
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical on-demand production of hydrogen peroxide in a modular flow reactor†
Thomas
Freese
 a,
Jelmer T.
Meijer
a,
Matteo
Miola
b,
Paolo P.
Pescarmona
b and
Ben L.
Feringa
*a
a,
Jelmer T.
Meijer
a,
Matteo
Miola
b,
Paolo P.
Pescarmona
b and
Ben L.
Feringa
*a
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bEngineering and Technology Institute Groningen (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 11th November 2024
Abstract
Hydrogen peroxide (H2O2) is a valuable green oxidant with a wide range of applications. Furthermore, it is recognized as a possible future energy carrier achieving safer operation, storage and transportation. The photochemical production of H2O2 serves as a promising alternative to the waste- and energy-intensive anthraquinone process. Following green and sustainable chemistry principles, we demonstrated a sustainable photocatalyst utilizing earth-abundant iron and biobased sources only. These iron oxide nanoparticles (FeOx NPs) facilitated effective H2O2 production under batch conditions. Here, through the design of a modular photo-flow reactor, we achieved continuous and enhanced production of H2O2 by minimizing Fenton degradation. After detailed investigation of Fenton chemistry, we designed a reactor tailored to optimize the performance of our catalyst system. Optimal reaction conditions balancing production and energy efficiencies allowed a remarkable increase in production of >14× and productivity by >3× when compared to batch conditions. The produced H2O2 was concentrated to 0.02 wt% via rotary evaporation, approaching commercially relevant concentrations. The reactor design also allowed other chemical transformations, such as photoclick chemistry, as well as the processing of biomass waste into valuable products.
Introduction
Hydrogen peroxide (H2O2) is a highly versatile and eco-friendly oxidant with widespread applications in the chemical industry, pulp and paper bleaching, wastewater treatment and disinfectants.1–6 In the past years H2O2 has also been proposed as an energy carrier in fuel cells, offering several advantages over hydrogen (H2) such as being a transportable liquid with full solubility in water and thus achieving safer operation, storage, and transportation.7–9 It is estimated that the global market for H2O2 will grow at a compound annual growth rate (CAGR) of 4.6% increasing to 5.7 million tons annual demand by 2028.10,11 Its characteristics as high-energy fuel and as environmentally-friendly oxidant, generating water (H2O) and oxygen (O2) as the only by-products, led to H2O2 being listed as one of the 100 most important chemicals on earth.12,13However, current production of H2O2via the anthraquinone process, comprising Pd-catalyzed hydrogenation of an alkyl-anthraquinone and consecutive oxidation in an organic solvent, involves high energy input and generates substantial volume of wastewater and solid waste.1,14 Hence, a growing demand for sustainable alternatives led to techniques such as electrocatalytic production of H2O2 through the oxygen reduction reaction (ORR) or water oxidation reaction (WOR),15–19 as well as the direct synthesis of H2O2 from H2 and O2.20–22 However, particularly in the direct synthesis, the high energy consumption and significant explosion risks associated with O2 and H2 gas mixtures hinder the industrial scale up of these methods.7,9 Harnessing green energy sources like solar energy for the photochemical production of H2O2 might offer a promising alternative for sustainable production of H2O2.2,4,9 While remarkable progress towards photochemical production of H2O2 has been made,23–31 especially metal-based photocatalysts often rely on noble and scarce metals, starting materials and solvents that are not biobased and renewable, and catalyst synthesis often requires special equipment or high temperatures.32,33
For future production processes of H2O2 to be a viable alternative, the process must align with the Sustainable Development Goals and the 12 Principles of Green Chemistry.34,35 Sustainable chemistry is at the core of the energy transition, feedstock transition and materials transition required for a circular economy.36–38
Based on these considerations, we previously established a strategy for the photochemical production of H2O2 catalyzed by earth-abundant iron.10 We developed several iron oxide (FeOx) nanoparticles (NPs) for successful photochemical oxygen reduction to H2O2 under visible light illumination (445 nm), achieving a selectivity for H2O2 of >99% (Fig. 1).10 Crucially, the FeOx NPs could be synthesized from biobased and abundant materials including metal, surfactant (i.e. capping agent, ligand) and solvent. Successful photochemical production of H2O2 was possible via real sunlight irradiation and in seawater.10
 | ||
| Fig. 1 Schematic representation of our previous study and the present work for photochemical production of H2O2. Batch and flow conditions are compared, where flow conditions allow for higher production. | ||
However, directly associated with our choice of metal the batch conditions were limiting the production through Fenton degradation of the produced H2O2 on the iron surface of our catalyst (9.4–14.8 mmol g−1 L−1, 0.136 μmol and 0.027 μmol h−1).10,39–41 Thus, after investigating the Fenton process in detail, we here designed a modular thin-film photo-flow reactor to increase the selective production of H2O2 by minimizing Fenton degradation and facilitating product H2O2 separation from the catalyst surface (Fig. 1). We successfully increased the production by >14× and productivity by >3× when compared to our original batch conditions (from 0.136 μmol to 1.938 μmol and from 0.027 μmol h−1 to 0.097 μmol h−1).
Through this continuous production of H2O2 we ultimately were able generate enough H2O2 to reach 0.02 wt% solutions through partial evaporation of water. The versatility of our flow reactor design was further demonstrated in other applications such as photoclick reactions and biomass valorization to renewable platform chemicals.
Results and discussion
In our previous work, we demonstrated a more sustainable synthesis of iron oxide (FeOx) nanoparticles terminated with different surfactants, where specifically FeOx NPs with specific geometry possessed photoactivity for oxygen reduction to hydrogen peroxide.10 An apparent quantum yield (AQY) of 0.11% was achieved for the photochemical oxygen reduction to H2O2 with visible light (445 nm) at ambient temperatures and pressures (9.4–14.8 mmol g−1 L−1, 0.136 μmol and 0.027 μmol h−1), corresponding to 1.7 ± 0.3 mmol g−1 L−1 h−1. The H2O2 yield could be increased by decreasing the pH, addition of cation exchangers and by production in biphasic systems (Heptane/DCM with Milli-Q water) (up to 19.5 ± 2.7 mmol g−1 L−1).10 FeOx nanoparticles with oleic acid (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as surfactant were successfully utilized in applications like wastewater treatment, polymerizations, and in situ oxidations. Production of H2O2 was possible via sunlight irradiation and in seawater.10
:1) as surfactant were successfully utilized in applications like wastewater treatment, polymerizations, and in situ oxidations. Production of H2O2 was possible via sunlight irradiation and in seawater.10
Sustainable storage solvent
Utilizing earth-abundant metals and biobased (co-)catalysts offered great potential for photocatalytic production of hydrogen peroxide as solar fuel. Generally, the nanoparticles were stable for at least 6 months without aging, agglomeration or changes in size (Tables S2 and S3†), which are crucial and remarkable features considering the magnetic properties of the catalyst.However, one remaining aspect that we improved through this research was the storage aspects of those nanoparticles: usually, the particles were suspended in dichloromethane (10 mL) and stored in nitrogen atmosphere and darkness at 5 °C. Recently though (30th April 2024), the United States Environmental Protection Agency (EPA) finalized a ban on most uses of methylene chloride (dichloromethane, DCM).42,43 Hence, for our catalyst system to stay relevant and especially to align with its goal for sustainable production of H2O2 we investigated greener solvent alternatives.44–50 The following three solvents were identified as successful candidates for replacing DCM (in this order): 2-methyltetrahydrofuran (2-MeTHF), tetrahydrofuran (THF), and methyl isobutyl ketone (MIBK) (Table S2†). Other solvents such as methyl tert-butyl ether (MTBE), methyl ethyl ketone (MEK), ethyl acetate (EtOAc), pentane and dimethyl carbonate were not able to stabilize the nanoparticle dispersion sufficiently. Having identified three green solvent alternatives for DCM with stable dispersions and no precipitation, we further evaluated particle sizes through dynamic light scattering. We observed tunable particle sizes depending on the storage solvent: 1.97 ± 0.31 nm (DCM), 9 ± 5 nm (2-MeTHF), 12 ± 7 nm (THF), and 10 ± 5 nm (MIBK). Ultimately, we recommend 2-MeTHF as future solvent for storage of FeOx NPs as it has higher sustainability character than THF.46–49
Fenton and photo-Fenton chemistry
While applying sustainable chemistry principles to catalyst design and synthesis, our choice of metal directly limited the batch conditions through Fenton degradation of product H2O2 on the iron surface of our catalyst.39The Fenton reaction is characterized by the interaction between peroxides and iron ions, resulting in the formation of reactive oxygen species capable of oxidizing both organic and inorganic compounds.40 The decomposition of peroxides over other metals are classified as Fenton-like reactions.51 Recently, the Fenton process has been effectively employed in the decomposition of hazardous organic substances in wastewater, for which the hydroxyl radical has been identified as critical species.40,52
The Fenton mechanism is represented by reactions (1)–(9) in Scheme 1, where reaction (1) is commonly referred to as the Fenton reaction. This involves the oxidation of ferrous (Fe2+) to ferric (Fe3+) ions to decompose hydrogen peroxide into hydroxyl radicals.40
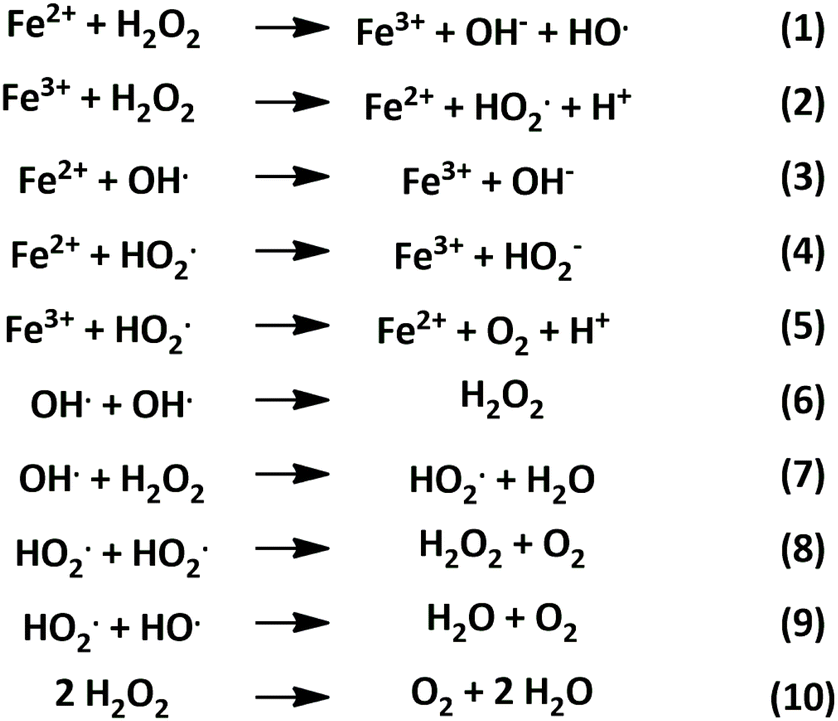 | ||
| Scheme 1 Degradation of hydrogen peroxide through Fenton chemistry and related reactions.40 | ||
Reactions (2)–(5) represent rate limiting steps within the Fenton mechanism, during which the hydrogen peroxide is consumed, and ferrous iron is regenerated. Reactions (5)–(9) are additional radical reactions occurring within the Fenton process. The overall decomposition of hydrogen peroxide by the Fenton process is depicted in reaction (10), which results in the formation of water and oxygen.40
Furthermore, when light of a specific wavelength irradiates a solution containing hydrogen peroxide and iron, the process can be expanded and termed as photo-Fenton chemistry.41 In photo-Fenton reactions, the formation of hydroxyl radicals is enhanced compared to dark-Fenton. In dark-Fenton reactions, the process stops with the accumulation of Fe3+ ions once all Fe2+ has been consumed (Scheme 2, reaction (1)). The increased degradation efficiency of photo-Fenton is attributed to the regeneration of Fe2+via the photoreduction of Fe3+ (Scheme 2, reaction (2)), allowing the continuous occurrence of reaction (1) and maintaining the catalytic cycle.41
 | ||
| Scheme 2 Enhanced degradation of hydrogen peroxide through photo-Fenton chemistry.41 | ||
While these aspects are advantageous for the degradation of organic pollutants in wastewater treatment, our designed photocatalyst was limited through the kinetics of production vs. degradation of hydrogen peroxide. Furthermore, the photolysis of hydrogen peroxide (reaction (3)) at wavelengths below 400 nm, contributes to the accelerated degradation of product peroxide.41
In our previous study we already circumvented Fenton degradation and enhanced production of H2O2 through biphasic solvent systems (aqueous/non-aqueous) in batch conditions.10 We were able to minimize Fenton degradation through separation of produced H2O2 and ˙O2− from the catalyst surface, as the catalyst suspended in organic solvents while H2O2 migrated to the water layer, thereby effectively performing an in situ extraction.10
Following up on these insights we further investigated Fenton properties of our catalyst system. In Fig. 2 the decomposition of a 1 mM solution (30 min nitrogenated) of hydrogen peroxide was followed at 240 nm (by UV-Vis) in darkness and light (without iron present). In comparison to the slow degradation in darkness (black), irradiation with light as depicted in orange accelerates H2O2 decomposition via photo-Fenton chemistry (445 nm). However, as soon as FeOx NPs were immobilized on the side of the cuvette and thereby exposed to a 1 mM hydrogen peroxide solution, the presence of iron drastically increased the decomposition of H2O2 in darkness (brown). Under light exposure, however, hydrogen peroxide was initially produced and subsequently decomposed at a slower rate (indicated in blue). Thus, Fenton chemistry is prominently active when utilizing these FeOx NPs, particularly in batch irradiation setups. While the production of H2O2 clearly outpaces the degradation of hydrogen peroxide initially, the whole system would benefit from flow chemistry to mitigate the influence of the Fenton chemistry. A modular photo-flow reactor was designed to allow the produced hydrogen peroxide to be continuously removed from the vicinity of the iron oxide photocatalyst and light source.
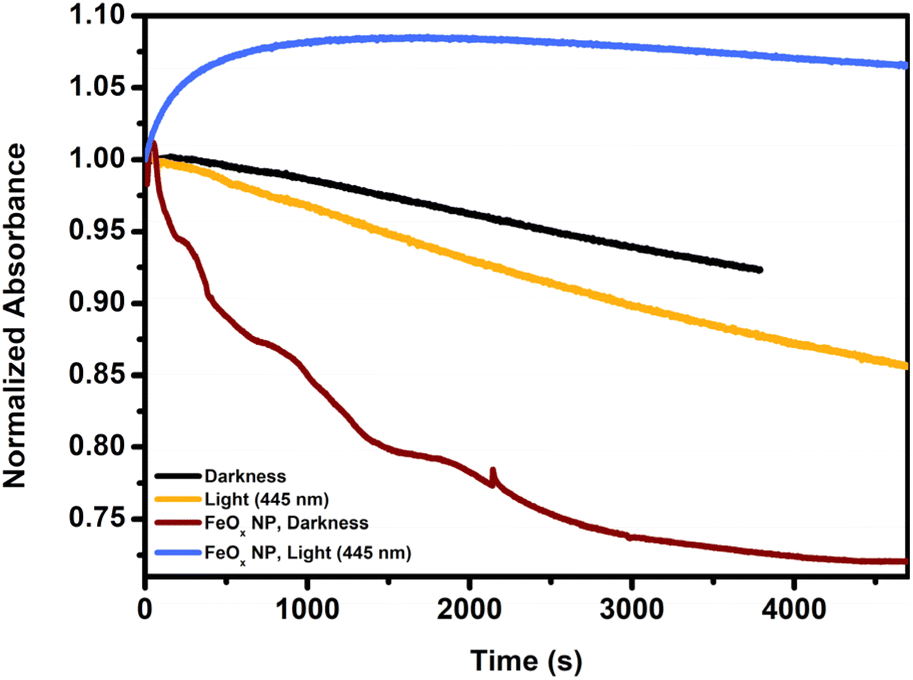 | ||
| Fig. 2 Fenton decomposition of hydrogen peroxide over FeOx NPs. UV-Vis absorbance of H2O2 followed at 240 nm. Initial concentration of hydrogen peroxide 1 mM, solution was 30 min nitrogenated. | ||
Photo-flow reactor design
In recent years, remarkable advances in photochemistry53 and electrochemistry54–63 have been combined with technological innovations in flow chemistry64,65 and high-throughput experimentation.66,67 Not only allowing enhanced organic synthesis,68,69 but also production of H2O2,70–72 we also gained valuable experience ourselves on applying electrochemistry73 and photochemistry in flow over the past years.74–77Having established the details on Fenton degradation for our catalytic system, we initially evaluated several reactor designs to produce H2O2 in flow. Crucially, through the hydrophobic properties of the catalyst, the material demonstrated a strong affinity for glass surfaces. This facilitated the immobilization of the catalyst without leaching within a batch- as well as flow-reactor. Thus, inspired by falling film reactors, we opted for a modular thin-film photo-flow design.
The reactor comprises multiple layers secured by screws (Fig. 3A). The upper (1) and lower (7) layers are constructed from stainless steel plates with screw holes. A window is cut into the center of these steel plates to allow light irradiation from the top and bottom. Adjacent to the upper steel layer (1) is a protective plastic support layer (2), which separates it from a quartz glass layer (3). Quartz is selected for its ability to facilitate reactions with UV light sources. The reaction occurs between two glass plates (3 and 5), with the lower glass plate (5) containing two holes that serve as the reactor's inlet and outlet. A rubber gasket (4) is positioned between the glass plates, serving as a mold for catalyst application on the glass. This gasket is made of H2O2-cured 1 mm thick ethylene propylene diene monomer (EPMD) that ensures chemical stability and a consistent flow pattern (which can be replaced with an alternative pattern if required).
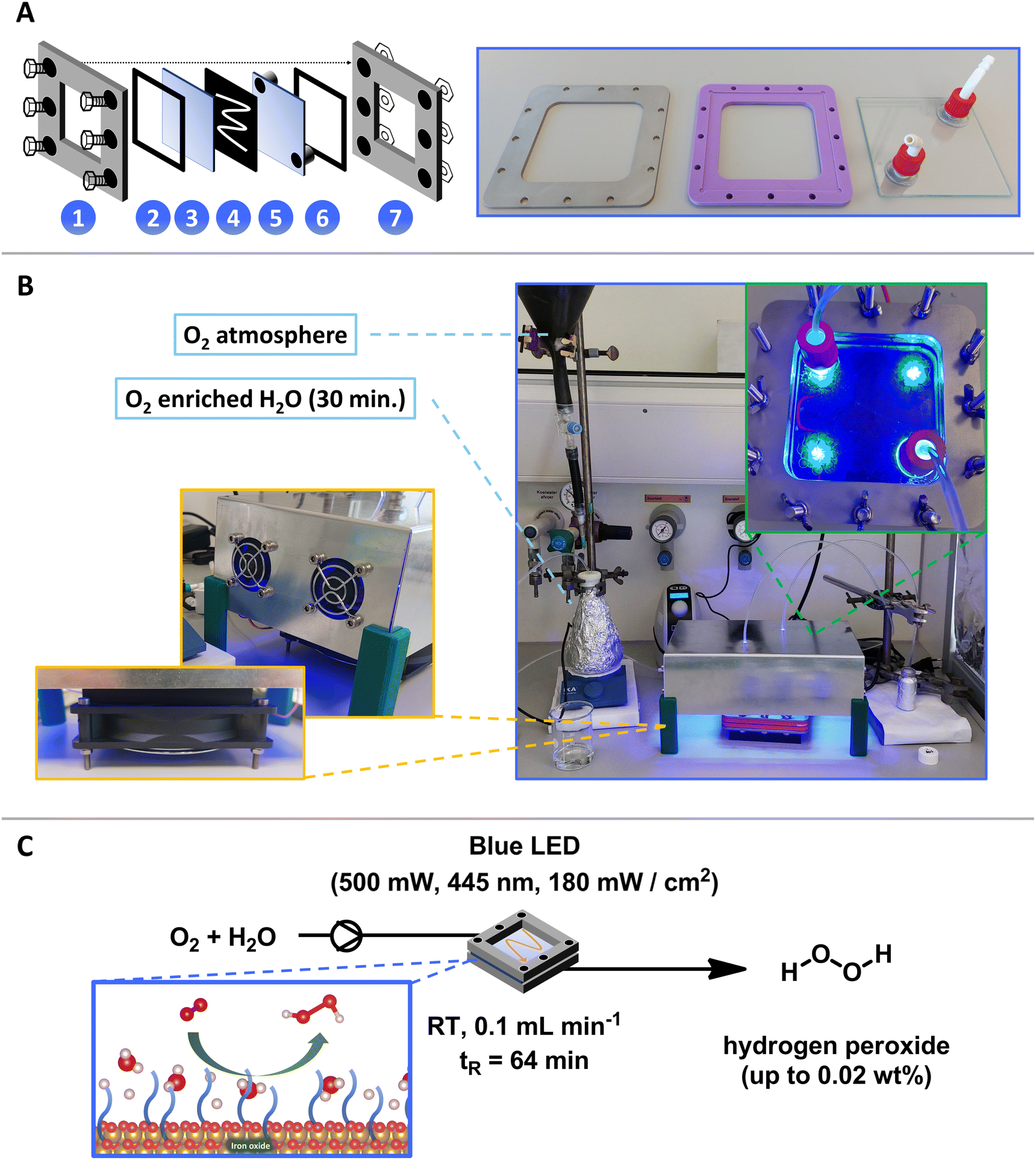 | ||
| Fig. 3 (A) Schematic representation and design of the parts of the modular photo-flow reactor. (B) Assembled photo-flow reactor to produce hydrogen peroxide including reactant solution (O2 atmosphere, blue), diaphragm liquid dosing pump (SIMDOS® 02 FEM 1.02 RC-P), reactor with LEDs (inlet green), light blocking box with cooling fans for temperature control (orange inlet), and product solution. (C) Reaction scheme and optimal conditions for the photochemical production of H2O2 in photo-flow reactor. Blue inlet: oxygen reduction reaction on the FeOx photocatalyst. Reaction conditions: 30 mg FeOx NPs, blue LED (500 mW, 445 nm, 180 mW cm−2), room temperature (without cooling), flow rate: 0.1 mL min−1 of oxygenated (30 min) Milli-Q water, residence time tR (64 min). | ||
The components of the proposed flow reactor were ultimately designed in accordance with the specifications outlined in Fig. 3A. The whole reactor design and size was determined through the standard dimensions of quartz glass (100 × 100 × 1 mm) at our institution. Within the borosilicate glass, two holes were precisely cut, to which G/L fittings were attached to establish inlet and outlet points. The plastic support and stainless-steel frame each incorporate three screw holes on opposite sides, ensuring uniform force distribution upon assembly. The positioning of the G/L fittings was carefully arranged to align within the window of the plastic and steel frame. Furthermore, a septum fabricated from silicone rubber was implemented to securely connect tubing to the reactor without risk of leakage. Integration of these components yields the fully operational and adaptable photo-flow reactor depicted in Fig. 3B. The inner dimensions of the designed flow reactor were 8 cm × 8 cm × 1 mm. Furthermore, the integration of various patterns of rubber gaskets was feasible. Here, elongated pathways enable increased reactor volume and offer the potential to accommodate a greater quantity of catalyst within the reactor.
Photo-flow production of H2O2
After the design and assembly of our reactor, we immobilized our FeOx NPs onto the glass within the flow reactor. These nanoparticles are usually suspended in organic solvents, allowing facile drop casting onto the glass surface. Following the evaporation of the solvent, the nanoparticles adhere to the glass surface. Subsequently, oxygenated Milli-Q water was flown over the catalyst without causing observable removal of particles, indicating successful immobilization. The Milli-Q water circulation within the reactor was facilitated by a diaphragm liquid dosing pump, reaching the photo-flow reactor window, where blue light irradiation initiates the oxygen reduction to H2O2, which is separated from the catalyst surface and then collected in the product flask (Fig. 3B). The schematic representation of the reaction and optimal conditions found for our photo-flow production of H2O2 are depicted in Fig. 3C.We initially attempted to produce H2O2 through recirculating the solvent by directing the product solution back into the reactant flask. However, this approach yielded non-quantifiable concentrations of H2O2 through Fenton degradation, as determined by peroxide test strips. Instead, a robust and successful method for hydrogen peroxide production was achieved, involving a single pass of the solution through the reactor at a considerable flow rate (0.1–0.3 mL min−1). In this configuration, the reactant flask containing oxygenated Milli-Q water was stirred continuously, while the introduction of a needle through the septum mitigated the development of under pressure arising from the pump. Utilizing perfluoro alkoxy alkane (PFA) tubing, the reactant was conveyed through the pump to the photoreactor, with another PFA tube serving as the outlet to the product flask.
After successful H2O2 production, experiments were systematically conducted by first operating the system in darkness to obtain a blank sample under light-free conditions. By peroxide test strips, the blank in darkness was confirmed to be negative and was subsequently used for normalization. Upon activation of the light source, aliquots were collected at hourly intervals over a period of four hours. Additionally, a fifth aliquot was collected after an overnight duration of 20 h. Consistent experimental procedures were maintained across all flow experiments, ensuring comparable results, as summarized in Table S5.† Kinetic analyses were conducted for the initial four hours of reaction under varying conditions to establish optimal reaction conditions. These kinetic profiles depict the cumulative production of hydrogen peroxide over time.
The initial catalyst amount was assessed through the immobilization of varying quantities of catalyst (20, 30, and 60 mg) on the glass surface of the flow reactor (Fig. 4A). It was observed that all catalyst loadings effectively covered the entire surface of the photoreactor. Through this investigation, it was determined that the highest production and productivity could be achieved with the immobilization of 30 mg of catalyst in the reactor. Conversely, a lower production rate was observed with 20 mg of photocatalyst immobilization, likely due to a reduced number of active sites or photon absorption. Interestingly, immobilization with 60 mg of photocatalyst resulted in intermediate production between the other catalyst amounts. This phenomenon could be attributed to the onset of Fenton degradation within the photoreactor when using 60 mg of FeOx NPs. However, it is plausible that this issue could be mitigated at higher flow rates. For subsequent studies, a catalyst loading of 30 mg was selected as the optimal condition.
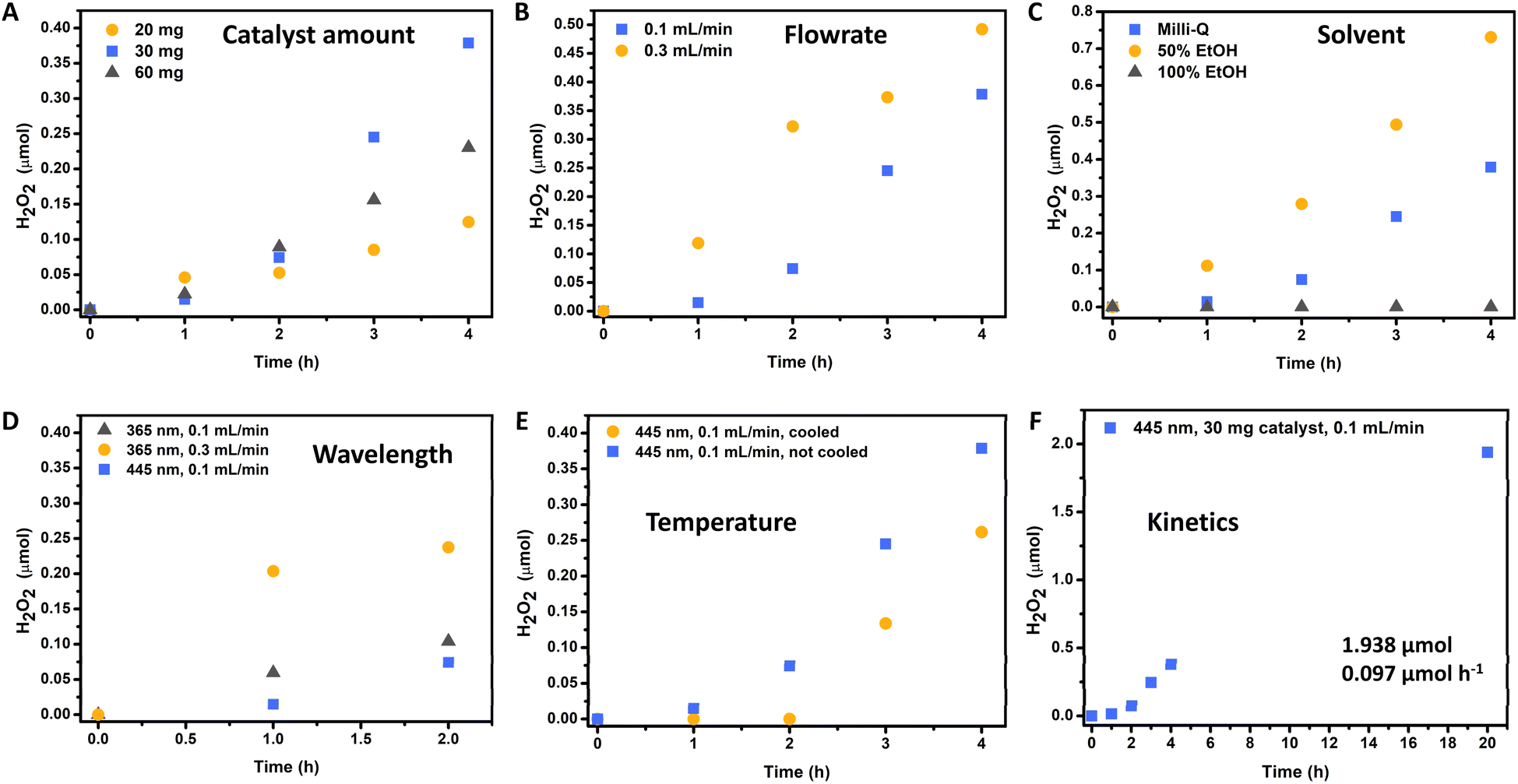 | ||
| Fig. 4 Evaluation of reaction conditions for the photochemical oxygen reduction to hydrogen peroxide in flow. (A) Effect of catalyst amount. (B) Effect of Milli-Q water flowrate. (C) Effect of solvent. (D) Effect of wavelength (UV light, 365 nm). (E) Effect of temperature. (F) Kinetics of H2O2 production under optimal conditions. Standard and optimal conditions: 30 mg FeOx NPs, Blue LED (500 mW, 445 nm, 180 mW cm−2), room temperature (without cooling), flowrate: 0.1 mL min−1 of oxygenated (30 min) Milli-Q water, residence time tR (64 min). | ||
The impact of Milli-Q water flow rate on hydrogen peroxide production is illustrated in Fig. 4B. Depending on the flowrate different residence times can be calculated correlated to the reactor volume of 6.4 mL (8 cm × 8 cm × 1 mm):
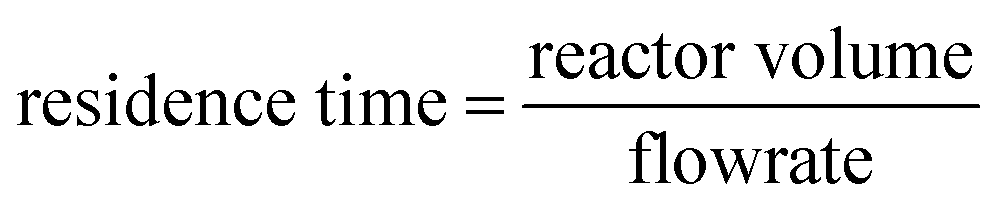 | (1) |
Thus, depending on the flowrate different residence times could be observed (flowrate 0.1 mL min−1 with residence time tR = 64 min; flowrate 0.3 mL min−1 with residence time tR = 21.3 min). It was found that a flow rate of 0.3 mL min−1 yielded a higher production of hydrogen peroxide over time. This outcome is attributed to the faster removal of produced hydrogen peroxide from the flow reactor, resulting in reduced contact with the immobilized photocatalyst. Consequently, the shorter duration of exposure to photo-Fenton degradation led to increased productivity and hydrogen peroxide production. However, for subsequent investigations, a flow rate of 0.1 mL min−1 was adopted, as it resulted in more positive peroxide test strips and higher concentrations of hydrogen peroxide. Although a flow rate of 0.3 mL min−1 produced the highest quantity of hydrogen peroxide, the product solution was more diluted, requiring additional energy for distillation in subsequent processing steps.
In the next step a solvent with superior oxygen solubility was selected, to enhance H2O2 production through ORR in our modular flow system. Previously, we already established that solvents such as ethanol, isopropanol or methanol were ineffective to serve as hole scavengers (i.e. electron donors) in the batch process.10 Thus, in this case we opted for ethanol as solvent due to its significantly higher oxygen solubilizing ability compared to water, varying between 5 to 7.5 times greater depending on the temperature.78,79 Through comparing pre-oxygenated solutions containing 0%, 50%, and 100% ethanol, we observed that no conversion could be obtained when utilizing pure ethanol as the nanoparticles were suspended over time (Fig. 4C). However, utilizing a 50% ethanol solution resulted in a substantial increase in hydrogen peroxide production, with a 92% improvement after 4 h compared to water as more oxygen could be dissolved. Additionally, the nanoparticles remained immobilized in the reactor and did not dissolve in the 50% ethanol solution. For further screenings, we opted for pure water as solvent for production of H2O2 to investigate the system without addition of organic solvents.
Subsequently, flow reactions using lower wavelengths irradiation were examined since the nanoparticles allowed higher UV-Vis absorption. Previously, it was found that the apparent quantum yield (AQY) to be higher at irradiation with 365 nm (0.08%) compared to 455 nm light (0.05%). Consequently, 365 nm irradiation was employed for H2O2 production at flow rates of 0.1 and 0.3 mL min−1. Fig. 4D illustrates an improved production rate under UV light irradiation. However, only two hours of production are depicted. After three hours of light exposure, no further conversion was obtained. We initially theorized thermal decomposition of the produced H2O2 as the UV light (365 nm) entered the reactor through a quartz glass plate but was fully absorbed by a borosilicate glass plate on the opposite side, leading to heat accumulation (from 22.2 °C to 32.4 °C within 2 h). Thus, we developed an air-cooling system for the reactor to allow stable temperatures (from 21.9 °C to stable 26.1 °C over 4 h). However, in both cases we were not able to increase the production of H2O2, indicating that heat accumulation was not the reason but probably enhanced photo-Fenton degradation with UV light (365 nm), facilitating product H2O2 decomposition. These results are in line with our previously reported temperature screening, where already in batch conditions higher temperatures allowed more rather than less H2O2 to be produced. In our flow reactor, while cooled production through UV light irradiation was not possible as photo-Fenton degradation kinetics were limiting the reaction, H2O2 production was observable at least for the first two hours in uncooled conditions with UV light irradiation before the degradation took over.
Knowing that 365 nm was not the optimal wavelength and that higher temperatures increase the production, we confirmed those results through our temperature screening (Fig. 4E): Through irradiation with 445 nm at uncooled conditions, the temperature increased from 21.3 °C (blank) to 25.8 °C (1 h), 27 °C (2 h), and 27.7 °C (3 h and 4 h). Here the production surpassed that under the cooled conditions, with which the temperature was stable (from 22.1 °C to stable 23.2 °C over 4 h).
We already proposed a mechanism for the photochemical production of H2O2 previously; however, despite extensive experimental efforts, the precise interaction between iron and oleic acid, whether as co-catalyst alone or involving additional sacrificial components, requires further investigation.
After these experiments, the optimal conditions to produce H2O2 in our flow system were: 30 mg FeOx NPs, blue LED (500 mW, 445 nm, 180 mW cm−2), room temperature (without cooling), flowrate: 0.1 mL min−1 of oxygenated (30 min.) Milli-Q water, residence time tR (64 min). The complete kinetic profile of these conditions is illustrated in Fig. 4F. After 20 h of irradiation, 1.938 ± 0.306 μmol of hydrogen peroxide was produced in flow. Compared to batch reactions (5 h), this represents a 14.25-fold increase in hydrogen peroxide production and a 3.59-fold increase in productivity (Fig. 5A). We were pleased to observe that the increased H2O2 production in our flow reactor demonstrates the effectiveness of circumventing photo-Fenton decomposition. This is accomplished by continuously removing the produced hydrogen peroxide from the reactor, thereby isolating it from the catalyst and light source.
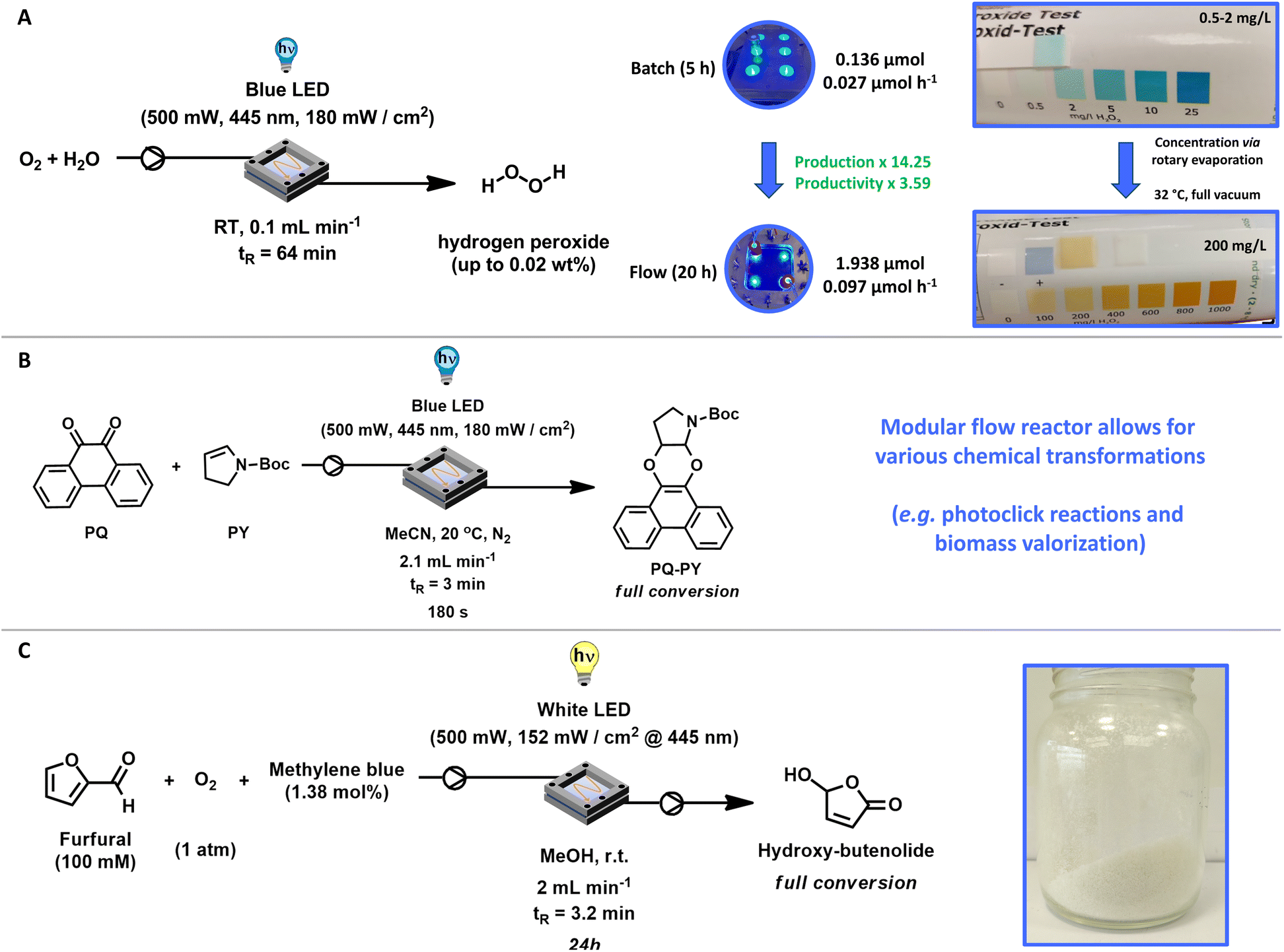 | ||
| Fig. 5 Final conditions for hydrogen peroxide production as well as other chemical transformations in our reactor design. (A) Optimal reaction condition and final improvements to produce H2O2 (up to 0.02 wt%) in our modular photo-flow reactor through the oxygen reduction reaction. (B) Photoclick reaction of 9,10-phenanthrenequinone with N-boc-2,3-dihydro-1H-pyrrole within 180 s. (C) Conversion of renewable feedstock and biomass derived furfural to commodity chemicals such as hydroxy-butenolide for applications in coatings, resins, and paints. | ||
Subsequently, to evaluate the robustness of our flow-reactor as well as catalyst system, we performed the production of H2O2 under those conditions in triplicate, without significant deviations (Table S5†). We produced several product solutions (18) with volumes between 50 mL and 200 mL. Generally, product solutions of 75 mL had a concentration of approximately 2 mg L−1. Following mild and fast rotary evaporation at 32 °C (to 50 °C) under full vacuum, the water volume was reduced, and the concentration thereby increased to 200 mg L−1. This corresponds to a hydrogen peroxide concentration of 0.02 wt% (Fig. 5A). Compared to batch production (0.5–2 mg mL−1) this represents a significant increase, allowing for concentrations despicable through percentage by weight nearing commercially viable concentrations. For instance, hydrogen peroxide has been demonstrated to effectively kill the SARS-COVID-19 virus at concentrations as low as 0.5% with a one-minute exposure.80 This method thus provides a viable approach towards on-demand and on-site hydrogen peroxide production, eliminating the need for transportation and mitigating the risks associated with the high concentrations typically involved in the anthraquinone process.
Applications in photoclick reactions and biomass valorisation
After having established the successful production of H2O2 through our modular flow-reactor and photocatalyst system, we investigated other chemical transformations.Photoclick reactions, also referred to as light-triggered cycloaddition reactions, combine the advantages of both photochemistry81 and classical click chemistry.82,83 Enabling chemoselective product formation, quantitative conversion, short reaction times, high functional group orthogonality, and mild reaction conditions with the potential for remarkable spatiotemporal control.77,84 These photoclick reaction systems have become indispensable as methodology for surface functionalization,85 polymer conjugation,86 photocrosslinking,87 protein labelling,88 and bioimiganing.77 We previously reported the light-induced photocycloaddition of 9,10-phenanthrenequinone (PQ) with electron-rich alkenes (ERA), known as the PQ-ERA reaction, which is a highly attractive photoclick reaction characterized by high selectivity, external non-invasive control with light and biocompatibility.89–91 In our modular flow reactor system, we improved the energy requirements of our previously reported system by utilizing blue LEDs (445 nm) instead of light with lower wavelength (395 nm). Product formation could be followed by UV-Vis (Fig. S27†) as well as high-pressure liquid chromatography (HPLC, Fig. S28†). Utilizing our modular photo-flow reactor we achieved full conversion of PQ with N-Boc-2,3-dihydro-1H-pyrrole (PY) to our desired product within 180 s, showcasing the versatility of the reactor setup (Fig. 5B).77,84
Another important research field in our group is the replacement of petrochemical-based chemicals92 and monomers like acrylates through sustainable alternatives.74,93 Envisioning applications such as coatings, resins, and paints, we developed several hydroxy- and alkoxybutenolides as bio-based alternatives for the formation of high-performance coatings.94,95 Our newly designed flow-reactor was also compatible with these transformations: starting from biomass-derived furfural, an environmentally benign photochemical conversion using visible light (white LED), oxygen and methylene blue in our flow reactor provided the desired hydroxybutenolide monomer with full conversion after 24 h (Fig. 5C).74,96 Here methylene blue is required as catalytic triplet photosensitizer for the generation of singlet oxygen (1O2), where the furan moiety of furfural undergoes a [4 + 2] cycloaddition with 1O2 and transform quantitatively into hydroxybutenolide.96 Through employing flow rates of 2 mL min−1 and thereby a residence time tR = 3.2 min, we successfully achieved the production of our desired product hydroxybutenolide (i.e. 5-hydroxy-2(5H)-furanone). Furthermore, as the flow reactor operated in a closed system, we were able to follow the kinetics of the Diels–Alder type reaction over time (Fig. S31†).96 Next to the product, the formation of methyl formate as well as the intermediate endoperoxide after Diels–Alder reaction with 1O2 could be observed (Fig. S31†).96 The conversion of renewable feedstock in our photo-flow reactor is another showcase of the versatility of the reactor design.
Conclusion
Through this research, we have successfully expanded the capabilities of our photocatalyst system in producing H2O2 from O2 through the oxygen reduction reaction. Initially, we developed a photocatalyst with strict adherence to the principles of green and sustainable chemistry, which now also omits dichloromethane as storage solvent after its recent prohibition.However, the utilization of earth-abundant iron in our FeOx NPs led to Fenton degradation of the produced H2O2, thereby limiting our productivity. By conducting detailed investigations into Fenton degradation and H2O2 production kinetics, we devised a modular photo-flow reactor to address this challenge. This reactor system enabled the application of our photocatalyst while circumventing Fenton degradation of the H2O2 product through continuous removal from the active site. The modular reactor allowed easy adjustments of flow patterns and catalyst amounts, and the incorporation of quartz glass enabled UV-light irradiation. Comparative studies under batch conditions revealed a remarkable increase in production of 14.25-fold and productivity by 3.59-fold. Notably, we obtained H2O2 solutions of 75 mL with concentrations of 2 mg mL−1, which could be concentrated to 0.02 wt% through rotary evaporation.
Furthermore, we demonstrated the versatility of our flow reactor by employing it in photoclick chemistry and in the conversion of biomass-derived furfural to acrylate replacements for paints and coatings.
In conclusion, our method provides a viable approach towards on-demand and on-site hydrogen peroxide production, eliminating the need for transportation and mitigating the associated risks of handling high concentrations typically encountered from the anthraquinone process.
Data availability
The data supporting this article have been included as part of the ESI.† This also includes the raw files for the design of the flow-reactor. Unprocessed raw data, such as 1H-NMR spectra of previously published compounds, will be made available by the corresponding author upon request.Author contributions
B. L. F. conceptualized the research project and coordinated it together with P. P. P. T. F. and J. T. M. conducted the research and its validation. M. M. provided scientific advice and designed the reactor together with T. F. and J. T. M. T. F. and B. L. F. prepared the manuscript with input from J. T. M., M. M., P. P. P. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is in collaboration with Shell and part of the Advanced Research Center for Chemical Building Blocks, ARC CBBC, which is cofounded and co-financed by the Netherlands Organization for Scientific Research (NWO, contract 736.000.000) and the Netherlands Ministry of Economic Affairs and Climate. We are grateful for the generous financial support to B. L. F. (the Ministry of Education, Culture and Science of the Netherlands Gravitation Programme no. 024.001.035)References
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS.
- T. Freese, J. T. Meijer, B. L. Feringa and S. B. Beil, Nat. Catal., 2023, 6, 553–558 CrossRef CAS.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- Y. Ding, S. Maitra, S. Halder, C. Wang, R. Zheng, T. Barakat, S. Roy, L. H. Chen and B. L. Su, Matter, 2022, 5, 2119–2167 CrossRef CAS.
- W. Yu, C. Hu, L. Bai, N. Tian, Y. Zhang and H. Huang, Nano Energy, 2022, 104, 107906 CrossRef CAS.
- Y. Guo, X. Tong and N. Yang, Nanomicro Lett., 2023, 15, 77 CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- Y. Xue, Y. Wang, Z. Pan and K. Sayama, Angew. Chem., Int. Ed., 2021, 60, 10469–10480 CrossRef CAS.
- Y. Sun, L. Han and P. Strasser, Chem. Soc. Rev., 2020, 49, 6605–6631 RSC.
- T. Freese, J. T. Meijer, M. B. Brands, G. Alachouzos, M. C. A. Stuart, R. Tarozo, D. Gerlach, J. Smits, P. Rudolf, J. N. H. Reek and B. L. Feringa, EES Catal., 2024, 2, 262–275 RSC.
- H. Cheng, J. Cheng, L. Wang and H. Xu, Chem. Mater., 2022, 34(10), 4259–4273 CrossRef CAS.
- R. L. Myers, The 100 Most Important Chemical Compounds: a Reference Guide, Greenwood Publishing Group, Westport, CT, 2007 Search PubMed.
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374–3381 CrossRef CAS PubMed.
- G. Goor, J. Glenneberg, S. Jacobi, J. Dadabhoy and E. Candido, Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2019, pp. 1–40 Search PubMed.
- S. C. Perry, D. Pangotra, L. Vieira, L. I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem, 2019, 3, 442–458 CrossRef CAS.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 RSC.
- X. Sheng, N. Daems, B. Geboes, M. Kurttepeli, S. Bals, T. Breugelmans, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2015, 176–177, 212–224 CrossRef CAS.
- J. S. Adams, M. L. Kromer, J. Rodríguez-López and D. W. Flaherty, J. Am. Chem. Soc., 2021, 143, 7940–7957 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- L. F. De Freitas, B. Puértolas, J. Zhang, B. Wang, A. S. Hoffman, S. R. Bare, J. Pérez-Ramírez, J. W. Medlin and E. Nikolla, ACS Catal., 2020, 10, 5202–5207 CrossRef.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 RSC.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed.
- Y. Isaka, Y. Kondo, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2018, 54, 9270–9273 RSC.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- D. Zheng, Y. Wu, X. Yang, S. Wang and Y. Fang, ChemSusChem, 2024, 2024, e202400528 CrossRef.
- Y. Xu, Y. Cao, L. Tan, Q. Chen and Y. Fang, J. Colloid Interface Sci., 2023, 633, 323–332 CrossRef CAS.
- Y. Xu, Y. Pan, W. Yahan, Y. Fang, M. Anpo, H. Yoshida and X. Wang, Appl. Catal., B, 2023, 331, 122701 CrossRef CAS.
- L. Yang, H. Chen, Y. Xu, R. Qian, Q. Chen and Y. Fang, Chem. Eng. Sci., 2022, 251, 117435 CrossRef CAS.
- P.-I. Dassie, R. Haddad, M. Lenez, A. Chaumonnot, M. Boualleg, P. Legriel, A. Styskalik, B. Haye, M. Selmane, D. P. Debecker, C. Sanchez, C. Chaneac and C. Boissiere, Green Chem., 2023, 25, 2800–2814 RSC.
- D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 599–603 CrossRef CAS.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 2000 Search PubMed.
- T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS.
- S. Mitchell, A. J. Martín, G. Guillén-Gosálbez and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2024, e202318676 CAS.
- A. Cannon, S. Edwards, M. Jacobs, J. W. Moir, M. A. Roy and J. A. Tickner, RSC Sustainability, 2023, 1, 2092–2106 RSC.
- H. J. H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC.
- A. Babuponnusami and K. Muthukumar, J. Environ. Chem. Eng., 2014, 2, 557–572 CrossRef CAS.
- G. Ruppert, R. Bauer and G. Heisler, J. Photochem. Photobiol., A, 1993, 73(1), 75–78 CrossRef CAS.
- Biden-Harris Administration Finalizes Ban on Most Uses of Methylene Chloride, Protecting Workers and Communities from Fatal Exposure, US EPA, https://www.epa.gov/newsreleases/biden-harris-administration-finalizes-ban-most-uses-methylene-chloride-protecting, accessed 9 June 2024.
- EPA bans most uses of methylene chloride, https://cen.acs.org/policy/chemical-regulation/EPA-bans-uses-methylene-chloride/102/web/2024/04, accessed 9 June 2024.
- T. Freese, N. Elzinga, M. Heinemann, M. M. Lerch and B. L. Feringa, RSC Sustainability, 2024, 2, 1300–1336 RSC.
- T. Freese, R. Kat, S. D. Lanooij, T. C. Böllersen, C. M. De Roo, N. Elzinga, M. Beatty, B. Setz, R. R. Weber, I. Malta, T. B. Gandek, A. Krikken, P. Fodran, R. Pollice and M. M. Lerch, ChemRxiv, 2023, 1–76 Search PubMed.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4(1), 1–24 CrossRef.
- L. Cseri, S. Kumar, P. Palchuber and G. Szekely, ACS Sustain. Chem. Eng., 2023, 11, 5696–5725 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- D. Meyerstein, Nat. Rev. Chem, 2021, 5, 595–597 CrossRef CAS.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- M. González-Esguevillas, D. F. Fernández, J. A. Rincón, M. Barberis, O. De Frutos, C. Mateos, S. García-Cerrada, J. Agejas and D. W. C. Macmillan, ACS Cent. Sci., 2021, 7, 1126–1134 CrossRef PubMed.
- E. Brillas, F. Alcaide and P. L. Cabot, Electrochim. Acta, 2002, 48, 331–340 CrossRef CAS.
- F. Yu, Y. Chen, Y. Pan, Y. Yang and H. Ma, Sep. Purif. Technol., 2020, 241, 116695 CrossRef CAS.
- M. Mohaghegh Montazeri, S. A. Hejazi and F. Taghipour, J. Environ. Chem. Eng., 2023, 11, 109636 CrossRef CAS.
- C. P. de León, Nat. Catal., 2020, 3(2), 96–97 CrossRef.
- E. Isarain-Chávez, C. de la Rosa, C. A. Martínez-Huitle and J. M. Peralta-Hernández, Int. J. Electrochem. Sci., 2013, 8, 3084–3094 CrossRef.
- X. Zhang, Y. Xia, C. Xia and H. Wang, Trends Chem., 2020, 2, 942–953 CrossRef CAS.
- D. Pangotra, L. I. Csepei, A. Roth, V. Sieber and L. Vieira, Green Chem., 2022, 24, 7931–7940 RSC.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu, K. Daasbjerg, T. Skrydstrup and N. Lock, Adv. Funct. Mater., 2020, 30, 1910408 CrossRef CAS.
- J. Creus, M. Miola and P. P. Pescarmona, Green Chem., 2023, 25, 1658–1671 RSC.
- M. Miola, X. M. Hu, R. Brandiele, E. T. Bjerglund, D. K. Grønseth, C. Durante, S. U. Pedersen, N. Lock, T. Skrydstrup and K. Daasbjerg, J. CO2 Util., 2018, 28, 50–58 CrossRef CAS.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- X. Fu, J. B. Pedersen, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Z. Andersen, A. Xu, N. H. Deissler, J. Bjarke, V. Mygind, C. Wei, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2023, 379, 707–712 CrossRef CAS PubMed.
- W. Debrouwer, W. Kimpe, R. Dangreau, K. Huvaere, H. P. L. Gemoets, M. Mottaghi, S. Kuhn and K. Van Aken, Org. Process Res. Dev., 2020, 24, 2319–2325 CrossRef CAS.
- J. Britton and T. F. Jamison, Nat. Protoc., 2017, 12(11), 2423–2446 CrossRef CAS PubMed.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castañón, A. H. Cherney, M. Christensen, D. B. Damon, J. Eugenio De Diego, S. García-Cerrada, P. García-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. Liu, P. C. Lobben, D. W. C. Macmillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, I. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, Org. Process Res. Dev., 2019, 23, 1213–1242 CrossRef CAS.
- A. Slattery, Z. Wen, P. Tenblad, J. Sanjosé-Orduna, D. Pintossi, T. Den Hartog and T. Noël, Science, 2024, 383, 1–12 CrossRef PubMed.
- H. B. Vibbert, C. Bendel, J. R. Norton and A. J. Moment, ACS Sustain. Chem. Eng., 2022, 10, 11106–11116 CrossRef CAS.
- A. Gopakumar, T. Zhang and S. Das, J. Flow Chem., 2023, 13, 185–192 CrossRef CAS.
- B. O. Burek, A. Sutor, D. W. Bahnemann and J. Z. Bloh, Catal. Sci. Technol., 2017, 7, 4977–4983 RSC.
- M. Miola, D. Chillé, G. Papanikolaou, P. Lanzafame and P. P. Pescarmona, Green Chem., 2023, 25, 1875–1883 RSC.
- J. G. H. Hermens, T. Freese, K. J. Van Den Berg, R. Van Gemert and B. L. Feringa, Sci. Adv., 2020, 6, 26–42 Search PubMed.
- J. G. H. Hermens, M. L. Lepage, A. Kloekhorst, E. Keller, R. Bloem, M. Meijer and B. L. Feringa, React. Chem. Eng., 2022, 7, 2280–2284 RSC.
- M. D. Edwards, M. T. Pratley, C. M. Gordon, R. I. Teixeira, H. Ali, I. Mahmood, R. Lester, A. Love, J. G. H. Hermens, T. Freese, B. L. Feringa, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2024, 28(5), 1917–1928 CrossRef CAS PubMed.
- Y. Fu, H. Helbert, N. A. Simeth, S. Crespi, G. B. Spoelstra, J. M. van Dijl, M. van Oosten, L. R. Nazario, D. van der Born, G. Luurtsema, W. Szymanski, P. H. Elsinga and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 10041–10047 CrossRef CAS.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Ind. Eng. Chem. Res., 2014, 53, 19331–19337 CrossRef CAS.
- R. Battino, T. R. Rettich and T. Tominaga, J. Phys. Chem. Ref. Data, 1983, 12, 163–178 CrossRef CAS.
- G. Kampf, D. Todt, S. Pfaender and E. Steinmann, J. Hosp. Infect., 2020, 104, 246–251 CrossRef CAS PubMed.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- Y. Fu, N. A. Simeth, R. Toyoda, R. Brilmayer, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202218203 CrossRef CAS PubMed.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2011, 133, 15730–15736 CrossRef CAS.
- W. Feng, L. Li, C. Yang, A. Welle, O. Trapp and P. A. Levkin, Angew. Chem., Int. Ed., 2015, 54, 8732–8735 CrossRef CAS PubMed.
- V. X. Truong, J. Bachmann, A. N. Unterreiner, J. P. Blinco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2022, 61, e202113076 CrossRef CAS PubMed.
- Z. Yu, Y. Pan, Z. Wang, J. Wang and Q. Lin, Angew. Chem., Int. Ed., 2012, 51, 10600–10604 CrossRef CAS PubMed.
- Y. Fu, K. Wu, G. Alachouzos, N. A. Simeth, T. Freese, M. Falkowski, W. Szymanski, H. Zhang and B. L. Feringa, Adv. Funct. Mater., 2023, 2306531 CrossRef CAS.
- Y. Fu, G. Alachouzos, N. A. Simeth, M. Di Donato, M. F. Hilbers, W. J. Buma, W. Szymanski and B. L. Feringa, Chem. Sci., 2023, 14, 7465–7474 RSC.
- Y. Fu, G. Alachouzos, N. A. Simeth, M. Di Donato, M. F. Hilbers, W. J. Buma, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2024, 63, e202319321 CrossRef CAS PubMed.
- T. Freese, B. Fridrich, S. Crespi, A. S. Lubbe, K. Barta and B. L. Feringa, Green Chem., 2022, 24, 3689–3696 RSC.
- J. G. H. Hermens, A. Jensma and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202112618 CrossRef CAS PubMed.
- J. G. H. Hermens, T. Freese, G. Alachouzos, M. L. Lepage, K. J. van den Berg, N. Elders and B. L. Feringa, Green Chem., 2022, 24, 9772–9780 RSC.
- M. L. Lepage, G. Alachouzos, J. G. H. Hermens, N. Elders, K. J. van den Berg and B. L. Feringa, J. Am. Chem. Soc., 2023, 145, 17211–17219 CrossRef CAS PubMed.
- B. L. Feringa, Recl. Trav. Chim. Pays-Bas, 1987, 106, 469–488 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01142b |
| This journal is © The Royal Society of Chemistry 2025 |