
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MoS2 quantum dot-decorated CNT networks as a sulfur host for enhanced electrochemical kinetics in advanced lithium–sulfur batteries†
Meng
Wei
 *ab,
Hanqing
Lu
a,
Zhen
Wang
a,
Baowen
Lu
a,
Pengtao
Wang
a,
Xinxin
Zhang
a,
Bingjie
Feng
a,
Yingjie
Xie
a,
Tao
Zhang
a,
Guanghui
Liu
a and
Song
Xu
*a
*ab,
Hanqing
Lu
a,
Zhen
Wang
a,
Baowen
Lu
a,
Pengtao
Wang
a,
Xinxin
Zhang
a,
Bingjie
Feng
a,
Yingjie
Xie
a,
Tao
Zhang
a,
Guanghui
Liu
a and
Song
Xu
*a
aSchool of Materials Science and Engineering, Zhengzhou University of Aeronautics, Zhengzhou 450046, China. E-mail: weimeng1005@zua.edu.cn; songxu@zua.edu.cn
bCollaborative Innovation Center of Aviation Economy Development, Henan Province, China
First published on 22nd October 2024
Abstract
The slow redox kinetics and shuttle effect of polysulfides severely obstruct the further development of lithium–sulfur (Li–S) batteries. Constructing sulfur host materials with high conductivity and catalytic capability is well acknowledged as an effective strategy for promoting polysulfide conversion. Herein, a well-designed MoS2 QDs-CNTs/S@Ni(OH)2 (labeled as MoS2 QDs-CNTs/S@NH) cathode was synthesized via a hydrothermal process, in which conductive polar MoS2 quantum dot-decorated carbon nanotube (CNT) networks coated with an ultrathin Ni(OH)2 layer acted as an efficient electrocatalyst. MoS2 QD nanoparticles with a high conductivity and catalytic nature can enhance the kinetics of polysulfide conversion, expedite Li2S nucleation, and decrease the reaction energy barrier. The thin outer Ni(OH)2 layer physically confines active sulfur and meanwhile provides abundant sites for adsorption and conversion of polysulfides. Benefiting from these merits, a battery using MoS2 QDs-CNTs/S@NH as the sulfur host cathode exhibits excellent electrochemical performances with rate capabilities of 953.7 mA h g−1 at 0.1C and 606.6 mA h g−1 at 2.0C. A prominent cycling stability of a 0.052% decay rate per cycle after 800 cycles is achieved even at 2C.
1 Introduction
Rapid global energy growth and a low-carbon economy have driven the vigorous development of higher energy-density devices.1,2 Almost every portable device, such as smart grids, portable electronics, and electric vehicles, that use electricity has benefited from the development of rechargeable lithium-ion batteries (LIBs).3,4 However, the most advanced commercial lithium-ion battery nearly reaches its theoretical limitation owing to the “Li+ intercalation” mechanism, which restricts its developing potential in competitive future application.5,6 In order to accomplish the goal of high energy density over 500 W h kg−1, it is an urgent need to explore new battery systems.7,8 In this case, lithium–sulfur (Li–S) batteries with a high theoretical energy density of 2600 W h kg−1, environmentally friendly nature and abundant reserves of sulfur have been regarded as the promising candidate for next generation batteries.9,10However, the application of Li–S batteries is still challenging. There are several technical problems that have not been solved, such as sluggish reaction kinetics, dielectric sulfur and its final product Li2S, soluble lithium polysulfides (LiPSs), and sulfur volume expansion.11,12 The most serious one is the dissolution of polysulfides and their shuttling effect (the so-called shuttle effect) caused by soluble lithium polysulfides (LiPSs), which can shuttle between the cathode and Li anode, resulting in the loss of active materials and low utilization of active sulfur.13,14 The dissolution of long-chain LiPSs not only increases the viscosity of the electrolyte, but also increases charge transfer resistance.15 Moreover, with the soluble LiPSs forming the nucleation barrier of solid Li2S2/Li2S, parts of them are lost from the electrode, leading to incapable transformation from liquid high-order lithium polysulfides (Li2Sn, 4 ≤ n ≤ 8) to solid lithium polysulfides (Li2Sn, 1 ≤ n ≤ 2) under high current density.16,17 Meanwhile, the converse transition of solid Li2S to dissolved LiPSs needs to overcome additional activation energy due to the aggregation of Li2S produced. Solid–liquid–solid phase transitions and their sluggish kinetic conversion problems sharply reduce the capacity, cycling life, and coulombic efficiency of Li–S batteries.18,19
Traditional physical confinement and chemisorption have been proven effective in dealing with the shuttle effect; however, they only alleviate the polysulfide shuttling to some extent and cannot solve the problem fundamentally.20,21 The root cause of the shuttle effect is the slow transformation of the liquid phase intermediate lithium polysulfide (Li2Sn, 4 ≤ n ≤ 8) to solid product Li2S2/Li2S, which continuously accumulates in the cathode and diffuses to the lithium anode driven by the concentration gradient and electric field.22,23 The electrochemical reaction kinetics can likely slow down inside the battery. Hence, a new perspective of electrocatalytic reaction kinetics that accelerates the sulfur “solid–liquid–solid” conversion process will be required.24–27
Enormous endeavors have been made to overcome this issue. Conductive materials (e.g., carbon-based materials,28–31 metal oxides,32,33 conductive polymers34) have been widely used as the host materials to construct a conductive network and weaken the dissolution of polysulfide. However, the low-polar carbon materials are limited to weak interactions with LiPSs and can only trap LiPSs by physical confinement and chemical adsorption, which cannot effectively suppress the shuttle effect and promote the conversion kinetics of the battery.35,36 In this case, polar metal compounds with strong adsorption capacity for LiPSs have been developed successively to promote ion adsorption and migration efficiently, such as VS2,37 TiS2,38 WS2,39 ReS2,40 TiO2,41 MnO2,42 VO2,43 VN,44,45 and so on. Among these, molybdenum disulfide (MoS2), with the change of lattice plane, high surface area and plentiful active sites, facilitates the strengthening of the overall absorption of Mo ions with negatively charged polysulfides.46 Meanwhile, MoS2 is a typical two-dimensional transition metal sulfide that forms zero-dimensional MoS2 quantum dots (MoS2 QDs) when the MoS2 QDs size is reduced to 10 nm and below. The advantages such as unsaturated bonds, strong binding energy, and abundant polar active sites are expected to greatly enhance the kinetics of polysulfides redox reactions.47
Herein, a novel architecture of MoS2 QDs-CNTs/S@NH was constructed to accommodate sulfur, followed by ultrathin Ni(OH)2 layer encapsulation. The MoS2 QDs-CNTs/S@NH cathode was prepared by a hydrothermal process, in which nanosized MoS2 QDs anchored on CNTs networks were used in both the sulfur host and multifunctional electrocatalysts to catalyze the LiPSs conversion. Benefiting from the synergy between conductive MoS2 QDs-CNTs framework and ultrathin Ni(OH)2 layer coating, the shuttle effect was effectively suppressed, and catalytic conversion of polysulfides showed a significant improvement. The MoS2 QDs-CNTs/S@NH-fabricated batteries with a high sulfur content of 70 wt% show excellent performance both in long cycling stability and rate capability. The MoS2 QDs-CNTs/S@NH cathode offers enhanced performance with an excellent capacity retention of 59.2% over 800 cycles at 2C. The improved electrochemical performance can be attributed to the design of the cathode, whose CNTs network provides fast electron/ion transfer and polar MoS2 QDs offering lots of active sites to adsorb polysulfides through chemical interactions. The thin Ni(OH)2 layer coats as a physically protective shield, not only confining active sulfur but also effectively inhibiting polysulfides shuttle through chemical interactions.
2 Experimental section
2.1 Materials
Molybdenum disulfide (Analytical pure, Shanghai Aladdin Reagent Co., Ltd.); nickel sulfate hexahydrate (AR, 99.9%), ammonium persulfate (AR, 98%) (all analytically pure, Shanghai Aladdin Biochemical Technology Co., Ltd.), ammonia solution (Analytically pure, Xi long Chemical Co., Ltd.), carbon nanotube multi-walled carbon nanotubes (ID: 5–10 nm, OD: 20–30 nm, length: 10–30 nm), sublimed sulfur, N-methyl pyrrolidone (NMP) (all analytically pure, Sinopharm Group Chemical Reagents Co., Ltd.); lithium sulfide, thioacetamide 1,3-dioxane (DOL), ethylene glycol dimethyl ether (DME) (Analytically pure, Shanghai Aladdin Reagent Co., Ltd.); LA133 Water system binder (Guangdong Candle New Energy Technology Co., Ltd.).2.2 Preparation of MoS2 QDs
Typically, methanol aqueous solution (40 vol%) and ethanol aqueous solution (45 vol%) were mixed in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. 100 mg of molybdenum disulfide powder was dispersed in 40 mL methanol/ethanol solution and ultrasonically treated at room temperature for 2 h. Then, the mixture was dispersed in an ultrasonic cell crusher for 2 h with low-temperature control. After centrifugation at 11000 rpm for 10 min 3 times, the MoS2 QDs were obtained and stored at 4 °C.
:1. 100 mg of molybdenum disulfide powder was dispersed in 40 mL methanol/ethanol solution and ultrasonically treated at room temperature for 2 h. Then, the mixture was dispersed in an ultrasonic cell crusher for 2 h with low-temperature control. After centrifugation at 11000 rpm for 10 min 3 times, the MoS2 QDs were obtained and stored at 4 °C.
2.3 Preparation of MoS2 QDs-CNTs
0.15 g of hydroxylated carbon nanotubes were dispersed in 50 mL distilled water and sonicated for 30 min. Subsequently, the molybdenum disulfide quantum dot (MoS2 QDs) solution was uniformly mixed into the CNTs dispersion and stirred for 45 min (mass ratio of 50:1 for CNTs to MoS2 QDs). The mixture was poured into a 100 mL hydrothermal reactor and reacted for 12 h at 180 °C. After cooling to room temperature, the resulting material is extracted and filtered, then washed several times with deionized water and ethanol to remove surface impurities. Finally, the experimental material was freeze-dried for 24 h to obtain a pure MoS2 QDs-CNTs sample.
2.4 Preparation of MoS2 QDs-CNTs/S
MoS2 QDs-CNTs/S composite material was prepared using a simple melt diffusion method. MoS2 QDs-CNTs and sublimed sulfur were mixed in a mass ratio of 3:7, and the mixture was ground for 40 min. Subsequently, the mixture was transferred to a stainless steel reactor and heated at 155 °C in an argon atmosphere for 12 h. After cooling to room temperature, the MoS2 QDs-CNTs/S composite material was obtained.
2.5 Preparation of MoS2 QDs-CNTs/S@NH
The Ni(OH)2 layer was coated on the surface of the MoS2 QDs-CNTs/S precursor using a surface chemical precipitation method. The mixed solution was formed by dispersing 4 mL nickel sulfate hexahydrate and 2.5 mL ammonium persulfate in 100 mL deionized water. Subsequently, 0.45 g MoS2 QDs-CNTs/S composite was dissolved in the mixed solution to form a stable solution. Then, 10 mL of concentrated ammonia solution was added, followed by stirring at room temperature for 30 min and standing for 20 min. The product was centrifuged at 5000 rpm for 10 min and washed several times. Finally, the MoS2 QDs-CNTs/S@NH material was obtained after drying in a vacuum oven at 60 °C for 12 h.2.6 Preparation of the MoS2 QDs-CNTs/S@NH cathode
The MoS2 QDs-CNTs/S@NH composite with conductive carbon black was mixed uniformly in a mass ratio of 8:1. Subsequently, the powder was added to the LA133 solution in a weight ratio of 9:1 and stirred for 12 h to prepare a homogeneous slurry. The prepared slurry was coated onto an aluminum foil with a thickness of 300 μm and subsequently dried in a vacuum drying oven for 12 h. After drying, the MoS2 QDs-CNTs/S@NH cathode was cut into disks with a diameter of 12 mm for assembly into CR2032 coin cells.
2.7 Material characterization
The crystallinity and structure of the samples were characterized using X-ray diffraction (XRD) with a Bruker D2 PHASER instrument. Scanning electron microscopy (SEM) utilizing the ZEISS Gemini 300 and transmission electron microscopy (TEM) employing the JEOL JEM-2100Plus were employed to characterize the morphology and structure of the samples. X-ray photoelectron spectroscopy (XPS) measurements were conducted to analyze the chemical states of elements and material composition. The sulfur content in the MoS2 QDs-CNTs/S@NH composite was characterized using a thermogravimetric analyzer (TG, TG 209F3). The concentration of Li2S6 in the solution was characterized using a UV-visible spectrophotometer (UV-vis, UV-5500PC), facilitating the analysis of lithium polysulfide adsorption properties in the samples.2.8 Electrochemical measurements
The MoS2 QDs-CNTs/S@NH composite, conductive superphosphate and binder LA133 were mixed in a ratio of 7:2:1 to form a uniform slurry. The slurry was cast onto an aluminum foil, vacuum-dried at 60 °C for 12 h and subsequently punched into discs with a diameter of 12 mm. The coin cells were assembled with MoS2 QDs-CNTs/S@NH as the cathode, Celgard 2500 as the separator, lithium foil as the anode and 1 M LiTFSI solution in a mixed solvent of 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME) in a volume ratio of 1:1 in a glove box filled with Ar (H2O, O2 < 0.1 ppm). The sulfur content of the whole cathode is approximately 70 wt%. The sulfur mass loading on the cathode ranges between 2 and 2.7 mg cm−2. Furthermore, the ratio of the cathode electrolyte to sulfur is 13 μL mg−1. The electrochemical performance of the system was characterized using the LAND testing system and CHI-604E electrochemical workstation. Cyclic voltammetry (CV) was tested at a scan rate of 0.1 mV s−1, and electrochemical impedance spectroscopy (EIS) was performanced by a frequency range of 100 kHz to 0.01 Hz.
3 Results and discussion
The synthetic process of the MoS2 QDs-CNTs/S@NH material is illustrated in Fig. 1. The MoS2 QDs-CNTs/S@NH shows an effective design with CNTs constructed as a conductor network and MoS2 QDs homogeneously and tightly anchored on the CNTs networks. The active sulfur is loaded on the surface of the CNTs network, and a thin Ni(OH)2 layer is coated outside as a protective shield.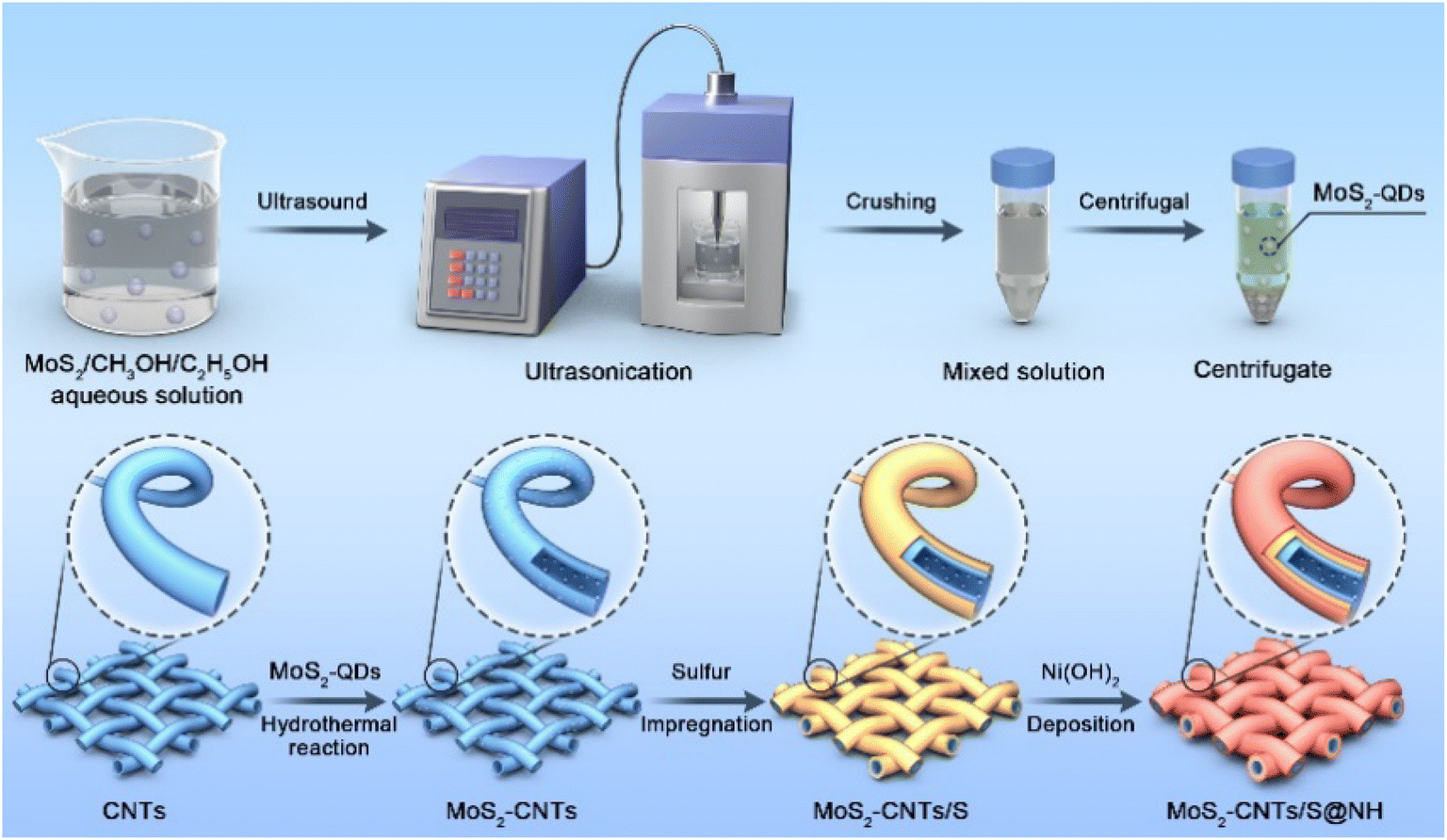 | ||
| Fig. 1 Schematic of the synthesis process of MoS2 QDs-CNTs/S@NH composites. | ||
Fig. 2(a)–(d) illustrates the morphological evolution process of the CNTs powder, MoS2 QDs-CNTs, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH, respectively. As shown in Fig. 2(a), the surface of naked carbon nanotubes (CNTs) is extremely smooth, with an outer diameter of approximately 30–40 nm+. Through the hydrothermal reaction process, MoS2 quantum dots (QDs) were successfully decorated onto the surface of carbon nanotubes, forming the MoS2 QDs-CNTs composite shown in Fig. 2(b). Compared to the bare carbon nanotubes, the MoS2 QDs-CNTs sample exhibits a relatively rough surface while preserving its initial one-dimensional structure. The MoS2 QDs-CNTs/S sample was prepared via the sulfur impregnation method, as shown in Fig. 2(c). The surface of MoS2 QDs-CNTs/S becomes slightly smoother than MoS2 QDs-CNTs, which can be attributed to the fact that MoS2 quantum dots (QDs) on the surface have been covered after sulfur loading. To further suppress the shuttle effect of polysulfides, the MoS2 QDs-CNTs/S composite was subsequently coated with a thin Ni(OH)2 layer. From Fig. 2(d) and the inset, a polar layer of Ni(OH)2 is clearly coated onto the surface of the MoS2 QDs-CNTs/S sample, achieving the successful construction of a core–shell structured MoS2 QDs-CNTs/S@NH cathode. Fig. 2(e) shows the high-resolution transmission electron microscope (HRTEM) image of MoS2 QDs-CNTs in which small MoS2 QDs (<5 nm) are evenly decorated on the surface of carbon nanotubes and some are encapsulated in narrow channels. The HRTEM is further proof of the coexistence of MoS2 QDs and CNTs. HRTEM images (Fig. 2(e)) show highly parallel and ordered lattice fringes, demonstrating the MoS2 QDs are well crystallized. The d-spacing of MoS2 QDs is 0.25 nm due to the (103) faces of MoS2 crystals. From the TEM image in Fig. 2(f) and the insertion, it is clear that the MoS2 QDs-CNTs nucleus is surrounded by a thin Ni(OH)2 shell. The high-magnification TEM image (Fig. 2(f)) confirms the nanosized MoS2 QDs decorated on the network structure of carbon nanotubes and coated by a thin Ni(OH)2 layer, which coincides with the SEM image in Fig. 2(d).
 | ||
| Fig. 2 SEM images of (a) CNTs, (b) MoS2 QDs-CNTs, (c) MoS2 QDs-CNTs/S, and (d) MoS2 QDs-CNTs/S@NH. (e) HRTEM image of MoS2 QDs-CNTs. (f and g) High-magnification TEM image of MoS2 QDs-CNTs/S@NH and the EDS mapping. | ||
The as-prepared MoS2 QDs-CNTs/S and its energy dispersive X-ray (EDX) analysis are also investigated (Fig. S1†). After coating with Ni(OH)2, the MoS2 QDs-CNTs/S@NH and corresponding EDS mappings are shown in Fig. 2(g). Fig. 2(g) depicts the C, Ni Mo, S and O elements detected in the MoS2 QDs-CNTs/S@NH composite material, which demonstrates a homogeneous distribution of elements (C, O, Mo, S) in composites and the presence of the Ni(OH)2 layer. The additional Ni(OH)2 layer not only prevents the migration of sulfur but also facilitates the polar chemical adsorption to immobilize the dissolved polysulfides.
X-ray diffraction (XRD) was performed to evaluate the purity and crystalline phases of the sample. To ensure the successful synthesis of MoS2 QDs-CNTs/S@NH, pure MoS2 QDs and Ni(OH)2 were both prepared and tested. As shown in Fig. 3(a), the diffraction peaks at 19.25°, 33.06° and 38.54° correspond to the (001), (100) and (101) planes of Ni(OH)2, confirming the successful preparation of nickel hydroxide.48 However, clear peaks related to molybdenum disulfide (MoS2) were not detected in the XRD spectrum of MoS2 QDs-CNTs. This can be attributed to the low mass ratio of MoS2 QDs as well as the incorporation of some MoS2 QDs into the lattice of carbon nanotubes without altering the crystal structure. The strong diffraction peaks of sublimed sulfur are detected in MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH composites, indicating the presence of a well-defined S8 crystal structure (S JCPDS 08-0247). While the Ni(OH)2 was coated, it was noted that no distinct diffraction peaks of Ni(OH)2 were observed in the MoS2 QDs-CNTs/S@NH sample, which could be attributed to either a low content of Ni(OH)2 or the overlapping of weak Ni(OH)2 peaks with the strong diffraction peaks of sulfur. The intensity of the sulfur peaks was relatively weaker in MoS2 QDs-CNTs/S@NH due to the Ni(OH)2 layer, which agrees with the Raman analysis. The sulfur content of the MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH samples was confirmed by thermogravimetric analysis (TGA). As shown in Fig. 3(b), a weight loss of 69.98% for the MoS2-CNTs/S sample can be observed when the temperature increases to 300 °C, which can be attributed to the evaporation of sulfur. For MoS2 QDs-CNTs/S@NH, approximately 69.16% sulfur loss was observed, slightly lower than that of MoS2 QDs-CNTs/S. This slight difference can be attributed to the decomposition of Ni(OH)2. Such low content of Ni(OH)2 is in accord with the fact that the diffraction peak of Ni(OH)2 was undetectable in the XRD analysis.
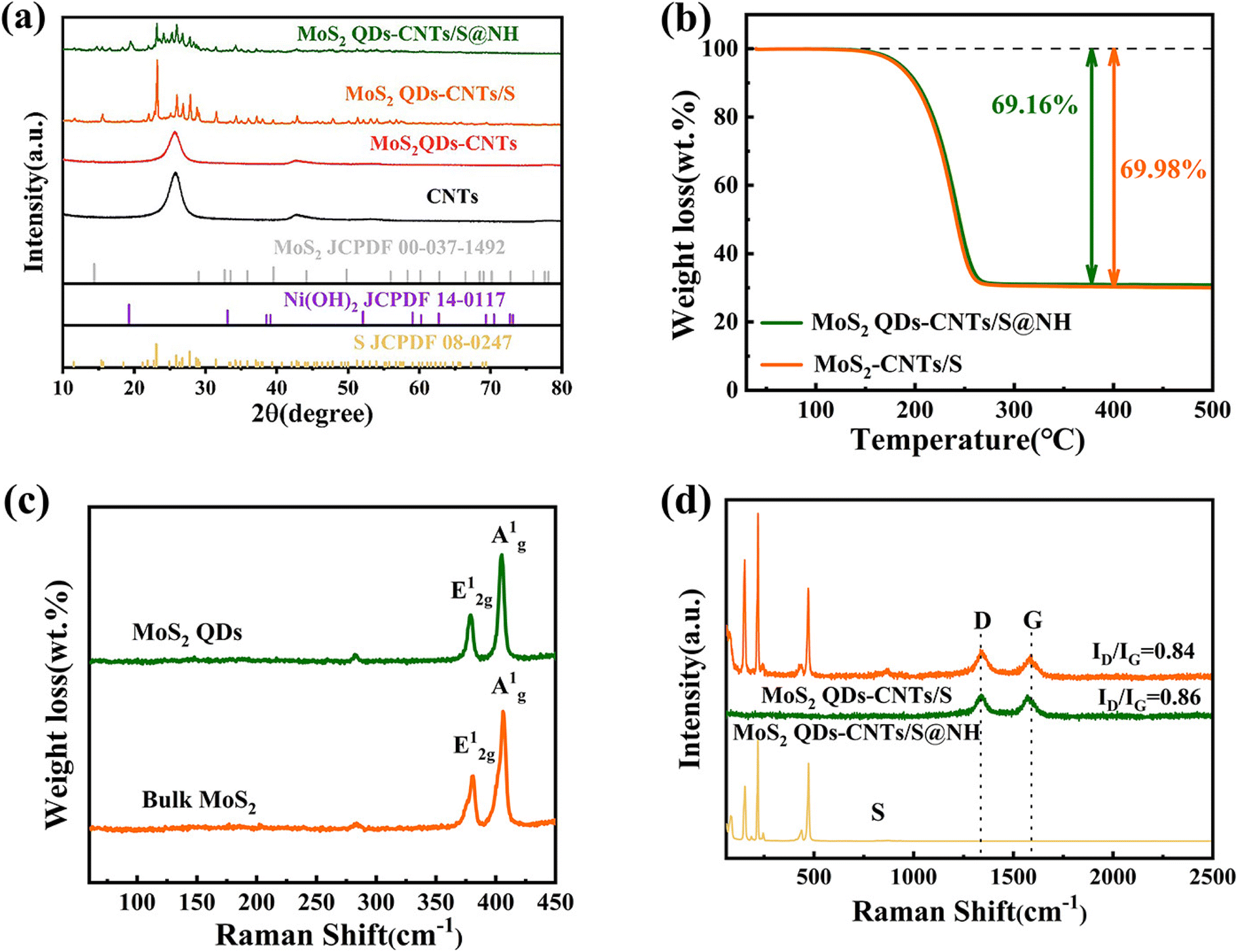 | ||
| Fig. 3 (a) XRD patterns of S, CNTs, MoS2 QDs-CNTs, Ni(OH)2, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH composite materials; (b) TG curves of MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH; (c) Raman spectra of MoS2 QDs and bulk MoS2. (d) Raman spectra of MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH. | ||
Raman spectroscopy was conducted to study the microstructure of bulk MoS2, MoS2 QDs, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH. In Fig. 3(c), the characteristic peaks Ag1 and E2g1 of MoS2 QDs were perfectly consistent with the peaks of bulk MoS2, which are located at 406.7 cm−1 and 381 cm−1.49,50 The Raman spectrum of MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH with two main modes, the D and the G band were located at 1337 cm−1 and 1573 cm−1, which reveals the disordered vibration of carbon atoms with defects and two-dimensional vibration in the hexagonal lattice of sp2-bonded carbon atoms (Fig. 3(d)). In general, the relative intensity ratio ID/IG is an indication of the carbon nanotube quality. The ID/IG value of MoS2 QDs-CNTs/S@NH (ID/IG 0.86) is smaller than that of MoS2 QDs-CNTs/S (ID/IG 0.84), indicating that combine with Ni(OH)2 does not affect the defects material after the sonication or hydrothermal process. The characteristic Raman peak of sulfur was observed on MoS2 QDs-CNTs/S, and three sharp peaks of the S/CNT/G film below 600 cm−1 could be assigned to the S–S bond in the composites. It is noteworthy that the Raman spectra of MoS2 QDs-CNTs/S@NH showed no sulfur peak, indicating that S8 molecules were well fixed in the Ni(OH)2 thin layer without significant molecular vibrations. This result further proves that the structure of Ni(OH)2 on the surface of sulfur can well limit the diffusion of sulfur, thereby improving the utilization rate and stability of sulfur.
X-ray photoelectron spectroscopy (XPS) was employed to further investigate the chemical composition and surface electric states of the elements in the MoS2 QDs-CNTs/S@NH composite. In the overall survey spectrum of Fig. 4(a), the presence of C, O, S, Mo and Ni identifies the existence of these elements in the composite. Fig. 4(b)–(f) display the high-resolution XPS spectra for C 1s, O 1s, Mo 3d, Ni 2p and S 2p of MoS2 QDs-CNTs/S@NH, respectively. As shown in Fig. 4(b), the C 1s spectrum can be deconvoluted into four peaks at 284.7, 285.59, 286.28 and 287.88 eV, which corresponded to the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O/C–S, C–O and O–CO bonds. The existence of S–C brings out the strong interactions between sulfur and the carbon matrix.51 In Fig. 4(c), the peak located at the binding energy of 531.38 eV in the O 1s spectrum can be ascribed to the formation of O–H bonds in Ni(OH)2. Fig. 4(d) displays the binding energies of Mo 3d5/2 and Mo 3d3/2 in the material at 227.78 eV and 232.08 eV, respectively. In Fig. 4(e), two prominent peaks of the Ni 2p XPS spectrum labeled as Ni 2p3/2 and Ni 2p1/2 are located at binding energies of 855.78 eV and 873.68 eV, and the satellite peaks are located at 861.48 eV and 879.38 eV. A change in the chemical valence of Ni indicates that polar Ni(OH)2 can serve as a medium or catalyst to transform polysulfides, which is beneficial for accelerating the reaction kinetics and enhancing the electrochemical performance. Fig. 4(f) displays the peaks of S 2p (corresponding to S 2p3/2 and S 2p1/2) located at 163.48 eV and 164.58 eV. The S 2p energy spectrum exhibits a high-energy peak at 168.48 eV, which can give credit to the formation of Ni–S chemical bonds, proving the highly efficient suppression of active sulfur by MoS2 QDs-CNTs/S@NH.
C, C–O/C–S, C–O and O–CO bonds. The existence of S–C brings out the strong interactions between sulfur and the carbon matrix.51 In Fig. 4(c), the peak located at the binding energy of 531.38 eV in the O 1s spectrum can be ascribed to the formation of O–H bonds in Ni(OH)2. Fig. 4(d) displays the binding energies of Mo 3d5/2 and Mo 3d3/2 in the material at 227.78 eV and 232.08 eV, respectively. In Fig. 4(e), two prominent peaks of the Ni 2p XPS spectrum labeled as Ni 2p3/2 and Ni 2p1/2 are located at binding energies of 855.78 eV and 873.68 eV, and the satellite peaks are located at 861.48 eV and 879.38 eV. A change in the chemical valence of Ni indicates that polar Ni(OH)2 can serve as a medium or catalyst to transform polysulfides, which is beneficial for accelerating the reaction kinetics and enhancing the electrochemical performance. Fig. 4(f) displays the peaks of S 2p (corresponding to S 2p3/2 and S 2p1/2) located at 163.48 eV and 164.58 eV. The S 2p energy spectrum exhibits a high-energy peak at 168.48 eV, which can give credit to the formation of Ni–S chemical bonds, proving the highly efficient suppression of active sulfur by MoS2 QDs-CNTs/S@NH.
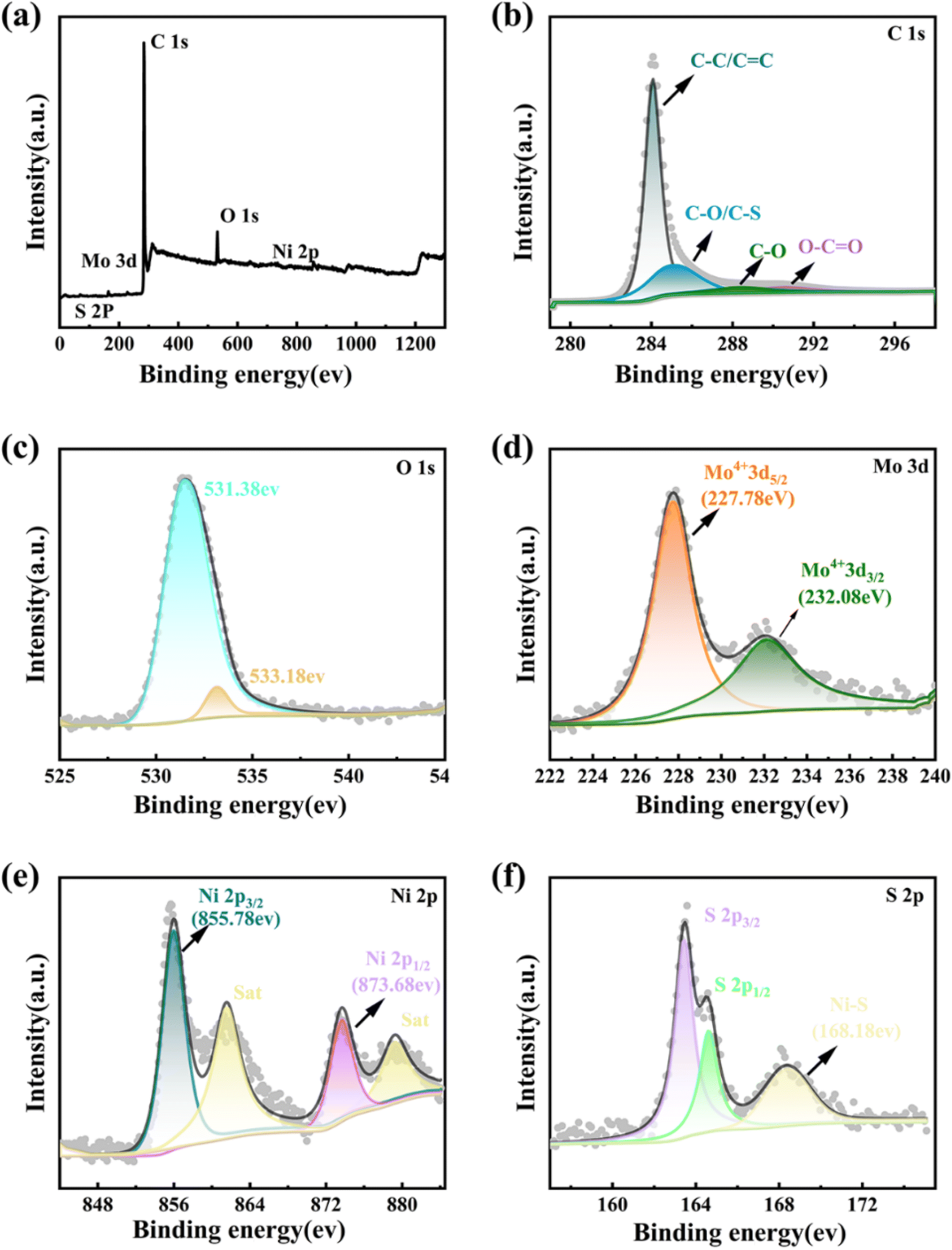 | ||
| Fig. 4 XPS spectra of the MoS2 QDs-CNTs/S@NH sample: (a) overall XPS spectrum, (b) C 1s, (c) O 1s, (d) Mo 3d, (e) Ni 2p, and (f) S 2p XPS spectra. | ||
In order to investigate the electrochemical improvements of the Li–S cells using CNTs/S, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH cathode, cyclic voltammetry (CV) tests were performed at a scan rate of 0.1 mV s−1. Fig. 5(a) illustrates the CV curves of cells using CNTs/S, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH as positive electrode materials and lithium foil as the negative electrode. For the three samples, one dominant anodic peak at around 2.45 V corresponds to the oxidation of Li2S to S8, and two reduction peaks were observed at around 2.29 V and 1.9 V during the discharge process, which correspond to the conversion of elemental sulfur (S8) into long-chain Li2Sx (4 < x < 8), followed by the further reduction of long-chain lithium polysulfides into short-chain lithium polysulfides, and ultimately to insoluble Li2S2 and Li2S.52 Comparatively, the reduction peak of MoS2 QDs-CNTs/S@NH shows a higher potential and the oxidation peak shows a lower potential, exhibiting the smallest potential difference between the reduction and oxidation peaks, which suggests the MoS2 QDs-CNTs/S@NH has a greater effect on promoting the kinetic reduction of long-chain LiPSs to Li2S2/Li2S and reducing the polarization of the Li–S battery. Meanwhile, the sharpness and intensity of the peaks are significantly higher, indicating faster LiPSs reaction kinetics and smaller electrochemical polarization (ΔV = 0.44 for MoS2 QDs-CNTs/S@NH; MoS2 QDs-CNTs/S ΔV = 0.448; CNTs/S ΔV = 0.526). These results are consistent with the previous results of Li2S6 adsorption experiments. Fig. 5(b) illustrates the cyclic voltammetry (CV) curves of MoS2 QDs-CNTs/S@NH for the first five cycles in the potential range of 1.5–3 V and a scanning rate of 0.1 mV s−1. It can be observed that the first five CV curves overlap well, and the oxidation/reduction peaks exhibit minimal changes, indicating the electrochemical reaction stability during the oxidation–reduction process.
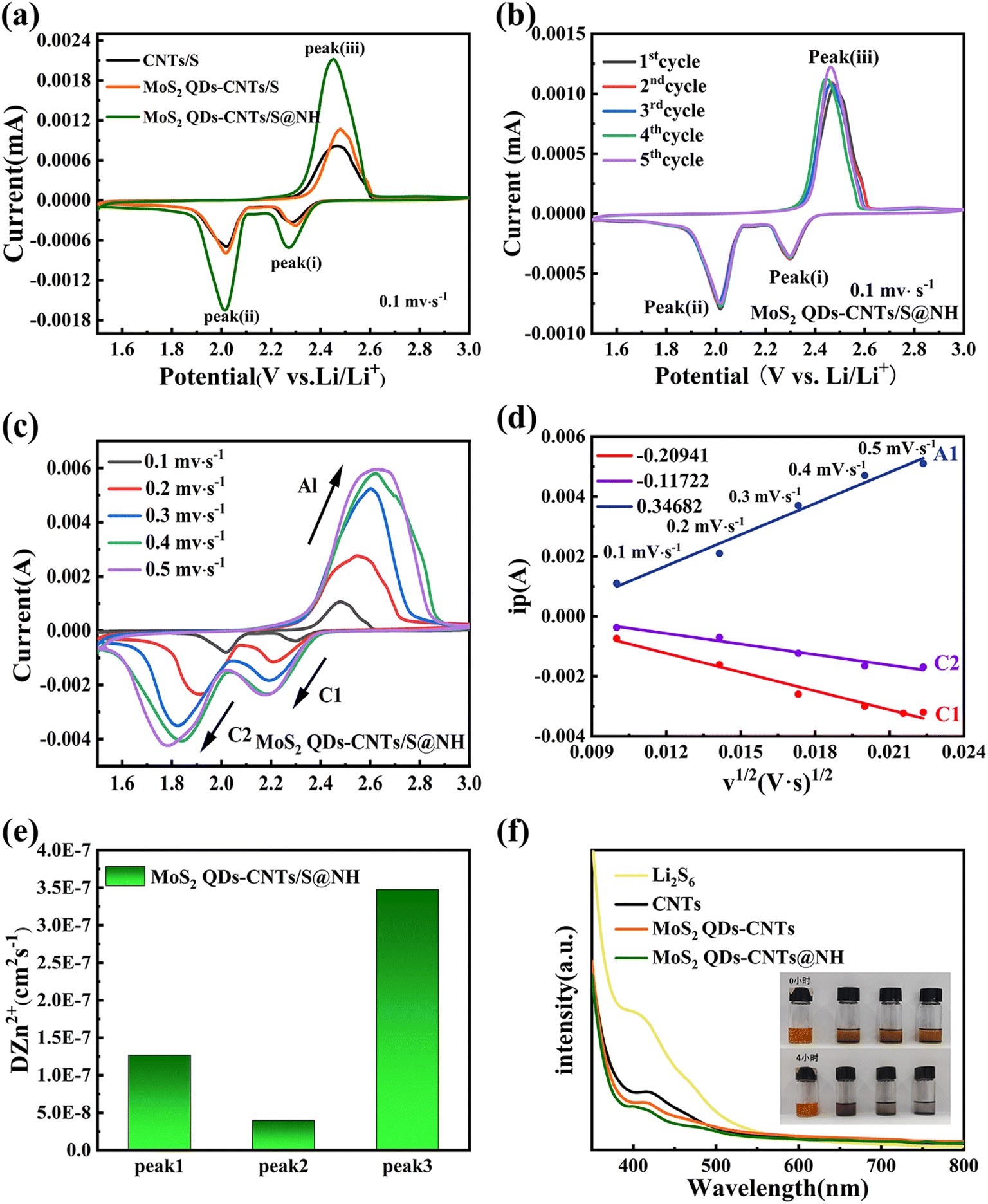 | ||
| Fig. 5 (a) CV curves of CNTs/S, MoS2 QDs-CNTs/S, and MoS2 QDs-CNTs/S@NH cathodes in the potential range of 1.5–3.0 V at 0.1 mV s−1; (b) CV curves at 0.1 mV s−1 for MoS2 QDs-CNTs/S@NH; (c) CV profiles at different scan rates of MoS2 QDs-CNTs/S@NH. (d) Plots of CV peak current vs. the square root of the scan rates for MoS2 QDs-CNTs/S@NH. (e) Impedance value of MoS2 QDs-CNTs/S@NH. (f) UV-vis spectra and variation in the color of Li2S6 solution adsorbed by CNTs/S, MoS2 QDs-CNTs/S, and MoS2 QDs-CNTs/S@NH for 4 h. | ||
To further investigate the importance of the acceleration of MoS2 QDs-CNTs/S@NH on Li+ diffusion, CV tests at different scan rates from 0.1 to 0.5 mV s−1 were performed. Fig. 5(c) and (d) presents the CV curves of MoS2 QDs-CNTs/S@NH at different scanning rates and the corresponding I–v1/2 fitting lines at each redox current peak. In Fig. 5(c), both the positive and negative electrode currents exhibit a more pronounced increasing trend for MoS2 QDs-CNTs/S@NH while the scan rate increases, proving the excellent catalytic activity of the MoS2 QDs-CNTs/S@NH structure due to the polar and catalytic MoS2 QDs decorated on CNTs and a thin Ni(OH)2 layer. As shown in Fig. 5(e), the peak current density of MoS2 QDs-CNTs/S@NH shows a good linear relationship with the square root of the scanning rate, illustrating that the reaction is a Li+ diffusion-controlled process. According to the Randles–Sevcik equation, the diffusion coefficient of lithium-ion (DLi+) could be calculated.53 The slopes of I–v1/2 of MoS2 QDs-CNTs/S@NH are 0.34682, −0.20941, −0.11722 for peaks A1, C1, C2, suggesting that the MoS2 QDs-CNTs/S@NH cathode has a much faster Li+ transport.
The CV results verify that the LiPSs have a high chance of being captured using Mo2S QDs-CNTs/S@NH as the sulfur host and high-efficiency catalyst. The Mo2S QD decorated CNTs network structure with a large surface area provides abundant surface active sites to achieve strong interaction with LiPSs. Meanwhile, Mo2S QDs take advantage of their full catalytic activity, which promotes the electron transfer of CNTs networks and then speeds up the conversion from LiPSs to insoluble Li2S. Moreover, a thin Ni(OH)2 layer coats as a physical barrier, not only restricting sulfur loss but also capturing LiPSs through strong chemical interactions. In addition, the Tafel plots of Mo2S QDs-CNTs/S@NH are also tested to demonstrate the enhanced conversion kinetics and electrocatalytic activity (Fig. S2†). The MoS2 QDs-CNTs/S@NH displays a much higher i0 than that of CNTs. The highest exchange current density is −3.647 mA cm−2 for the MoS2 QDs-CNTs/S@NH electrode. Hence, it turns out that the QDs-CNTs/S@NH can effectively accelerate the reversible electrochemical conversion toward LiPSs.
To further showcase this strong interaction between MoS2 QDs-CNTs/S@NHs and polysulfides, a visualized adsorption experiment in Li2S6 solution (0.01 M) was carried out. Fig. 5(f) displays the photograph of the electrolytes from the three types of samples. The electrolyte using CNTs and MoS2 QDs-CNTs cathode exhibited a pale-yellow color. In contrast, the electrolytes used with the MoS2 QDs-CNTs@NH cathode remain colorless within 2 h, which indicates that the Li2S6 in the electrolyte is almost absorbed by MoS2 QDs-CNTs@NH. The comparative experiments show that MoS2 QDs-CNTs@NH exhibits stronger adsorption capacity and good consistency than CNTs and MoS2-CNTs. This is because the MoS2 QDs-CNTs@NH can suppress LiPSs dissolution during charge/discharge cycling. As shown in Fig. 5(f), the pure Li2S6 solution has a significant adsorption peak at 400 nm, which can be attributed to the S62− species. The adsorption strength of UV-vis in the solution with MoS2 QDs-CNTs@NH was significantly reduced, which confirmed that the chemisorption capacity was greatly enhanced by MoS2 QDs and Ni(OH)2 coating.49
In order to further investigate the solid–liquid conversion and Li2S deposition kinetics of polysulfides at the electrolyte/electrode interface, the constant potential discharge curves of CNTs and MoS2 QDs CNTs surfaces were measured using Li2S6 as the active substance. As shown in the current time curve in Fig. 6, the dark areas near the X-axis and Y-axis represent the liquid–liquid reduction reactions of Li2S6 and Li2S8, respectively, while the light areas represent the liquid–solid conversion reactions of Li2S deposition. In Fig. 6(b), it is worth noting that the MoS2 QDs-CNTs have a higher Li2S deposition (364.7 mA h g−1) than that of CNTs (329.7 mA h g−1) (Fig. 6(a)). The results indicated that MoS2 QDs were beneficial in achieving efficient Li2S precipitation. The oxidative deposition of Li2S was further investigated by using a potentiostat, and the dissolved capacity of MoS2 QDs-CNTs was much higher than that of CNTs during charging at 2.05 V, revealing the effective oxidation of Li2S on the surface of MoS2 QDs-CNTs. The catalytic effect of MoS2 QDs was demonstrated by the results of the potentiostatic discharge curves of CNTs and MoS2 QDs-CNTs.
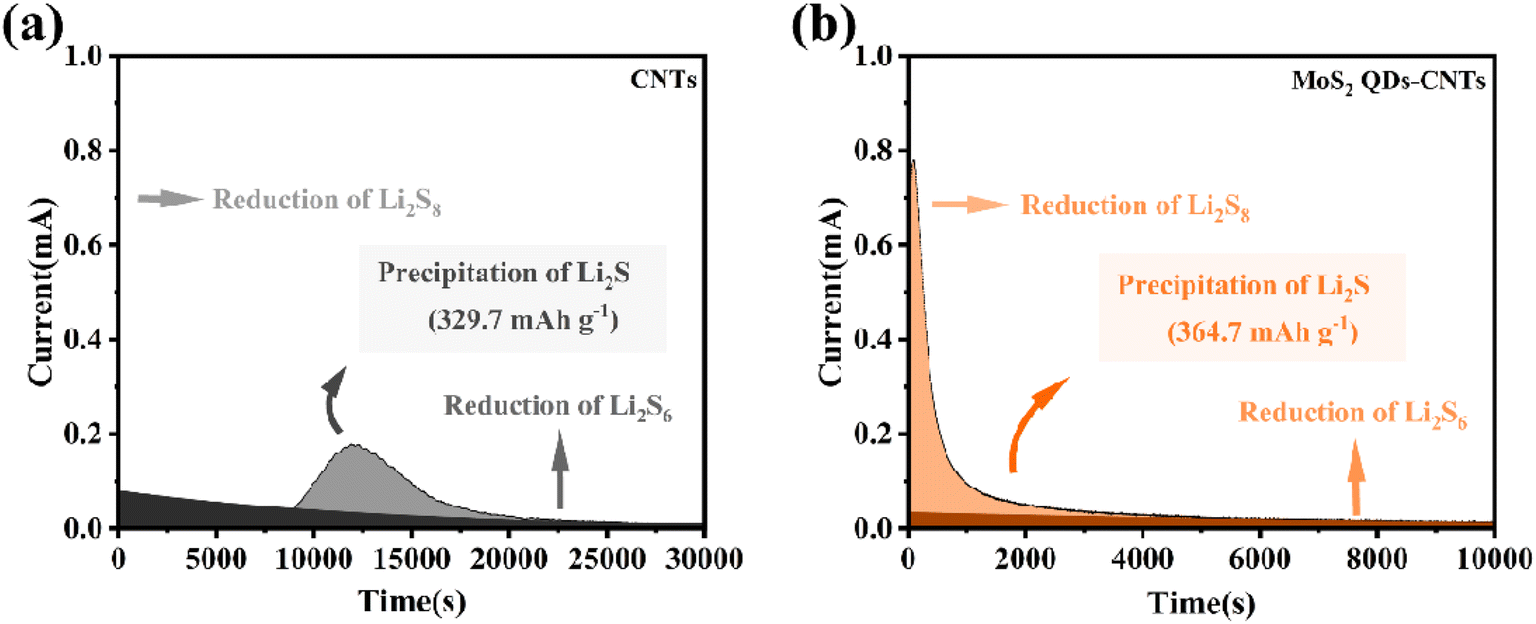 | ||
| Fig. 6 Potentiostatic discharge curves of CNTs and MoS2 QDs-CNTs. | ||
The cyclic performance of CNTs/S, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH at 0.5C is shown in Fig. 7(a). For MoS2 QDs-CNTs/S@NH, the initial discharge capacity of 1141.4 mA h g−1 and reversible capacity of 884.6 mA h g−1 are obtained after 200 cycles with 77.5% retention. In comparison, the initial discharge capacities of MoS2 QDs-CNTs/S and CNTs/S are 719.5 and 555.6 mA h g−1. After 200 cycles, their values decrease to 424.1 and 309.8 mA h g−1 (retention rates of 58.9% and 55.7%). The poor cycling stability of CNTs/S is attributed to the lack of an effective barrier for polysulfides and relatively slow sulfur redox reactions. Here, the improved cycling stability of MoS2 QDs-CNTs/S can be attributed to the designed structure. The CNTs network not only offers rapid electron/ion pathways but also buffers volume changes. The significant number of unsaturated bonds from MoS2 QDs efficiently anchor polysulfides, while the external Ni(OH)2 shell not only physically confines the active material but also acts as a barrier layer to suppress the diffusion of polysulfides. In addition, the coulombic efficiency of MoS2 QDs-CNTs/S@NH during cycling is approximately 99%, which is higher than that of MoS2 QDs-CNTs/S and CNTs/S.
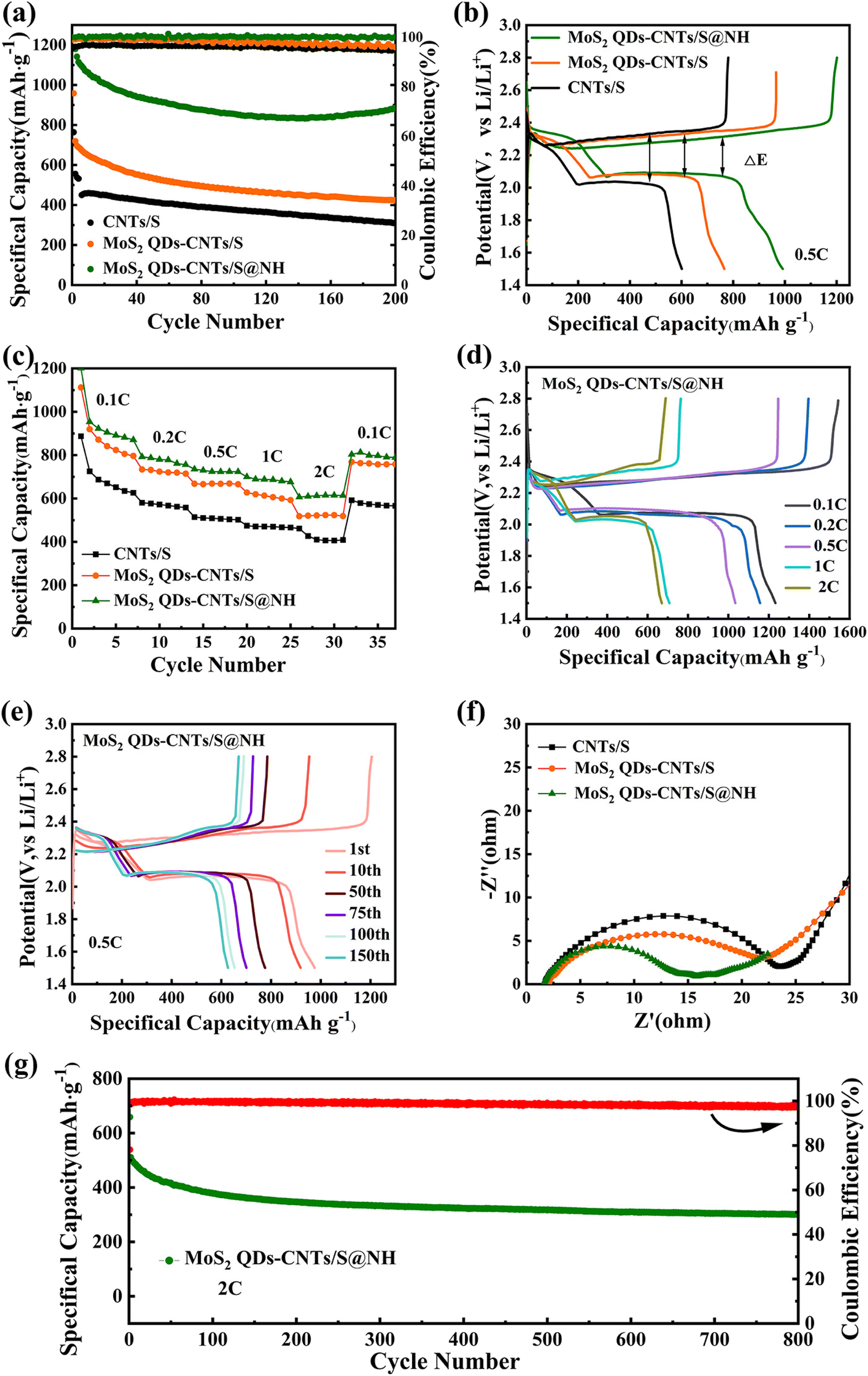 | ||
| Fig. 7 The electrochemical performance of CNTs/S, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH. (a) Cycling performance and coulombic efficiency at 0.5C during 200 cycles, (b) initial discharging/charging curves, and (c) rate capabilities of CNTs/S, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH. (d) Galvanostatic charge–discharge profiles at different rates. (e) Galvanostatic charge–discharge profiles at different cycles of MoS2 QDs-CNTs/S@NH. (f) Nyquist plots of CNTs/S, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH electrodes. (g) The long-term cycling performance of the MoS2 QDs-CNTs/S@NH cathode at 2.0C. | ||
Fig. 7(b) presents the initial galvanostatic discharge/charge curves of CNTs/S, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH at 0.2C. Clearly, MoS2 QDs-CNTs/S@NH exhibits the longest discharge/charge plateau among the three materials, while MoS2 QDs-CNTs/S displays a moderate but longer discharge/charge plateau compared to CNTs/S. These curves exhibit similar shapes at different current densities, clearly showing two distinct discharge voltage plateaus and one charge voltage plateau. In the discharge curves, the two plateaus at approximately 2.31 V and 2.04 V correspond to the two-stage reactions of sulfur to long-chain polysulfides and then to short-chain sulfides. In the charge–discharge plateaus, the two sloping charge plateaus correspond to the oxidation of Li2S/Li2S2 to L2S8/S, consistent with the analysis from the CV curves. Additionally, MoS2 QDs-CNTs/S@NH demonstrates a higher reduction peak potential and a lower oxidation peak potential (ΔE = 247.8 mV). As a result, the electrochemical kinetics in MoS2 QDs-CNTs/S@NH are greatly enhanced, and the polarization is significantly reduced. Due to the minimal polarization, during the extended discharge plateau, sulfur can be completely converted to insoluble Li2S2/Li2S, thus improving the utilization of active materials and achieving high specific capacity.
Fig. 7(c) displays the rate performance of CNTs/S, MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH cathode at different currents. For MoS2 QDs-CNTs/S@NH in Fig. 7(d), the reversible discharge/charge capacities provided by the MoS2 QDs-CNTs/S@NH cathode are 1061.1/1539.2, 893.7/1201.5, 768.1/1053.9, 697.9/103 and 614.4/941.1 at 0.1, 0.2, 0.5, 1.0 and 2.0C, respectively. When the current density returns to 0.1C, the discharge capacity of the MoS2 QDs-CNTs/S@NH recovers to 797.4 mA h g−1, demonstrating good reversible capacity. In contrast, both MoS2 QDs-CNTs/S and CNTs/S exhibit significantly lower capacities at the same current rate. Clearly, MoS2 QDs-CNTs/S@NH displays the best rate performance. The superior rate performance of MoS2 QDs-CNTs/S@NH can be primarily attributed to the dual suppression effect, and the dissolution and diffusion of polysulfides are dually suppressed by the internal MoS2 QDs and the external Ni(OH)2 shell.
For more information about the kinetics of charge transfer and ion diffusion of CNTs/S, MoS2 QDs-CNTs/S, and MoS2 QDs-CNTs/S@NH cathodes, electrochemical impedance spectroscopy (EIS) was performed. The corresponding results are displayed in Fig. 7(f). The intersection of the plots on the real axis represents the equivalent series resistance (Rs), which includes the electrolyte resistance, the contact resistance and the resistance of the electrode. All three EIS spectra consist of a single semicircle in the high-frequency region and an inclined line in the low-frequency region. The high-frequency semicircle represents the charge transfer resistance (Rct), and the diagonal line in the low-frequency region is attributed to the Warburg impedance caused by Li+ diffusion in the electrode. Part of the irreversible Li2S caused the capacity loss, so the Rs can represent the effectiveness of the redox reaction in the cathode. Clearly, CNTs/S exhibits a relatively high Rs value of 24 Ω, while MoS2 QDs-CNTs/S and MoS2 QDs-CNTs/S@NH demonstrate significantly lower Rs values. Thus, the charge transfer resistance (Rct) of MoS2 QDs-CNTs/S@NH (16 Ω) is lower than that of MoS2 QDs-CNTs/S (21 Ω) and CNTs/S (24 Ω). The minimum Rct implies the highest electronic conductivity and ionic migration rate between the SEI and electrolyte. These results indicate that constructing a unique core–shell structure of MoS2 QDs-CNTs/S@NH can accelerate charge transfer kinetics and inhibit the diffusion of polysulfides.
The long cycling performance of the MoS2 QDs-CNTs/S@NH cathode at high current densities was also investigated. As shown in Fig. 7(g), the MoS2 QDs-CNTs/S@NH cathode exhibits a capacity of 511.2 mA h g−1 and a reversible capacity of 302.2 mA h g−1 after 800 cycles at 2C. On average, it retains a capacity retention rate of 59.2%, and a negligible capacity decay of 0.051% per cycle. In addition, the average coulombic efficiency is higher than 97.6%, proving that the shuttling effect has been efficiently suppressed. The results indicate that the remarkable cycling stability of MoS2 QDs-CNTs/S@NH at high current densities benefit from the well-designed structures assembled by the number of MoS2 QD decorated on CNTs interwoven networks with ultrathin Ni(OH)2 coating. This structure can efficiently capture polysulfides through synergistic adsorption and catalysis, which ensures high electrode conductivity, accommodates volume changes, and suppresses shuttle effects. Nanosizing MoS2 is the most feasible method to increase its specific surface area and the number of active sites. Due to the fact that the active sites in MoS2 are mainly located at the edge sites, reasonable control of its morphology and size is necessary to obtain MoS2 nanomaterials rich in edge sites and maximize the exposure of active sites. Many studies have also demonstrated that MoS2 nanostructures exhibit good binding strength with polysulfides. As an activated catalyst, it not only promotes the redox kinetics of polysulfides but also facilitates the effective decomposition of lithium sulfide.
4 Conclusion
In summary, a new nanocomposite of MoS2 QDs-CNTs/S@NH was ingeniously designed as both a sulfur host and catalyst to enhance electrochemical kinetics and trapped LiPSs for Li–S batteries. Combining the advantage of polar and catalytic MoS2 QDs with the highly conductive CNTs network, the LiPSs would probably be adsorbed by a large number of unsaturated bonds of MoS2 QDs and physical adsorption of conductive CNTs. The thin Ni(OH)2 coating forms a protective layer, which not only prevents sulfur loss but also effectively captures LiPSs through chemical interactions. The Li–S battery containing MoS2 QDs-CNTs/S@NH as cathode shows outstanding long-term cycling stability and good rate performance. During 800 cycles at 2C, a capacity decay rate of 0.051% per cycle was obtained. This work provides a reasonable design of a sulfur host and shows its potential for application in Li–S batteries.Data availability
Data and research materials will be available on request. The data supporting this article have been included in the article and its ESI.†Author contributions
Meng Wei: conceptualization, writing – review & editing; Hanqing Lu: investigation; Zhen Wang: formal analysis; Baowen Lu, Pengtao Wang: resources; Xinxin Zhang, Bingjie Feng, Yingjie Xie: validation, Tao Zhang, Guanghui Liu, Song Xu: methodology.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Science and Technology Project of Henan Province (No. 232102241002); Aeronautical Science Foundation of China (No. 2022Z040055001); 2023 Henan University Research Teaching project; the Natural Science Foundation of Henan Province (222300420576); Graduate Education Innovation Program Fund of Zhengzhou University of Aeronautics (Grant No. 2023CX77).References
- C. Zhao, Y. Huang, B. Jiang, Z. Chen, X. Yu, X. Sun, H. Zhou, Y. Zhang and N. Zhang, Adv. Energy Mater., 2023, 14, 2302586 CrossRef.
- X. Kang, T. He, R. Zou, S. Niu, Y. Ma, F. Zhu and F. Ran, Small, 2023, 20, 2306503 CrossRef PubMed.
- P. Salimi, E. Venezia, S. Taghavi, S. Tieuli, L. Carbone, M. Prato, M. Signoretto, J. Qiu and R. Proietti Zaccaria, Energy Environ. Mater., 2023, 100316 Search PubMed.
- J. Li, Z. Wang, K. Shi, Y. Wu, W. Huang, Y. Min, Q. Liu and Z. Liang, Adv. Energy Mater., 2023, 2303546 Search PubMed.
- Y. Kong, L. Wang, M. Mamoor, B. Wang, G. Qu, Z. Jing, Y. Pang, F. Wang, X. Yang, D. Wang and L. Xu, Adv. Mater., 2023, 36, 2310143 CrossRef PubMed.
- X. Zhu, T. Bian, X. Song, M. Zheng, Z. Shen, Z. Liu, Z. Guo, J. He, Z. Zeng, F. Bai, L. Wen, S. Zhang, J. Lu and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202315087 Search PubMed.
- Z. Li, I. Sami, J. Yang, J. Li, R. V. Kumar and M. Chhowalla, Nat. Energy, 2023, 8(1), 84–93 CrossRef CAS.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5(4), 299–308 CrossRef CAS.
- Y. Yan, P. Zhang, Z. Qu, M. Tong, S. Zhao, Z. Li, M. Liu and Z. Lin, Nano Lett., 2020, 20(10), 7662–7669 CrossRef CAS PubMed.
- J. S. Yeon, S. Yun, J. M. Park and H. S. Park, ACS Nano, 2019, 13(5), 5163–5171 CrossRef CAS PubMed.
- M. Jana, R. Xu, X.-B. Cheng, J. S. Yeon, J. M. Park, J.-Q. Huang, Q. Zhang and H. S. Park, Energy Environ. Sci., 2020, 13(4), 1049–1075 RSC.
- S. Wang, Z. Wang, F. Chen, B. Peng, J. Xu, J. Li, Y. Lv, Q. Kang, A. Xia and L. Ma, Nano Res., 2023, 16(4), 4438–4467 CrossRef CAS.
- W. Zhang, H. Pan, N. Han, S. Feng, X. Zhang, W. Guo, P. Tan, S. Xie, Z. Zhou, Q. Ma, X. Guo, A. Vlad, M. Wübbenhorst, J. Luo and J. Fransaer, Adv. Energy Mater., 2023, 13(43), 2301551 CrossRef CAS.
- L. Ren, K. Sun, Y. Wang, A. Kumar, J. Liu, X. Lu, Y. Zhao, Q. Zhu, W. Liu, H. Xu and X. Sun, Adv. Mater., 2024, 36, 2310547 CrossRef CAS PubMed.
- P. R. Adhikari, E. Lee, L. Smith, J. Kim, S. Shi and W. Choi, RSC Adv., 2023, 13(14), 9402–9412 RSC.
- Z. Song, L. Wang, W. Jiang, M. Pei, B. Li, R. Mao, S. Liu, T. Zhang, X. Jian and F. Hu, Adv. Energy Mater., 2023, 14, 2302688 CrossRef.
- S. Liu, X. Xia, S. Deng, D. Xie, Z. Yao, L. Zhang, S. Zhang, X. Wang and J. Tu, Adv. Mater., 2018, 31(3), 1806470 CrossRef PubMed.
- M. S. Md Zaini, N. F. Anuar, S. A. M. Al-Junid and S. S. A. Syed-Hassan, Mater. Sci. Energy Technol., 2023, 6, 205–225 CAS.
- X. Meng, S. Chen, S. Hong, L. Zheng, X. Liu, G. Shi, C. W. Bielawski and J. Geng, Chem. Eng. J., 2024, 486, 150241 CrossRef CAS.
- X. Jiao, J. Hu, Y. Zuo, J. Qi, W. Yan and J. Zhang, Nano Energy, 2024, 119, 109078 CrossRef CAS.
- J. Qin, R. Wang, P. Xiao and D. Wang, Adv. Energy Mater., 2023, 13(26), 2300611 CrossRef CAS.
- Y. Huang, L. Zhang, J. Ji, C. Cai and Y. Fu, Energy Storage Mater., 2024, 64, 103065 CrossRef.
- B. Liu, H. Gu, J. F. Torres, Z. Yin and A. Tricoli, Energy Environ. Sci., 2024, 17, 1073–1082 RSC.
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu, H. Ji and L.-J. Wan, J. Am. Chem. Soc., 2019, 141(9), 3977–3985 CrossRef CAS PubMed.
- J. Billaud, F. Bouville, T. Magrini, C. Villevieille and A. R. Studart, Nat. Energy, 2016, 1(8), 1–6 Search PubMed.
- X. Meng, X. Liu, X. Fan, X. Chen, S. Chen, Y. Meng, M. Wang, J. Zhou, S. Hong, L. Zheng, G. Shi, C. W. Bielawski and J. Geng, Adv. Sci., 2021, 9(3), 2103773 CrossRef PubMed.
- J. Zhou, X. Chen, W. Gong, X. Meng, C. Chen, X. Zhou, M. Wang, K. N. Hui and J. Geng, J. Energy Storage, 2024, 75, 109505 CrossRef.
- X. Gu, S. Zhang and Y. Hou, Chin. J. Chem., 2016, 34(1), 13–31 CrossRef CAS.
- J. Castillo, A. Soria-Fernández, S. Rodriguez-Peña, J. Rikarte, A. Robles-Fernández, I. Aldalur, R. Cid, J. A. González-Marcos, J. Carrasco, M. Armand, A. Santiago and D. Carriazo, Adv. Energy Mater., 2023, 14(1), 2302378 CrossRef.
- J. K. Kim, Y. Choi, E. D. Jeong, S. J. Lee, H. G. Kim, J. M. Chung, J.-S. Kim, S.-Y. Lee and J.-S. Bae, Nanomaterials, 2022, 12(20), 3605 CrossRef CAS PubMed.
- Y. Chang, Y. Ren, L. Zhu, Y. Li, T. Li and B. Ren, Electrochim. Acta, 2022, 420, 140454 CrossRef CAS.
- X. Zhang, IOP Conf. Ser.: Earth Environ. Sci., 2021, 781(4), 042051 CrossRef.
- R. Mori, J. Solid State Electrochem., 2023, 27(4), 813–839 CrossRef CAS.
- F. Shi, L. Zhai, Q. Liu, J. Yu, S. P. Lau, B. Y. Xia and Z.-L. Xu, J. Energy Chem., 2023, 76, 127–145 CrossRef CAS.
- S. Zeng, G. M. Arumugam, X. Liu, Y. Yang, X. Li, H. Zhong, F. Guo and Y. Mai, Small, 2020, 16(39), 2001027 CrossRef CAS PubMed.
- T. T. Zhang, C. Y. Yang, J. Qu, W. Chang, Y. H. Liu, X. Z. Zhai, H. J. Liu, Z. G. Jiang and Z. Z. Yu, Chem.–Eur. J., 2022, 28(31), e202200363 CrossRef CAS PubMed.
- P. Zeng, Z. Zhou, B. Li, H. Yu, X. Zhou, G. Chen, B. Chang, M. Chen, H. Shu, J. Su and X. Wang, ACS Appl. Mater. Interfaces, 2022, 14(31), 35833–35843 CrossRef CAS PubMed.
- W. Zhao, L.-C. Xu, R. Li, Y. Guo, Z. Yang, R. Liu and X. Li, Mater. Today Commun., 2022, 30, 103196 CrossRef CAS.
- X. Liu, H. Rao, K. Sun, H. Gou, T. Lu and Y. Qian, Appl. Surf. Sci., 2022, 599, 154022 CrossRef CAS.
- J. Tang, C. Jin, L. Huo, S. Du, X. Xu, Y. Yan, K. Jiang, L. Shang, J. Zhang, Y. Li, Z. Hu and J. Chu, ACS Appl. Mater. Interfaces, 2022, 14(45), 50870–50879 CrossRef CAS PubMed.
- Y. Liu, T. Lei, Y. Li, W. Chen, Y. Hu, J. Huang, J. Chu, C. Yan, C. Wu and C. Yang, J. Power Sources, 2023, 556, 232501 CrossRef CAS.
- J. Zu, W. Jing, X. Dai, Z. Feng, J. Sun, Q. Tan, Y. Chen and Y. Liu, J. Alloys Compd., 2023, 933, 167767 CrossRef CAS.
- C. Zhao, Y. Zhou, T. Shi, H. Yin, C. Song, L. Qin, Z. Wang, H. Shao and K. Yu, J. Alloys Compd., 2023, 934, 167975 CrossRef CAS.
- Y. Lu, M. Zhao, Y. Yang, M. Zhang, N. Zhang, H. Yan, T. Peng, X. Liu and Y. Luo, Nanoscale Horiz., 2022, 7(5), 543–553 RSC.
- L. Sun, W. Gong, J. Zhou, J. Zhang, C. Chen, X. Meng, X. Han, H. Mai, C. W. Bielawski and J. Geng, J. Colloid Interface Sci., 2024, 653, 1694–1703 CrossRef CAS PubMed.
- G. Wen, X. Zhang, Y. Sui, K. Rao, J. Liu, S. Zhong and L. Wu, Chem. Eng. J., 2022, 430, 133041 CrossRef CAS.
- X. Yang, S. Chen, W. Gong, X. Meng, J. Ma, J. Zhang, L. Zheng, H. D. Abruña and J. Geng, Small, 2020, 16(48), 2004950 CrossRef CAS PubMed.
- F. Li, J. Li, Z. Cao, X. Lin, X. Li, Y. Fang, X. An, Y. Fu, J. Jin and R. Li, J. Mater. Chem. A, 2015, 3(43), 21772–21778 RSC.
- H. Wei, Y. Ding, H. Li, Q. Zhang, N. Hu, L. Wei and Z. Yang, Electrochim. Acta, 2019, 327, 134994 CrossRef CAS.
- Y. Xia, H. Zhong, R. Fang, C. Liang, Z. Xiao, H. Huang, Y. Gan, J. Zhang, X. Tao and W. Zhang, J. Power Sources, 2018, 378, 73–80 CrossRef CAS.
- B. Yu, A. Huang, D. Chen, K. Srinivas, X. Zhang, X. Wang, B. Wang, F. Ma, C. Liu, W. Zhang, J. He, Z. Wang and Y. Chen, Small, 2021, 17(23), 2100460 CrossRef CAS PubMed.
- H. Zhang, L. Yang, P. Zhang, C. Lu, D. Sha, B. Yan, W. He, M. Zhou, W. Zhang, L. Pan and Z. Sun, Adv. Mater., 2021, 33(21), 2008447 CrossRef CAS PubMed.
- H. Wang, S.-A. He, Z. Cui, C. Xu, J. Zhu, Q. Liu, G. He, W. Luo and R. Zou, Chem. Eng. J., 2021, 420, 129693 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00068d |
| This journal is © The Royal Society of Chemistry 2024 |