was detected by a charge-coupled device (CCD, 2048 × 512 pixels) cooled to −120 °C. The spectral resolution of the spectrometer was 1 cm−1. For Raman experiments, 70 mg of catalyst was placed in a CCR 1000 reactor (Linkam Scientific Instruments) equipped with a CaF2 window (Korth Kristalle GmbH). A fluidized bed reactor was employed to avoid laser-induced damage, allowing the use of a laser power of 9 mW at the location of the sample. Data processing included cosmic ray removal and background subtraction. The spectra were further analyzed by a least-squares fitting analysis using four Lorentzian functions (see Fig. S5†). Operando spectra were measured after dehydration for 1 h in 12.5% O2/He and further pre-treatment in 7.5% H2/Ar and 12.5% CO2/He at 550 °C to start from a partially oxidized state. Afterwards, the catalyst was exposed to reactive conditions (12.5% CO2/12.5% C3H8/He) and regenerated under oxidizing conditions (12.5% CO2/He) with a total flow rate of 40 mln min−1 for each gas phase. The spectra were further analyzed by a least-squares fitting analysis using Lorentzian functions after normalization to the F2g peak of ceria.2.5. Vis-Raman spectroscopy
Visible (Vis) Raman spectroscopy was performed at 514 nm excitation, emitted from an argon ion gas laser (Melles Griot). The light was focused onto the sample, gathered by an optical fiber and dispersed by a transmission spectrometer (Kaiser Optical, HL5R). The dispersed Raman radiation was subsequently detected by an electronically cooled CCD detector (−40 °C, 1024 × 256 pixels). The spectral resolution was 5 cm−1 with a wavelength stability of better than 0.5 cm−1. For Raman experiments, 70 mg of catalyst was filled into a CCR 1000 reactor (Linkam Scientific Instruments) equipped with a quartz window (Linkam Scientific Instruments). The laser power at the sample location was 4 mW. Data analysis of the Raman spectra included cosmic ray removal and an auto new dark correction. Operando spectra were measured after dehydration for 1 h in 12.5% O2/He and further pre-treatment in 7.5% H2/Ar and 12.5% CO2/He at 550 °C to start from a partially oxidized state. Afterwards, the catalyst was exposed to reactive conditions (12.5% CO2/12.5% C3H8/He) and regenerated under oxidizing conditions (12.5% CO2/He) with a total flow rate of 40 mln min−1 for each gas phase. The spectra were normalized to the F2g peak of ceria. The F2g position was determined by a least-square fitting analysis. The nuclearity distribution was determined from the vanadyl peaks by performing a least-square fitting analysis using five Lorentzian functions at distinct spectral positions.362.6. Diffuse reflectance UV-Vis spectroscopy
Diffuse reflectance (DR) UV-Vis spectra were recorded on a Jasco V-770 UV-Vis spectrometer. Dehydrated BaSO4 was used as the white standard. For each experiment, 90 mg of catalyst was placed in the commercially available reaction cell (Praying Mantis High Temperature Reaction Chamber, Harrick Scientific) equipped with transparent quartz glass windows. Operando spectra were measured after dehydration for 1 h in 12.5% O2/He and further pre-treatment in 7.5% H2/Ar and 12.5% CO2/He at 550 °C to start from a partially oxidized state. Afterwards, the catalyst was exposed to reactive conditions (12.5% CO2/12.5% C3H8/He) and regenerated under oxidizing conditions (12.5% CO2/He) with a total flow rate of 40 mln min−1 for each gas phase. The band gap energies were determined using Tauc plots, while the reduction peak area was quantified by a least-squares fitting analysis of the spectra using Gaussian–Lorentzian (70/30) product functions. Product functions were used to account for the large contribution of natural line broadening to the overall line-shape (Lorentzian), caused by the short life-time of the electronically excited states.452.7. Diffuse reflectance infrared Fourier transform spectroscopy
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was performed using a Vertex 70 spectrometer (Bruker). A liquid nitrogen-cooled mercury cadmium telluride (MCT) detector was used, operating at a resolution of 1 cm−1. Dehydrated potassium bromide was employed as an infrared transparent sample for the background spectrum. For each experiment, 90 mg of catalyst was placed in the commercially available reaction cell (Praying Mantis High Temperature Reaction Chamber, Harrick Scientific) equipped with transparent KBr windows.For quasi in situ measurements the samples were treated in 12.5% O2/He, 7.5% H2/Ar, 12.5% CO2/He, 12.5% CO2/12.5% C3H8/He, and finally regenerated in 12.5% CO2/He for 1 h at 550 °C, respectively. For each gas phase the sample was cooled rapidly to room temperature (200 °C min−1) in pure helium for the measurement before heating it back to 550 °C for the next gas treatment.
Data processing consisted of background removal by subtracting a baseline formed by 12 anchor points. To quantify the adsorbate peaks in the Ce–OH region, the spectra were fitted using Lorentzian functions employing the Levenberg–Marquardt algorithm implemented in OriginLab 2022b. Finally, to quantify the total amount of carbonates on the surface, the region between 1200 and 1800 cm−1 was integrated.
3. Results and discussion
First, the catalytic activity of the sample will be discussed. Due to the plethora of possible reactions over the catalyst, an analysis of the reaction network will be performed, followed by an operando and quasi in situ spectroscopic investigation. Since the samples used for this study are identical to those in our previous studies on the VOx/CeO2 system, structural characterization will not be performed in this study. For the detailed characterization results, please refer to our previous work.33,36,463.1. Activity data and reaction network analysis
Fig. 1 shows the temperature-dependent propane conversions and propylene selectivities of bare ceria and vanadia-loaded samples during ODH (12.5% CO2/12.5% C3H8/He) and DDH conditions (12.5% C3H8/He) between 225 and 550 °C. Exemplary product amounts of CO and ethylene produced during the ODH and DDH in the same temperature range are given in the ESI (see Fig. S1†).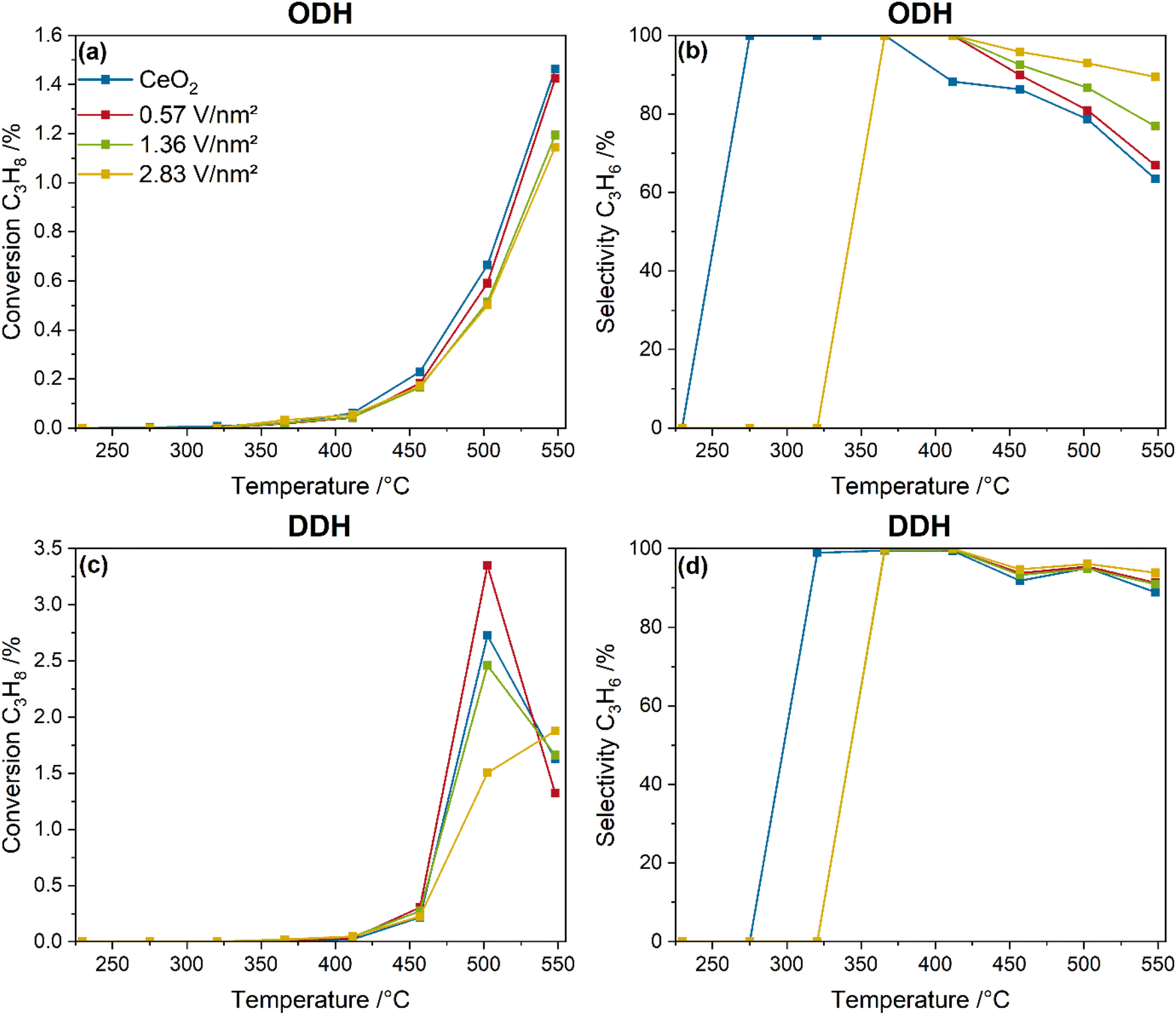 | ||
| Fig. 1 Comparison between the temperature-dependent conversions (a and c) and selectivities (b and d) of bare ceria and vanadia-loaded samples between 225 and 550 °C under ODH (12.5% CO2/12.5% C3H8/He) and DDH (12.5% C3H8/He) conditions. | ||
As shown in Fig. 1a, the temperature-dependent propane conversion of bare ceria and vanadia-loaded samples is characterized by an exponential increase. The background conversion caused by the Linkam reactor is already removed from the data presented. Ceria shows the highest conversion at all temperatures, while the conversion for the vanadia-loaded samples decreases with increasing amount of vanadia. However, the conversion stays below 1.5% for all samples at all temperatures. The propylene selectivities, shown in Fig. 1b, start at 100% for low temperatures where the conversions are <0.1%, and start to decrease at 365 °C for ceria, gradually decreasing to 60%. The propylene selectivities of the vanadia-loaded samples stay above those of bare ceria and increase with the vanadia loading for all temperatures. Most of these differences in the propylene selectivities are likely caused by the different conversions, which show an inverse trend. A temperature of 550 °C was chosen for the spectroscopic investigation of the system to maximize the conversions, thus enhancing the visibility of spectroscopic changes. The catalytic activity of the VOx/CeO2 system is similar to VOx/SiO2 catalysts for bare ceria and lower vanadia loadings, but better for the highest loading, while the selectivity is slightly lower.23 In comparison to VOx/In2O3 it reaches only ∼50% of the conversion21,22 and compared to VOx/TiO2 about 30%.24 However, the VOx/CeO2 system is still of great interest to obtain a better understanding of supported VOx catalysts in the CO2-assisted propane ODH in general. It might also be of use for the development of multi-oxide catalysts, as was done for the VOx/SiO2 system by adding WOx, significantly improving the catalytic activity.20
In comparison, Fig. 1c shows the propane conversion of bare ceria and the vanadia-loaded samples under DDH conditions. The conversion starts to be detected at significantly higher temperatures than under ODH conditions, then shows a stronger increase for all samples at 500 °C to 1.5–3.5%, and finally decreases sharply. This effect is attributed to the depletion of ceria lattice oxygen, giving a first important hint towards involvement of ceria lattice oxygen in the reaction mechanism, while CO2 is required to stabilize the catalytic performance. The propylene selectivities under DDH conditions, shown in Fig. 1d, also show significantly higher values, staying above 85% for all temperatures. This emphasizes that the DDH reaction exhibits significantly higher conversions and selectivities but has no stable performance. This is further emphasized by the molar product amounts of CO and ethylene for both reaction conditions (see Fig. S1†). First, the amount of CO produced during ODH conditions is increased by 1.5 orders of magnitude. The formation of CO during DDH conditions indicates that some total oxidation or propane dry reforming occurs during the DDH. However, the significantly higher amount of CO during ODH conditions indicates the consumption of CO2 towards CO, possibly regenerating the ceria lattice, thereby stabilizing the catalytic activity. A similar behavior can be observed for ethylene that is one order of magnitude higher under ODH conditions. This indicates that much more cracking occurs under ODH conditions, which is likely to be one reason for the lower propylene selectivities. Comparison between the ODH and DDH data reveals that introduction of CO2 into the gas feed diminishes the reaction performance significantly but is required for a stable performance of VOx/CeO2 catalysts. Therefore, only ODH conditions will be considered in the following.
Fig. 2 shows the influence of the pre-treatment conditions on the catalytic performance of the VOx/CeO2 system under ODH conditions. Two different pre-treatment conditions were tested. First, switching to ODH conditions directly from a fully oxidized catalyst (12.5% O2/He), and second, ODH conditions after catalyst treatment in 12.5% O2/He, 7.5% H2/Ar and 12.5% CO2/He, which ensures a catalyst in a pre-reduced state that is more representative of reaction conditions. As can be seen in Fig. 2, the pre-treatment conditions have no significant influence on the measured propylene selectivities. However, the loading-dependent propane conversions are significantly influenced. The conversions decrease from bare ceria to the 2.83 V nm−2 sample for only oxidative pre-treatment conditions, but increase when the reaction is started from a partially reduced state. The propane conversion of bare ceria decreases from 1.6 to 0.7%, while the conversion of the 2.83 V nm−2 sample increases from 1.1 to 1.7%, which is the highest conversion overall. The conversion of the 0.57 and 1.36 V nm−2 samples is similar but is slightly lower for the 1.36 V nm−2 sample. When starting from a partially reduced state, both the conversion and selectivity increase with increasing vanadia loading, which indicates that the vanadia loading influences the selectivity not by only changing the conversion but by an actual change in the reaction pathway. The influence of the pre-treatment conditions on the mechanism is therefore of relevance to understand the changes in the conversion trends.
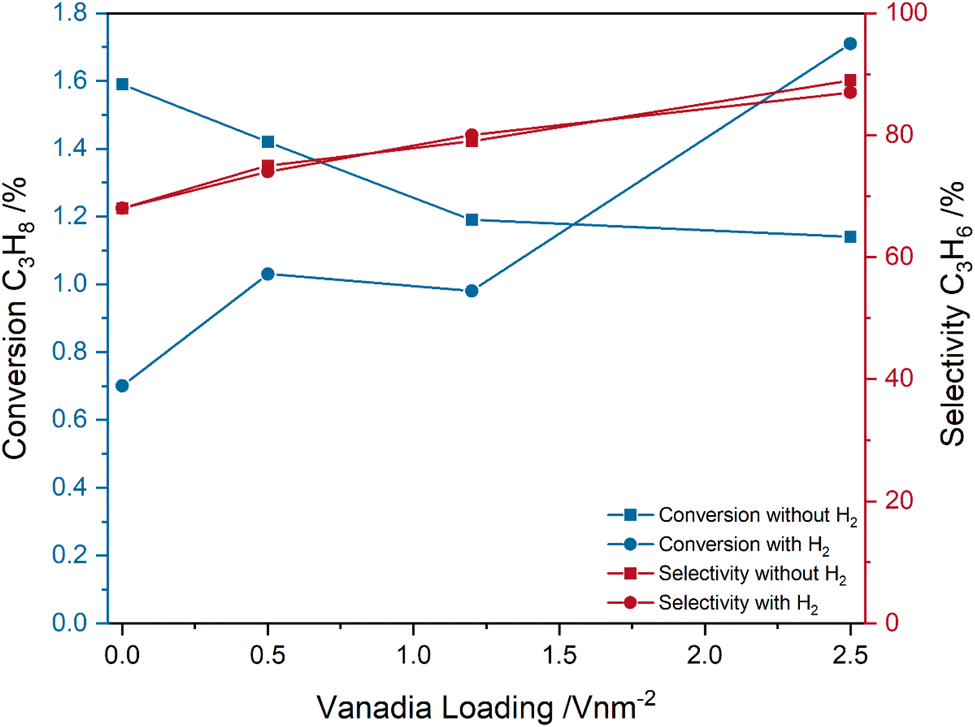 | ||
| Fig. 2 Comparison between the conversions and selectivities of bare ceria and vanadia-loaded samples after different pre-treatments (fully oxidized with O2 and partially reduced with H2 and subsequently regenerated with CO2) under ODH conditions (12.5% CO2/12.5% C3H8/He) at 550 °C. | ||
To understand the reaction network in more detail, Fig. 3 depicts the analysis of the reaction products based on the obtained product distributions shown in the ESI (see Fig. S2†), including the propane and CO2 conversions, the propylene selectivity, and the C3H6/CO, C2H4/H2, and created CO/converted CO2 ratio, as indicators for the ODH, cracking and propane dry reforming (PDR) reactions, respectively. In addition, the ESI shows the CO/H2 ratio, as an indicator for the contribution from the reverse water-gas shift reaction (RWGSR, see Fig. S3†).
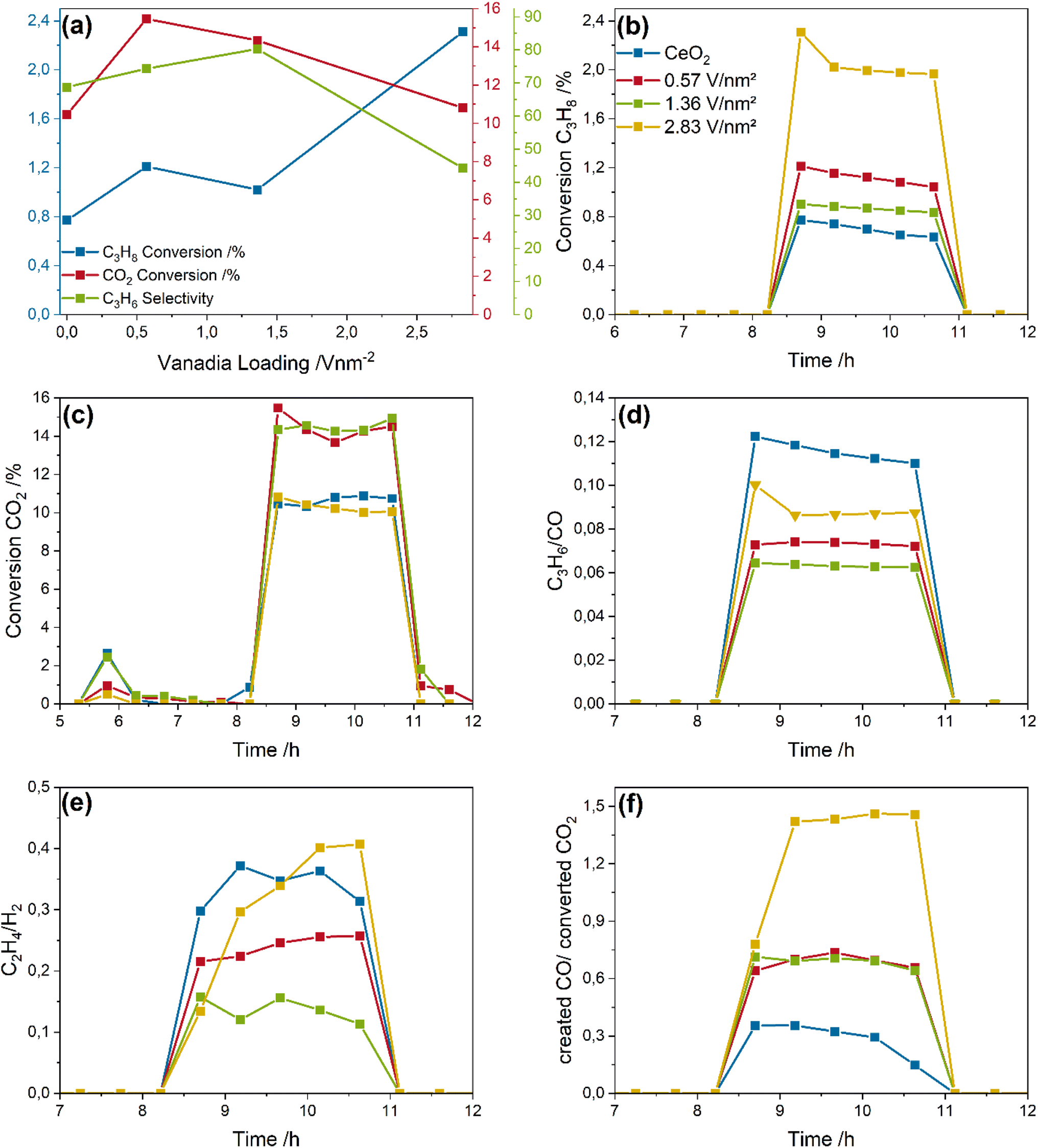 | ||
| Fig. 3 Analysis of the CO2-assisted propane ODH reaction network over bare ceria and vanadia-loaded samples: (a) loading-dependent propane and CO2 conversions and propylene selectivities, (b) propane and (c) CO2 conversions, and (d) propylene/CO, (e) ethylene/H2 and (f) created CO/converted CO2 ratios. | ||
Fig. 3a gives an overview of the initial (first 30 minutes of the reaction phase) propane and CO2 conversions and the propylene selectivity. The propane conversion slightly increases from 0.8% for bare ceria to 1.2 and 1% for the 0.57 and 1.36 V nm−2 samples but significantly increases to 2.3% for the 2.83 V nm−2 sample. The selectivity shows a linear increase from 65 to 80% with increasing vanadia loading up to 1.36 V nm−2. For the 2.83 V nm−2 sample, the propylene selectivity drops significantly to 42% concurrently to the sharp increase in propane conversion observed. The selectivity values include the cracking and PDR reaction determined in the discussion below. Importantly, the total amount of propylene produced stays constant for all four samples (see Fig. S2†). The selectivity decrease caused by propane total oxidation is not included, since the exact amount cannot be determined due to the different reaction routes in which CO and CO2 are involved. In comparison, the CO2 conversion (10–16%) is higher than that of propane by about one order of magnitude, indicating the occurrence of side reactions or processes unrelated to the ODH reaction. Since the stoichiometry of propane and CO2 during CO2-assisted propane ODH is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the difference between the propane and CO2 conversion can be understood as the total amount of side reactions occurring. However, since different reaction pathways can be responsible for CO2 consumption and CO formation, this is only a qualitative trend.
:1, the difference between the propane and CO2 conversion can be understood as the total amount of side reactions occurring. However, since different reaction pathways can be responsible for CO2 consumption and CO formation, this is only a qualitative trend.
The propane conversions (see Fig. 3b) change only slightly over time, indicating a stable reaction performance within the investigated time frame. Only the 2.83 V nm−2 sample shows a slightly higher conversion within the first 30 minutes (2.3%) compared to the stable conversion of 2% afterwards, which might be indicative of surface species that are highly reactive towards propane but cannot be regenerated during reaction, leading to a stable but lowered conversion. This is different from the reaction behavior of VOx/TiO2 catalysts under the same reaction conditions, which exhibited a significant deactivation behavior unless more CO2 was present.24
Fig. 3c depicts the CO2 conversion of the catalysts, showing only small variations. The small consumption of CO2 at ∼6 h is caused by the regeneration of the material after the reduction in 7.5% H2/Ar and is indicative of a partial regeneration of VOx/CeO2.42 It is particularly notable that the trend in the CO2 conversions does not follow that of the propane conversions, as bare ceria and the 2.83 V nm−2 sample exhibit the lowest CO2 conversions. Therefore, the CO2 conversion goes through a maximum with increasing vanadia loading, indicating that CO2 is used in other processes than the ODH reaction that are, in part, not related to propane conversion due to the different loading-dependent reactivity behavior towards propane and CO2.
To disentangle the observed reactivity behavior, the C3H6/CO, C2H4/H2, and created CO/converted CO2 ratio is formed from the molar product distributions shown in the ESI (see Fig. S2†), serving as an indicator for the ODH, cracking and propane dry reforming (PDR) reactions, respectively (see Fig. 3d–f). The RWGSR is probed by the CO/H2 ratio shown in Fig. S3.† The C3H6/CO ratio decreases from a value of 0.12 for bare ceria to a value of 0.06 for the 1.36 V nm−2 sample before increasing again for the 2.83 V nm−2 sample. If the ODH reaction was the only relevant reaction, CO and propylene would be produced in a 1:1 ratio. The observed ratios, however, clearly show, that much more CO is produced than would be expected for the ODH reaction, pointing to the occurrence of additional reaction pathways like PDR and total oxidation. It is also apparent, that the relative amount of produced propylene goes through a minimum for the 1.36 V nm−2 sample, which might be caused by the changes in nuclearities of surface vanadia species.36
Fig. 3e shows the C2H4/H2 ratio, which is an indicator for the prevalence of cracking. The values range between 0.13 for the 1.36 V nm−2 sample and 0.4 for the bare ceria and 2.83 V nm−2 sample, indicating that much more H2 is produced than ethylene. During the cracking reaction, propane is consumed and ethylene, methane and H2 are produced in a stoichiometric ratio.47,48 Therefore, the higher amount of detected hydrogen is indicative of further side reactions, e.g. PDR, which can produce hydrogen and also CO, which would additionally explain the observed C3H6/CO ratios. The loading-dependent trend in the ratios is similar to that observed in Fig. 3d; hence, much more CO and H2 is detected than ethylene and propylene. This behavior points to a significant proportion of PDR occurring, while the amount of cracking seems to be negligible. Notably, the C2H4/H2 ratio increases sharply for the 2.83 V nm−2 sample, which may indicate structural changes favouring cracking over PDR.
To obtain further insight into the reactivity behavior of the VOx/CeO2 samples, Fig. 3f shows the created CO/converted CO2 ratio, which is indicative of the amount of PDR. Here, only the 2.83 V nm−2 sample shows a value larger than one, which is indicative of PDR, since additional CO must be produced from propane. This is fully consistent with the observed propane conversions and propylene selectivities (see Fig. 3a). The overall propylene yield therefore does not benefit from the increased propane conversion.49 For the remaining samples, the ratio is below one, which indicates that more CO2 is converted than CO is produced. The additional converted CO2 will be used in other reactions like surface adsorbate formation (see below). The other two samples are in between the behavior of bare ceria and the 2.83 V nm−2 sample, with a value of almost one. This might indicate that dehydrogenation is dominant. However, the much higher CO2 conversion compared to propane and the very low C3H6/CO ratios indicate that significant amounts of CO2 and CO need to be additionally produced, which would be consistent with a mix of PDR, cracking and total oxidation. The total oxidation is especially well suited to produce these gases, since propane is converted to COx, consuming more lattice oxygen than selective oxidation. These need to be regenerated by additional CO2 creating additional CO. Since the total oxidation can produce both CO and CO2 but the regeneration of lattice oxygen can only lead to CO, this affects the created CO/converted CO2 ratio.
At last, Fig. S3† shows the CO/H2 ratio, which has been considered as an indicator of the ratio between ODH and DDH + RWGSR. However, due to many other pathways towards H2 and especially CO, this becomes overlayed by other effects, as reflected in the detected values, which vary between 5 and 30. Therefore, the two dehydrogenation pathways cannot be differentiated. Table S1† summarizes the carbon balances. The values range from 6.2 to 7.2%, which is comparably low for reactions that create significant amounts of carbon, indicating that carbon formation on the catalyst surface is negligible.
In summary, the catalyst samples exhibit ODH activity under 12.5% CO2/12.5% C3H8/He conditions at 550 °C. While under DDH conditions higher activities and selectivities are observed, only the ODH conditions lead to a stable reaction behavior. Additionally, the pre-treatment conditions are of importance, and a consecutive pre-treatment in hydrogen and CO2 facilitates the ODH reaction. Regarding the side reactions, a nuclearity-dependent behavior is observed, as determined from the analysis of the reaction network. Bare ceria shows the smallest contribution from PDR but has the highest prevalence of total oxidation. With increasing vanadia loading, the relative contribution from total oxidation of propane decreases while that of PDR increases. The 2.83 V nm−2 sample shows the lowest conversion of CO2 despite its higher propane conversion. This indicates the least prevalence of side reactions overall, despite its lower selectivity compared to the other samples, stemming from the high PDR rate. The total amount of consumed CO2 can be used as an indicator for the total amount of side reactions, since cracking was determined to be negligible, and the amount of dehydrogenation seems to stay constant.
3.2. Operando and in situ spectroscopic analysis
To understand the reactivity behavior in more detail, the catalysts are investigated by employing different operando and in situ spectroscopies as well as diffraction. The overall structural characterization of the catalysts was performed in great detail previously and will not be discussed again here.33,36 However, the relevant peaks of each spectrum will be briefly assigned for better readability and crucial information about the system will be given at the relevant points. For VOx/CeO2, selective resonance Raman enhancement by multi-wavelength excitation can be achieved, by using 385 and 514 nm excitation, as described previously.33 385 nm UV excitation was used at the position of the band gap absorption of ceria, leading to a significant and selective intensity increase of ceria-related features, allowing for their dedicated analysis. 514 nm Vis excitation is used to enhance the vanadia-based features enabling the detection of the vibrational vanadyl fine structure related to the vanadia nuclearities.To exclude the formation of crystalline CeVO4 due to the elevated temperatures and reducing conditions, in situ XRD was performed under 12.5% CO2/12.5% C3H8/He for the 2.83 V nm−2 sample between room temperature and 700 °C. Samples with lower loadings were not investigated since CeVO4 formation is favored at high vanadium contents.34,39,40 Fig. S4† shows that signals from CeVO4 are only observable starting at 700 °C. Therefore, the formation of crystalline CeVO4 has no relevance for the following study. However, the possibility of formation of nanodispersive CeVO4 remains, which can be observed via Raman spectroscopy.37,39
Fig. 4 depicts UV-Raman spectra (385 nm excitation) of bare ceria and the 1.36 V nm−2 sample recorded under different gas conditions at 550 °C. The spectra were normalized to the F2g peak. Here, a selective intensity enhancement of the ceria peaks is expected due to resonance effects caused by the absorption of ceria (see also Fig. 7). The observed intensity changes between the spectra recorded during initial CO2 and ODH conditions were quantified by a peak-fitting analysis and the results are summarized in Fig. 4c and d. The UV-Raman spectra for the 0.57 and 2.83 V nm−2 sample are given in the ESI (see Fig. S5†). An exemplary fit of the UV-Raman spectrum of the 1.36 V nm−2 sample under 12.5% O2/He is shown in the ESI (see Fig. S6†).
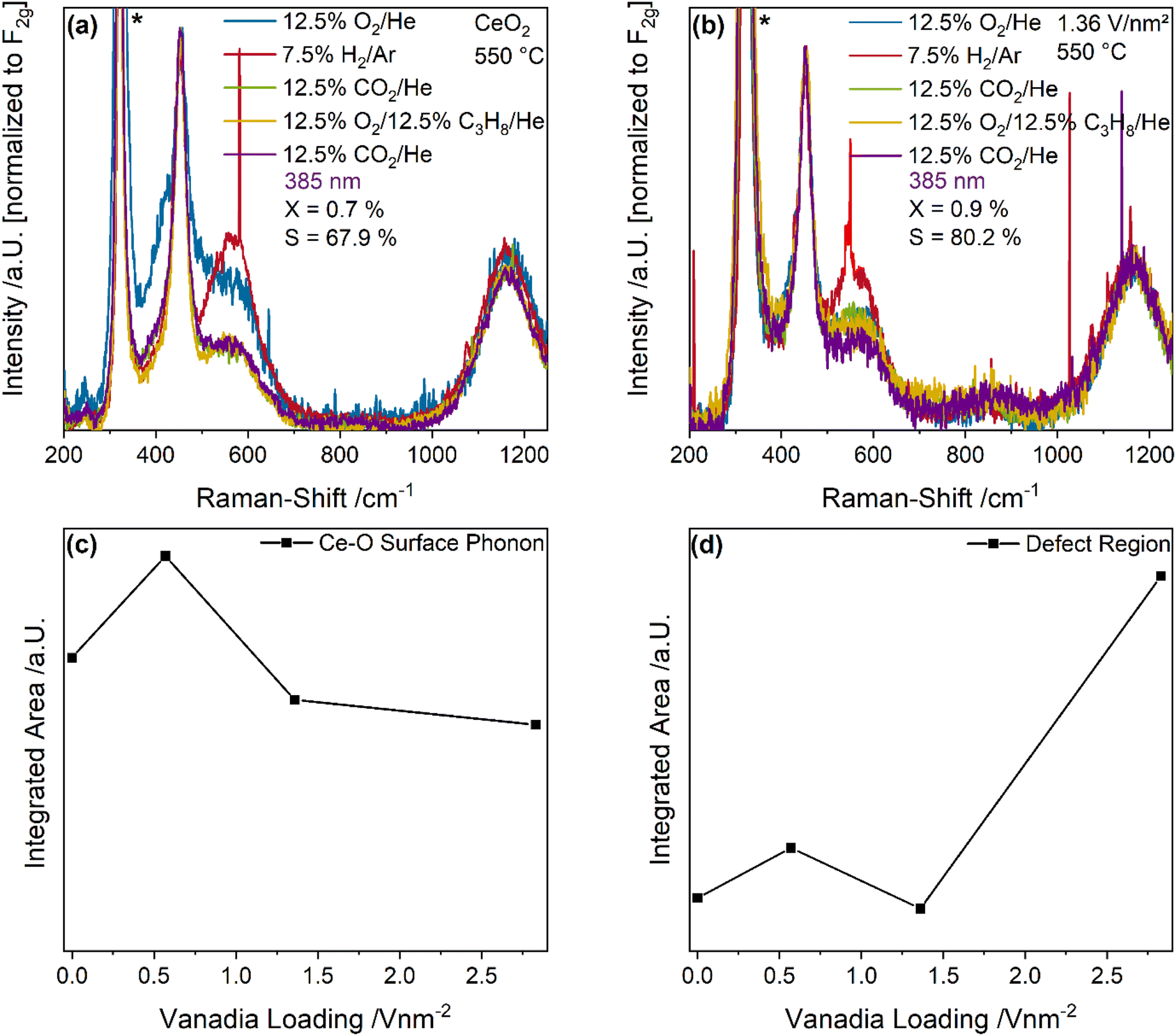 | ||
| Fig. 4 UV-Raman spectra (385 nm excitation) of (a) CeO2 and (b) the 1.36 V nm−2 sample recorded under the indicated gas feeds at 550 °C. Spectra were normalized to the F2g peak and the CaF2 window signal is marked with an asterisk. Quantification of the (c) Ce–O surface phonon and (d) defect region changes between spectra recorded under 12.5% CO2/He and 12.5% CO2/12.5% C3H8/He. | ||
The UV-Raman spectra of bare ceria under different gas conditions are characterized by peaks at 250, 405, 455, 590 and 1170 cm−1, which are caused by the longitudinal and transversal Ce–O surface phonon, the F2g mode, the defect region and the 2LO peak, respectively (see Fig. 4a).43,50 At the 250 cm−1 position, the longitudinal surface phonon is overlapped by the 2TA signal of ceria. The reduction and re-oxidation behavior of the sample was discussed in detail before.46 Briefly, under reducing conditions, the defect region increases in intensity, while the surface Ce–O oxygen signals show a decrease, consistent with the consumption of lattice oxygen and formation of vacancies. When CO2 is introduced for re-oxidation after H2/Ar treatment, the surface phonon regenerates very slightly, while the defect region decreases in intensity significantly below the initial intensity under O2 conditions. This was previously explained by the formation of carbonates which block surface oxygen sites, while regenerating vacancies.42 Upon switching to ODH conditions, similar changes as upon the switch from O2 to H2 can be observed but to a significantly smaller extent, which are regenerated in the second CO2 containing feed. This indicates that under reaction conditions some additional surface oxygen can be used despite carbonate formation, or alternatively that surface carbonates can act as an oxygen source for the reaction.
As shown in Fig. 4b, UV-Raman spectra of the 1.36 V nm−2 sample at 550 °C under different gas feeds do not exhibit additional peaks compared to the spectra of bare ceria. A similar behavior was previously observed for the VOx/TiO2 system under the same gas feeds and at the same temperature.24 It is caused by the high temperature decreasing the overall intensity together with the increased absorption of the support material which leads to resonance enhancement of the support and can diminish the vanadia signal. Comparison of all samples reveals the same structural dynamics when switching to reaction conditions, the extent of which decreases with increasing amounts of vanadia. This might be caused by vanadia blocking surface oxygen sites since vanadia is anchored to these sites, and as a consequence, less carbonates can be formed, which seems likely since all changes in the spectra are caused by ceria lattice oxygen. Notably, neither for bare ceria nor for the 1.36 V nm−2 sample a signal of the D- and G-bands originating from amorphous carbon is observed, consistent with the low carbon balances detected by GC (see Table S1†).
To fully understand the observed behavior, the changes in the UV-Raman spectra between the initial CO2 exposure and ODH conditions were quantified by a fitting analysis. An exemplary fit is shown in the ESI (see Fig. S6†). The results for the transversal surface phonon at 405 cm−1 and the defect region at 590 cm−1 are shown in Fig. 4c and d, respectively. The intensity difference of the Ce–O surface phonon increases from bare ceria to the 0.57 V nm−2 sample and then decreases again for higher vanadia loadings. The observed maximum for the 0.57 V nm−2 sample is in agreement with the increased propane and CO2 conversion at this loading, the latter representing the highest overall amount of CO2 being converted. The decrease at a loading of 1.36 V nm−2 can be explained by the lower conversion of propane as well as the decreased prevalence of the total oxidation reaction. It stays almost constant afterwards, since the total oxidation rate decreases again, while the PDR rate increases sharply. Overall, the behavior of the Ce–O surface phonon indicates that the surface may be mostly deactivated by CO2 due to carbonate formation caused by the pretreatment, but that the remaining surface oxygen atoms can still participate in the reaction and seem to provide the oxygen for all occurring reactions, correlating with the overall conversion of all reactants.
Fig. 4d shows the corresponding intensity changes of the defect region, which correlate well with the propane conversion, indicating that the conversion of propane leads to the formation of new defects in the material, which occurs concurrently with the consumption of Ce–O surface oxygen. However, the determined trends are very different: while the Ce–O surface phonon intensity changes correlate with the conversion of all molecules, the defect region only correlates with the propane conversion. Therefore, some reactions might lead to the consumption of clean surface Ce–O oxygen without vacancy formation, i.e. carbonate/formate formation, further emphasizing its importance. It is apparent that newly created vacancies are relevant in the conversion of propane but might be regenerated by CO2, making them unavailable for further propane conversion due to possible adsorbate formation. This will be discussed in more detail in the DRIFTS section of this manuscript (see Fig. 8 and discussion). In addition, the defect region may be influenced by the presence of monomeric vanadia species due to their known interaction with ceria surface vacancies.33,38,51 These would exhibit the most significant effect at low loadings due to their prominence, which is discussed in more detail below (see Fig. 6 and discussion).
To investigate the changes in the ceria and vanadia structure in more detail Fig. 5 shows Vis-Raman spectra (514 nm excitation) of bare ceria and the 1.36 V nm−2 sample recorded under different gas conditions at 550 °C. At this wavelength a selective intensity enhancement of the vanadia-related signals is expected due to resonance conditions (see also Fig. 7). The F2g shift and the change in hydride area between the initial CO2 and ODH conditions were quantified by a peak-fitting analysis and integration, respectively, (for details see experimental section), as shown in Fig. 5c and d. The Vis-Raman spectra of the 0.57 and 2.83 V nm−2 samples are given in the ESI (see Fig. S7†).
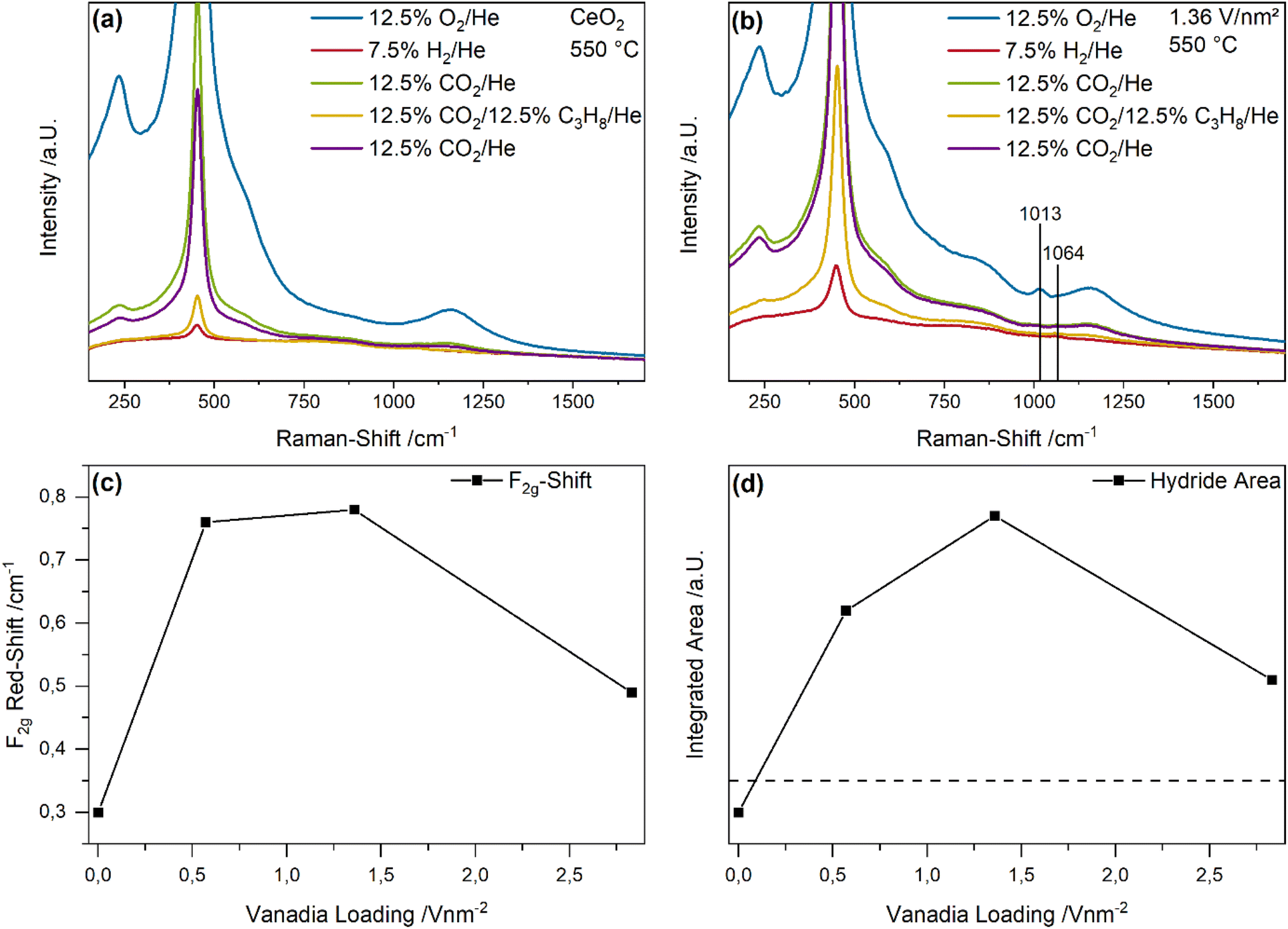 | ||
| Fig. 5 Vis-Raman spectra (514 nm excitation) of (a) bare ceria and (b) the 1.36 V nm−2 samples under different gas feeds recorded at 550 °C. (c) The F2g shift and (d) the hydride areas were determined by fitting and integration. The dashed line in panel d indicates no intensity change. For details see text. | ||
Fig. 5a shows the Vis-Raman spectra of bare ceria under different gas feeds at 550 °C. The spectrum under oxidizing conditions generally exhibits the same peaks observed in the UV-Raman spectra but with different relative intensities. Here, as a result of the different reductive properties of the gas feeds and the high temperatures, the overall intensity of the spectra and the background varies significantly, hindering intensity trends to be clearly determined. However, the F2g red-shift is a good indicator for subsurface/bulk reduction of ceria, caused by the formation of Ce3+ ions, which, due to their larger radius than Ce4+ ions distort the lattice, thus leading to a mode softening.52 In addition, after the exposure to H2, a peak at 1064 cm−1 can be observed that has previously been assigned to Ce–H bulk hydrides and may be of importance regarding hydrogen transfer during the reaction.46,53
In comparison, the Vis-Raman spectra of the 1.36 V nm−2 sample show similar changes with respect to the F2g position and the Ce–H peak at 1064 cm−1 (see Fig. 5b). In addition, the fine structure of the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration is detected between 1000 and 1050 cm−1, showing a blue-shift of the maximum with increasing vanadia loading (see Fig. 6a), allowing for the discrimination between different nuclearities (see Fig. 6b).33,35,37,54 The vanadyl peak decreases to zero under both reductive gas feeds but is regenerated in the first and second CO2 containing atmosphere, indicating its participation in the reaction. However, as previously shown, depending on the nuclearity, only some vanadia species fully regenerate to V5+ species, which are often described as the catalytically most active species.41 Since V4+ and V3+ species cannot be observed in Raman spectroscopy due to their small scattering cross sections,55–57 these species will be discussed in the UV-Vis section (see Fig. 7 and Discussion). The vanadyl fine structure will also be discussed in more detail below (see Fig. 6 and Discussion).
O stretching vibration is detected between 1000 and 1050 cm−1, showing a blue-shift of the maximum with increasing vanadia loading (see Fig. 6a), allowing for the discrimination between different nuclearities (see Fig. 6b).33,35,37,54 The vanadyl peak decreases to zero under both reductive gas feeds but is regenerated in the first and second CO2 containing atmosphere, indicating its participation in the reaction. However, as previously shown, depending on the nuclearity, only some vanadia species fully regenerate to V5+ species, which are often described as the catalytically most active species.41 Since V4+ and V3+ species cannot be observed in Raman spectroscopy due to their small scattering cross sections,55–57 these species will be discussed in the UV-Vis section (see Fig. 7 and Discussion). The vanadyl fine structure will also be discussed in more detail below (see Fig. 6 and Discussion).
 | ||
| Fig. 6 (a) Vanadyl region from the Vis-Raman spectra (514 nm excitation) between 1000 and 1045 cm−1 for bare ceria and vanadia-loaded samples during 12.5% O2/He exposure at 550 °C together with the results of peak-fit analyses. (b) Loading-dependent nuclearity distribution based on the analyses shown in (a). For details see text. | ||
 | ||
| Fig. 7 UV-Vis spectra of (a) bare ceria and (b) the 1.36 V nm−2 sample under different gas feeds, recorded at 550 °C. (c) Band gap energies as determined using Tauc plots and (d) the reduction peak area based on the results of a fitting analysis. | ||
The F2g shift between the initial CO2 exposure and the subsequent ODH conditions serves as an indicator for the ceria reduction upon reaction (see Fig. 5c). The F2g shifts range between a minimum of 0.3 cm−1 for bare ceria, and a maximum of 0.75 and 0.8 cm−1 for the 0.57 and 1.36 V nm−2 samples, while the 2.83 V nm−2 sample is characterized by a shift of 0.5 cm−1. These values are comparably small,46 which might be caused by the overall high oxygen mobility at this temperature, leading to an additional regeneration route via diffusion of bulk oxygen.58 The observed behavior indicates that the participation of ceria lattice oxygen varies significantly with the vanadia loading. It correlates well with the conversion of all reactants with a significant increase between bare ceria and the 0.57 and 1.36 V nm−2 samples, as observed for the propane and CO2 conversions, indicating that ceria lattice oxygen is an important driver for the occurring reactions. In comparison, the 2.83 V nm−2 sample shows a significantly different behavior, which despite similar conversions of both propane and CO2 as the other samples is characterized by a F2g red-shift, suggesting that ceria lattice oxygen is replaced as active oxygen site by vanadia oxygen, provided by oligomeric vanadia species present primarily at higher loadings. Such a change in active site would also explain the sudden shift in reactivity behavior when the loading is increased from 1.36 to 2.83 V nm−2.
Finally, the intensity change in the region between 1050 and 1100 cm−1, referred to as hydride area, was quantified by integration (see Fig. 5d). The dashed line corresponds to no intensity change. Hence, for bare ceria, the amount of subsurface/bulk hydrides decreases from the initial CO2 to the ODH conditions, while a strong increase is observed for the 0.57 and 1.36 V nm−2 samples that decreases again for the 2.83 V nm−2 sample. These changes might be caused by the ability of vanadia to transfer hydrogen from propane to the ceria surface. Due to the high temperature at which the CO2-assisted ODH is performed, a strong diffusion of hydrogen into the bulk, which was already observed at temperatures ∼300 °C, is likely.46 Overall, the observed intensity trend for the bulk hydrides is similar to the determined propylene selectivity, which might be one way by which vanadia increases the propylene selectivity since the hydrogen transfer from propane to ceria is increased, leading to the formation of bulk hydrides. This effect is not observed for bare ceria, indicating that it is caused by the presence of surface vanadia. However, as bare ceria still exhibits a propylene selectivity of 65%, also other routes towards selective oxidation must be operating. The amount of hydrides formed by the 2.83 V nm−2 sample decreases significantly in line with the propylene selectivity, which indicates that in this case hydride formation plays only a minor role, further highlighting a nuclearity-dependent catalytic behavior. Therefore, the quantification of the nuclearity distribution is crucial to understand the reactivity behavior in more detail.
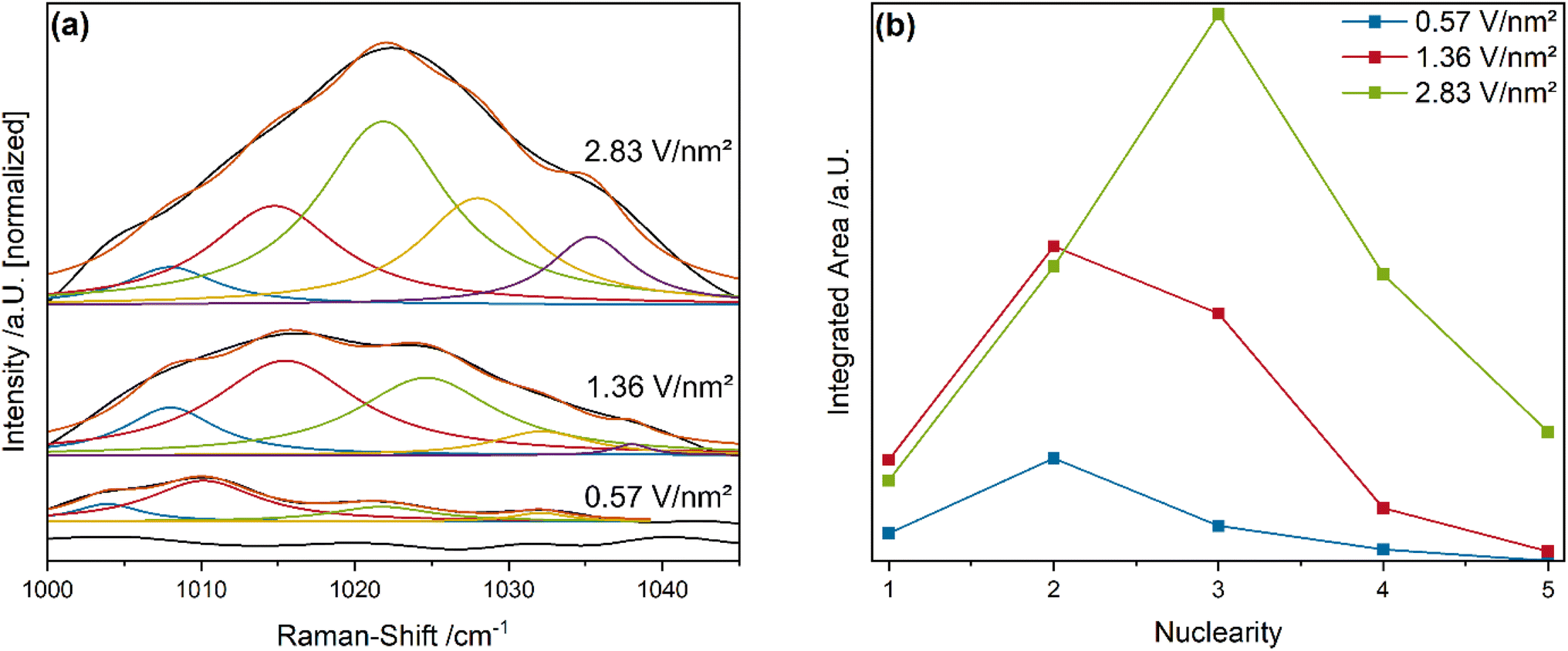
Fig. 6a shows the vanadyl stretching region of the vanadia-loaded samples, compared to bare ceria as a reference, recorded during 12.5% O2/He exposure using Vis-Raman spectroscopy (514 nm excitation), together with the results of peak-fitting analyses.
In the following, it is assumed that the chain length does not change significantly with the gas phase, but only with the temperature through sintering.59 As expected, bare ceria does not show any relevant signals in this region. For the VOx/CeO2 samples, the overall intensity increases with increasing vanadia loading and the maximum gradually shifts towards higher Raman-shifts. This behavior is caused by the increasing prevalence of higher vanadia nuclearities, which facilitate dipole–dipole coupling between VO groups of the same chain, leading to a blue-shift.33,35,36 As a result, the fine structure consists of five separable peaks that are shifted with respect to each other in a small range, due to different local chemical environments.
Fitting this fine structure with five Lorentz functions at restricted positions allows to extract a nuclearity distribution for each loading (see Fig. 6b). Note that this distribution differs from that obtained in our previous study at 275 °C using O2 as the oxidizing agent,33,36 due to the significantly different temperature. Since the clusters are more mobile on the ceria surface at 550 °C a higher relative contribution from stable nuclearities (i.e. trimers) is expected.38,60 The 0.57 V nm−2 sample exhibits the strongest contributions from dimers, with significant contributions from monomers and trimers and a small tetrameric contribution. No oligomeric nuclearities are present. In comparison, the 1.36 V nm−2 sample shows significantly higher contributions from all nuclearities as expected for the higher loading. The relative contribution shifts in part from monomers to trimers, with dimers still being the largest fraction. However, the relative amount of each nuclearity is similar for both the 0.57 and 1.36 V nm−2 samples, which is in good agreement with their similar reactivity behavior. Functions of the individual nuclearities during propane ODH were previously assigned based on ME-DRIFTS results.36 Monomeric species can inhibit the oxygen mobility in the ceria lattice by interaction with oxygen vacancies due to their structural relaxation into them.33 However, due to the elevated temperatures the mobility of the monomeric species is expected to increase the ability to move out of the vacancy.38 Such facilitated mobility is consistent with the fact that the 0.57 V nm−2 sample with the highest relative amount of monomers shows the strongest F2g shift as well as the overall highest amount of CO2 conversion with significant total oxidation contributions. Trimeric species, due to their highly stable ring configuration, were determined to be observer species with no catalytic function in the reaction.36,38 Dimeric species were proposed to transfer hydrogen atoms from propane to the ceria surface, facilitating the propane ODH reaction by catalyzing the rate-determining step.36 The amount of dimeric species present on the surface is similar to the amount of bulk hydrides and appears to be related, indicating that dimers might transfer hydrogen, resulting in bulk hydride formation, which seems to be crucial for the selective conversion of propane to propylene (see Fig. 5d). The formation of additional Ce–OH groups from the transferred hydrogen might also be viable and will be discussed in more detail in the DRIFTS section (see Fig. 8 and Discussion).
 | ||
| Fig. 8 Quasi in situ DRIFT spectra of (a) CeO2 and (b) the 1.36 V nm−2 sample recorded after treatment under the indicated gas feeds at 550 °C and subsequent cooling to room temperature in helium. The inset shows an enlarged view of the Ce–OH region. (c) Carbonate areas during the first CO2/He and the reactive gas phase and (d) relevant Ce–OH peaks, which show a significant change in intensity upon switching from 12.5% CO2/He to 12.5% CO2/12.5% C3H8/He. | ||
The 2.83 V nm−2 samples contains the highest amount of trimers and a lower relative contribution from the other nuclearities. In addition, it is the only sample that exhibits a significant fraction of oligomeric vanadia species. Besides dimers, oligomeric vanadia species were previously shown to transfer hydrogen from propane to the ceria surface during propane ODH.36 Since the reactivity and spectroscopic behavior of this sample differs in many ways, including its higher PDR activity, less hydride formation and a shift from ceria to vanadia oxygen as the active oxygen site, oligomeric species seem to play an important role in this regard. In fact, oligomeric vanadia species in VOx/CeO2 during CO2-assisted propane ODH behave similar to oligomeric species in VOx/TiO2 systems during regular propane ODH, where they deliver oxygen to facilitate the reactions but exhibit a lower propylene selectivity, indicating their high reducibility.24 The VO group of the oligomeric species might still be able to transfer the hydrogen, however, the target oxygen may not be available on the ceria surface. To investigate the reduced vanadia species and the bulk reduction of ceria in more detail, operando UV-Vis spectroscopy was performed.
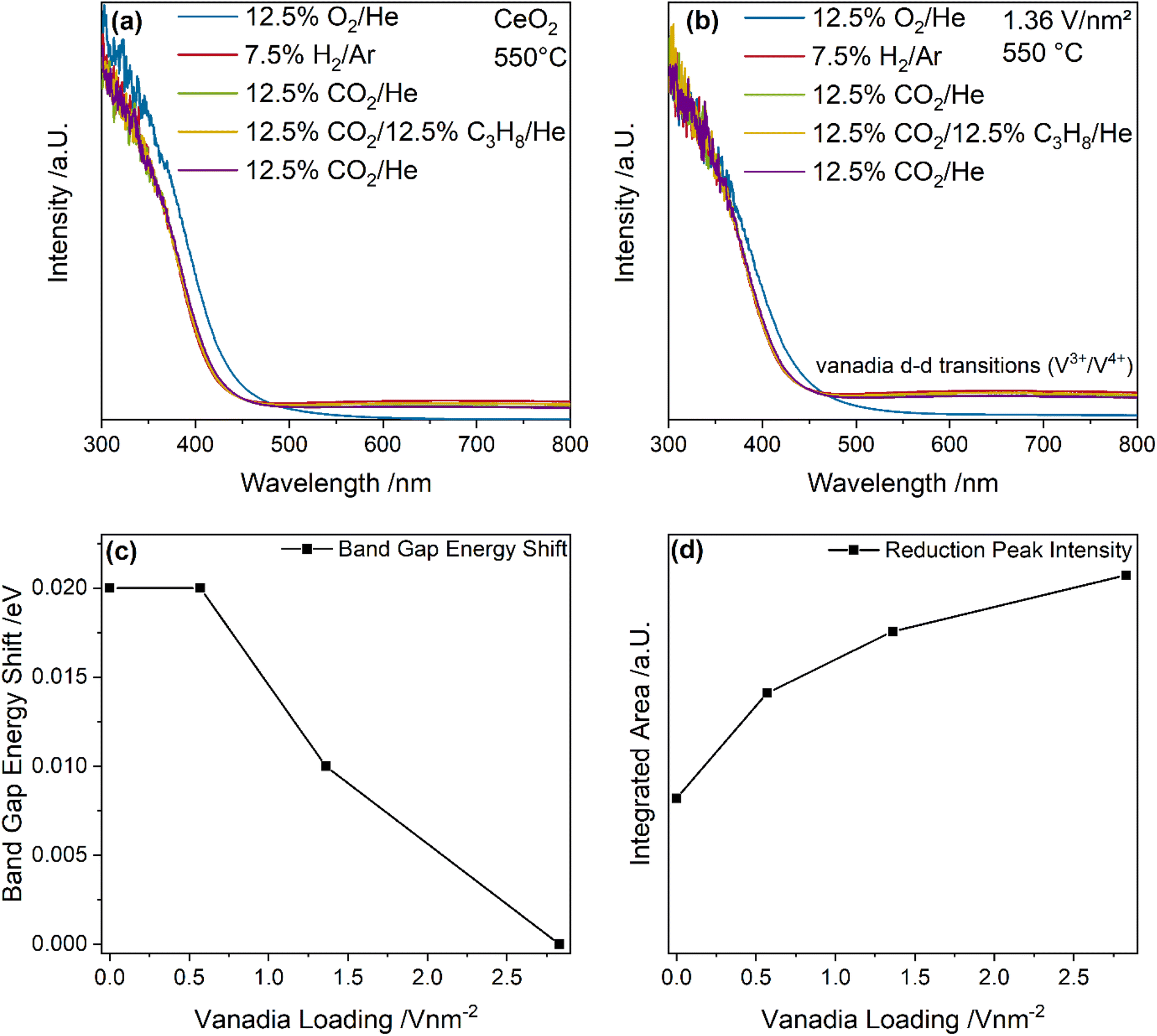
Fig. 7 shows UV-Vis spectra of bare ceria and the 1.36 V nm−2 sample recorded under different gas conditions at 550 °C. Based on these spectra, the band gap energy shift and the change in reduction peak area between the initial CO2 and the ODH conditions were determined (for details see experimental section), as shown in Fig. 7c and d. UV-Vis spectra of the 0.57 and 2.83 V nm−2 samples are given in the ESI (see Fig. S8†), as well as an exemplary fit of a UV-Vis spectrum (see Fig. S9†). The exact fitting procedure was described in one of our previous studies.46
As shown in Fig. 7a, the UV-Vis spectra of bare ceria are dominated by strong band gap absorption caused by O2p → Ce4f transitions.61,62 Upon switching from oxidizing to reductive conditions, two changes in the spectra become apparent. First, the band gap shifts, which is caused by the introduction of additional states in the band gap when the sample is reduced, lowering the energy required for a transition, which red-shifts the band gap.43,62 However, in this case, the band gap shows a blue-shift during this initial switch, which is typically associated with an oxidation of ceria.43,62 This was previously related to the newly appearing absorption at 633, which changes the spectra shape significantly, thus influencing the graphical analysis via Tauc plots.46 However, for small changes in the absorption region above 500 nm (as observed for the other gas-phase changes), the results of the Tauc analysis are not significantly affected and the differences between the initial CO2 exposure and ODH conditions can be analyzed. Second, a peak at 633 nm is observed, which is caused by a Ce3+ → Ce4+ charge transition, which is also caused by the reduction of ceria introducing Ce3+ states into the lattice.63,64 Both are suitable descriptors for the bulk reduction of ceria. As can be seen in Fig. 7b, UV-Vis spectra of the 1.36 V nm−2 sample under different gas feeds at 550 °C are also dominated by the ceria absorption but also show overlapping d–d transitions from reduced (V3+, V4+) vanadia, which depend on the vanadia species.65,66 Shorter vanadia nuclearities absorb at lower wavelengths while higher nuclearities absorb at higher wavelengths.67 In the following, the intensity of the region above 500 nm will be referred to as the reduction peak, describing the combined contributions from reduced ceria and vanadia. Therefore, both the bulk reduction of ceria as well as the prevalence of reduced vanadia states can be probed.
Fig. 7c summarizes the band gap red-shifts between the initial CO2 and the ODH conditions for bare ceria and the vanadia-loaded samples. The band gap is most red-shifted for bare ceria and ceria loaded with 0.57 V nm−2, indicating that these samples exhibit the highest degree of bulk reduction, which differs from the trend deduced from the F2g mode. This indicates that the degree of reduction in the ceria differs between the surface, subsurface and the bulk. The surface is influenced by the anchored vanadia and possibly by the presence of adsorbates. The subsurface exhibits a trend that follows the involvement of ceria lattice oxygen and is influenced by the overall reactivity. It can be regenerated by both gas phase CO2 and subsequent oxygen diffusion into the subsurface or through oxygen diffusion from the bulk. The F2g red-shift decreases for the highest vanadia loading (see Fig. 5c), which is in agreement with vanadia oxygen instead of ceria oxygen becoming the active site. This is likely caused by oligomers, only significantly present for the 2.83 V nm−2 sample. This sample also shows no shift of the band gap anymore, indicating that the reduction of ceria in this sample is limited to the surface shifting away from a significant ceria participation. Notably, the differences between the samples are comparably small, and since the error of the Tauc method is estimated to be ±0.01 eV, this trend needs to be supported by additional data. The small shift might originate from a small bulk contribution to the reaction, which mostly occurs in the surface/subsurface regions.
Fig. 7d quantifies the reduction peak area for bare ceria and vanadia-loaded samples including contributions from ceria and vanadia reduction. The reduction peak area shows an increase with increasing vanadia loading, while the slope shows a stepwise decrease. Since the vanadia-loaded samples are characterized by a significantly higher absorption than bare ceria this behavior indicates, that the peak includes reduced vanadia species, showing their participation in the reaction. However, this trend does not correlate with any specific reaction pathway, which may be caused by the fact that both ceria and vanadia are involved in a plethora of reactions, which all show a nuclearity-dependent behavior. Nevertheless, it still supports the previous interpretations that both vanadia and ceria reduction are crucial for the reaction mechanism.
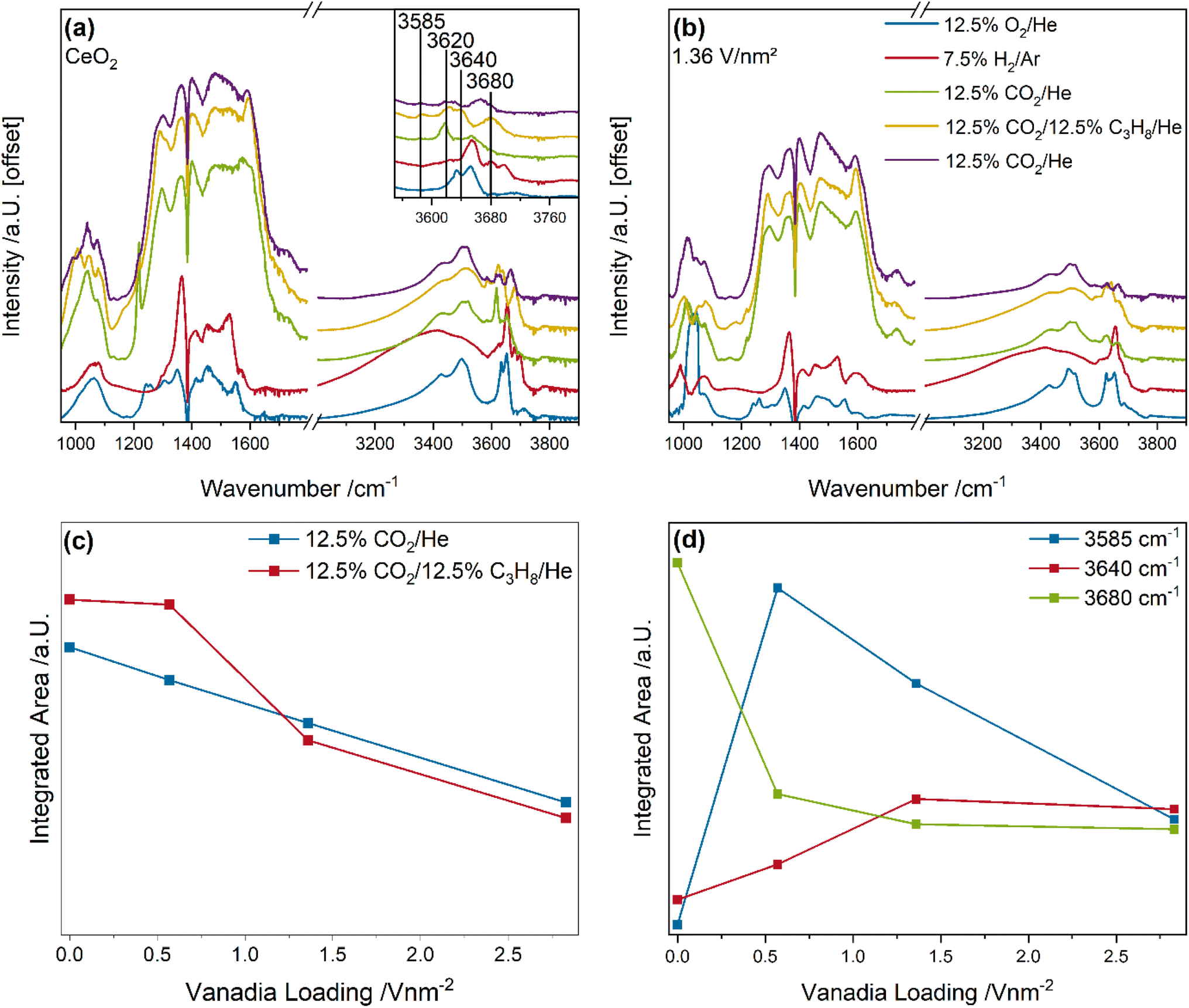
Fig. 8 shows the quasi in situ DRIFT spectra of bare ceria and the 1.36 V nm−2 sample recorded after pre-treatment under different gas conditions and cooling to room temperature in pure helium. The carbonate area was quantified by integration and the Ce–OH region by a peak-fitting analysis (for details see experimental section), as shown in Fig. 8c and d. The quasi in situ DRIFT spectra of the 0.57 and 2.83 V nm−2 sample are given in the ESI (see Fig. S10†), while an exemplary fit of the Ce–OH region is shown in Fig. S11.†
The quasi in situ DRIFT spectra of bare ceria and the 1.36 V nm−2 sample show significant changes in the carbonate and Ce–OH regions between the different gas phases but to a different extent (see Fig. 8a and b). Especially the carbonate region shows a sharp increase in the overall intensity after the initial CO2 exposure, which is indicative of the formation of ubiquitous amounts of carbonate adsorbates (which can deactivate surface sites),68 while no significant contribution of formates was observed. This behavior might explain the significant differences in the activity of the samples after differing pre-treatment conditions and between ODH and DDH conditions.
To analyze these changes in more detail, the carbonate region and the Ce–OH region of the bare ceria and vanadia-loaded samples were quantified. In the carbonate region (see Fig. 8c), the intensity decreases linearly with increasing vanadia loading from bare ceria to the 2.83 V nm−2 during the initial CO2 exposure. This is explained by the anchoring of vanadia to surface oxygen atoms, which cannot be reduced under H2 conditions. Therefore, less surface sites are available for reduction and subsequent adsorbate formation. When the conditions are switched from CO2 to ODH conditions, a nuclearity-dependent behavior is observed. For bare ceria and the 0.57 V nm−2 sample an increase in the carbonate intensity is observed, while for the 1.36 and 2.83 V nm−2 samples, an intensity decrease is observed, indicating a fundamental difference in the reaction behavior. The increase in the carbonate region is explained by the further reduction of surface oxygen sites due to the transfer of hydrogen from propane to ceria, leading to oxygen vacancy creation, which can be regenerated by gas phase CO2. This is supported by the fact that the intensity of the entire carbonate region increases for these samples. In comparison, the intensity for the 1.36 and 2.83 V nm−2 samples decreases at ∼1750 and ∼1210 cm−1, which is indicative of monodentate carbonates.68 This might indicate that hydrogen can be transferred to an oxygen atom of monodentate carbonate species in proximity of vanadia species. These can react to CO and H2O via RWGSR, leaving a clean surface site, which can then react with propane and create new adsorbates or vacancies. This also explains the more significant intensity increase in the defect region in the UV-Raman spectra (see Fig. 4) under ODH conditions due to the possible depletion of surface adsorbates. This effect seems to occur to a similar extent for both samples, which indicates that it is facilitated by monomers or dimers, which are present in similar amounts for both samples. However, since monomers cannot be fully regenerated by CO2, dimers seem to be the most likely nuclearity. Since the total amount of monomers and dimers is much higher in the 1.36 and 2.83 V nm−2 samples compared to the 0.57 V nm−2 sample, this seems to be likely. This effect seems to occur only for monodentate carbonate species, while bidentate carbonate species may be too stable to participate in this reaction.
At last, the changes in the intensity of the Ce–OH region between the initial CO2 and ODH conditions were quantified (see Fig. 8d). Three peaks are observed at 3585, 3640, and 3680 cm−1, which are assigned to vibrations of V–OH, Ce–OH II-B, and Ce–OH II-A, respectively.69,70 The peak at 3620 cm−1 (compare Fig. S11†) is likely caused by the formation of H2O and is therefore not further discussed. The V–OH bond exhibits an intensity of zero for bare ceria, increases significantly for the 0.57 V nm−2 sample, and then decreases gradually towards the 2.83 V nm−2 sample. This indicates, that the V–OH species is less prevalent at higher loadings, where the shift towards the direct participation of vanadia oxygen occurs, indicating a very fast transfer to other oxygen sites at higher loadings. In comparison, the peak located at 3640 cm−1 originates from a doubly bridged Ce–OH species in proximity to oxygen vacancies. Since the amount of created oxygen vacancies during ODH conditions increases with increasing vanadia loading, these species also increase in concentration. Their intensity does not correlate with the prevalence of PDR, by comparison with Fig. 3f. Therefore, it seems likely that this Ce–OH species is involved in different selective oxidation routes in addition to Ce–H formation and the reaction with monodentate carbonates. At last, the peak at 3680 cm−1 originates from a doubly bridged Ce–OH species on the clean ceria surface with no vacancies in proximity. This species is especially reactive towards further hydrogen,69 which makes it a target for the quick hydrogen transfer of hydrogen from the total oxidation reaction. It can then be reduced from Ce–OH2 to a vacancy and water. This is in agreement with its intensity, which decreases with increasing vanadia loading, in good agreement with the decrease in total oxidation rate.
In summary, the spectroscopic results show that different reactions occur on the VOx/CeO2 surface (see Fig. 9), exhibiting different reaction paths depending on the vanadia nuclearities. First, after the pre-treatment, the catalyst surface is saturated with mostly carbonate adsorbates and little surface oxygen remains exposed. The main hydrogen transfer route towards propylene is the hydrogen transfer to monodentate carbonates or doubly bridged Ce–OH species in proximity of an oxygen vacancy and a bulk Ce–H. The transfer is facilitated by the VO group of dimeric vanadia and ceria lattice oxygen is used as the active oxygen site for the reaction. The combination of Ce–H and Ce–OH that absorb the hydrogen, eventually leads to the formation of H2O and oxygen vacancies which can be regenerated by CO2, which is shown schematically in Fig. 9. The extent to which side reactions occur is nuclearity-dependent and different reaction routes are involved. Due to the reduction and exposure to CO2, most of the catalyst surface is deactivated by either stable carbonate species blocking surface oxygen or vacancies. However, vacancies that are created on the vanadia can be regenerated by the ceria lattice via oxygen spill-over and vacancies in the ceria lattice can be regenerated via CO2via carbonate species. For more details on the regeneration and CO2 activation mechanism of the reduced system, please refer to the previous study on this system published by our group.42Fig. 9 summarizes the major findings in a mechanistic scheme of the propane ODH reaction over the 1.36 V nm−2 sample.
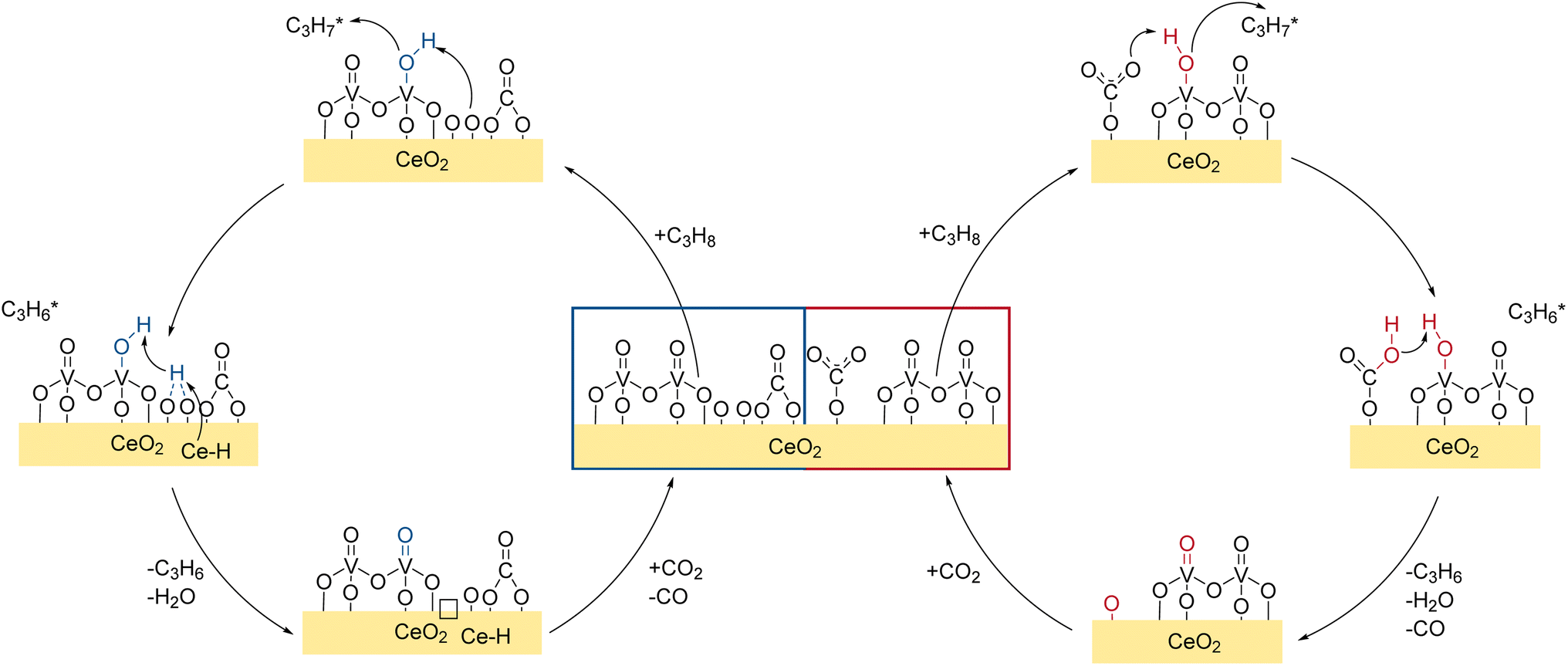 | ||
| Fig. 9 Mechanistic scheme of the CO2-assisted propane ODH over VOx/CeO2. Details regarding the role of the bare support and side reactions are given in the text. | ||
4. Conclusion
In this study, we addressed the CO2-assisted ODH of propane over VOx/CeO2 catalysts, by analyzing the complex reaction network, identifying important structural features for the different reactions occurring and elucidating a mechanistic scheme by combining multiple operando and in situ methods. To determine the influence of vanadia nuclearities on the catalytic performance, differently loaded vanadia catalysts were employed (0–2.5 V nm−2).The catalytic activity of the samples under DDH conditions is significantly higher and more selective than under ODH conditions but is not stable, leading to catalyst deactivation. Under ODH conditions, a stable performance is obtained, which depends strongly on the pre-treatment conditions. A pre-treatment in H2 and CO2 results in a partially reduced state of the catalyst, which leads to the highest conversions for vanadia-loaded catalysts. The reaction network is significantly influenced by the vanadia surface density of the catalyst, while the CO2 conversions are significantly higher for all samples than the propane conversions, caused by a significant amount of side reactions. On bare ceria, the total oxidation of propane dominates, while small amounts of cracking are observed. With increasing vanadia loading, less total oxidation but more cracking and PDR occurs. An especially sharp increase is observed for the 2.83 V nm−2 sample, which exhibits the highest PDR rates but the lowest total contribution of side reactions.
As a result of the mechanistic analysis, different pathways towards propylene and different active sites towards side reactions were identified. For bare ceria, the total oxidation was identified to occur via surface lattice oxygen, while the conversion of propane is initiated by its adsorption onto the catalyst, creating defect sites. Hydrogen is transferred directly to the surface, and the subsequent transfer path of hydrogen determines the reaction pathway. Bridged Ce–OH species are highly reactive towards further hydrogen, leading to total oxidation, while the transfer to monodentate carbonates can lead to the selective conversion towards propylene. Bidentate carbonates are not affected by the hydrogen transfer. In comparison, vanadia-loaded catalysts exhibit additional pathways compared to bare ceria that lead to propylene formation, increasing the selectivity. First, vanadia is able to transfer hydrogen from propane to ceria, forming relatively stable bulk hydrides, thereby removing hydrogen from the surface and decreasing the prevalence of the total oxidation reaction. In addition, vanadia is anchored to ceria surface oxygen, poisoning some of the oxygen sites responsible for total oxidation. At the 2.83 V nm−2 loading, a sharp increase in the PDR rate can be observed, indicating a clear loading dependence of the catalytic behavior. In addition, a shift from ceria oxygen to vanadia oxygen atoms as the active site in propane conversion was observed via the F2g red-shift. At last, the reduced catalyst can be regenerated by CO2, which is crucial for the stability of the catalytic performance. This regeneration occurs via carbonate formation from CO2 and an oxygen vacancy on the surface, leading to lattice oxygen regeneration. However, due to the high stability of the carbonates and the high CO desorption barrier, surface sites are blocked, explaining the reactivity difference between ODH and DDH conditions.
To disentangle the nuclearity distribution and assign the individual nuclearities to catalytic functions, the vibrational vanadyl fine structure was analyzed. Monomeric vanadia, which was previously described to interact with ceria oxygen vacancies,33 is able to move out of the vacancies at the elevated temperatures required for CO2-assisted propane ODH, leading to a less significant decrease in oxygen mobility compared to lower temperatures, increasing the prevalence of total oxidation. Furthermore, at 550 °C, an increased amount of trimers are expected to be present on the surface due to their high energetic stability.38 Based on this stability, they were previously described to be observer species, which do not participate in the reaction, which was confirmed for the CO2-assisted propane ODH.36 Dimers instead of oligomers are the main hydrogen transfer site during selective conversion and are able to transfer hydrogen into the bulk, forming hydrides, and to transfer hydrogen to monodentate carbonates, leading to RWGSR and propylene formation. The presence of oligomers was shown to lead to a significant contribution of PDR and a shift towards vanadia oxygen as the active oxygen site. This is likely caused by the increased reducibility of these samples.60
Our results highlight the potential of combining multiple operando and in situ methods to obtain detailed understanding of the reaction mechanism of the CO2-assisted propane ODH, which is an important reaction regarding CO2 activation and usage in chemical industry. While the VOx/CeO2 catalyst shows moderate CO2-ODH performance, its reaction mechanism as well as the complex interplay between different vanadia nuclearities, the ceria surface and the bulk, as well as their relevance for the occurrence of selective oxidation or side reactions were understood in detail for the first time. This is of great importance for future developments, since ceria is a promising material for the activation of CO2, while vanadia can be highly active for propane activation. This catalyst is envisioned to be part of a multi-oxide system that reduces catalyst deactivation through carbonate formation, thereby leading to significantly higher activities. Our methodical approach is readily transferable to other systems of interest in the context of CO2 activation and contributes to a knowledge-based design of vanadia materials to replace highly toxic CrOx for CO2-assisted ODH reactions.
Data availability
The presented data is available at https://doi.org/10.5281/zenodo.13970461.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge Lea Totzauer for performing XRD experiments and analysis. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, HE 4515/11-1, HE 4515/11-2).References
- I. Amghizar, L. A. Vandewalle, K. M. van Geem and G. B. Marin, New Trends in Olefin Production, Engineering, 2017, 3(2), 171–178, DOI:10.1016/j.eng.2017.02.006.
- F. Cavani, N. Ballarini and A. Cericola, Oxidative dehydrogenation of ethane and propane: How far from commercial implementation?, Catal. Today, 2007, 127(1–4), 113–131, DOI:10.1016/j.cattod.2007.05.009.
- A. V. Lavrenov, L. F. Saifulina, E. A. Buluchevskii and E. N. Bogdanets, Propylene production technology: Today and tomorrow, Catal. Ind., 2015, 7(3), 175–187, DOI:10.1134/S2070050415030083.
- T. K. Phung, T. M. Le Pham, K. B. Vu and G. Busca, (Bio)Propylene production processes: A critical review, J. Environ. Chem. Eng., 2021, 9(4), 105673, DOI:10.1016/j.jece.2021.105673.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, Critical Literature Review of the Kinetics for the Oxidative Dehydrogenation of Propane over Well-Defined Supported Vanadium Oxide Catalysts, ACS Catal., 2014, 4(10), 3357–3380, DOI:10.1021/cs5003417.
- J. H. Carter, T. Bere, J. R. Pitchers, D. G. Hewes, B. D. Vandegehuchte, C. J. Kiely, S. H. Taylor and G. J. Hutchings, Direct and oxidative dehydrogenation of propane: from catalyst design to industrial application, Green Chem., 2021, 23(24), 9747–9799, 10.1039/D1GC03700E.
- J. Baek, H. J. Yun, D. Yun, Y. Choi and J. Yi, Preparation of Highly Dispersed Chromium Oxide Catalysts Supported on Mesoporous Silica for the Oxidative Dehydrogenation of Propane Using CO2: Insight into the Nature of Catalytically Active Chromium Sites, ACS Catal., 2012, 2(9), 1893–1903, DOI:10.1021/cs300198u.
- F. M. Baena-Moreno, M. Rodríguez-Galán, F. Vega, B. Alonso-Fariñas, L. F. Vilches Arenas and B. Navarrete, Carbon capture and utilization technologies: a literature review and recent advances, Energy Sources, Part A, 2019, 41(12), 1403–1433, DOI:10.1080/15567036.2018.1548518.
- A. Al-Mamoori, A. Krishnamurthy, A. A. Rownaghi and F. Rezaei, Carbon Capture and Utilization Update, Energy Technol., 2017, 5(6), 834–849, DOI:10.1002/ente.201600747.
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Climate change mitigation potential of carbon capture and utilization in the chemical industry, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(23), 11187–11194, DOI:10.1073/pnas.1821029116.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, CO2 Activation over Catalytic Surfaces, ChemPhysChem, 2017, 18(22), 3135–3141, DOI:10.1002/cphc.201700782.
- M. Jacquemin, A. Beuls and P. Ruiz, Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism, Catal. Today, 2010, 157(1–4), 462–466, DOI:10.1016/j.cattod.2010.06.016.
- T.-q. Lei, Y.-h. Cheng, C.-x. Miao, W.-m. Hua, Y.-h. Yue and Z. Gao, Silica-doped TiO2 as support of gallium oxide for dehydrogenation of ethane with CO2, Fuel Process. Technol., 2018, 177, 246–254, DOI:10.1016/j.fuproc.2018.04.037.
- M. A. Botavina, G. Martra, Y. A. Agafonov, N. A. Gaidai, N. V. Nekrasov, D. V. Trushin, S. Coluccia and A. L. Lapidus, Oxidative dehydrogenation of C3–C4 paraffins in the presence of CO2 over CrOx/SiO2 catalysts, Appl. Catal., A, 2008, 347(2), 126–132, DOI:10.1016/j.apcata.2008.05.037.
- F. Gashoul Daresibi, A. A. Khodadadi, Y. Mortazavi, S. Huotari and M. Ritala, Highly dispersed atomic layer deposited CrOx on SiO2 catalyst with enhanced yield of propylene for CO2–mediated oxidative dehydrogenation of propane, Mol. Catal., 2022, 526(80), 112396, DOI:10.1016/j.mcat.2022.112396.
- P. Michorczyk, P. Pietrzyk and J. Ogonowski, Preparation and characterization of SBA-1–supported chromium oxide catalysts for CO2 assisted dehydrogenation of propane, Microporous Mesoporous Mater., 2012, 161, 56–66, DOI:10.1016/j.micromeso.2012.05.011.
- H. Wang, T. D. Nguyen and G. Tsilomelekis, Propane oxidative dehydrogenation using CO2 over CrOx/Fe–CeO2 catalysts, Catal. Sci. Technol., 2023, 13(8), 2360–2369, 10.1039/D2CY01563C.
- Z.-Y. Wang, Z.-H. He, L.-Y. Li, S.-Y. Yang, M.-X. He, Y.-C. Sun, K. Wang, J.-G. Chen and Z.-T. Liu, Research progress of CO2 oxidative dehydrogenation of propane to propylene over Cr-free metal catalysts, Rare Met., 2022, 41(7), 2129–2152, DOI:10.1007/s12598-021-01959-y.
- M. L. Balogun, S. Adamu, M. S. Ba-Shammakh and M. M. Hossain, Promotional effects of CO2 on the oxidative dehydrogenation of propane over mesoporous VOX/γAl2O3 catalysts, J. Ind. Eng. Chem., 2021, 96(20), 82–97, DOI:10.1016/j.jiec.2020.12.022.
- I. Ascoop, V. V. Galvita, K. Alexopoulos, M.-F. Reyniers, P. van der Voort, V. Bliznuk and G. B. Marin, The role of CO2 in the dehydrogenation of propane over WOx–VOx/SiO2, J. Catal., 2016, 335, 1–10, DOI:10.1016/j.jcat.2015.12.015.
- X. Jiang, B. M. Lis, S. C. Purdy, S. Paladugu, V. Fung, W. Quan, Z. Bao, W. Yang, Y. He, B. G. Sumpter, K. Page, I. E. Wachs and Z. Wu, CO2-Assisted Oxidative Dehydrogenation of Propane over VOx/In2O3 Catalysts: Interplay between Redox Property and Acid–Base Interactions, ACS Catal., 2022, 12(18), 11239–11252, DOI:10.1021/acscatal.2c02099.
- X. Jiang, B. M. Lis, Y. Wu, I. E. Wachs and Z. Wu, Effect of the Molecular Structure of Surface Vanadia on Activity and Regenerability of VOx/In2O3 Catalysts for CO2-Assisted Oxidative Dehydrogenation of Propane, J. Phys. Chem. C, 2023, 127(13), 6311–6320, DOI:10.1021/acs.jpcc.3c00183.
- S. Rogg and C. Hess, CO2 as a soft oxidant for propane oxidative dehydrogenation: A mechanistic study using operando UV Raman spectroscopy, J. CO2 Util., 2021, 50(9), 101604, DOI:10.1016/j.jcou.2021.101604.
- L. Schumacher; K. Hofmann; C. HessInsight into the Reaction Mechanism and Deactivation during CO2-Assisted Propane ODH over VOx/TiO2 Catalysts: An Operando Spectroscopic Study, 2023, DOI:10.26434/chemrxiv-2023-rnfrd.
- A. Dinse, B. Frank, C. Hess, D. Habel and R. Schomäcker, Oxidative dehydrogenation of propane over low-loaded vanadia catalysts: Impact of the support material on kinetics and selectivity, J. Mol. Catal. A: Chem., 2008, 289(1–2), 28–37, DOI:10.1016/j.molcata.2008.04.007.
- B. Beck, M. Harth, N. G. Hamilton, C. Carrero, J. J. Uhlrich, A. Trunschke, S. Shaikhutdinov, H. Schubert, H.-J. Freund, R. Schlögl, J. Sauer and R. Schomäcker, Partial oxidation of ethanol on vanadia catalysts on supporting oxides with different redox properties compared to propane, J. Catal., 2012, 296, 120–131, DOI:10.1016/j.jcat.2012.09.008.
- M. Ziemba and C. Hess, Influence of gold on the reactivity behaviour of ceria nanorods in CO oxidation: combining operando spectroscopies and DFT calculations, Catal. Sci. Technol., 2020, 10(11), 3720–3730, 10.1039/D0CY00392A.
- L. Lin, S. Yao, Z. Liu, F. Zhang, N. Li, D. Vovchok, A. Martínez-Arias, R. Castañeda, J. Lin, S. D. Senanayake, D. Su, D. Ma and J. A. Rodriguez, In Situ Characterization of Cu/CeO2 Nanocatalysts for CO2 Hydrogenation: Morphological Effects of Nanostructured Ceria on the Catalytic Activity, J. Phys. Chem. C, 2018, 122(24), 12934–12943, DOI:10.1021/acs.jpcc.8b03596.
- M. Ziemba, J. Weyel and C. Hess, Elucidating the mechanism of the reverse water–gas shift reaction over Au/CeO2 catalysts using operando and transient spectroscopies, Appl. Catal., B, 2022, 301, 120825, DOI:10.1016/j.apcatb.2021.120825.
- P. Ober, S. Rogg and C. Hess, Direct Evidence for Active Support Participation in Oxide Catalysis: Multiple Operando Spectroscopy of VOx/Ceria, ACS Catal., 2020, 10(5), 2999–3008, DOI:10.1021/acscatal.9b05174.
- Y. Li, Z. Wei, F. Gao, L. Kovarik, C. H. F. Peden and Y. Wang, Effects of CeO2 support facets on VOx/CeO2 catalysts in oxidative dehydrogenation of methanol, J. Catal., 2014, 315, 15–24, DOI:10.1016/j.jcat.2014.04.013.
- J.-L. Sánchez-García, B. E. Handy, Á. G. Rodríguez, M. M. González-Chávez, R. García de León and M.-G. Cardenas-Galindo, Relating the Synthesis Method of VOx/CeO2/SiO2 Catalysts to Red-Ox Properties, Acid Sites, and Catalytic Activity for the Oxidative Dehydrogenation of Propane and n-Butane, Top. Catal., 2022, 65(13–16), 1408–1418, DOI:10.1007/s11244-022-01661-y.
- L. Schumacher and C. Hess, The active role of the support in propane ODH over VOx/CeO2 catalysts studied using multiple operando spectroscopies, J. Catal., 2021, 398(13), 29–43, DOI:10.1016/j.jcat.2021.04.006.
- M. V. Martínez-Huerta, G. Deo, J. L. G. Fierro and M. A. Bañares, Changes in Ceria-Supported Vanadium Oxide Catalysts during the Oxidative Dehydrogenation of Ethane and Temperature-Programmed Treatments, J. Phys. Chem. C, 2007, 111(50), 18708–18714, DOI:10.1021/jp0772225.
- M. Baron, H. Abbott, O. Bondarchuk, D. Stacchiola, A. Uhl, S. Shaikhutdinov, H. -J. Freund, C. Popa, M. V. Ganduglia-Pirovano and J. Sauer, Resolving the Atomic Structure of Vanadia Monolayer Catalysts: Monomers, Trimers, and Oligomers on Ceria, Angew. Chem., Int. Ed., 2009, 121(43), 8150–8153, DOI:10.1002/ange.200903085.
- L. Schumacher, J. Weyel and C. Hess, Unraveling the Active Vanadium Sites and Adsorbate Dynamics in VOx/CeO2 Oxidation Catalysts Using Transient IR Spectroscopy, J. Am. Chem. Soc., 2022, 144(32), 14874–14887, DOI:10.1021/jacs.2c06303.
- Z. Wu, A. J. Rondinone, I. N. Ivanov and S. H. Overbury, Structure of Vanadium Oxide Supported on Ceria by Multiwavelength Raman Spectroscopy, J. Phys. Chem. C, 2011, 115(51), 25368–25378, DOI:10.1021/jp2084605.
- C. Penschke, J. Paier and J. Sauer, Oligomeric Vanadium Oxide Species Supported on the CeO2 (111) Surface: Structure and Reactivity Studied by Density Functional Theory, J. Phys. Chem. C, 2013, 117(10), 5274–5285, DOI:10.1021/jp400520j.
- M. V. Martínez-Huerta, G. Deo, J. L. G. Fierro and M. A. Bañares, Operando Raman-GC Study on the Structure−Activity Relationships in V5+/CeO2 Catalyst for Ethane Oxidative Dehydrogenation: The Formation of CeVO4, J. Phys. Chem. C, 2008, 112(30), 11441–11447, DOI:10.1021/jp802827t.
- M. Martínez-Huerta, J. M. Coronado, M. Fernández-García, A. Iglesiaz-Juez, G. Deo, J. L. G. Fierro and M. A. Banares, Nature of the vanadia-ceria interface in V5+/CeO2 catalysts and its relevance for the solid-state reaction toward CeVO4 and catalytic properties, J. Catal., 2004, 225(1), 240–248, DOI:10.1016/j.jcat.2004.04.005.
- H.-X. Fan, L.-F. Zhen, A. Rajendran, J. Feng and W.-Y. Li, Crucial role of H and O spillover in VOx/CeO2 catalysts reduction and re-oxidation during the ODH reaction, Appl. Surf. Sci., 2023, 626, 157250, DOI:10.1016/j.apsusc.2023.157250.
- L. Schumacher, M. Ziemba and C. Hess, Investigation of the CO2 Activation and Regeneration of Reduced VOx/CeO2 Catalysts Using Multiple In Situ Spectroscopies, ChemCatChem, 2024 DOI:10.1002/cctc.202301409.
- A. Filtschew, K. Hofmann and C. Hess, Ceria and Its Defect Structure: New Insights from a Combined Spectroscopic Approach, J. Phys. Chem. C, 2016, 120(12), 6694–6703, DOI:10.1021/acs.jpcc.6b00959.
- P. S. Waleska and C. Hess, Oligomerization of Supported Vanadia: Structural Insight Using Surface-Science Models with Chemical Complexity, J. Phys. Chem. C, 2016, 120(33), 18510–18519, DOI:10.1021/acs.jpcc.6b01672.
- L. Antonov and D. Nedeltcheva, Resolution of overlapping UV–Vis absorption bands and quantitative analysis, Chem. Soc. Rev., 2000, 29(3), 217–227, 10.1039/A900007K.
- L. Schumacher, M. Ziemba, K. Brunnengräber, L. Totzauer, K. Hofmann, B. J. M. Etzold, B. Albert and C. Hess, Understanding the Reduction Behavior of VOx/CeO2 on a Molecular Level: Combining Temperature-Programmed Reduction with Multiple In-Situ Spectroscopies and X-ray Diffraction, J. Phys. Chem. C, 2023, 127(12), 5810–5824, DOI:10.1021/acs.jpcc.3c00622.
- A. G. Buekens and G. F. Froment, Thermal Cracking of Propane. Kinetics and Product Distributions, Ind. Eng. Chem. Process Des. Dev., 1968, 7(3), 435–447, DOI:10.1021/i260027a022.
- E. Gomez, S. Kattel, B. Yan, S. Yao, P. Liu and J. G. Chen, Combining CO2 reduction with propane oxidative dehydrogenation over bimetallic catalysts, Nat. Commun., 2018, 9(1), 1398, DOI:10.1038/s41467-018-03793-w.
- A. Siahvashi and A. A. Adesina, Hydrogen production via propane dry reforming: Carbon deposition and reaction-deactivation study, Int. J. Hydrogen Energy, 2018, 43(36), 17195–17204, DOI:10.1016/j.ijhydene.2018.07.118.
- C. Schilling, A. Hofmann, C. Hess and M. V. Ganduglia-Pirovano, Raman Spectra of Polycrystalline CeO2: A Density Functional Theory Study, J. Phys. Chem. C, 2017, 121(38), 20834–20849, DOI:10.1021/acs.jpcc.7b06643.
- A. Iglesias-Juez, M. V. Martínez-Huerta, E. Rojas-García, J.-M. Jehng and M. A. Bañares, On the Nature of the Unusual Redox Cycle at the Vanadia Ceria Interface, J. Phys. Chem. C, 2018, 122(2), 1197–1205, DOI:10.1021/acs.jpcc.7b09832.
- C. Schilling and C. Hess, Real-Time Observation of the Defect Dynamics in Working Au/CeO2 Catalysts by Combined Operando Raman/UV–Vis Spectroscopy, J. Phys. Chem. C, 2018, 122(5), 2909–2917, DOI:10.1021/acs.jpcc.8b00027.
- Z. Li, K. Werner, L. Chen, A. Jia, K. Qian, J.-Q. Zhong, R. You, L. Wu, L. Zhang, H. Pan, X.-P. Wu, X.-Q. Gong, S. Shaikhutdinov, W. Huang and H.-J. Freund, Interaction of Hydrogen with Ceria: Hydroxylation, Reduction, and Hydride Formation on the Surface and in the Bulk, Chemistry, 2021, 27(16), 5268–5276, DOI:10.1002/chem.202005374.
- P. Waleska, S. Rupp and C. Hess, Operando Multiwavelength and Time-Resolved Raman Spectroscopy: Structural Dynamics of a Supported Vanadia Catalyst at Work, J. Phys. Chem. C, 2018, 122(6), 3386–3400, DOI:10.1021/acs.jpcc.7b10518.
- L. J. Burcham, G. Deo, X. Gao and I. E. Wachs, In situ IR, Raman, and UV-Vis DRS spectroscopy of supported vanadium oxide catalysts during methanol oxidation, Top. Catal., 2000, 11–12(1–4), 85–100, DOI:10.1023/A:1027275225668.
- P. Shvets, O. Dikaya, K. Maksimova and A. Goikhman, A review of Raman spectroscopy of vanadium oxides, J. Raman Spectrosc., 2019, 50(8), 1226–1244, DOI:10.1002/jrs.5616.
- Z. Wu, H.-S. Kim, P. C. Stair, S. Rugmini and S. D. Jackson, On the structure of vanadium oxide supported on aluminas: UV and visible Raman spectroscopy, UV-visible diffuse reflectance spectroscopy, and temperature-programmed reduction studies, J. Phys. Chem. B, 2005, 109(7), 2793–2800, DOI:10.1021/jp046011m.
- J. M. López, A. L. Gilbank, T. García, B. Solsona, S. Agouram and L. Torrente-Murciano, The prevalence of surface oxygen vacancies over the mobility of bulk oxygen in nanostructured ceria for the total toluene oxidation, Appl. Catal., B, 2015, 174–175, 403–412, DOI:10.1016/j.apcatb.2015.03.017.
- P. Granger, H. W. Siaka and S. B. Umbarkar, What News in the Surface Chemistry of Bulk and Supported Vanadia Based SCR-Catalysts: Improvements in their Resistance to Poisoning and Thermal Sintering, Chem. Rec., 2019, 19(9), 1813–1828, DOI:10.1002/tcr.201800092.
- C. Penschke, J. Paier and J. Sauer, Vanadium Oxide Oligomers and Ordered Monolayers Supported on CeO2 (111): Structure and Stability Studied by Density Functional Theory, J. Phys. Chem. C, 2018, 122(16), 9101–9110, DOI:10.1021/acs.jpcc.8b01998.
- B. Huang, R. Gillen and J. Robertson, Study of CeO2 and Its Native Defects by Density Functional Theory with Repulsive Potential, J. Phys. Chem. C, 2014, 118(42), 24248–24256, DOI:10.1021/jp506625h.
- N. V. Skorodumova, R. Ahuja, S. I. Simak, I. A. Abrikosov, B. Johansson and B. I. Lundqvist, Electronic, bonding, and optical properties of CeO2 and Ce2O3 from first principles, Phys. Rev. B, 2001, 64(11), 548, DOI:10.1103/PhysRevB.64.115108.
- C. Binet, A. Badri and J.-C. Lavalley, A Spectroscopic Characterization of the Reduction of Ceria from Electronic Transitions of Intrinsic Point Defects, J. Phys. Chem., 1994, 98(25), 6392–6398, DOI:10.1021/j100076a025.
- C. W. M. Castleton, J. Kullgren and K. Hermansson, Tuning LDA+U for electron localization and structure at oxygen vacancies in ceria, J. Chem. Phys., 2007, 127(24), 244704, DOI:10.1063/1.2800015.
- J. Liu, Z. Zhao, C. Xu, A. Duan and G. Jiang, CeO2-supported vanadium oxide catalysts for soot oxidation: the roles of molecular structure and nanometer effect, J. Rare Earths, 2010, 28(2), 198–204, DOI:10.1016/S1002-0721(09)60080-6.
- R. J. Deeth, Electronic structures and d-d spectra of vanadium(IV) and VO2+ complexes: discrete variational Xα calculations, J. Chem. Soc., Dalton Trans., 1991, 65(6), 1467–1477, 10.1039/DT9910001467.
- D. Nitsche and C. Hess, Structure of Isolated Vanadia and Titania: A Deep UV Raman, UV-Vis, and IR Spectroscopic Study, J. Phys. Chem. C, 2016, 120(2), 1025–1037, DOI:10.1021/acs.jpcc.5b10317.
- G. N. Vayssilov, M. Mihaylov, P. St. Petkov, K. I. Hadjiivanov and K. M. Neyman, Reassignment of the Vibrational Spectra of Carbonates, Formates, and Related Surface Species on Ceria: A Combined Density Functional and Infrared Spectroscopy Investigation, J. Phys. Chem. C, 2011, 115(47), 23435–23454, DOI:10.1021/jp208050a.
- A. Badri, C. Binet and J.-C. Lavalley, An FTIR study of surface ceria hydroxy groups during a redox process with H2, Faraday Trans., 1996, 92(23), 4669, 10.1039/FT9969204669.
- J. E. Sambeth, M. A. Centeno, A. Paúl, L. E. Briand, H. J. Thomas and J. A. Odriozola, In situ DRIFTS study of the adsorption–oxidation of CH3OH on V2O5, J. Mol. Catal. A: Chem., 2000, 161(1–2), 89–97, DOI:10.1016/S1381-1169(00)00152-7.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information and results on the reactivity and operando spectroscopic behavior. See DOI: https://doi.org/10.1039/d4su00527a |
| This journal is © The Royal Society of Chemistry 2024 |