
Graphitic nanoflakes modulate the structure and binding of human amylin†
Alexa
Kamboukos
 a,
Billy J.
Williams-Noonan
ab,
Patrick
Charchar
a,
Irene
Yarovsky
a and
Nevena
Todorova
*a
a,
Billy J.
Williams-Noonan
ab,
Patrick
Charchar
a,
Irene
Yarovsky
a and
Nevena
Todorova
*a
aSchool of Engineering, RMIT University, Melbourne, Victoria 3001, Australia. E-mail: nevena.todorova@rmit.edu.au
bSchool of Science, RMIT University, Melbourne, Victoria 3001, Australia
First published on 26th August 2024
Abstract
Human amylin is an inherently disordered protein whose ability to form amyloid fibrils is linked to the onset of type II diabetes. Graphitic nanomaterials have potential in managing amyloid diseases as they can disrupt protein aggregation processes in biological settings, but optimising these materials to prevent fibrillation is challenging. Here, we employ bias-exchange molecular dynamics simulations to systematically study the structure and adsorption preferences of amylin on graphitic nanoflakes that vary in their physical dimensions and surface functionalisation. Our findings reveal that nanoflake size and surface oxidation both influence the structure and adsorption preferences of amylin. The purely hydrophobic substrate of pristine graphene (PG) nanoflakes encourages non-specific protein adsorption, leading to unrestricted lateral mobility once amylin adheres to the surface. Particularly on larger PG nanoflakes, this induces structural changes in amylin that may promote fibril formation, such as the loss of native helical content and an increase in β-sheet character. In contrast, oxidised graphene nanoflakes form hydrogen bonds between surface oxygen sites and amylin, and as such restricting protein mobility. Reduced graphene oxide (rGO) flakes, featuring lower amounts of surface oxidation, are amphiphilic and exhibit substantial regions of bare carbon which promote protein binding and reduced conformational flexibility, leading to conservation of the native structure of amylin. In comparison, graphene oxide (GO) nanoflakes, which are predominantly hydrophilic and have a high degree of surface oxidation, facilitate considerable protein structural variability, resulting in substantial contact area between the protein and GO, and subsequent protein unfolding. Our results indicate that tailoring the size, oxygen concentration and surface patterning of graphitic nanoflakes can lead to specific and robust protein binding, ultimately influencing the likelihood of fibril formation. These atomistic insights provide key design considerations for the development of graphitic nanoflakes that can modulate protein aggregation by sequestering protein monomers in the biological environment and inhibit conformational changes linked to amyloid fibril formation.
1. Introduction
Human islet amyloid polypeptide (amylin) is a 37-residue inherently disordered peptide (IDP) hormone that is co-secreted with insulin in pancreatic β-cells, with biological significance in blood glucose regulation. Under certain pathological conditions, the functional form of amylin is prone to abnormal unfolding and aggregation, leading to the formation of insoluble amyloid fibrils,1,2 a process linked to β-cell dysfunction and the onset of type II diabetes (T2D).3,4 Structural characterisation of the early and later stages of amyloid fibril formation is important for developing targeted strategies for the treatment and/or prevention of T2D and many other debilitating amyloid-related diseases.5,6 Extensive experimental and computational studies have aided in structurally characterising amylin in its native, aqueous state. The conformational preferences of amylin in solution show a predominant α-helix in the N-terminal residues 5–20, and a less structured C-terminus involving turns and coiled motifs.7–9 Amyloid fibril formation involves the self-association of misfolded amylin monomers, leading to β-sheet rich oligomers, protofibrils and finally mature fibrils. Due to the disordered nature of amylin, it is unclear which specific residues are most influential in amyloid formation, emphasising the need for further structural characterisations. While residues 20–29 are traditionally known to form β-sheet structures that template peptide–peptide association,10–12 other studies identify amyloidogenic regions with β-sheet propensity that spans across residues 8–20 and 30–37.13,14 Membrane insertion of amylin fibrils and intermediate oligomers is linked to membrane disruption and toxic effects due to pancreatic β-cell dysfunction and death.15,16 Currently, there are limited therapeutic strategies to prevent and/or reverse protein aggregation, prompting urgency for the design of therapeutic agents capable of intervening at different stages of the fibril formation pathway.In recent years, there has been a growing interest in exploring the influence of graphitic nanoparticles (NPs) within biological environments, particularly in their potential role in modulating amyloid diseases.17–19 Pristine graphene (PG) is a two-dimensional (2D) layer of covalently bonded aromatic sp2 carbon with excellent thermal stability, electrical conductivity, and mechanical strength, but is insoluble in aqueous media due to its high hydrophobicity. Surface modifications on PG can impart biorelevant attributes such as improved solubility, dispersibility, biocompatibility, and low cytotoxicity. Through surface oxidation, PG becomes a hydrophilic graphene derivative known as graphene oxide (GO). Compared to PG, the chemical heterogeneity of GO enhances its dispersibility and stability in biological environments, while the introduction of oxygenated functional groups facilitates selective hydrogen bonding with biomolecules. Subsequent chemical reduction of GO decreases the surface oxygen concentration to produce reduced graphene oxide (rGO), a more hydrophobic but still soluble GO derivative. The advantageous chemical, mechanical, and physical properties of GO and rGO make them prominent materials for the development of biomedical devices. Experimental evidence suggests that these graphitic NPs may be beneficial in modulating the process of amyloid fibril formation.20–22
Research efforts investigating the influence of carbon NPs on amylin aggregation have highlighted the potential of these emerging NPs to interfere with amyloid formation and act as therapeutic agents for managing T2D (see ref. 17 and references therein). Guo et al. showed that graphene and single-walled carbon nanotubes (SWCNTs) can impede the early stages of amylin aggregation via strong protein–substrate binding and preventing β-sheet formation, whereas highly curved fullerene (C60) NPs were found to inhibit β-sheet formation to a lesser extent.23 Others reported that GO can lower amylin aggregation and protect β-cells,24 graphene quantum dots (GQDs) can supress amylin fibrillation,25 and fluorine functionalised GQDs can inhibit fibril formation through stabilising the native structure of amylin to prevent the development of β-sheets.26 Despite these research advancements, few studies probe the influence GO has on amylin aggregation due to the infancy of its discovery.24,27,28 Improving the design of GO-based therapeutic agents requires careful consideration into how amylin aggregation is influenced by GO surface attributes (e.g., size, functionalisation, curvature and charge), as these properties are well-known to dictate the fibril-promoting or inhibiting action of NPs.29 Atomically resolved structures of amylin interacting with GO-based nanosurfaces are therefore necessary to assist the design of novel biomedical devices, with applications including drug delivery, biosensing, and therapeutics.30,31
Recent advancements in computational power have broadened the scope of atomistic simulation techniques capable of investigating the dynamic behaviour of proteins interfaced with (in)organic surfaces to support, rationalise, and complement experimental findings.32–34 Previous computational modelling studies investigating amylin in the presence of pristine carbonaceous NPs have provided insights into the structure and binding of amylin onto hydrophobic surfaces. Studies investigating the adsorption behaviour of short C-terminal amylin peptide fragments (NFGAILS) onto PG with sheet sizes of approximately 4–5 nm found amylin fragments lie flat on PG with all residues tightly adsorbed, while hydrophobic and aromatic stacking interactions with the surface drive the peptide adsorption.23,35 PG surface-mediated adsorption also promoted coiled peptide structures.23 Overall, PG bound amylin exhibited fewer β-rich structures when compared against in-solution structures, due to strong adsorption onto the surface, and this was also shown for SWCNTs.23 Molecular dynamics (MD) simulations of amylin dimers on C60 showed that dimers mostly adopt disordered coil structures with a low propensity of short β-sheet components. Helical regions were identified to span across residues 4–36, with residues 5–7 containing the highest helical propensity.36 Similar to amylin fragments adsorbed to PG,23,35 the presence of C60 facilitated aromatic stacking and hydrophobic interactions, as well as cation–π interactions between charged Arg11 and aromatic rings on C60.
Simulation studies investigating the binding of amylin to oxidised carbon and other hydrophilic NPs have also facilitated understanding into the influence of surface hydrophilicity on amylin structure and interactions. In a combined experimental and simulation study, GO of ∼3–4 nm in size was found to diminish amylin's α-helicity by approximately half of its initial helical content.24 Similarly, α-helical content reduction and associated coil increase was observed during the adsorption of amylin onto other types of oxidised carbon NPs.25,36–38 Compared to amylin in solution, β-sheet formation decreased when adsorbed onto functionalised carbon NPs,36–38 consistent with PG.23 The inclusion of oxygen containing functional groups on pristine carbon NPs introduces opportunities for hydrogen bonding and electrostatic interactions in addition to inherent aromatic stacking and hydrophobic carbon-based interactions.24,25,36–38 The accumulative effect of these interactions was shown by simulation to induce wrapping of monomeric amylin around 3.6 nm hydroxylated GDQs, leading to complete reduction of ordered protein secondary structure.25 Similarly, an MD study of amylin monomer/fibril adsorption onto citrate-capped gold (Au) NPs revealed distinct binding behaviours. The C-terminal adsorption onto bare Au regions partially unfolded the C-terminal α-helix, while N-terminal adsorption onto hydrophilic citrate regions stabilised the N-terminal helix through electrostatic interactions between charged residues and the anionic citrate layer.39
The above examples show that atomistic MD simulations can complement and explain experimental data by providing fundamental insights into the binding behaviour of amyloidogenic proteins on NPs. However, using computational modelling to accurately describe complex systems with many degrees of freedom in all-atom detail requires substantial statistical sampling of the potential energy landscape to correctly elucidate structural, kinetic, and thermodynamic information. In “brute force” or spontaneous molecular dynamics (SPON-MD) simulations, a common bottleneck is the prohibitive computational costs associated with adequately sampling conformational transitions across high energy barriers within reasonable timescales.40,41 It is therefore common in protein simulations, and especially for IDPs, to apply enhanced sampling techniques in order to overcome the energy barriers between states and accelerate conformational sampling.42 Variations of enhanced sampling algorithms capable of bypassing high energy barriers and accessing longer timescales are becoming increasingly present in the field. These techniques include, but are not limited to, umbrella sampling,43 replica exchange-molecular dynamics (REMD,44 or REST45/REST246) and metadynamics (MetaD).47,48 MetaD has been extensively applied to study peptide/surface binding phenomena, with its implementation to model bio–nano systems forecasted to steadily increase.49 In this approach, small repulsive Gaussian potentials are added along a predefined collective variable (CV) at defined time increments within the simulation. This technique reduces the dimensionality of the system to accelerate the transition between states. The history-dependent nature of the bias penalises the re-entry into previously sampled CV space (conformations). Within the MetaD variant methods, the bias-exchange (BE-MetaD)50 approach involves running replicate simulations at the same temperature on different CVs, with an optional neutral replica (no bias). BE-MetaD circumvents the high computational cost of needing to compute many CVs with standard MetaD. Studies have shown the superior performance of MetaD compared to SPON-MD and other sampling methods when modelling the conformational preferences of proteins in solution, including IDPs, and specifically, amylin.50–53 However, applications of MetaD to study the adsorption of amyloidogenic proteins such as amylin onto surfaces/interfaces have been limited.
In this work, we apply BE-MetaD simulations to study the adsorption and structure of monomeric amylin onto graphitic nanoflakes (NFs) varying in size (3, 5 and 7 nm) and degree of surface functionalisation (PG, rGO, GO). This research builds upon previous work by Peng et al.9 which benchmarked all-atom forcefields and conformational sampling methods for modelling monomeric amylin in-solution and found CHARMM22* forcefield provided results most aggregable with experimental findings. In another preceding study, Peng et al.54 modelled graphitic NFs varying in size and surface oxidation in aqueous medium to understand the influence of the NF chemical composition on its curvature, surface roughness and hydration properties in solution, mimicking the biological environment. Following the previous works,9,54 here we explore the interactions between amylin and the graphitic NFs varying in size and oxidation to provide mechanistic insights into tailoring graphitic NF design to optimise the binding preferences of amylin.
2. Methodology
2.1 System and simulation details
To explore the interaction of graphitic nanoflakes with amylin, we employed water-equilibrated nanoflake models from our previous work54 based on pristine graphene (PG) nanoflakes of sizes 3 × 3 nm2, 5 × 5 nm2, and 7 × 7 nm2. A symmetrical square shape for the graphene nanoflakes was used to simplify the geometry and facilitate direct size comparison. This nanoflake shape is commonly used in MD simulations to study the fundamental behaviours of the material and its interactions with biomolecules.24,54–56 Functional groups (hydroxyl, epoxy, and carboxyl) were randomly distributed on each PG surface to generate graphene oxide (GO) and reduced graphene oxide (rGO) nanoflake models, following the chemical composition described in experimental literature.57 In all models, unfunctionalised carbon atoms on the flake edges were capped with hydrogen atoms. The carbon-to-oxygen (C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O) ratio of GO and rGO was maintained at 5:1 and 10:1, respectively, to be consistent with experimental ratios.58,59 In total, we investigated nine graphitic NF models, labelled PG3, PG5, PG7, rGO3, rGO5, rGO7, GO3, GO5 and GO7, based on their surface size (3, 5 and 7 nm) and oxidation degree (PG, rGO and GO) (Fig. S1b†).
:O) ratio of GO and rGO was maintained at 5:1 and 10:1, respectively, to be consistent with experimental ratios.58,59 In total, we investigated nine graphitic NF models, labelled PG3, PG5, PG7, rGO3, rGO5, rGO7, GO3, GO5 and GO7, based on their surface size (3, 5 and 7 nm) and oxidation degree (PG, rGO and GO) (Fig. S1b†).
Using BIOVIA Materials Studio60 and Visual Molecular Dynamics (VMD)61 programs, each amylin–NF complex was constructed by adding an amylin monomer to the periodic simulation cell of the various water-equilibrated NF structures. The initial configuration of amylin was taken as the most preferred (sampled) conformation of amylin in solution, as determined in our previous study.9 This conformation comprises a predominant N-terminal α-helix and a disordered C-terminus involving turns and coiled motifs (Fig. S1a†). Amylin was placed at a minimum nearest atom distance of 0.8–1 nm from the basal plane of the NF surfaces, and randomly rotated to generate distinct starting orientations. The amylin–NF complexes were centred in periodic cubic unit-cells to allow at least 1.2 nm separation from the box edge. Each simulation box was solvated with ∼1 g cm−3 water, a 0.15 mol L−1 NaCl salt concentration to mimic physiological conditions, and additional Na+ and Cl− counterions to achieve charge neutrality. The net charge of amylin and the graphitic NFs was consistent with our preceding studies.9,54 The charge of amylin was +2, and the partial atomic charges of GO were determined using the QEq algorithm62 based on the experimental zeta-potential of GO at neutral pH (−44 mV),63 resulting in a total surface charge of −0.04e nm−2. The rGO NFs were modelled as neutral particles due to their significantly lower zeta-potential and carbon-to-oxygen ratio compared to GO,63 and the need for the NF to have a whole charge value for system charge neutralization. The PG NFs were also modelled as neutral particles for similar reasons. A total of nine amylin–NF complexes were generated.
Simulations were conducted using the GROMACS (2020.3)64,65 software and the PLUMED v266 plugin. The CHARMM22* forcefield, CGenFF67,68 parameters for the graphitic NFs, and TIP3P water,69 were used throughout as previously benchmarked for modelling amylin and graphitic NF structures in solution.9,54 The Verlet cutoff scheme was used for neighbour searching as well as the particle mesh Ewald (PME) summation70 for calculating long-range electrostatic interactions. PME order was set to 4 and a grid FFT spacing of 0.16. van der Waals and short-range electrostatics were cut-off at a value of 1.4 nm. Constant temperature of 300 K was achieved by using the v-rescale method using τT = 0.1 ps.71 Constant pressure was achieved by coupling the system to a Parrinello–Rahman barostat72 with an isotropic pressure treatment, compressibility of 4.5 × 10−5, coupling constant of 2.0 ps and reference pressure of 1.0 bar. The LINCS algorithm73 was applied to constrain all bond lengths to their respective equilibrium values, allowing for a simulation timestep of 2 fs. Prior to molecular dynamics, each complex was subjected to energy minimisation using the steepest descent algorithm. Initially, the peptide and the nanoflake were position restrained during a 2 ns solvent equilibration at constant temperature and volume (NVT ensemble). Position restraints were then removed to allow the protein and nanoflakes to freely diffuse in the simulation cell and the amylin–NF systems were subjected to well-tempered BE-MetaD50,74 at a constant temperature and pressure (NPT ensemble).
Three collective variables were used in the BE-MetaD simulations to bias the amylin–NF complexes. These CVs included: amylin radius of gyration (CV1); amylin-to-NF centre of mass (COM) distance (CV2); and the orientation angle of amylin relative to the NF, defined as the angle between the COMs of the NF, amylin N-terminal residue, and C-terminal residue (CV3) (Fig. S2†). The final replicate was unbiased and used for all analysis. The CVs were chosen to account for the conformation of amylin (CV1), the binding pathway between amylin and the NF (CV2), and the binding orientation (CV3). A Gaussian hill height of 1 kJ mol−1 was deposited every 2500 timesteps, with a bias factor of 40. An exchange between replicates was attempted every 2.5 ps. Full parameters of Gaussian width for CV1–3 are described in ESI (Table S1†).
Variations in the system free energy were calculated using a systematic approach to establish convergence (Fig. S6†). This approach involved extracting the free energy surface (FES) for each CV for increasing increments of 100 timesteps by integrating hill height using the sum_hills function in PLUMED. Only low-energy structures from the FES with an energy below 3 kT from the lowest energy state (minimum) were considered to be biologically relevant, representing approximately 95% of the observed phase space in an unbiased distribution. Full details describing convergence results are provided in the ESI.†
Data analysis was performed on the same length of simulation time for each of the amylin–NF complexes to maintain consistency across all systems. The final 650 ns of the unbiased BE-MetaD simulations was used for analysis, with structures outputted at a frequency of 1 frame every 4 ps, producing a total of 5.85 μs of BE-MetaD simulation data across all systems.
Comparative spontaneous MD simulations (SPON-MD) were also conducted to evaluate the sampling efficiency of the BE-MetaD method for modelling protein–nanoparticle complexes. The SPON-MD simulations were performed using identical starting amylin–NF complexes as the BE-MetaD simulations, along with additional amylin–NF starting structures that feature alternative initial orientations of amylin relative to the NF. The two sets of independent SPON-MD simulations were combined to produce an ensemble trajectory (2 μs) for each amylin–NF system. Further details of the SPON-MD simulation approach and results are presented in the ESI.†
2.2 Analysis details
To investigate the NF surface-adsorbed behaviour of amylin, we filtered the total ensemble of simulated frames into either adsorbed or desorbed states based on the geometric criterion of any amylin atom being within 0.4 nm of any NF atom. This distance cut-off is in line with previous modelling studies that developed ways to classify the adsorbed states of proteins on surfaces.75–77 Analysis was performed using standard GROMACS, VMD, and PLUMED analysis tools on all recorded adsorbed states, and separately, on the most frequently sampled (low energy) states. This approach enabled a more comprehensive understanding of the binding mechanisms and interactions of amylin with NFs of varying size and degree of oxidation. The conformational variance of surface-adsorbed amylin was investigated using RMSD cluster analysis. The contact probability of amylin on each NF atom was used to generate contact maps illustrating the probability of each NF atom being in contact with amylin. Two-dimensional free energy maps based on the amylin radius of gyration and amylin–NF contact area were created to understand the conformational free energy landscape of amylin adsorbed onto the graphitic NFs. RMSD cluster analysis and secondary structure analysis was performed on the identified low energy conformations (hotspot regions) in the 2D free energy maps. The driving forces of amylin adsorption onto the graphitic NFs were examined by calculating the minimum distance of individual protein residues to the NF atoms as well as analysing hydrogen bond formation between amylin and the oxidised NFs. Further analysis details are outlined in the ESI.†3. Results and discussion
3.1 Amylin–PG
The PG NFs simulations offer valuable insights into the structure and binding of amylin on hydrophobic graphitic surfaces. We utilise backbone RMSD cluster analysis to categorise the amylin–NF structures sampled in the simulations, grouping similar surface-bound amylin conformations and separating distinct conformations into different clusters. The results reveal a high degree of conformational diversity in amylin upon adsorption (less than 0.4 nm) onto PG NFs, as indicated by the large number of low population clusters shown in Fig. 1a. This conformational diversity is more pronounced as NF size increases, suggesting backbone fluctuations increase in the presence of larger PG particles. We also generated 2D contact probability maps to determine the location of amylin when adsorbed onto the NFs. The contact maps demonstrate that the likelihood of surface-bound amylin being anchored to a single location on PG decreases as the NF size increases, supporting the trend that amylin is more conformationally dynamic when adsorbed onto larger PG NFs (Fig. 1b). Amylin is found to preferentially interact with the central planar regions of the NFs and tends to avoid the hydrogen-terminated edges and corners of the PG flakes. This may be due to the increased NF curvature of the graphene edges restricting binding compared to the planar central NF region.54,78 In the time-evolved SPON-MD simulations of the amylin–PG systems, there is no distinct binding location of amylin on the NFs, further indicating the protein remains dynamic upon adsorption (Fig. S9†). These findings suggest that PG surfaces encourage non-specific protein adsorption, leading to high protein mobility and lateral diffusion, which is most significant on the larger PG surfaces. In the context of fibrillation, surfaces that induce considerable translational mobility have been suggested to facilitate the assembly of monomers into aggregates by allowing the peptides to diffuse along the surface and attach to fibril ends.79–81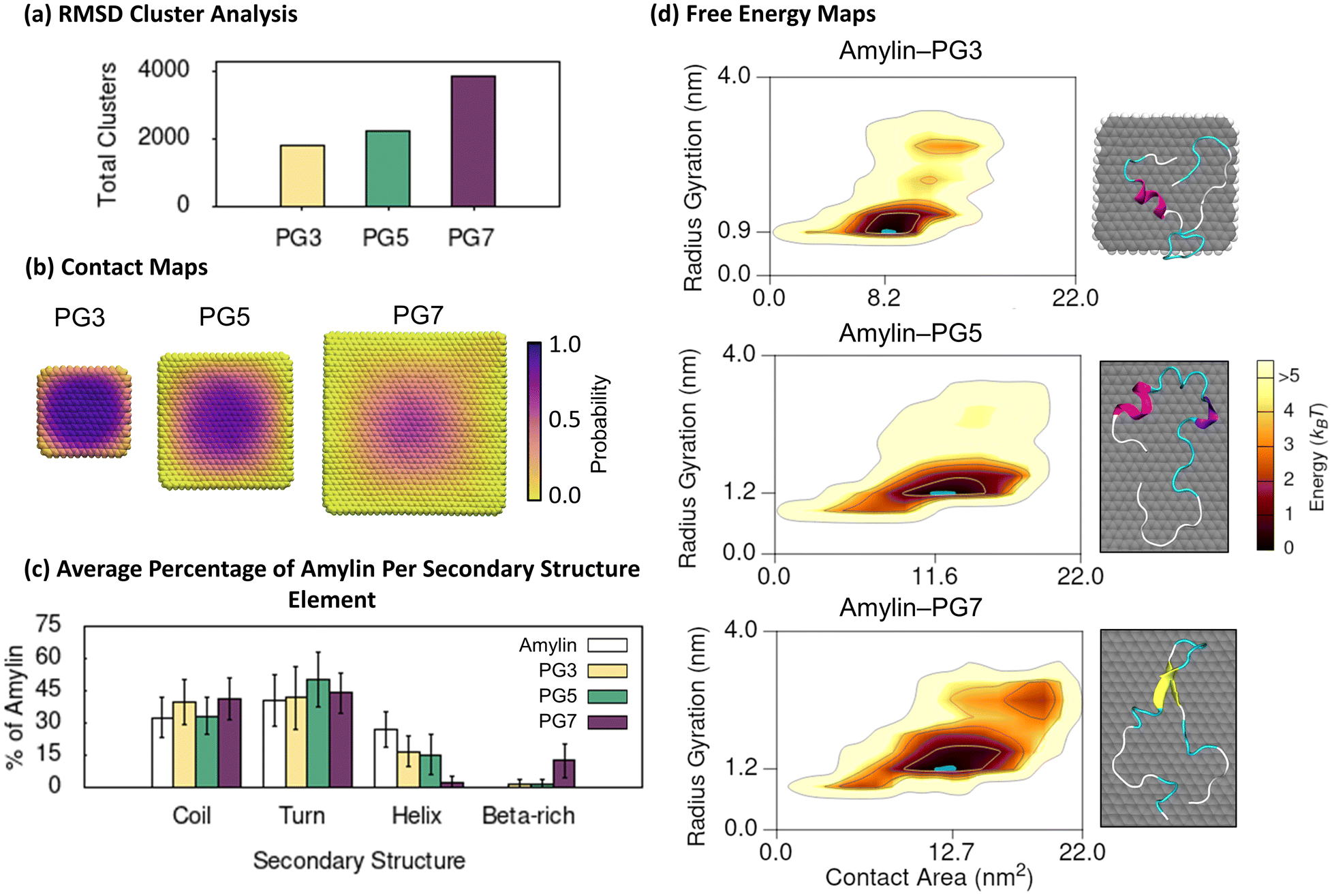 | ||
| Fig. 1 Amylin conformational preferences and contacts with pristine graphene. (a) Total number of unique surface-bound amylin conformations identified by RMSD backbone cluster analysis from the amylin–PG simulations. (b) Contact probability maps showing the relative proportion of simulation time each PG atom maintains close association (less than 0.4 nm) with amylin. The probability values are coloured with a perceptually linear, sequential colour scale to identify the dynamics of amylin adsorption and persistent binding locations. (c) Average percentage of amylin adopting different type of secondary structure within the hotspot regions with errorbars representing standard deviation. The average secondary structure of amylin in-solution9 is provided in white bars for reference. (d) Amylin–PG free energy maps showing the relationship between amylin radius gyration and amylin–NF contact area. The blue highlight corresponds to the lowest energy hotspot region. Inset images illustrate representative structures from the given hotspots, as determined by RMSD backbone cluster analysis, and are coloured based on their typical secondary structure features: coils (white), turns (cyan), α-helix (magenta), 310-helix (violet), extended conformation (yellow), and isolated bridge (orange). Solvent hidden for clarity. | ||
To explore the relationship between protein shape/compactness (amylin radius of gyration) and the extent of protein adsorption onto the PG NFs (amylin–NF contact area), we utilise 2D free energy maps. The maps show that irrespective of NF size, amylin adsorption onto PG is characterised by a single high-density ‘hotspot’ (containing low energy amylin conformations) (Fig. 1d). On larger NFs, the amylin radius of gyration and amylin–NF contact area both increased, suggesting stronger protein–NF interactions and more conformational restructuring.
Secondary structure analysis of amylin's low energy states demonstrates adsorption onto PG leads to a loss of native helical content and the emergence of β-like character (Fig. 1c). Relative to the solution state,9 the number of helical residues in PG-adsorbed amylin significantly decreases as the particle size increases. This reduction is ∼40% for both PG3 and PG5, and ∼90% for PG7. The low energy conformations of amylin–PG5 and amylin–PG7 demonstrate adsorption onto larger PG substrates facilitates protein unfolding and extension over the surface (Fig. 1d and 2d). In contrast, on the smaller PG3 flake, amylin adsorption and structural extension are less prominent (Fig. 1d and 2c). These findings are qualitatively in agreement with previous studies that report helical disruption or absence following the adsorption of amylin onto graphitic NPs, such as PG23,35 and C60,36 as well as other studies that show a correlation between graphene size increase and protein unfolding (or helix reduction).82,83 While amylin on PG3 and PG5 NFs features negligible isolated β-bridge structures (Fig. 1c and S15†), amylin on PG7 exhibits extended β-sheet conformations formed across C-terminal residues 15–30, with a turn present between residues 20–24 (Fig. 1 and S15†). Collectively, the results of RMSD clustering and secondary structure analysis (Fig. 1a and c) indicate that adsorbed amylin undergoes greater backbone fluctuations and conformational changes on the larger PG7 surface, including complete loss of helical elements and the formation of β-sheet structures. In contrast, when amylin is adsorbed on PG5, only partial helical loss is observed, and there are no residues in β-sheet conformation. The different conformational preferences may be related to the variation in size and curvature of the PG NFs, whereby the larger size and increased curvature of PG754 compared to PG5 helps facilitate a more substantive surface diffusion and conformational changes of amylin. Similar β-rich conformations induced by PG7 NF, termed ‘β-hairpins’, have been extensively observed during simulations of full-length amylin in solution,8,9,84–87 and are proposed to accelerate fibril formation via templating of the flat β-hairpin segments. As such, we postulate that the increased β-rich conformations induced by PG7 NF may indicate a propensity for larger PG flakes to nucleate fibril formation. This is in line with previous experiments and simulations that demonstrated the presence of graphene or graphite can both stabilise and promote β-sheet structures in other peptides.77,88–90 Recently, it was also demonstrated that β-sheet-featuring amyloid forms on surfaces when proteins with α-helices unfold following interfacial adhesion.91
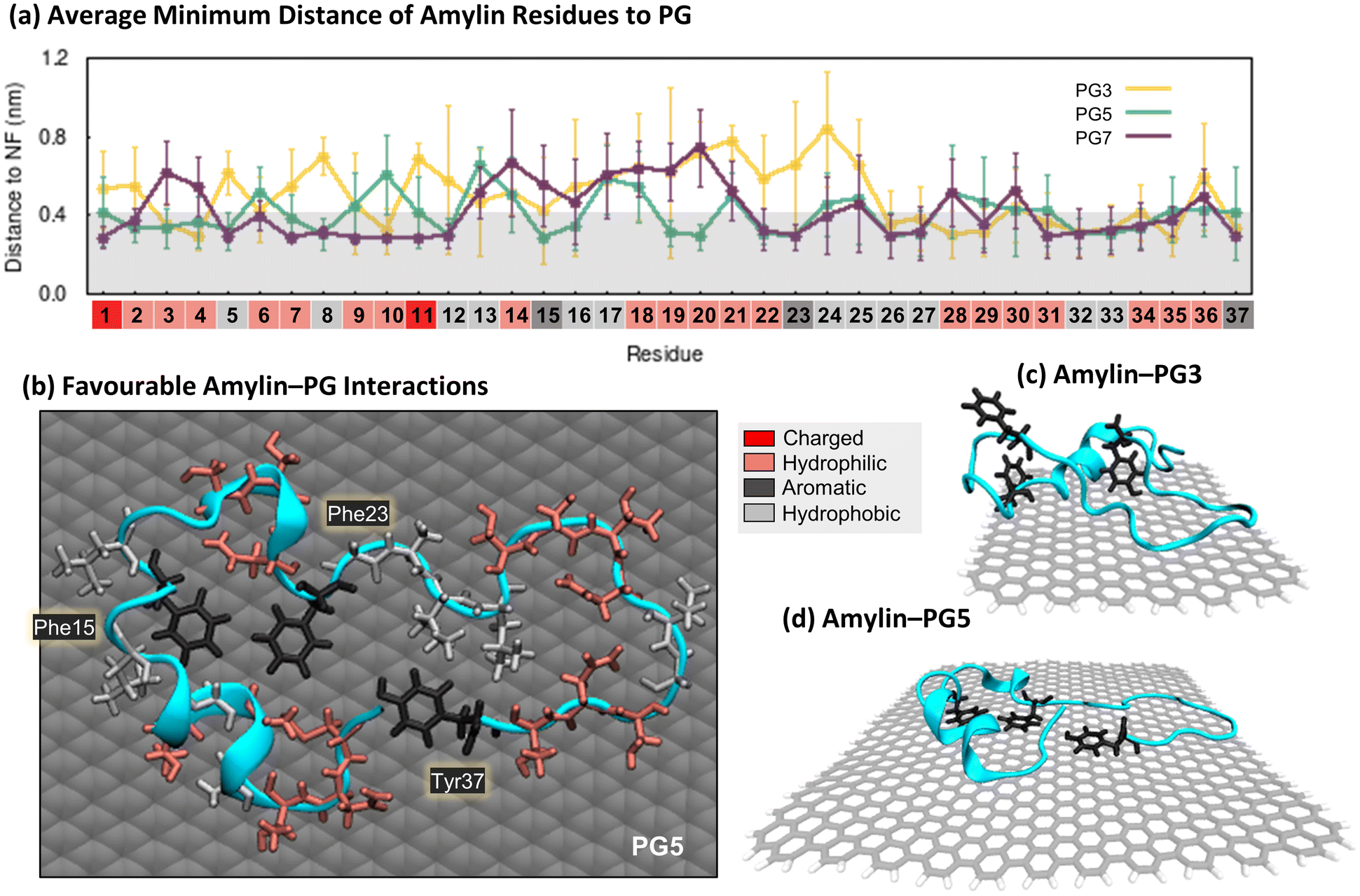 | ||
| Fig. 2 Specific interactions between amylin and pristine graphene. (a) Average minimum distances measured between individual amylin residues and the PG3, PG5, and PG7 surfaces. Calculated on the lowest energy states of each system (shown in Fig. 1), with errorbars representing standard deviation. Shaded grey on the plot is used to indicate residue–NF contacts, defined as distances less than 0.4 nm. Residue numbers are coloured by sidechain physicochemical properties: dark red – charged (Lys and Arg); light red – hydrophilic (Asn, Thr, Gln, His, Ser and Cys); light grey – hydrophobic (Ala, Leu, Val, Gly and Ile); and dark grey – aromatic (Phe and Tyr). (b)–(d). Representative snapshots that highlight favourable amylin–PG interactions. The amylin backbone is shown in cyan cartoon representation, residue sidechains are drawn in licorice atomic detail and coloured based on their properties (i.e., charged, hydrophilic, hydrophobic, and aromatic), and graphene carbon atoms are shown in grey with either space-filling (b) or licorice (c and d) representations. Water and ions are hidden for clarity. (b) Top-view of amylin–PG5 showing residues within 0.4 nm of the NF. (c), (d). Side-view of amylin interacting with PG3 and PG5, emphasising the orientation of aromatic residues relative to the substrates. | ||
The minimum distance between individual protein residues to the NF provides information about the driving forces behind amylin adsorption on the PG NFs (Fig. 2a). Analysis of the low energy states of amylin adsorbed onto the PG NFs reveals that numerous residues across the protein exhibit direct contact (<0.4 nm) with the substrate, consistent with previous studies23,35,92 (Fig. 2a). Regardless of the physicochemical properties of their sidechains (hydrophobic, hydrophilic, or charged), all types of residues favourably interact with PG, suggesting there is non-specific surface adsorption. This observation aligns with other studies that also show graphene interacts strongly with all naturally occurring amino acids.93,94 As the PG NF size increases, more protein residues are in contact with the NF and these contacts become more stable (Fig. 2a). Aromatic residues (Phe15, Phe23 and Tyr37) preferentially interact with the PG surface, however, the larger 5 and 7 nm NFs better facilitate the planar arrangement of aromatic residues, guided by π–π stacking interactions (Fig. 2b and d). This type of aromatic stacking encourages sidechain extension over the PG surface, contributing to amylin unfolding. In comparison, the limited surface area for aromatic and other contacts on PG3 impedes amylin structural rearrangements and results in the protein adopting more compact configurations (Fig. 2c). Fig. 2a also shows that Phe15 is at a closer distance to PG5 than PG7. This finding is due to amylin adopting extended β-sheet conformations spanning residues 15–30 on PG7, causing residues 14–20 to be positioned farther from the surface compared to residues not in a β-sheet conformation, such as those observed on PG5 (Fig. 1c and d).
The BE-MetaD simulation results presented in Fig. 1 and 2 are in general agreement with the SPON-MD simulation findings (Fig. S9 and S10†). The results from the different simulation methods showed common trends, such as enhanced amylin conformational mobility, reduced helical content, and increased surface adsorption, as a function of increasing PG NF size. However, the preferred (hotspot) surface-bound amylin conformations attained from the two simulation methods showed nuanced differences in their secondary structures. The SPON-MD simulations sampled protein states with, on average, approximately more than double the helical content compared to the BE-MetaD hotspots. Notably, the SPON-MD amylin–PG3 and amylin–PG5 simulations exhibited conformations with higher helical residue content than native amylin due to an additional C-terminal helix being formed by residues 21–36 (Fig. S16†). Further, the SPON-MD low energy structures for amylin bound to PG7 showed negligible β-rich character (Fig. S9†), in contrast to the BE-MetaD simulations where amylin featured β-sheet characteristics in approximately 14% of its residues (Fig. 1). These conformational variations quantitatively manifest as differences in the minimum distance of protein residues to PG obtained by the different simulation methods (Fig. 2a and S10†). These findings indicate that SPON-MD leads to amylin being kinetically trapped in states exhibiting more helical structures compared to BE-MetaD. Furthermore, for all amylin–PG NF systems, BE-MetaD noticeably explores a larger region of phase space compared to the SPON-MD ensemble generated within the simulation timeframes, as indicated by the free energy maps (Fig. S9†). Previous MD simulation studies involving protein–surface interactions have highlighted the challenges in effectively sampling processes such as helical breakdown, α-helix to β-sheet transitions, and protein movement across structured water layer for complete adsorption, when relying solely on SPON-MD.95–97 The differences found between the two applied methodologies may also be associated to the different cut-off values used for calculating van der Waals and electrostatics (see ESI†). Despite these variabilities, the observed amylin–NF interactions in both simulation methods were qualitatively similar, as expected from simulations employing the same forcefield.
Overall, based on the above simulation results, we postulate that larger PG NFs (>5 nm) may be capable of nucleating and accelerating the formation of amyloid fibrils on the surface. The potential fibril-accelerating capacity of these surfaces is ascribed to the extensive and planar surface providing an ideal binding substrate for peptides, promoting lateral diffusion, and facilitating the adoption of fibril-prone elongated and β-rich structures. Consequently, our results suggest larger PG NFs have a superior capacity to serve as templates that mediate peptide self-assembly and fibrillation. In contrast, smaller PG NFs (e.g., PG3) have a reduced surface area that is expected to limit the adsorption and assembly of peptides on the surface. While biological application of PG NFs is limited by their low solubility, these simulation findings indicate the potential for utilising larger PG surfaces to fabricate well-structured protein assemblies and functional amyloid fibres for the development of bio-inspired materials.49
3.2 Amylin–rGO
Through studying the adsorption of amylin on rGO NFs, we investigate the effect of low surface oxidation (C:O ratio of 10:1) on the structure and binding properties of amylin. Compared to the amylin–PG simulations, rGO significantly reduces the conformational dynamics of surface-bound amylin, with less than half the number of unique rGO-bound amylin conformations identified by RMSD clustering (Fig. 1a and 3a). This suggests backbone fluctuations are decreased when the graphene surface is functionalised. Moreover, unlike PG where amylin's conformational diversity systematically increases with NF size (Fig. 1a), backbone fluctuations are less impacted by rGO NF size (Fig. 3a). The 3 nm and 5 nm amylin–rGO simulations produce a comparable number of RMSD clusters, thus having a similar influence on the conformational dynamics of surface–bound amylin, while the 7 nm rGO NF increases the number of backbone conformations by ca. 30% (Fig. 3a). Contact maps showing the probability each NF atom has for being in contact (less than 0.4 nm) with amylin demonstrate that the decreased backbone flexibility is caused by directed binding of amylin on rGO (Fig. 3b). The inclusion of oxygenated functional groups on the surface increases the probability for contact between specific atoms/regions of the NF and amylin. For example, amylin is found to exhibit more edge contacts on rGO NFs than on PG NFs, largely facilitated through surface–amylin hydrogen bonding (described below). The consequence of specific surface sites on rGO is a more laterally restricted amylin binding with reduced translational mobility. The increased translational mobility of amylin adsorbed on the PG NFs compared to the oxidised NFs may also be related to the lower surface roughness of the pristine surfaces due to the absence of functional groups, as quantified in the previous study.54 Surfaces that facilitate monomer adsorption and entrapment while inhibiting the lateral diffusion of peptides along the surface are believed to discourage peptide assembly.79–81
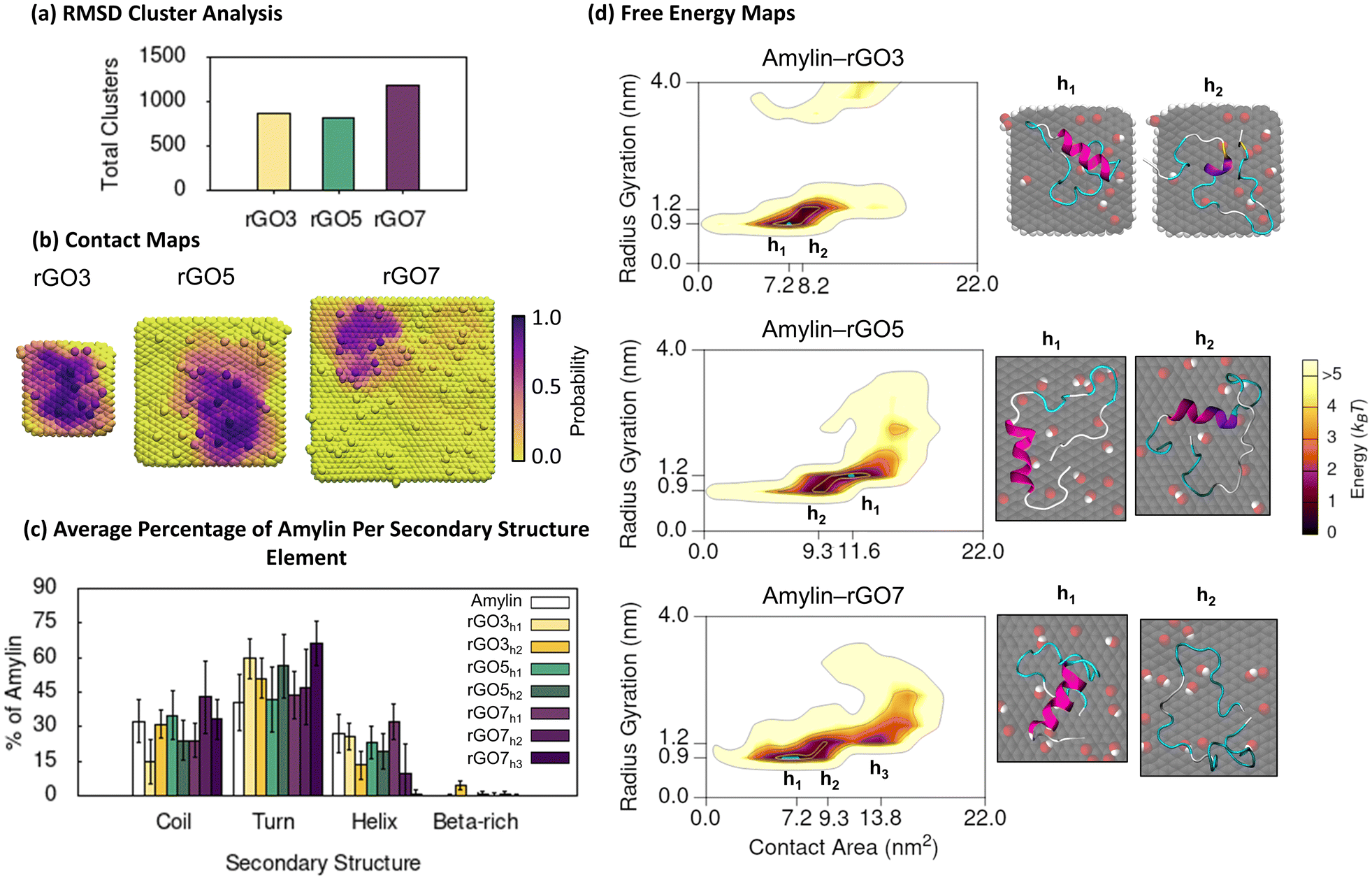 | ||
| Fig. 3 Amylin conformational preferences and contacts with reduced graphene oxide. (a) Total number of unique surface-bound amylin conformations identified by RMSD backbone cluster analysis from the amylin–rGO simulations. (b) Contact probability maps showing the relative proportion of simulation time each rGO atom maintains close association (less than 0.4 nm) with amylin. (c) Average percentage of amylin adopting different type of secondary structure within the hotspot regions with errorbars representing standard deviation. The average secondary structure of amylin in-solution9 is provided in white bars for reference. (d) Amylin–rGO free energy maps showing the relationship between amylin radius gyration and amylin–NF contact area. Hotspots (h) are sequentially labelled numerically based on their free energy and the blue highlight corresponds to the lowest energy hotspot region. | ||
The 2D free energy maps correlating the amylin radius gyration and amylin–NF contact area provide understanding into the adsorbed conformations of amylin on rGO NFs (Fig. 3d). Compared to the amylin–PG simulations of equal NF size, the amylin–rGO simulations sample adsorbed states that have a lower amylin–NF contact area, emphasising that the adsorption onto rGO results in more stringent protein binding (Fig. 1d and 3d). Furthermore, while the amylin–PG free energy maps converge to one main hotspot region (Fig. 1d), amylin–rGO adsorption has a more complex 2D free energy landscape with multiple distinct low energy states (Fig. 3d). The rGO maps reveal that amylin prefers to adsorb with a larger amylin–NF contact area on the rGO5 NF compared to the rGO3 and rGO7 NFs, potentially suggesting the intermediate-sized NF is most conducive for strong adhesion. The larger rGO5 contact area is also consistent with the atomic contact probability depicted in Fig. 3b being more widespread on rGO5, and may explain the fewer backbone fluctuations when amylin is bound to rGO5 due to stronger anchoring inhibiting protein dynamics (Fig. 3a).
Exploring the interaction of amylin with rGO NFs in more detail reveals patterns in protein structure and potential implications for fibril inhibition. In the most stable states of amylin adsorbed onto rGO3 and rGO7 (hotspots rGO3h1 and rGO7h1), the middle residues 5–19 of amylin are least likely to make surface contact (Fig. 4a). The minimal binding of these residues helps to preserve amylin's solution-state helical structure and even promotes an increase in the helical content on rGO7 (Fig. 3c and S17†). On rGO5, closer contacts are formed between amylin's central residues and the NF (hotspot rGO5h1), leading to a ∼15% reduction in the native helical content of amylin (Fig. 3c and S17†). Interestingly, rGO3 promotes a less populated, ancillary hotspot (rGO3h2) where structures exhibit lower helical content and a persistent, though singular, isolated β-bridge linking Leu12 to Asn35 (Fig. 3c and S17†). For all other hotspots, amylin's adsorption results in low energy conformations devoid of β-character (Fig. 3d and S17†), consistent with previous studies.23,36–38 It is noteworthy to emphasise that the 7 nm rGO NF does not induce β-conformations, whereas the same size PG promotes extended β-sheet conformations. Compared to the amylin–PG simulations (Fig. 1c), our results show that rGO more effectively maintains helices, inhibits protein extension, and prevents β-sheet formation on the NFs (Fig. 3c). Given that graphene with a low level of surface oxidation slows down the rate of amylin unfolding, it can be suggested that rGO may potentially serve as a fibril inhibitor.29
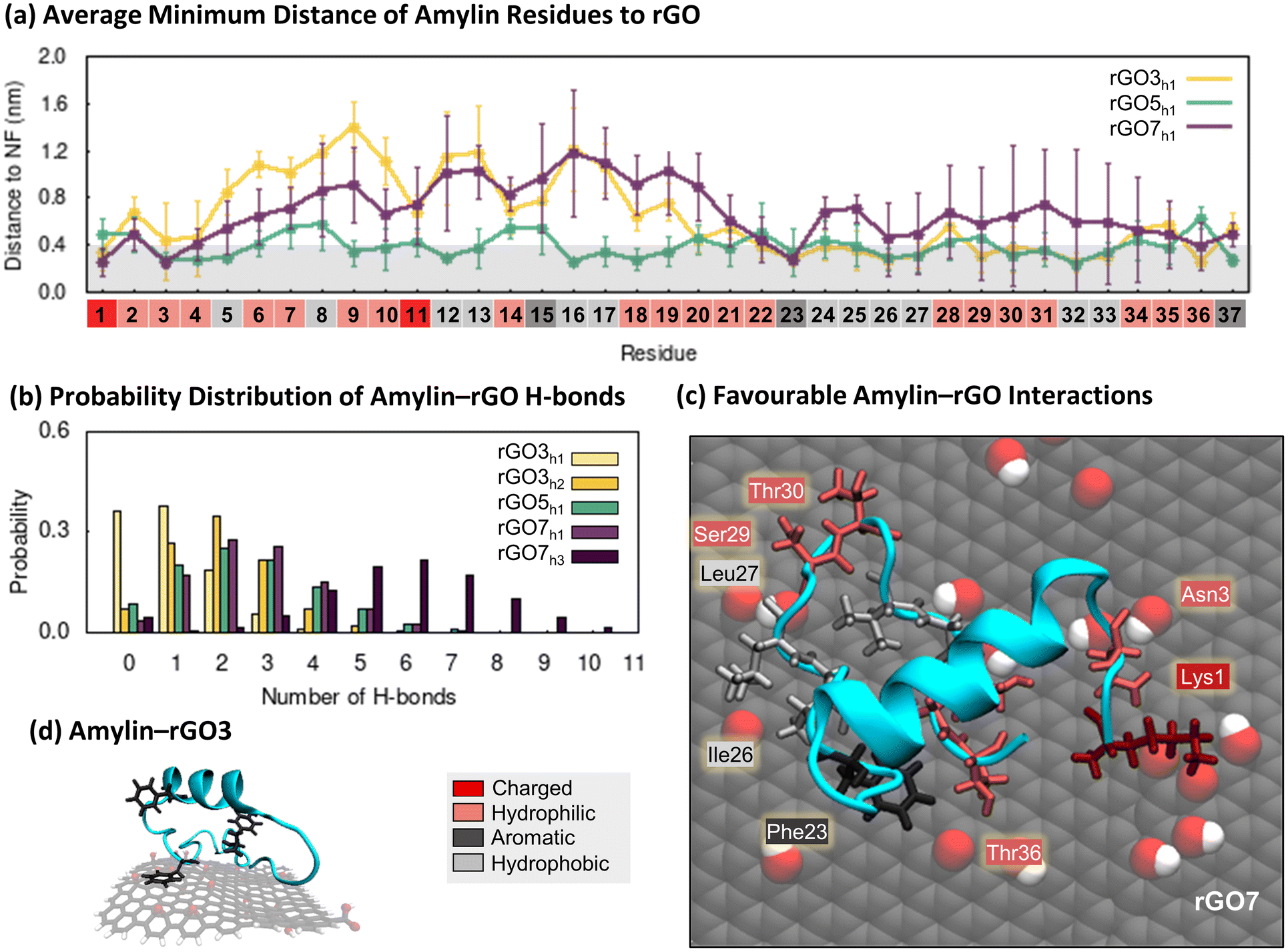 | ||
| Fig. 4 Specific interactions between amylin and reduced graphene oxide. (a) Average minimum distances measured between individual amylin residues and the rGO3, rGO5, and rGO7 surfaces, with errorbars representing standard deviation. Shaded grey on the plot is used to indicate residue–NF contacts, defined as distances less than 0.4 nm. Residue numbers are coloured by sidechain physicochemical properties. (b) Probability distribution of hydrogen bonds formed between amylin and rGO in the amylin–rGO hotspots. (c) and (d). Representative snapshots that highlight favourable amylin–rGO interactions. The amylin residue sidechains are drawn in licorice atomic detail and coloured based on their properties. (c) Top-view of amylin–rGO7 showing residues within 0.4 nm of the NF. (d) Side-view of amylin interacting with rGO3, emphasising the orientation of aromatic residues relative to the substrate. | ||
On rGO7, less populated but still stable states (i.e., ancillary hotspot rGO7h2) are observed that are predominantly unfolded or denatured, likely due to their more stable adsorption on the NFs (Fig. S22†). Previous studies have shown that the adsorption of amylin and amyloid-β to functionalised carbon-based NPs can induce a higher degree of protein adsorption, resulting in complete helical breakdown.25,98 When multiple peptide units were introduced, strong protein binding to the oxidised carbon-based NPs was found to hinder and disrupt the intra- and inter-chain interactions necessary for protein aggregation.24,25 Other studies examining the influence of GO size on amyloid-β fibrillation have demonstrated that larger GO sizes more effectively reduce fibrillation by facilitating enhanced monomer trapping and stronger interactions with peptides, leading to the suppression of β-sheet secondary structures and discouragement of peptide–peptide interactions.21,99 In light of these previous works, our simulation results propose that larger rGO NF sizes (>5 nm) are potentially more effective in preventing fibril formation compared to smaller rGO3 NFs.
To examine the factors influencing amylin adsorption onto rGO NFs, we monitored distances between protein residues and the surfaces, the formation of amylin–NF hydrogen bonds, and water structuring, using the low energy amylin conformations identified earlier (Fig. 4). The presence of oxygenated functional groups on rGO introduces the opportunity for amylin to form hydrogen bonds with the graphitic surfaces (Fig. 4b). Amylin residues also competitively interact with the hydrophilic and hydrophobic regions on the rGO substrates. Analogous to the amylin–PG simulations, aromatic residues (Phe15, Phe23 and Tyr37) favourably interact with exposed carbon patches via π–π stacking (Fig. 4a, c and d). Water molecules are observed to structure around the NF oxygenated functional groups, leaving unfunctionalised graphene regions dehydrated (Fig. S21†). The considerable area of exposed carbon patches, due to both fewer oxygenated functional groups and lower surface hydration, encourages more protein residues to adhere to the bare carbon compared to the oxygenated regions of the NFs. These protein–surface binding trends are highlighted in the representative amylin–rGO7 conformation shown in Fig. 4c. In this low energy state, the charged Lys1 residue can be seen forming persistent (electrostatic) contacts with an oxygen-rich NF region, while aromatic/hydrophobic residues Phe23, Ile26 and Leu27 interact with exposed graphene regions. Hydrophilic residues competitively interact with both the oxygen and carbon atoms of the NF (Fig. 4c). The comparison of amylin binding on PG and rGO suggests that the interactions with both oxygen-rich and carbon-rich regions of the substrate work synergistically to decrease the conformational flexibility and lateral mobility of amylin upon rGO adsorption. This is likely because the sidechains of adsorbed residues are more effectively ‘locked’ in position on the amphiphilic rGO surface.
The BE-MetaD simulation results shown in Fig. 3 and 4 are mostly consistent with our SPON-MD simulations (Fig. S11 and S12†). Both simulation methods showed similar amylin–rGO interactions, a reduction in amylin conformational mobility, and some common low energy surface-bound amylin conformations. The preferred amylin conformations sampled by SPON-MD on rGO5 and rGO7 exhibit negligible reduction in helical content relative to native amylin in-solution. In the SPON-MD simulations of amylin binding to rGO5 and rGO7, some less populated low energy states are sampled that are predominantly unfolded due to extensive protein adsorption (SPON-MD hotspots rGO5h2 and rGO7h2 in Fig. S11 and S22†). Denatured states also emerge in the BE-MetaD simulations of amylin on rGO7 (i.e., hotspot rGO7h2 in Fig. 3), further confirming the ability of larger rGO NFs to strongly bind amylin and unfold its secondary structure. There are some notable differences in the amylin–rGO binding between the simulation methods. The contact probability maps generated from the SPON-MD simulations (Fig. S11b†) show amylin explores different, less localised, positions on the rGO NFs compared to the BE-MetaD simulations. This contrasting behaviour originates from the two independent SPON-MD trajectories, each with a unique initial orientation of amylin relative to the NF, exploring different areas of the conformational free-energy landscape of amylin. In the rGO5 and rGO7 SPON-MD simulations, one simulation trajectory conserved more of the native structure of amylin, while the other led to helical breakdown and increased protein–NP interactions. In SPON-MD, the initial orientation and randomised atomic velocities of the protein can bias simulation evolution. Additionally, given the heterogenous distribution of oxygen on the rGO surface, the specific location where amylin adsorbs on the NF impacts the protein–surface interactions and conformational changes that transpire. Other MD simulation studies exploring protein–graphene interactions also report that initial protein orientation influences protein adsorption, further emphasising the challenge and complexity in modelling protein–surface interactions.77,88 Our simulations highlight the advantages of running MD simulations with diverse initial protein orientations in protein–NP systems that feature heterogenous surfaces, to circumvent insufficient sampling where proteins adsorb and remain trapped at specific locations on the surface. However, in practice, this approach requires substantial computational time and resources, especially when using enhanced sampling techniques such as BE-MetaD. Despite these challenges, our RMSD clustering results and the 2D free energy maps (Fig. 3 and S11†) show that BE-MetaD with a single initial protein orientation provided an appreciable improvement in conformational sampling relative to SPON-MD.
In summary, compared to the PG surfaces of equal sizes, rGO substrates may inhibit protein aggregation due to their amphiphilic nature, which decreases protein mobility and restricts protein unfolding. We find that larger rGO sizes (>5 nm) in particular, may be more effective fibril inhibitors, owing to their enhanced ability to prevent β-sheet secondary structures and facilitate more stable adsorption. We hypothesise that oxidised graphitic surfaces could effectively sequester protein monomers from solution and anchor the absorbed monomers in place, thereby preventing the assembly of peptide monomers into larger fibrillar aggregates.
3.3 Amylin–GO
Finally, we simulated amylin adsorption on GO NFs that had a high surface oxygen concentration (C:O ratio of 5:1, twice the oxygenation on rGO). RMSD cluster analysis of amylin on GO, showed an approximate 10–100% increase in the number of unique structures relative to the simulations of amylin on equivalent sizes of rGO NF (Fig. 3a and 5a). This implies that the amylin backbone is more conformationally dynamic when the graphitic surface is more oxidised, but not quite as dynamic as on pristine graphene surfaces (Fig. 1a). These findings are consistent with previous computational modelling studies that note GO promotes more structural and/or conformational transitions in lysozyme and Aβ monomers compared to rGO.100,101 In the context of GO size, our results show an increase in NF size leads to a reduction in total number of unique amylin conformations (Fig. 5a), suggesting that larger GO NFs discourage protein mobility.
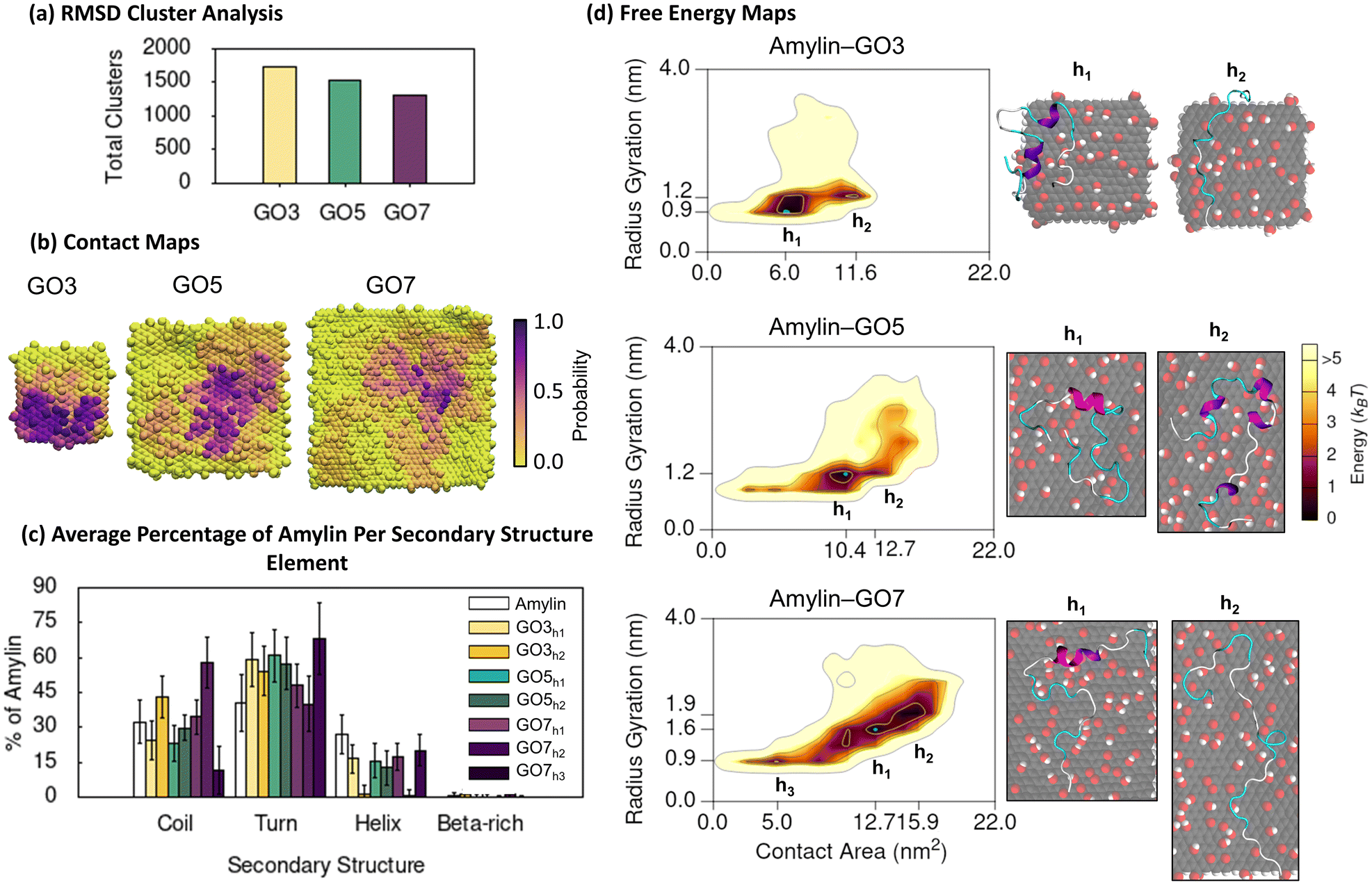 | ||
| Fig. 5 Amylin conformational preferences and contacts with graphene oxide. (a) Total number of unique surface-bound amylin conformations identified by RMSD backbone cluster analysis from the amylin–GO simulations. (b) Contact probability maps showing the relative proportion of simulation time each GO atom maintains close association (less than 0.4 nm) with amylin. (c) Average percentage of amylin adopting different type of secondary structure within the hotspot regions with errorbars representing standard deviation. The average secondary structure of amylin in-solution9 is provided in white bars for reference. (d) Amylin–GO free energy maps showing the relationship between amylin radius gyration and amylin–NF contact area. Hotspots (h) are sequentially labelled numerically based on their free energy and the blue highlight corresponds to the lowest energy hotspot region. | ||
The contact probability maps demonstrate that GO presents specific surface sites for amylin binding (Fig. 5b), akin to that on the rGO surfaces (Fig. 3b). Therefore, like rGO, the oxygen sites on GO reduce the lateral diffusion of surface-bound amylin to potentially inhibit fibril formation through monomer adsorption and entrapment.79–81 However, compared to rGO (Fig. 3b), amylin–GO adsorption occurs over a wider NF surface area with fewer NF atoms having a high contact probability (Fig. 5b), resulting in amylin being more conformationally versatile and dynamic on GO. It is worth mentioning that the time-evolved MD simulations sampled a few short-lived amylin desorption events near the start of the simulations on GO3 and GO7 NFs, which may imply a weaker initial protein binding. The increased oxygenation of GO also generates a more extensive hydration layer around the NF (Fig. S21†), in agreement with our previous graphitic NFs simulations.54 Other modelling studies also consistently demonstrate that a weaker protein/biomolecule adsorption occurs on GO compared to more hydrophobic surfaces, likely due to extensive hydration of the GO substrate.93,101–104 This is further supported by experimental data of amylin adsorption onto self-assembled monolayers (SAMs), describing that hydrophilic-terminated SAMs adsorb monomers weaker than surfaces that are more hydrophobic.105
Considering NF size, the contact probability maps demonstrate amylin interacts with a greater capacity as NF surface area increases (Fig. 5b). The greater contact area may be associated to a more stable adsorption of amylin on the larger GO NFs and may clarify the reduction in protein backbone fluctuations with increasing GO size (Fig. 5a). Chen et al.99 also found larger GO sizes increased the contact surface area with Aβ peptides, which in turn resulted in a more effective prevention of fibril formation. We observe that the size of the GO NF also influences the location of amylin binding on the NF. While amylin consistently binds to both the central and edge regions of GO, as the GO size decreases, there is a preference for more edge binding (Fig. 5b). For the small GO3 NF, the increased NF edge interactions lead to some amylin conformations that wrap around the NF and contact both basal planes simultaneously (Fig. 6d). A similar ‘wrapped’ conformation of amylin on GO3 has been reported computationally during the adsorption of amylin onto GQDs of similar dimensions.25
 | ||
| Fig. 6 Specific interactions between amylin and graphene oxide. (a) Average minimum distances measured between individual amylin residues and the GO3, GO5, and GO7 surfaces, with errorbars representing standard deviation. Shaded grey on the plot is used to indicate residue–NF contacts, defined as distances less than 0.4 nm. Residue numbers are coloured by sidechain physicochemical properties. (b) Probability distribution of hydrogen bonds formed between amylin and GO in the amylin–GO hotspots. (c) and (d) Representative snapshots that highlight favourable amylin–GO interactions. The amylin residue sidechains are drawn in licorice atomic detail and coloured based on their properties. (c) Top-view of amylin–GO7 showing residues within 0.4 nm of the NF. (d) Side-view of amylin interacting with GO3, emphasising the orientation of aromatic and charged residues relative to the substrate. | ||
Consistent with the amylin–rGO free energy maps (Fig. 3d), the adsorption of amylin on the GO NFs induces a variety of protein conformations, as demonstrated by the multiple hotspot regions in the 2D free energy maps (Fig. 5d). Like the amylin–PG and amylin–rGO systems, contact area increases as the size of the proximal NF increases, inferring that stronger protein–NF interactions establish when adsorbed to the larger GO nanoparticles. Consistent with earlier amylin–GO adsorption studies,24 we find GO disrupts the structure of amylin's helices, leading to a ∼40% reduction in the solution-state helical content (hotspots GO3h1, GO5h1 and GO7h1). The remaining helical residues that are preserved are found to have minimal NF contact (Fig. 6a and S19†). Across all NF sizes, amylin favourably adsorbs to GO with a higher contact area, less compact configuration, and a greater helical reduction, compared to the equivalent sized rGO NFs (Fig. 3c, d and 5c, d). These simulation findings agree with a recent theoretical study investigating the adsorption of amyloid-β peptide (Aβ42) onto carbon nanotubes (CNTs) varying in surface functionalisations.98 They documented increased protein-CNT contact area and protein unfolding on surfaces exhibiting higher levels of oxidation. Similar to the amylin–rGO simulations and previous studies,23,36–38 GO does not encourage β-conformations (Fig. 5c and S19†), but as is the case with rGO, GO NFs may induce some predominantly unfolded/denatured states. Apart from the ‘wrapped’ configuration on GO3 mentioned earlier (Fig. 6d), another conformation is observed on the larger GO7 NF where amylin structurally extends to its limits (hotspots GO7h2). The larger particle surface area on GO7 accommodates the elongation of amylin along a single side of the planar surface (Fig. 5d). While both rGO7 (hotspots rGO7h2) and GO7 (hotspots GO7h2) induced adsorbed states with complete helical breakdown, amylin–GO structures are completely extended across the NF (i.e., less compact) and the loss of helical character is more pronounced (Fig. 3d and 5d). Our simulation results corroborate previous studies showing that the adsorption of amylin onto highly hydrophilic surfaces can induce extensive protein adsorption and structural denaturation.25,98,99 These denatured states involve strong amylin–GO binding, which may disrupt the intra- and inter-chain interactions needed for protein aggregation.24,25,99
To elucidate the impact of NF oxygen concentration on the mechanism of amylin adsorption, we again monitored the minimum distance of protein residues from the surface and examined the formation of amylin–NF hydrogen bonds (Fig. 6). As expected, increasing the NF oxygen concentration (compared to rGO) results in the formation of more amylin–NF hydrogen bonds (Fig. 4b and 6b). The amylin backbone, along with the sidechains of hydrophilic and charged residues (i.e., Lys1, Thr30, Asn31, and Ser34), demonstrate there is a preference for selective hydrogen bonding to the oxygenated functional groups on GO (Fig. 6c). Aromatic residues (Phe23 and Tyr37) engage in planar π–π stacking on exposed carbon regions that are surrounded by a boundary of NF oxygen atoms (Fig. 6a and c), highlighting the preference for graphatic π–π stacking even at high NF oxygen concentrations. While the sidechains of bound hydrophobic residues readily align over carbon regions of the NF, the hydration of the surface (Fig. S21†) and limited availability of exposed carbon patches restricts the number of NF carbon–amylin contacts that can form (Fig. 6c). These binding preferences emphasise that amylin conformations are governed by the surface nanopatterning that emerges from the number and location of oxygenated functional groups on the graphitic nanoflakes. In comparison to the rGO surfaces, there is a greater number of NF oxygen–protein contacts compared to NF carbon–protein contacts. There is also a more prominent hydration layer leaving fewer unfunctionalised carbon regions exposed (Fig. S21†). This water structuring around the NF mediates amylin binding to the GO surface. Overall, the above findings demonstrating the types of interactions formed between amylin and the oxidised surfaces, such as hydrogen bonding, hydrophobic interactions and aromatic stacking interactions, are in accordance with previous studies.24,25,36–38 The differences in how specific amino acids prefer either carbon or oxygen sites on rGO and GO are in line with the findings of Baweja et al.101 They reported that the adsorption of Aβ on GO was primarily driven by electrostatic interactions, whereas adsorption of Aβ on rGO had contributions from both electrostatic and van der Waals interactions due to the larger hydrophobic regions of unfunctionalised carbon. Baweja et al. and others, also describe that the larger carbon areas of rGO promote stronger Aβ monomer and fibril binding compared to GO.101,106
The BE-MetaD (Fig. 5 and 6) and corresponding SPON-MD (Fig. S13 and S14†) simulations of amylin–GO consistently showed that larger GO NF sizes generally correlate with decreased amylin conformational diversity. The RMSD cluster analysis from the SPON-MD simulations shows a slight deviation from this trend, revealing that there is ∼55% fewer unique surface-bound amylin conformations on GO5 compared to GO7. However, both GO surfaces larger than 5 nm still produce significantly fewer amylin conformations than on GO3. This could suggest undersampling by SPON-MD on GO5, especially since amylin did not undergo significant conformational changes upon binding, unlike in all the other amylin–NF simulations (Fig. S8†). Comparing to BE-MetaD, the SPON-MD simulation of amylin–GO5 and amylin–GO7 systems explored fewer unique amylin backbone conformations, but in the amylin–GO3 system, SPON-MD identified a threefold increase in the number of RMSD clusters. This suggests that BE-MetaD more effectively samples amylin undergoing conformation transitions into low energy adsorbed states that have a larger amylin–NF contact area (Fig. 5d and S13†). Although there are subtle differences in the preferred amylin–GO binding locations between the two methods (e.g., amylin wraps around different edges on the GO3 NF, SPON-MD hotspot GO3h2, Fig. S13†), the SPON-MD simulations generally produce protein states with a smaller amylin–NF contact area and greater helical content compared to the BE-MetaD simulations (Fig. S13†). This indicates that BE-MetaD more effectively explores the adsorption process, aligning with previous studies demonstrating that enhanced sampling techniques accelerate the exploration of protein adsorption, leading to the formation of greater protein–surface interactions.95,97 While both methods show the majority of low energy conformations have a negligible presence of β-rich character (Fig. S13, S19 and S20†), SPON-MD of amylin–GO5 revealed a persistent isolated β-bridge forming between Ala25 and Ser34 (Fig. S19†). This is likely due to amylin forming fewer interactions on the GO5 NF during the SPON-MD simulations, as previously mentioned. Moreover, the 2D free energy maps show BE-MetaD explores a larger region of phase space than SPON-MD for all amylin–GO NF systems (Fig. 5d and S13†), underscoring the value of BE-MetaD in predicting the structure and binding of amylin on GO NFs. Discrepancies between the two methodologies may be associated to the different cut-off value used for calculating van der Waals and electrostatics (see ESI†), but reassuringly, similar amylin–NF interactions were consistently observed by both methods (Fig. S14†).
There is substantial experimental evidence indicating that GO NFs inhibit fibril formation.20–22 Our simulations provide a molecular-level understanding of how oxidised graphitic NFs might be achieving this inhibition. We propose that larger surfaces may potentially prevent fibril formation through two mechanisms: (1) by sequestering peptide monomers from solution, thereby decreasing the likelihood of proteins aggregating in solution; and (2) by promoting adsorbed amylin to adhere with minimal β-sheet secondary structure, which could deter and disrupt the intra- and inter-chain interactions that contribute to protein aggregation on surfaces. When considering the optimal oxidation level for NFs to function as fibril inhibitors, the simulations suggest that a C:O ratio of 5:1 (i.e., GO) might be less effective in preventing fibril formation than a 10:1 ratio (i.e., rGO). This is indicated by the GO substrates facilitating enhanced amylin conformational dynamics, increased unfolding, and more significant destruction of secondary structure compared to rGO surfaces. The simulations reveal that the conformations of amylin are influenced by nanopatterning. In contrast to rGO where the large regions of unfunctionalised carbon effectively ‘locked’ adsorbed amylin in place, on GO the increased surface oxygen concentration encourages conformational dynamics and the formation of amylin–NF hydrogen bonds. This leads to the extension of amylin over the GO NF surface, in accordance with the oxygen patterning, and driven by a preference for hydrogen bonding and aromatic stacking interactions. Therefore, controlling the size of the hydrophobic/hydrophilic NF regions is critical in order to exploit the preference for amylin–NF interactions that preserve the native in-solution structure of amylin. MD simulations have shown nanoscale surface heterogeneity can dictate the adsorption behaviour of proteins to either promote or prevent binding based on the matching between the surface and protein functionality.75,107–109 Recent experimental studies have highlighted the importance of achieving an appropriate balance between hydrophilic and hydrophobic domains in determining the inhibitory effects of engineered nanochaperones against amylin fibrillation.110 Specifically, hydrophobic domains facilitate the adsorption of amylin monomers, while hydrophilic domains act as a boundary to prevent self-aggregation. Our simulation findings support this concept and previous studies,101 by demonstrating how the oxygen concentration and distribution of GO surfaces may be tailored for selective and strong binding with amylin to effectively inhibit amyloid formation.
4. Conclusions
Using atomistic molecular dynamics simulations we explored the adsorption of amylin on graphitic nanoflakes varying in size and degree of surface oxidation. We found amylin adopted a variety of surface-bound conformations depending on the size and nanopatterns of oxygenated functional groups on the graphene nanoflakes. The hydrophobic pristine graphene nanoflakes promoted non-specific protein adsorption resulting in increased protein mobility, especially on the relatively large nanoflakes, over 5 nm. The extent of structural changes, such as loss of native helical content and β-sheet formation increased with an increase in the nanoflake size. The oxidised graphitic nanoflakes provided specific binding sites for hydrogen bonding with amylin. The surface oxygen concentration and nanopatterning were observed to control the bound conformations of amylin. The higher oxygen concentration on the GO surfaces promoted substantial water structuring and induced conformational dynamics which led to protein unfolding and partial helical breakdown due to the extension of the amylin structure over the NF surface. In contrast, reduced graphene oxide having substantial number of bare carbon regions and subsequently lower surface hydration, promoted reduced protein flexibility and conformational dynamics, which is important for maintaining the native in-solution structure of amylin. These results indicate that graphitic nanoflakes can be engineered to effectively sequester amylin monomers in the biological environment, inhibit conformational changes associated with amyloid fibril formation and ultimately prevent peptide–peptide association. In this study we demonstrate that tailoring the oxygen concentration and surface patterning of GO can result in specific and robust protein binding, influencing the likelihood of fibril formation. Such molecular insights offer important considerations for the design of graphitic nanoflakes that can contribute to improved treatments for amyloid-related disorders.Data availability
The data supporting this article including convergence evaluation and additional results of the BE-META and spontaneous molecular dynamics (SPON-MD) simulations have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the high-performance computing resources provided by the Australian Government through the National Computational Infrastructure (NCI) Sustainable Australian Supercomputing Flagship Scheme (project e87 and e90) and Pawsey Supercomputing Research Centre. I. Y. acknowledges funding from the Australian Research Council under the Discovery Project scheme (Grants No. DP170100511, DP190102290, DP230100709). A. K. acknowledges the RTP Stipend Scholarship (RSS-SC) from the Australian Government. The authors acknowledge Justin Walmsley, Darcy Griffiths and Ausaid Mahmood for their contributions to preliminary data collection.References
- O. Sumner Makin and L. C. Serpell, J. Mol. Biol., 2004, 335, 1279–1288 CrossRef CAS PubMed.
- M. Cecchini, R. Curcio, M. Pappalardo, R. Melki and A. Caflisch, J. Mol. Biol., 2006, 357, 1306–1321 CrossRef CAS PubMed.
- D. Milardi, E. Gazit, S. E. Radford, Y. Xu, R. U. Gallardo, A. Caflisch, G. T. Westermark, P. Westermark, C. L. Rosa and A. Ramamoorthy, Chem. Rev., 2021, 121, 1845–1893 CrossRef CAS PubMed.
- M. Alrouji, H. M. Al-Kuraishy, A. I. Al-Gareeb, A. Alexiou, M. Papadakis, H. M. Saad and G. E.-S. Batiha, Diabetol. Metab. Syndr., 2023, 15, 101 CrossRef CAS PubMed.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fandrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- S. J. Moore, K. Sonar, P. Bharadwaj, E. Deplazes and R. L. Mancera, Molecules, 2018, 23, 2142 CrossRef PubMed.
- J. R. Cort, Z. Liu, G. M. Lee, K. N. Huggins, S. Janes, K. Prickett and N. H. Andersen, Protein Eng., Des. Sel., 2009, 22, 497–513 CrossRef CAS PubMed.
- C. Wu and J. E. Shea, PLoS Comput. Biol., 2013, 9, e1003211 CrossRef CAS PubMed.
- E. Peng, N. Todorova and I. Yarovsky, PLoS One, 2017, 12, e0186219 CrossRef PubMed.
- G. G. Glenner, E. David Eanes and C. A. Wiley, Biochem. Biophys. Res. Commun., 1988, 155, 608–614 CrossRef CAS PubMed.
- P. Westermark, U. Engström, K. H. Johnson, G. T. Westermark and C. Betsholtz, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5036–5040 CrossRef CAS PubMed.
- L. E. Buchanan, E. B. Dunkelberger, H. Q. Tran, P. N. Cheng, C. C. Chiu, P. Cao, D. P. Raleigh, J. J. de Pablo, J. S. Nowick and M. T. Zanni, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19285–19290 CrossRef CAS PubMed.
- M. R. Nilsson and D. P. Raleigh, J. Mol. Biol., 1999, 294, 1375–1385 CrossRef CAS PubMed.
- L. A. Scrocchi, K. Ha, Y. Chen, L. Wu, F. Wang and P. E. Fraser, J. Struct. Biol., 2003, 141, 218–227 CrossRef CAS PubMed.
- K. Sasahara, Biophys. Rev., 2018, 10, 453–462 CrossRef CAS PubMed.
- L. Khemtémourian, J. A. Killian, J. W. Höppener and M. F. Engel, Exp. Diabetes Res., 2008, 2008, 421287 CrossRef PubMed.
- S. Szunerits, A. Abderrahmani and R. Boukherroub, Acc. Chem. Res., 2022, 55, 2869–2881 CrossRef CAS PubMed.
- C. Li and R. Mezzenga, Nanoscale, 2013, 5, 6207–6218 RSC.
- P. C. Ke, E. H. Pilkington, Y. Sun, I. Javed, A. Kakinen, G. Peng, F. Ding and T. P. Davis, Adv. Mater., 2020, 32, e1901690 CrossRef PubMed.
- M. Mahmoudi, O. Akhavan, M. Ghavami, F. Rezaee and S. M. Ghiasi, Nanoscale, 2012, 4, 7322–7325 RSC.
- J. Wang, Y. Cao, Q. Li, L. Liu and M. Dong, Chemistry, 2015, 21, 9632–9637 CrossRef CAS PubMed.
- S. Bag, A. Sett, S. DasGupta and S. Dasgupta, RSC Adv., 2016, 6, 103242–103252 RSC.
- J. Guo, J. Li, Y. Zhang, X. Jin, H. Liu and X. Yao, PLoS One, 2013, 8, e65579 CrossRef CAS PubMed.
- P. Nedumpully-Govindan, E. N. Gurzov, P. Chen, E. H. Pilkington, W. J. Stanley, S. A. Litwak, T. P. Davis, P. C. Ke and F. Ding, Phys. Chem. Chem. Phys., 2016, 18, 94–100 RSC.
- M. Wang, Y. Sun, X. Cao, G. Peng, I. Javed, A. Kakinen, T. P. Davis, S. Lin, J. Liu, F. Ding and P. C. Ke, Nanoscale, 2018, 10, 19995–20006 RSC.
- M. Yousaf, H. Huang, P. Li, C. Wang and Y. Yang, ACS Chem. Neurosci., 2017, 8, 1368–1377 CrossRef CAS PubMed.
- S. Li, D. Lin, X. Hu and X. Yang, Nanotechnology, 2018, 29, 495102 CrossRef PubMed.
- X. Zhou, C. Cao, Q. Chen, Q. Yu, Y. Liu, T. Yin and J. Liu, J. Mater. Chem. B, 2015, 3, 7055–7067 RSC.
- S. Wang, J. Zheng, L. Ma, R. B. Petersen, L. Xu and K. Huang, Biochim. Biophys. Acta, Gen. Subj., 2022, 1866, 130061 CrossRef CAS PubMed.
- F. De Leo, A. Magistrato and D. Bonifazi, Chem. Soc. Rev., 2015, 44, 6916–6953 RSC.
- A. J. Makarucha, N. Todorova and I. Yarovsky, Eur. Biophys. J., 2011, 40, 103–115 CrossRef CAS PubMed.
- T. Casalini, V. Limongelli, M. Schmutz, C. Som, O. Jordan, P. Wick, G. Borchard and G. Perale, Front. Bioeng. Biotechnol., 2019, 7, 268 CrossRef PubMed.
- M. Ozboyaci, D. B. Kokh, S. Corni and R. C. Wade, Q. Rev. Biophys., 2016, 49, e4 CrossRef PubMed.
- N. Todorova and I. Yarovsky, Aust. J. Chem., 2019, 72, 574–584 CrossRef CAS.
- J. Liu, Z. Yang, H. Li, Z. Gu, J. A. Garate and R. Zhou, J. Chem. Phys., 2014, 141, 22D520 CrossRef PubMed.
- C. Bai, Z. Lao, Y. Chen, Y. Tang and G. Wei, Front. Chem., 2020, 8, 51 CrossRef CAS PubMed.
- C. Bai, D. Lin, Y. Mo, J. Lei, Y. Sun, L. Xie, X. Yang and G. Wei, Phys. Chem. Chem. Phys., 2019, 21, 4022–4031 RSC.
- Y. Mo, S. Brahmachari, J. Lei, S. Gilead, Y. Tang, E. Gazit and G. Wei, ACS Chem. Neurosci., 2018, 9, 2741–2752 CrossRef CAS PubMed.
- S. T. Wang, Y. Lin, N. Todorova, Y. Xu, M. Mazo, S. Rana, V. Leonardo, N. Amdursky, C. D. Spicer, B. D. Alexander, A. A. Edwards, S. J. Matthews, I. Yarovsky and M. M. Stevens, Chem. Mater., 2017, 29, 1550–1560 CrossRef CAS PubMed.
- L. Yang, C.-W. Liu, Q. Shao, J. Zhang and Y. Q. Gao, Acc. Chem. Res., 2015, 48, 947–955 CrossRef CAS PubMed.
- A. S. Kamenik, S. M. Linker and S. Riniker, Phys. Chem. Chem. Phys., 2022, 24, 1225–1236 RSC.
- S. Bhattacharya and X. Lin, Biomolecules, 2019, 9, 146 CrossRef CAS PubMed.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- P. Liu, B. Kim, R. A. Friesner and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13749–13754 CrossRef CAS PubMed.
- L. Wang, R. A. Friesner and B. J. Berne, J. Phys. Chem. B, 2011, 115, 9431–9438 CrossRef CAS PubMed.
- G. Bussi, A. Laio and M. Parrinello, Phys. Rev. Lett., 2006, 96, 090601 CrossRef PubMed.
- A. Laio and F. L. Gervasio, Rep. Prog. Phys., 2008, 71, 126601 CrossRef.
- B. J. Williams-Noonan, A. Kamboukos, N. Todorova and I. Yarovsky, Chem. Phys. Rev., 2023, 4, 021304 CrossRef CAS.
- S. Piana and A. Laio, J. Phys. Chem. B, 2007, 111, 4553–4559 CrossRef CAS PubMed.
- T. N. Do, W. Y. Choy and M. Karttunen, J. Chem. Theory Comput., 2014, 10, 5081–5094 CrossRef CAS PubMed.
- W. Zhao, Z. Xu, Q. Cui and N. Sahai, Langmuir, 2016, 32, 7009–7022 CrossRef CAS PubMed.
- L. Li, T. Casalini, P. Arosio and M. Salvalaglio, J. Chem. Theory Comput., 2022, 18, 1915–1928 CrossRef CAS PubMed.
- E. Peng, N. Todorova and I. Yarovsky, ACS Omega, 2018, 3, 11497–11503 CrossRef CAS PubMed.
- E. Puigpelat, J. Ignés-Mullol, F. Sagués and R. Reigada, Langmuir, 2019, 35, 16661–16668 CrossRef CAS PubMed.
- Z.-S. Yan, X.-L. Li, Y.-Q. Ma and H.-M. Ding, Langmuir, 2022, 38, 13972–13982 CrossRef CAS PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- BIOVIA Materials Studio 6.1, Dassault Systemes, San Diego, CA, USA, 2012 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- A. K. Rappe and W. A. Goddard III, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef CAS.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- K. Vanommeslaeghe and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- M. Penna, K. Ley, S. Maclaughlin and I. Yarovsky, Faraday Discuss., 2016, 191, 435–464 RSC.
- M. J. Penna, M. Mijajlovic, C. Tamerler and M. J. Biggs, Soft Matter, 2015, 11, 5192–5203 RSC.
- J. Guo, X. Yao, L. Ning, Q. Wang and H. Liu, RSC Adv., 2014, 4, 9953–9962 RSC.
- X. Yin, B. Li, S. Liu, Z. Gu, B. Zhou and Z. Yang, RSC Adv., 2019, 9, 10094–10099 RSC.
- A. Keller and G. Grundmeier, Appl. Surf. Sci., 2020, 506, 144991 CrossRef CAS.
- K. Shezad, K. Zhang, M. Hussain, H. Dong, C. He, X. Gong, X. Xie, J. Zhu and L. Shen, Langmuir, 2016, 32, 8238–8244 CrossRef CAS PubMed.
- T. V. Sønderby, Y. Zou, P. Wang, C. Wang and D. E. Otzen, Biophys. J., 2022, 121, 3422–3434 CrossRef PubMed.
- L. Li-jun, W. Qi, W. Tao, S. Jia-wei and K. Yu, Chin. J. Chem. Phys., 2009, 22, 627 CrossRef.
- M. Zhou, Q. Shen, J.-W. Shen, L. Jin, L. Zhang, Q. Sun, Q. Hu and L. Liang, Colloids Surf., B, 2019, 174, 575–581 CrossRef CAS PubMed.
- A. S. Reddy, L. Wang, S. Singh, Y. L. Ling, L. Buchanan, M. T. Zanni, J. L. Skinner and J. J. de Pablo, Biophys. J., 2010, 99, 2208–2216 CrossRef CAS PubMed.
- Q. Qiao, G. R. Bowman and X. Huang, J. Am. Chem. Soc., 2013, 135, 16092–16101 CrossRef CAS PubMed.
- N. F. Dupuis, C. Wu, J.-E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2009, 131, 18283–18292 CrossRef CAS PubMed.
- K. Q. Hoffmann, M. McGovern, C.-C. Chiu and J. J. De Pablo, PLoS One, 2015, 10, e0134091 CrossRef PubMed.
- Y. Zhu, J. Guo, A. Zhang, L. Li, X. Liu, H. Liu and X. Yao, Environ. Res., 2019, 171, 1–10 CrossRef CAS PubMed.
- X. Yu, Q. Wang, Y. Lin, J. Zhao, C. Zhao and J. Zheng, Langmuir, 2012, 28, 6595–6605 CrossRef CAS PubMed.
- X. Wang, J. K. Weber, L. Liu, M. Dong, R. Zhou and J. Li, Nanoscale, 2015, 7, 15341–15348 RSC.
- Y. Zhang, Y. Liu, Y. Liu, P. Zuo, S. Miao, B. Hu, Y. Kang, W. Liu, Q. Yang, H. Ren and P. Yang, J. Am. Chem. Soc., 2023, 145, 17125–17135 CrossRef CAS PubMed.
- N. Todorova, A. J. Makarucha, N. D. Hine, A. A. Mostofi and I. Yarovsky, PLoS Comput. Biol., 2013, 9, e1003360 CrossRef PubMed.
- M. Barria-Urenda, A. Ruiz-Fernandez, C. Gonzalez, C. Oostenbrink and J. A. Garate, J. Chem. Inf. Model., 2023, 63, 6642–6654 CrossRef CAS PubMed.
- Z. E. Hughes and T. R. Walsh, J. Mater. Chem. B, 2015, 3, 3211–3221 RSC.
- C. Mucksch and H. M. Urbassek, PLoS One, 2014, 8, e64883 CrossRef PubMed.
- L. Ou, Y. Luo and G. Wei, J. Phys. Chem. B, 2011, 115, 9813–9822 CrossRef CAS PubMed.
- C. Liao and J. Zhou, J. Phys. Chem. B, 2014, 118, 5843–5852 CrossRef CAS PubMed.
- J. Wang, H. Dong, T. Leng, Y. Yu and Y. Li, Phys. Chem. Chem. Phys., 2022, 24, 4270–4279 RSC.
- Y. Chen, Z. Chen, Y. Sun, J. Lei and G. Wei, Nanoscale, 2018, 10, 8989–8997 RSC.
- Y. Bai, Z. Ming, Y. Cao, S. Feng, H. Yang, L. Chen and S.-T. Yang, Colloids Surf., B, 2017, 154, 96–103 CrossRef CAS PubMed.
- L. Baweja, K. Balamurugan, V. Subramanian and A. Dhawan, J. Mol. Graphics Modell., 2015, 61, 175–185 CrossRef CAS PubMed.
- M. Saeedimasine, E. G. Brandt and A. P. Lyubartsev, J. Phys. Chem. B, 2021, 125, 416–430 CrossRef CAS PubMed.
- S. A. Malik, Z. Mohanta, C. Srivastava and H. S. Atreya, Nanoscale Adv., 2020, 2, 1904–1912 RSC.
- L. Baweja, K. Balamurugan, V. Subramanian and A. Dhawan, Langmuir, 2013, 29, 14230–14238 CrossRef CAS PubMed.
- R. Hajiraissi, M. Hanke, Y. Yang, B. Duderija, A. Gonzalez Orive, G. Grundmeier and A. Keller, Langmuir, 2018, 34, 3517–3524 CrossRef CAS PubMed.
- Z. He, J. Li, S. H. Chen and R. Zhou, J. Phys. Chem. B, 2019, 123, 9098–9103 CrossRef CAS PubMed.
- A. Hung, S. Mwenifumbo, M. Mager, J. J. Kuna, F. Stellacci, I. Yarovsky and M. M. Stevens, J. Am. Chem. Soc., 2011, 133, 1438–1450 CrossRef CAS PubMed.
- A. Hung, M. Mager, M. Hembury, F. Stellacci, M. M. Stevens and I. Yarovsky, Chem. Sci., 2013, 4, 928–937 RSC.
- M. Penna and I. Yarovsky, Nanoscale, 2020, 12, 7240–7255 RSC.
- H. Niu, X. Hou, Y. Zhang, X. Wu, F. Deng, F. Huang, L. Shi and R. Ma, ACS Macro Lett., 2021, 10(6), 662–670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01315h |
| This journal is © The Royal Society of Chemistry 2024 |