DOI:
10.1039/D4NR01683A
(Paper)
Nanoscale, 2024,
16, 13019-13028
Shape-tunable two-dimensional assemblies from chromophore-conjugated crystallizable poly(L-lactides) with chain-length-dependent photophysical properties†
Received
17th April 2024
, Accepted 31st May 2024
First published on 11th June 2024
Abstract
This work reports temperature-dependent shape-changeable two-dimensional (2D) nanostructures by crystallization-driven self-assembly (CDSA) from a chromophore-conjugated poly(L-lactide) (PLLA) homopolymer (PTZ-P1) that contained a polar dye, phenothiazine (PTZ), at the chain-end of the crystallizable PLLA. The CDSA of PTZ-P1 in a polar solvent, isopropanol (iPrOH), by an uncontrolled heating–cooling process, majorly generates lozenge-shaped 2D platelets via chain-folding-mediated crystallization of the PLLA core, leading to the display of the phenothiazines on the 2D surface that confers colloidal stability and orange-emitting luminescent properties to the crystal lamellae. Isothermal crystallization at 60 °C causes a morphological change in PTZ-P1 platelets from lozenge to truncated-lozenge to perfect hexagon under different annealing times, while no shape change was noticed in the structurally similar PTZ-P2 polymer with a longer PLLA chain under similar conditions. This study unveils the complex link between the 2D platelet morphologies and degree of polymerization (DP) of PLLA and the corona-forming dye character. Further, the co-assembly potential of PTZ-P1 with hydrophobic pyrene-terminated PLLAs of varying chain lengths (PY-P1, PY-P2, and PY-P3) was examined, as these two dyes could form a Förster Resonance Energy Transfer (FRET) pair on the 2D surface. The impact of the length of the crystallizable PLLA on the photophysical properties of the surface-occupied chromophores revealed crucial insights into interchromophoric interactions on the platelet surface. A reduction in the propensity for π-stacking with increasing chain-folding in longer PLLAs is manifested in the chain-length-dependent FRET efficiencies and excimer emission lifetimes within the resultant monolayered 2D assemblies. The unconventional “butterfly-shaped” molecular architecture of the tested phenothiazine, combined with its varied functional features and polar character, adds a distinctive dimension to the underdeveloped field of CDSA of chromophore-conjugated poly(L-lactides), opening future avenues for the development of advanced nanostructured biodegradable 2D materials with programmable morphology and optical functions.
Introduction
Ultrathin organic two-dimensional (2D) materials1 of π-conjugated systems have attracted widespread attention due to their potential applications ranging from biomaterials2a to electronic devices.2b Due to the lack of effective and generally applicable strategies, controlling the shape, size, and molecular packing of many π-systems in two dimensions over a mesoscopic length scale represents a key challenge that dictates their potential functional output. A promising strategy for creating various hierarchical and anisotropic structures, including 2D arrangements in semicrystalline block copolymers (BCPs), involves the use of an efficient “crystallization-driven self-assembly” (CDSA)3 approach. In this process, chain-folding-mediated crystallization of the core-forming polymeric block in a corona-selective solvent compels the terminal corona-forming segment to locate on the 2D surface and provide colloidal stability. Thus far, the primary focus in this area has been to either customize the shapes of the nanostructures by playing with the relative volume fractions of the core- and/or corona-forming polymeric blocks or to precisely control the dimensions by a seeding approach called “living” CDSA,3 which is analogous to the emerging living supramolecular polymerization (LSP)4 technique recently explored in functional π-systems. Manipulating the functional output from crystalline polymeric 2D nanostructures has captured the attention of the scientific community only in very recent times. Recently, we reported CDSA as an unconventional supramolecular bottom-up strategy for the fabrication of monolayered 2D assemblies of π-systems from different chromophore-conjugated poly(L-Lactide) (PLLA) homopolymers.5 By judicially incorporating soluble corona-forming polar dyes such as naphthalene monoimide (NMI) or merocyanine (MC) at the chain-end of the crystallizable PLLA core in place of non-functional polymeric segments, we were successful in creating well-defined lozenge (diamond)-shaped crystalline lamellae with excellent colloidal stability and fluorescent properties in isopropanol (iPrOH). This is because the bulk solvent, due to its polar character, could effectively disperse the ordered 2D array of the polar dyes located on the platelet surface.5 Furthermore, Förster resonance energy transfer (FRET)5 properties could be achieved from the 2D surface of those crystalline nanosheets by incorporating suitable donor and acceptor FRET pairs. Despite our previous success in achieving predictable monolayered 2D assemblies of different π-systems, we were unable to control their shapes, unlike achievable in crystallizable BCPs, possibly due to the kinetically-driven crystallization process in the absence of a long solubilizing polymeric corona, leading to rigid and frozen structures. We anticipated that systematic variation in the polymer chain length in chromophore-appended PLLAs may offer exciting possibilities to tune their 2D morphology, as well as the photophysical properties of the surface-occupied terminal dyes by influencing their π-stacking, which is expected to be dependent on the extent of chain folding. In this work, we focus on investigating the role of different parameters such as temperature, chain length variation, and the character of the terminal dye on the crystallization kinetics, lamellae morphology, and photophysical properties in CDSA of chromophore-conjugated PLLA homopolymers.
We designed a new PLLA homopolymer (PTZ-P1) that featured a terminal phenothiazine (PTZ) dye as a soluble corona-forming moiety for the crystallizable PLLA core. Phenothiazine, a tricyclic heteroarene,6 has been widely employed in organic transistors,7 photovoltaics,8 and light-emitting diodes.9 It offers many advantages, such as extremely high hole mobility,10 chemical stability,11 and ease of functionalization at multiple positions.12 Extensive research has focused on the solid-state properties of phenothiazine derivatives, particularly their unique optical and electronic properties and flexibility of functionalization. Unlike conventional planar π-scaffolds, the hierarchical self-assembly of phenothiazine in solutions,13 particularly in two dimensions, is limited by its unique non-planar conformation characterized by a “butterfly” or bent-type structure.6 Herein, we addressed this gap by leveraging the CDSA technique, which offered a tunable platform for studying phenothiazine's self-assembled properties. PTZ-P1 could self-assemble into orange-emitting lozenge and truncated-lozenge-shaped 2D platelets by CDSA in iPrOH with monolayered thickness.14 We demonstrated morphological transformation from diamond to regular hexagon by optimizing the crystallization temperature and annealing time, maintaining a narrow dispersity of 1.02 for PTZ-P1. Building on these findings, we extended our investigation on co-assemblies of PTZ-P1 (acceptor) with pyrene-terminated PLLAs (donor) of different chain lengths (PY-P1, PY-P2, and PY-P3) to analyze their distance-dependent FRET properties. Our results reveal that the length of the donor chain has an unprecedented effect on the FRET response on the 2D surface. An increment in the FRET efficiency with a reduction in the donor chain length was observed, resulting in the highest achievable efficiency of ∼87% in PTZ-P1 + PY-P1 co-assembly.
Results and discussion
Synthesis of 2D platelets from phenothiazine-conjugated Poly(L-lactides)
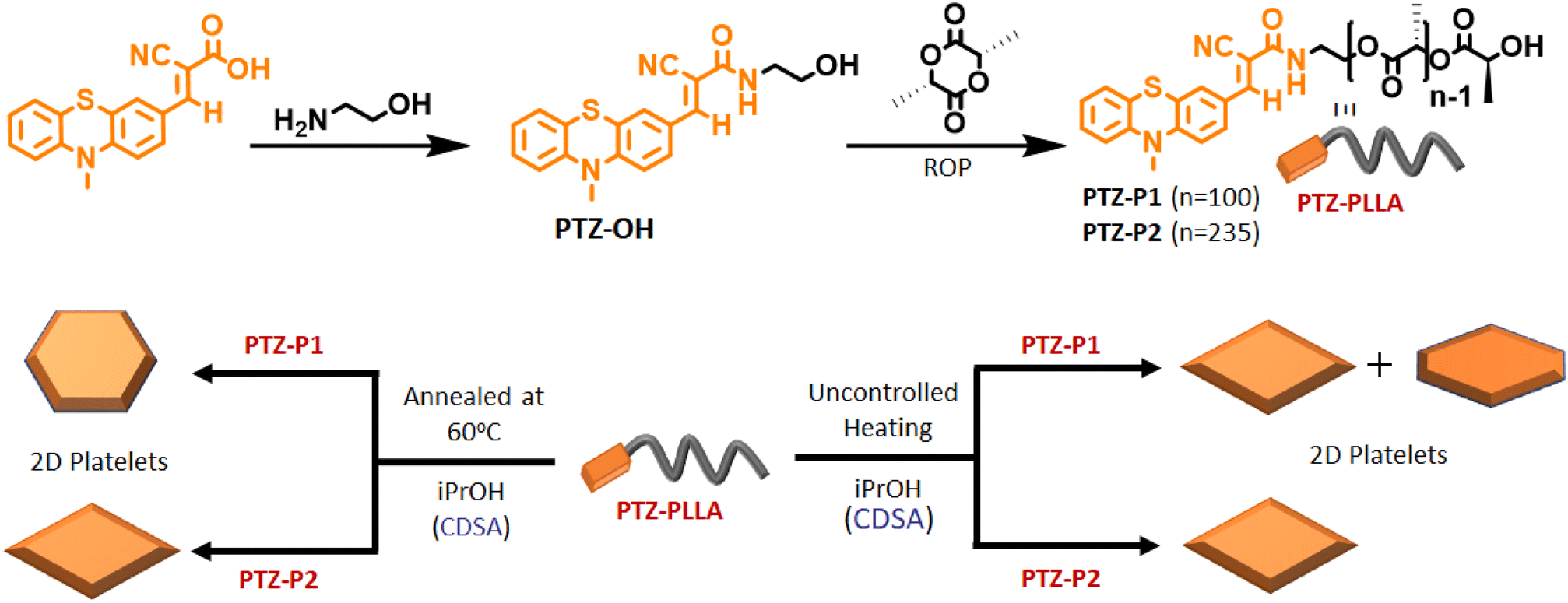
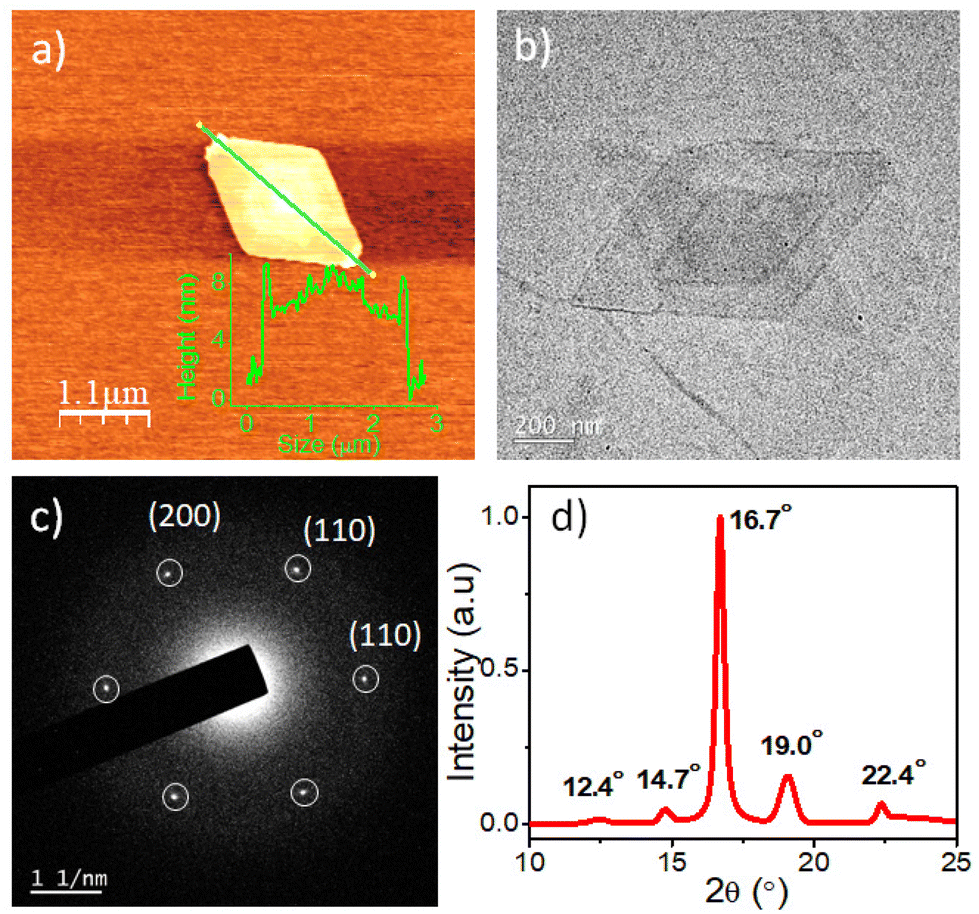
In this study, a hydroxyl-functionalized phenothiazine (PTZ-OH) (Scheme 1 and S1†) serves as a potent initiator in the synthesis of poly(L-lactide), PTZ-P1 by ring-opening polymerization (ROP). The size exclusion chromatography (SEC) showed a monomodal peak for PTZ-P1 (Fig. S1†). All the characteristic peaks of PTZ could be seen in the 1H-NMR spectrum of PTZ-P1 in CDCl3, which confirms the incorporation of the dye at the chain end of the homopolymer (Fig. S20 and Table S1†). From the end-group analysis, the degree of polymerization (DP) was determined to be 100, complying with the theoretical value. Next, the self-assembly of PTZ-P1 was studied in iPrOH. An unimer solution of PTZ-P1 in CHCl3 was slowly evaporated to obtain a thin film. Subsequent addition of iPrOH and vigorous heating of the solution with a heat gun followed by spontaneous cooling and aging for 30 minutes directly resulted in discrete lozenge (diamond)-shaped and truncated-lozenge-shaped 2D structures with an average size of ≈2.34 μm, as shown in the atomic force microscopy (AFM) images (Fig. 1a and S2a†). The height profile showed a ∼8 nm thickness of the 2D platelets, closely corresponding to ∼32 repeating units.15 This suggests the formation of monolayered crystalline lamellae, which is in agreement with previous reports where crystallization-driven self-assembly of PLLA is a contributing factor.14,15b No visual precipitation and structural homogeneity suggest appreciably high colloidal stability of the 2D platelets of PTZ-P1 in iPrOH. This is conferred by the surface-occupied PTZ dye, which, by its intramolecular electronic conjugation, exhibits a high polar character that facilitates its solvation in the polar solvent iPrOH,5a where the PLLA core is insoluble. Transmission electron microscopy (TEM) analysis showed the formation of similar 2D platelets with comparable size (Fig. 1b). Furthermore, the highly crystalline nature of the diamond platelets was confirmed by the selected area electron diffraction (SAED) analysis (Fig. 1c).14 The diffraction patterns reveal four (110) growth planes with d-spacings of 0.551 nm and two (200) planes with d-spacings of 0.557 nm. Similar diffraction spots have been previously reported for the solution-grown single crystals of PLLA and assigned to the orthorhombic α-form of PLLA.14 The wide-angle X-ray scattering (WAXRD) analysis (Fig. 1d) of the PLLA powder obtained by a slow removal of the solvent revealed distinct peaks at 2θ = 12.4° (103), 14.7° (010), 16.7° (110/200), 19.0° (203), and 22.4° (015), in accordance with the α-form of PLLA crystals.5,14,16, Additionally, the differential scanning calorimetry (DSC) thermogram from solid PTZ-P1 mass after removal of iPrOH, displays a single endothermic peak at 160 °C, corresponding to the melting point of the PLLA homocrystals (Fig. S3†).5b,15,16d
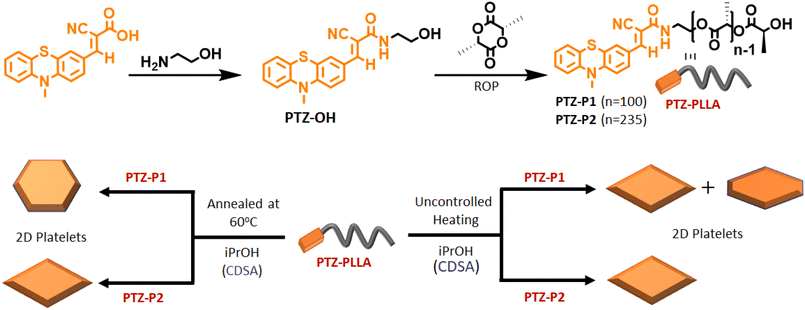 |
| | Scheme 1 Top: schematic representation of the synthesis of PTZ-PLLA by ROP; bottom: crystallization-driven self-assembly (CDSA) of poly(L-lactide) homopolymers, PTZ-P1 and PTZ-P2 in isopropanol (iPrOH) under different conditions, and morphological modulation in PTZ-P1 2D platelets from a mixture of orange-emitting lozenge (diamond) and truncated-lozenge to hexagon. | |
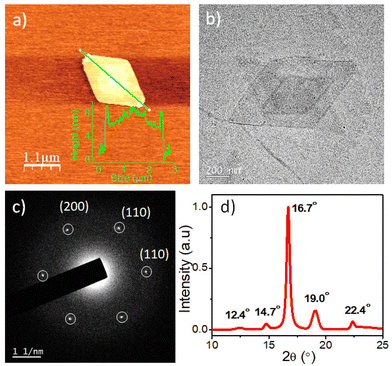 |
| | Fig. 1 (a) AFM image (inset: showing height profile of the 2D platelet) and (b) TEM image of the self-assembled diamond-shaped platelet of PTZ-P1; C = 0.05 mg mL−1; (c) Selected area electron diffraction (SAED) patterns obtained from the diamond platelet of PTZ-P1 in iPrOH; C = 0.05 mg mL−1; (d) WAXRD of PTZ-P1. The powdered sample was prepared by slowly drying its self-assembled dispersion in iPrOH. | |
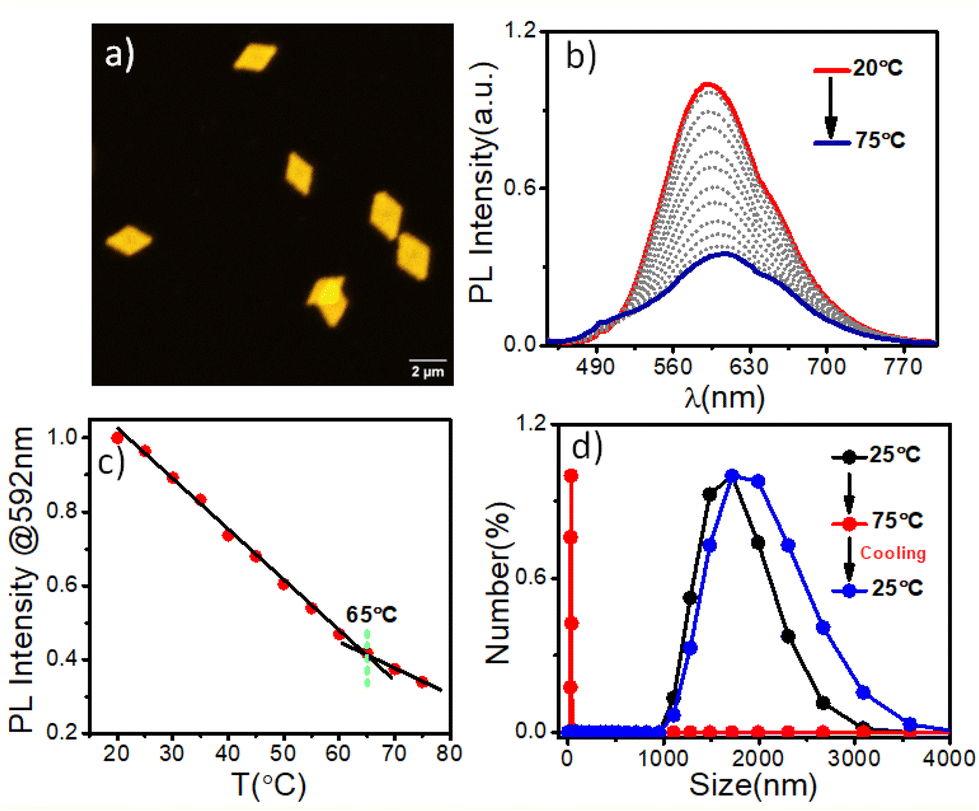
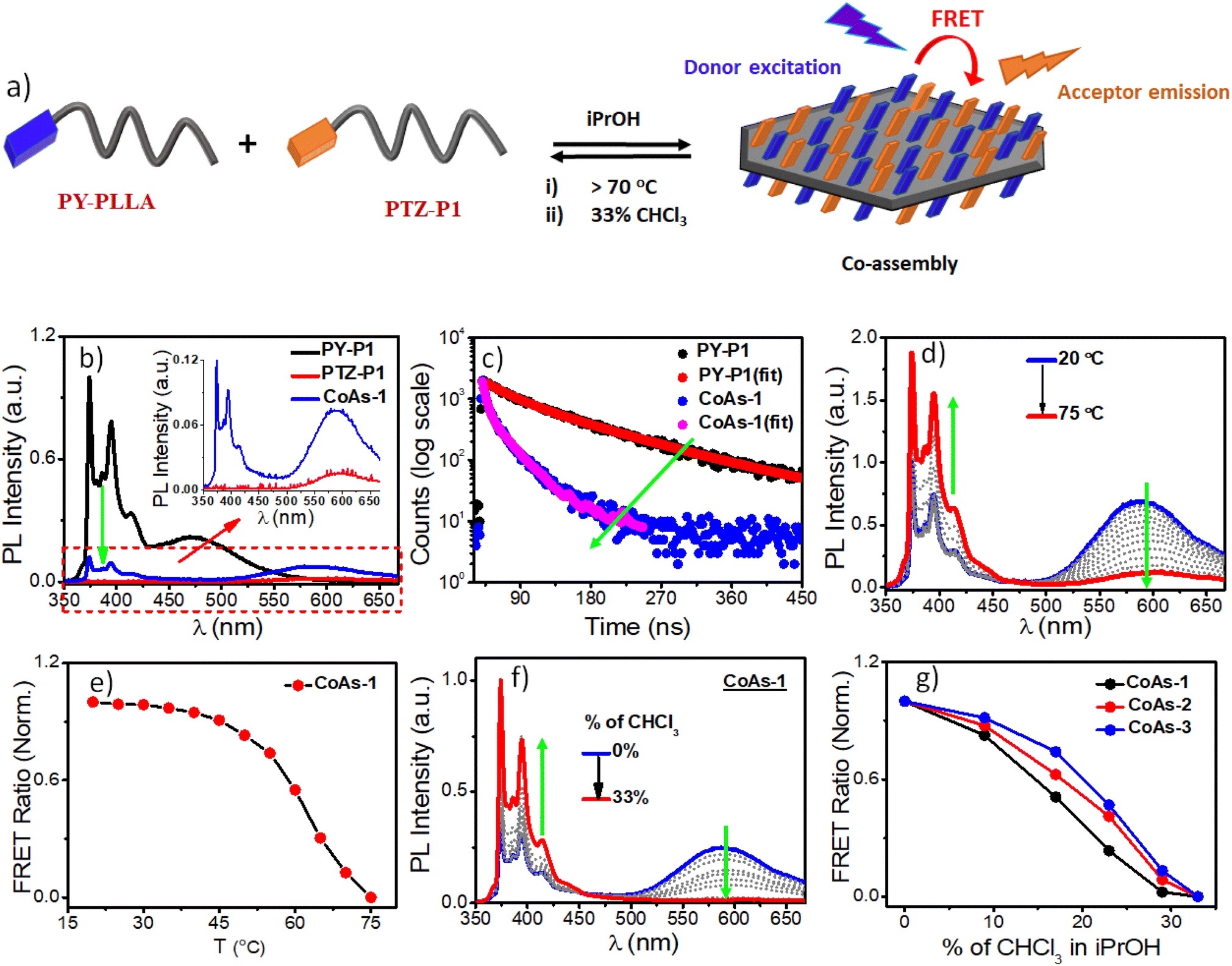
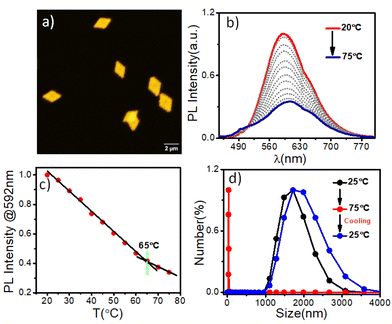
The terminally attached PTZ dye's fluorescent character additionally conferred luminescent properties to the crystalline PLLA platelets. Orange-emitting lozenge-shaped platelets, along with some truncated structures, could be clearly visualized by confocal laser scanning microscopy (CLSM) analysis (Fig. 2a and S2b†). The weight-average area (Aw) and number-average area (An) were measured to be 2.95 μm2 and 2.86 μm2, respectively, with a narrow dispersity of Aw/An = 1.03 (Fig. 2a and S2b†).5,14e,f Corroborating with the CLSM analysis, PTZ-P1 in iPrOH showed an emission band at λem = 592 nm (Fig. 2b), which is a result of high conjugation of the PTZ-chromophore due to its push–pull nature. The variable-temperature (VT) photoluminescence (PL) studies (Fig. 2b) demonstrate a sharp decrease in emission intensity with a rise in temperature, signifying the disassembly behavior of the 2D platelets. This trend continues until near saturation, marked by an inflection point at approximately 65 °C (Fig. 2c), denoted as the critical disassembly temperature of the 2D platelets. This is consistent with our recent findings on the disintegration temperature of other chromophore-functionalized PLLA diamond platelets in iPrOH.5a The enhanced emission at lower temperatures is attributed to the restricted motion of the fluorescent dyes in their confined 2D array. The dynamic light scattering (DLS) data (Fig. 2d) revealed a significant reduction in the aggregate size from ∼2 μm to ∼32 nm as the solution temperature was raised from 25 °C to 75 °C. This size reduction is consistent with the anticipated disassembly from the changes observed in the VT-PL studies. Interestingly, DLS analysis shows that the original size of the crystals can be restored upon cooling the solution back to 25 °C (Fig. 2d), indicating the reversible nature of the thermal disintegration process of the 2D platelets. The thermal disintegration of the 2D structures was further illustrated by capturing the CLSM images at 65 °C and 70 °C, which showed partial and complete disassembly, respectively (Fig. S5c–f†).
 |
| | Fig. 2 (a) CLSM image of self-assembled PTZ-P1 in iPrOH; C = 0.05 mg mL−1; (b) Variable-temperature (VT) photoluminescence (PL) studies of PTZ-P1 (λex = 430 nm); excitation and emission slit = 2.5 nm/2.5 nm; (c) PL-Intensity vs. temperature plot from (b); (d) VT-DLS data of PTZ-P1; C = 0.1 mg mL−1. | |
Morphological transformation
Next, we investigated the self-assembly of PTZ-P1 crystallized isothermally at a higher temperature under different annealing conditions. For that, PTZ-P1 in iPrOH was heated for 2 hours, 3 hours, and 4 hours at a constant temperature of 60 °C, subsequently cooled to room temperature, and then aged for 18 hours to allow the growth of single crystals. Notably, after annealing for 2 hours, the originally generated lozenge-shaped 2D platelets transformed into quasi hexagons and, after 3 hours and 4 hours, converted to nearly perfect hexagons with a very narrow dispersity of Aw/An = 1.02 (Fig. 3a–c and S4a–c†).14a,c,17 Separately, we have observed that the lozenge-shaped platelets that were initially obtained from an uncontrolled heating–cooling process can also transform into hexagonal platelets upon annealing at 60 °C for 4 hours (Fig. S4d†). The AFM image (Fig. 3d) confirmed a monolayered hexagonal structure that was comparable in thickness (∼7 nm) to the diamond-shaped crystalline lamellae generated from an uncontrolled heating–cooling process (Fig. 1a). However, the lamellae size (length between the two opposite vertices) increased from 2.5 μm to 4 μm with the change in the crystal morphology (Fig. 1a and 3d). The SAED data collected from the hexagonal platelet (Fig. 3f) revealed comparable diffraction patterns to the lozenge-shaped platelets (Fig. 1c), indicating similar crystal structures.14a,c As a result, the hexagonal structures of PTZ-P1 in iPrOH exhibit emission properties much like their diamond-shaped counterparts (Fig. S5a†), and both morphologies shared analogous disassembly temperatures as revealed from their VT-PL spectra (Fig. 2b and S5b†). Although less common, hexagonal-shaped crystals of PLLA are known to pack using both {110} and {100} growth planes and possess pseudo-hexagonal symmetry.14a,c,17 In contrast, the growth faces of the lozenge-shaped crystals are the four {110} planes with chain-folding along the {110} direction. The competition between growth rates along the {100} and {110} planes determines the ultimate morphology of polymer crystals of PLLA. The emergence of the truncated-face {100 plane} in PTZ-P1 crystals occurred during the uncontrolled heating–cooling crystallization process, and for truncated-lozenge structures, the {110} crystal planes grew faster than the {100} crystal planes. However, during isothermal crystallization at 60 °C, the truncation along the {100} crystal plane became active while the growth rate along the four {110} crystal planes diminished, and this process became more pronounced with increasing annealing time.14a–c,17c When the growth rates along the {100} and {110} crystal planes approached comparability, regular hexagons were formed from PTZ-P1, as seen after 3–4 hours of annealing. Consequently, the angles between all adjacent edges appear to be close to 120°. Thus, PLLA chains in hexagonal crystals correspond to those of the α-form with orthorhombic packing similar to its lozenge-shaped crystals.14a–c,17c This is also evidenced by the presence of comparable WAXRD (Fig. S6†) and SAED (Fig. 3f) patterns of PTZ-P1 in these two unique 2D nanostructures. Such temperature-driven morphological transitions associated with polymer crystals are known to be caused by the diffusion-controlled change in chain mobility,18 amongst many other parameters.19 We have synthesized another PTZ-functionalized PLLA (PTZ-P2) with a longer chain length (DP = 235) while keeping other parameters unaltered. A dilute solution of PTZ-P2 in iPrOH also produced diamond-shaped platelets by uncontrolled heating–cooling method, while its annealed sample at 60 °C for 4 hours did not exhibit any signs of truncation (Fig. S7a–c and S8a†). No morphological change in the case of the longer chain-length PTZ-P2 (DP = 235) is explained by its slower diffusion-controlled mobility at 60 °C as compared to the shorter chain-length PTZ-P1 (DP = 100) (Fig. S8a†). Our hypothesis was supported by the fact that when PTZ-P2 was further heated to 80 °C for 4 hours, it gained some chain mobility to grow bigger crystals that show truncated faces (Fig. S8b–d†). However, after 20 hours of prolonged heating at 80 °C (Fig. S8e–g†), smaller-sized diamond platelets were regenerated, possibly resulting from the dissolution of the PLLA chains from the edges of the initially formed bigger crystals. This suggests an optimal annealing temperature and time are essential for crystal growth and its morphological transition.
 |
| | Fig. 3 CLSM images of single crystals of PTZ-P1 homopolymer in iPrOH obtained at a constant nucleation temperature (60 °C) and varying annealing time as indicated in the figures: (a) t = 2 h; (b) t = 3 h; (c) t = 4 h; (d) AFM image and (e) TEM image of 4 h annealed sample of PTZ-P1 in iPrOH at 60 °C; C = 0.05 mg mL−1; (f) Selected area electron diffraction (SAED) patterns obtained from the hexagonal platelet of PTZ-P1 in iPrOH; C = 0.05 mg mL−1. | |
Chain-length variation: impact on 2D morphology and photophysical properties of pyrene-conjugated poly(L-lactides)
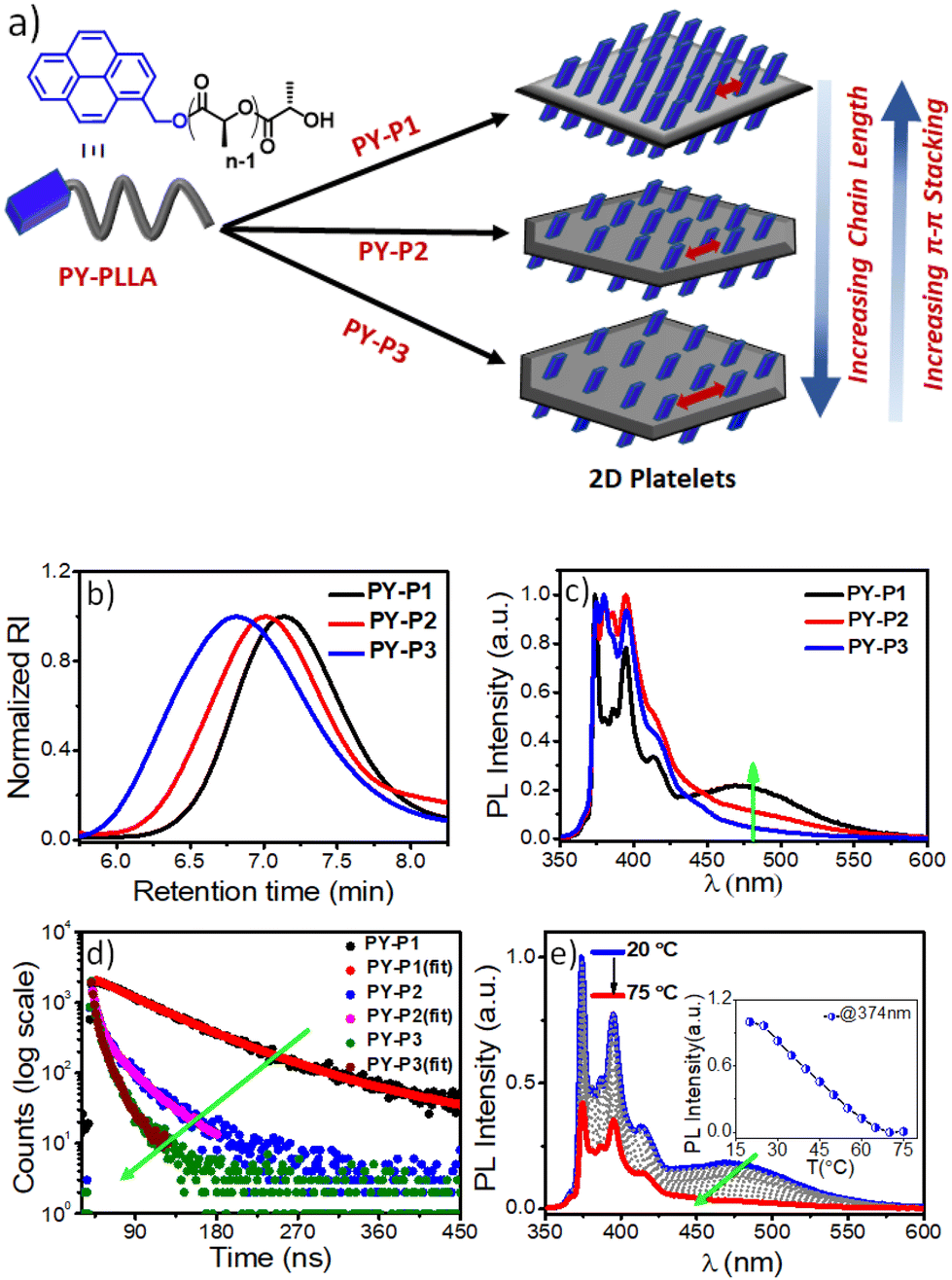
We envisaged that the CDSA approach offers a unique advantage in investigating the chain-length-dependent FRET responses from the 2D surface of the co-assembled nanostructures, which had not been investigated earlier. We anticipated that pyrene could participate as an efficient FRET donor for the tested phenothiazine acceptor due to their required spectral overlap (Fig. S9†). With this objective, we synthesized pyrene-appended PLLAs, i.e., PY-P1, PY-P2, and PY-P3, with DPs of 93, 245, and 800, respectively (Table S1†), by ROP of L-lactide using PY-OH as an initiator. A monomodal peak was observed for all the pyrene derivatives in THF in size-exclusion chromatography (SEC) with a moderate dispersion of 1.3–1.4 (Fig. 4b).5a Before investigating the FRET properties in the co-assemblies, we first studied the impact of chain-length variation on the self-assembly of PY-P1, PY-P2, and PY-P3. All pyrene-conjugated PLLAs produced 2D platelets that show poor colloidal stability and tend to precipitate in iPrOH after a few hours, in contrast to the stable and uniform structures seen in the case of PLLAs chain-terminated with polar dyes like phenothiazine and previously studied NMI or merocyanines.5a A dilute solution of PY-P1, PY-P2, and PY-P3 produced crystalline lamellae of diamond, truncated-diamond, and pseudo-hexagon shapes (Fig. S10†), respectively, indicating that longer pyrene-end-capped PLLA chains are more prone to truncation due to the faster growth along {100} planes.15b It is interesting to note that the morphology of the 2D platelets generated from CDSA of the phenothiazine-functionalized PLLAs showed a reverse trend as compared to the previous literature knowledge on block copolymers of enantiopure polylactides15b and our own findings with hydrophobic pyrene-end-capped PLLAs, i.e. the platelet morphology of PTZ-P2 with longer PLLA chains shows no sign of truncation at ambient temperature or even at 60 °C, unlike the shorter one. In CDSA of block copolymers, the ultimate morphology is governed by many factors like polymer composition, structure of the corona, solvent-corona interactions, the presence of cosolvent, and different self-assembly conditions.3,14 For CDSA of the chromophore-conjugated PLLAs with small molecule-based corona (where the steric component is minimal), under a given condition in a particular solvent, the crystallization kinetics will be dictated by both the corona solvation and degree of core crystallization, which are likely to be influenced by the nature of the terminal chromophore and the crystallizable polymer chain length, respectively. As the extent of interaction between the corona-forming dye and the polar solvent (iPrOH) will be different for phenothiazine (polar) and pyrene (nonpolar), the repeating unit variation does not exert the same impact on the polymer crystallization kinetics in the CDSA of these two types of chromophore-conjugated PLLAs. Thus, our results unravel the remarkable role that a single corona-forming π-scaffold has in CDSA, which goes beyond stabilizing the 2D structure and conferring it with distinct surface properties. The terminal dye plays a pivotal role in the crystallization process and crystal shape, which is comparable to the impact of solvent-selective non-crystallizable polymeric segments in the CDSA of block copolymers.
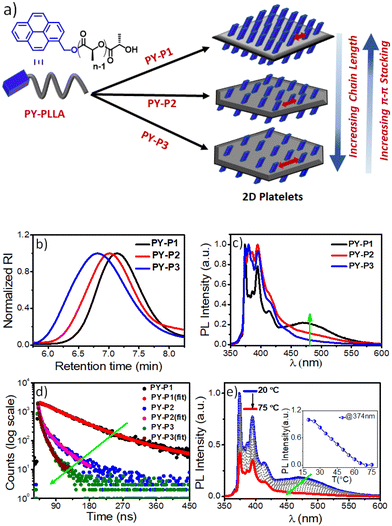 |
| | Fig. 4 (a) Schematic diagram of homocrystals of PY-PLLA with three different chain length; (b) SEC plots of PY-P1, PY-P2 and PY-P3 in THF; (c) Photoluminescence (PL) spectra (normalized) of PY-P1, PY-P2 and PY-P3 in iPrOH; λex = 337 nm, C = 0.1 mg mL−1; (d) Time-resolved fluorescence decay profiles of PY-P1, PY-P2 and PY-P3 at 472 nm (λex = 337 nm); (e) Changes in the emission spectra of self-assembled PY-P1 in iPrOH as a function of temperature; Inset: plot of the PY-P1 emission intensity (λem = 374 nm) vs. temperature, C = 0.1 mg mL−1, pathlength = 10 mm, excitation and emission slit = 1 nm/1 nm; λex = 337 nm. | |
Pyrene, a spatially sensitive probe, exhibits an excimer band at ∼472 nm when two fluorophores are in close spatial proximity, in addition to monomeric fluorescence emission peaks (373–405), and the excimer band intensity depends on the distance of separation between the pyrenes and the π–π overlap.20 As the density of the corona-forming segment on the platelet surface is dependent on the length of the polymer chain, we anticipated that the increased number of chain-folding in higher DP PLLA will reduce the surface density of pyrene, leading to less probable spatial proximity of the chromophores for π-stacking interactions on the 2D surface as compared to the PLLA with shorter chain length. This can be manifested by comparing the excimer/monomer (e/m) ratio in the three polymers PY-P1, PY-P2, and PY-P3. The emission spectrum of PY-P1 exhibited a broad excimer band at 472 nm (Fig. 4c), attributed to π-stacking interactions among the terminally-linked pyrenes.20 The excimer band was notably reduced in PY-P2 and became almost negligible in PY-P3, suggesting a chain length-dependent phenomenon. This is apparent from the e/m ratio that follows the trend PY-P1 (0.23) > PY-P2 (0.12) > PY-P3 (0.05).21 Time-correlated single photon counting (TCSPC) studies further confirmed this, revealing a drastic reduction in the excimer band lifetime22 from 100.00 ns to 25.85 ns to 8.40 ns for PY-P1, PY-P2, and PY-P3 respectively (Fig. 4d and Table S3†), indicative of insignificant π–π-interactions in PY-P2 and PY-P3. To validate our hypothesis and eliminate any changes caused by intrinsic differences in the dye concentration in these three polymers, we conducted a control experiment with the free dye (PY-OH) at a concentration equivalent to the pyrene concentration in PY-P1 (Fig. S11†). Under the same conditions, no excimer band was observed from the dye without the polymer chain (Fig. S11†), emphasizing that the π-stacking of pyrene in iPrOH and subsequent excimer band formation were an outcome of the CDSA. In further investigations, we conducted a dilution experiment in iPrOH using chloroform (CHCl3) as a good solvent for disassembly of the preformed 2D platelets of PY-P1. The gradual addition of CHCl3 resulted in a reduction in the intensity of the excimer band. Notably, with 33% CHCl3, the excimer emission completely disappeared, affirming that it originates from the 2D platelets obtained through CDSA (Fig. S12†). VT-photoluminescence studies reveal similar results. As temperature ascends from 20 °C to 75 °C, the excimer band progressively diminishes for PY-P1. The monomer band intensity (λ = 374 nm) vs. temperature plot reveals a consistent disassembly temperature of 70 °C for PY-P1 (Fig. 4e), closely matching the disassembly temperature observed for PY-P2 and PY-P3 (Fig. S13b–d†), as well as PTZ-P1 (Fig. 2c). This synchronized disassembly behavior across different dye-appended PLLAs underscores the thermal sensitivity of these 2D structures across varying tested chain lengths.
Chain-length variation: impact on FRET response in two-component 2D assemblies
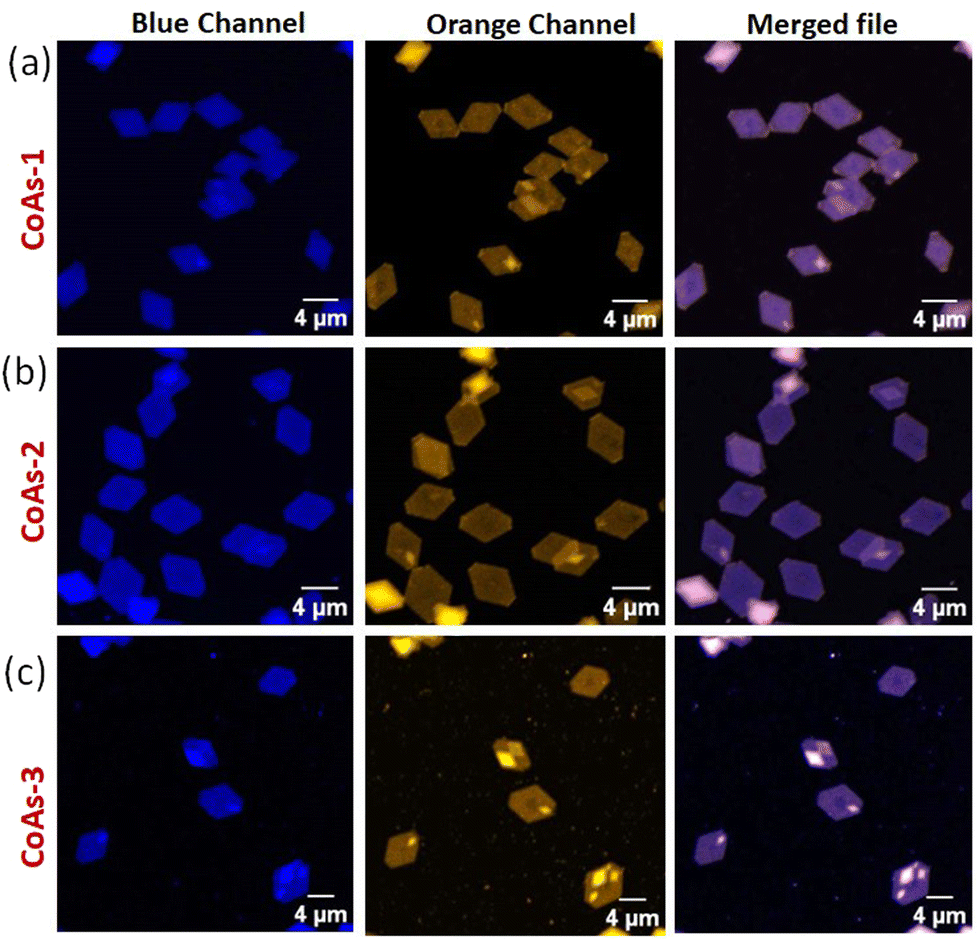
Next, we wanted to explore the impact of polymer chain-length variation on the FRET properties in two-component CDSA of PTZ-P1 polymer with pyrene-functionalized PY-P1, PY-P2, and PY-P3 having different repeating units. Two-dimensional co-assemblies of PY-P1, PY-P2, and PY-P3 with PTZ-P1 prepared by following the previously mentioned uncontrolled heating–cooling process in iPrOH were assigned as CoAs-1, CoAs-2, CoAs-3 respectively. The CLSM images (Fig. 5) unveiled majorly truncated lozenge-shaped platelets with morphological consistency across all tested co-assembled structures (CoAs-1, CoAs-2, and CoAs-3) and emitting in blue and orange under selective excitation at their respective wavelengths. Intriguingly, the perfect overlay of the two distinct colors in the merged image revealed dual emission (Fig. 5) from the crystalline 2D lamellae. This transformation provided clear evidence of the co-localization of the two dyes (PY and PTZ) on the platelet surface. While the CLSM investigation offered visual confirmation of the presence and co-localization of the two chromophores on the surface of the 2D crystallites, we relied upon FRET studies to obtain valuable information about the spatial interaction between the donor and acceptor dyes. Remarkably, we noted a substantial overlap between the absorption spectrum of PTZ-P1 and the emission spectrum of PY-P1, establishing them as a FRET pair (Fig. S13a†), laying the foundation for further exploration of the chain-length dependent FRET properties within the three studied systems, i.e., CoAs-1, CoAs-2, and CoAs-3. As FRET is highly sensitive to the distance between the two communicating dyes (which has to be within 10 nm),22a,23 we envisaged that for the same FRET pair, the energy transfer efficiency can be tuned by varying the distance between the dyes through alteration in the PLLA chain length. Upon photoexcitation of CoAs-1 at the absorption wavelength of the donor PY-P1 (λex = 337 nm), we observed quenching in the pyrene emission (λem = 374 nm) with complete disappearance of the excimer band (Fig. 6b). Intriguingly, this was accompanied by a 5-fold enhancement in the emission intensity of the acceptor PTZ-P1 (λem = 592 nm) compared to the direct acceptor emission at λex = 337 nm (Fig. 6b). An impressively high FRET efficiency (E = 1 − IDA/ID) of 88% was observed (Fig. 6b), where IDA and ID represent the emission intensity of PY-P1 (λem = 374 nm) in the presence and absence of the PTZ-P1 acceptor, respectively. This was validated by the fluorescence lifetime measurement by time-correlated single photon counting (TCSPC) experiments (Fig. 6c).22 The average lifetime of the donor dye in PY-P1 (λex = 280 nm) was determined to be τD = 109.91 ns. Significantly, its lifetime was markedly reduced to 14.64 ns (τDA) within the CoAs-1 (Table S2†), providing clear evidence of energy transfer from PY-P1 to PTZ-P1. The energy transfer efficiency (E = 1 − τDA/τD) from the lifetime measurements (Fig. 6c) yielded a value of ∼87% complementing with the steady-state data, where τDA and τD represent the lifetime of pyrene in PY-P1 (λem = 374 nm) in the presence and absence of the PTZ-P1 polymer, respectively. To further probe that the FRET originating from the 2D surface is a consequence of the CDSA of the PLLA chain, we conducted a dilution experiment in CoAs-1 using a corona-selective cosolvent, CHCl3. With the gradual addition of CHCl3 (v/v) in iPrOH, the emission intensity of PTZ-P1 in CoAs-1 decreased concurrently with an increase in the PY-P1 emission (Fig. 6f and g). The disappearance of FRET, indicated by the alteration in emission intensities, upon the addition of CHCl3 further confirmed the sensitivity of these crystalline lamellae to the presence of a good solvent, leading to the disruption of the organized chromophore assembly. Additionally, we explored the effect of temperature on the FRET response, which is expected to be dependent on the thermal stability of the 2D structures. With a gradual increase in temperature from 25 °C to 75 °C, FRET started diminishing in CoAs-1 and completely ceased above 70 °C, suggesting temperature-dependent disassembly of the 2D platelets (Fig. 6d and e). This study further underscores the occurrence of nonradiative energy transfer from the donor to the acceptor polymer on the surface of the 2D co-platelet with reasonably high thermal stability. To investigate any effect of variation in the donor polymer chain length on the FRET response, similar studies were performed with CoAs-2 and CoAs-3 (Fig. S14 and S15†). A reduction in the average lifetime of PY-P2 in CoAs-2 from 74.65 ns to 22.52 ns and PY-P3 in CoAs-3 from 73.10 ns to 33.00 ns (Fig. S14a, b and Table S2†) validated similar energy transfer processes occurring in CoAs-2 (∼70%) and CoAs-3 (∼55%), however, with reduced efficiencies as compared to CoAs-1 (∼87%). A reverse correlation of the FRET efficiency and excimer formation (Fig. 4a and c) with respect to the ascending donor chain length going from PY-P1 to PY-P2 to PY-P3 can be attributed to the diminishing interchromophoric interactions on the 2D surface. The extended spatial distance induced by longer polymer chains creates a less favorable environment for close proximity and intimate interaction between the donor and acceptor chromophores, resulting in a decreased probability of energy transfer events in CoAs-2 and CoAs-3. We conducted further assessments by measuring the FRET distances24 in CoAs-1 (1.98 nm), CoAs-2 (2.23 nm), and CoAs-3 (2.46 nm), which align with the proposed hypothesis. Further, the temperature- and cosolvent-dependent FRET studies reveal that all three donor–acceptor co-assembled 2D platelets exhibit comparable thermal stability and complete disassembly above 70 °C (Fig. S15c†) or ∼33% CHCl3 addition (Fig. 6g). However, the tolerance for the cosolvent becomes slightly higher as the donor chain length increases in the two-component 2D platelets (Fig. 6g).
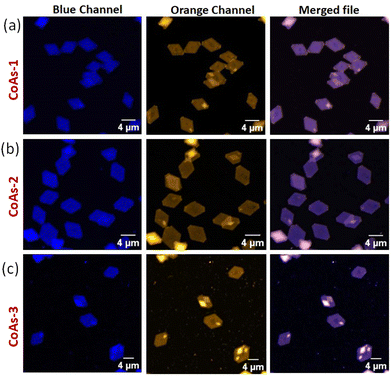 |
| | Fig. 5 CLSM images of co-assembly of (a) PY-P1 + PTZ-P1 (CoAs-1); (b) PY-P2 + PTZ-P1 (CoAs-2), and (c) PY-P3 + PTZ-P1 (CoAs-3); [PY-P1] = [PY-P2] = [PY-P3] = [PTZ-P1] = 0.05 mg mL−1. | |
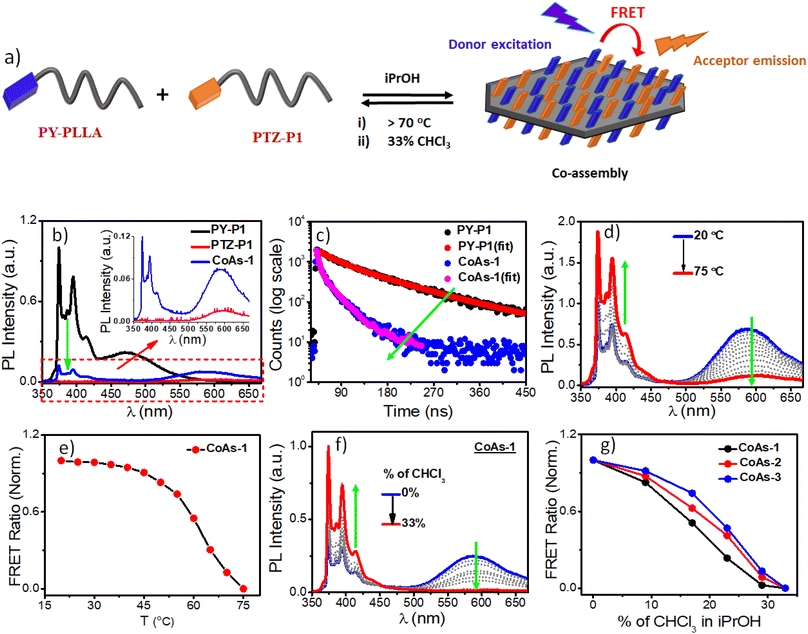 |
| | Fig. 6 (a) A schematic demonstration of Förster Resonance Energy Transfer (FRET) in crystallization-driven co-assembly of PY-PLLA and PTZ-P1 on the surface of 2D platelets; (b) Emission spectra of PY-P1, PTZ-P1 and their 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-assembly in iPrOH; λex = 337 nm, slit = 1 nm/1 nm; (c) Time-resolved fluorescence decay profiles of PY-P1 and (1:1) PY-P1 + PTZ-P1 co-assembly (CoAs-1); (d) Variable-Temperature (VT) PL studies of CoAs-1 (λex = 337 nm, slit = 1.5 nm/1.5 nm) from 20 °C to 75 °C, 5° interval; (e) Plot of FRET ratio of CoAs-1 as a function of temperature (normalized); (f) Changes in the emission spectra of 1:1 PY-P1 + PTZ-P1 co-assembly (CoAs-1) in iPrOH as a function of CHCl3 addition; λex = 337 nm, slit = 1.5 nm/1.5 nm; (g) Plot of FRET ratio (IA/IA + ID) vs. % of CHCl3 in iPrOH, where IA and ID represent emission of PTZ-P1 (λem = 592 nm) and PY-P1, PY-P2, and PY-P3 (λem = 374 nm), respectively. :1 co-assembly in iPrOH; λex = 337 nm, slit = 1 nm/1 nm; (c) Time-resolved fluorescence decay profiles of PY-P1 and (1:1) PY-P1 + PTZ-P1 co-assembly (CoAs-1); (d) Variable-Temperature (VT) PL studies of CoAs-1 (λex = 337 nm, slit = 1.5 nm/1.5 nm) from 20 °C to 75 °C, 5° interval; (e) Plot of FRET ratio of CoAs-1 as a function of temperature (normalized); (f) Changes in the emission spectra of 1:1 PY-P1 + PTZ-P1 co-assembly (CoAs-1) in iPrOH as a function of CHCl3 addition; λex = 337 nm, slit = 1.5 nm/1.5 nm; (g) Plot of FRET ratio (IA/IA + ID) vs. % of CHCl3 in iPrOH, where IA and ID represent emission of PTZ-P1 (λem = 592 nm) and PY-P1, PY-P2, and PY-P3 (λem = 374 nm), respectively. | |
Conclusions
In summary, crystallization-driven self-assembly (CDSA) from dipolar phenothiazine end-capped poly(L-lactide) (PLLA) homopolymers (PTZ-P1/PTZ-P2) has been demonstrated in iPrOH. This results in colloidally stable discrete and monolayered, lozenge-shaped, and truncated-lozenge-shaped 2D platelets with surface-decorated phenothiazine dyes, leading to previously unexplored orange emission from the 2D structures. Notably, a temperature-dependent morphological transition from lozenge-shaped 2D platelets to perfect hexagons could be achieved with structural precision and narrow dispersion (1.02) in the shorter chain length PTZ-P1. Furthermore, we examined the potential effects of introducing pyrene (donor), a second dye with a FRET relationship with the phenothiazine (acceptor) moiety being studied, on the surface properties of the resulting crystalline 2D platelets. An important understanding of the correlation between the crystallizable PLLA chain length and the spatial proximity of the surface-occupied chromophores for π-stacking was gained by studying the effects of varying degrees of polymerization in PY-P1, PY-P2, and PY-P3 on the excimer emissions in their respective homoplatelets and FRET efficiencies in the co-platelets (CoAs-1, CoAs-2, and CoAs-3) with PTZ-P1, which is a significant new development in CDSA of chromophore-conjugated crystallizable polymers. Furthermore, we showed that the 2D platelet morphology depends not only on external factors like temperature and annealing times in a specific crystallizable solvent (iPrOH) but also on intrinsic structural parameters like the length of the PLLA chain and the nature of the corona-forming terminal π-scaffold, which potentially influence the unimer-to-nuclei ratio through varying degrees of corona-solvent interactions. This knowledge enriches our toolbox for generating distinct hierarchical 2D assemblies of π-conjugated systems with consistent properties by controlling different crystallization parameters in PLLA scaffolds. Overall findings of this work advance our understanding of the intricate correlation between chemical structure, supramolecular assembly, and resulting photophysical attributes in CDSA of chromophore-conjugated crystallizable poly(L-lactides), demonstrating its potential as a versatile tool for the design and engineering of functional biodegradable 2D materials with customized optical behaviors from structurally diverse π-systems.
Author contributions
C. C. carried out the synthesis and characterization of the polymers and conducted the experiments. C. C. and A. R. analyzed the data. All authors contributed to the manuscript preparation. A. D. conceptualized the project, raised research funding, and supervised the entire work. All authors have approved the final version of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
C. C. thanks UGC-India for fellowship. A. R. thanks CSIR-India for fellowship. A. D. thanks BRNS, DAE (grant no. 58/20/10/2022-BRNS/37054), SERB CRG (grant no. CRG/2022/003069), and the Technical Research Centre (TRC) of IACS for funding. The authors thank Central Scientific Services (CSS) at IACS for the instrumental facilities.
References
-
(a) G. Fernández, F. García, F. Aparicio, E. Matesanz and L. Sánchez, Chem. Commun., 2009, 7155 RSC;
(b) F. Yang, S. Cheng, X. Zhang, X. Ren, R. Li, H. Dong and W. Hu, Adv. Mater., 2018, 30, 1702415 CrossRef;
(c) A. D. Merg, E. van Genderen, A. Bazrafshan, H. Su, X. Zuo, G. Touponse, T. B. Blum, K. Salaita, J. P. Abrahams and V. P. Conticello, J. Am. Chem. Soc., 2019, 141, 20107 CrossRef CAS;
(d) R. R. Liang, S. Y. Jiang, R. Han A and X. Zhao, Chem. Soc. Rev., 2020, 49, 3920 RSC.
-
(a) X. Zhang, C. Gong, O. U. Akakuru, Z. Su, A. Wu and G. Wei, Chem. Soc. Rev., 2019, 48, 5564 RSC;
(b) M. C. Lemme, D. Akinwande, C. Huyghebaert and C. Stampfer, Nat. Commun., 2022, 13, 1392 CrossRef CAS.
-
(a) S. Agbolaghi, S. Abbaspoor and F. Abbasi, Prog. Polym. Sci., 2018, 81, 22 CrossRef CAS;
(b) S. Ganda and M. H. Stenzel, Prog. Polym. Sci., 2020, 101, 101195 CrossRef CAS;
(c) L. MacFarlane, C. Zhao, J. Cai, H. Qiu and I. Manners, Chem. Sci., 2021, 12, 4661 RSC;
(d) C. Yang, Z.-X. Li and J.-T. Xu, J. Polym. Sci., 2022, 60, 2153 CrossRef CAS;
(e) Y. Cha, C. Jarrett-Wilkins, M. A. Rahman, T. Zhu, Y. Sha, I. Manners and C. Tang, ACS Macro Lett., 2019, 8, 835 CrossRef CAS;
(f) X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644 CrossRef CAS;
(g) H. Qiu, Y. Gao, C. E. Boott, O. E. Gould, R. L. Harniman, M. J. Miles, S. E. Webb, M. A. Winnik and I. Manners, Science, 2016, 352, 697 CrossRef CAS;
(h) T. Xia, Z. Tong, Y. Xie, M. C. Arno, S. Lei, L. Xiao, J. Y. Rho, C. T. J. Ferguson, I. Manners, A. P. Dove and R. K. O'Reilly, J. Am. Chem. Soc., 2023, 145, 25274 CrossRef CAS;
(i) Xu Zhang, G. Chen, L. Liu, L. Zhu and Z. Tong, Macromolecules, 2022, 55, 8250 CrossRef CAS.
-
(a) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188 CrossRef CAS;
(b) R. D. Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241 CrossRef CAS;
(c) J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646 CrossRef CAS;
(d) W. Wagner, M. Wehner, V. Stepanenko, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 16008 CrossRef CAS;
(e) S. Yagai, Y. Kitamoto, S. Datta and B. Adhikari, Acc. Chem. Res., 2019, 52, 1325 CrossRef CAS;
(f) J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730 CrossRef CAS;
(g) G. Ghosh, S. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757 RSC;
(h) B. Adelizzi, N. J. Van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110 CrossRef CAS;
(i) A. Sarkar, R. Sasmal, C. Empereur-mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, B. Sundaram, S. S. Agasti, G. M. Pavan and S. J. George, J. Am. Chem. Soc., 2020, 142, 7606 CrossRef CAS;
(j) P. Khanra, A. K. Singh, L. Roy and A. Das, J. Am. Chem. Soc., 2023, 145, 5270 CrossRef CAS;
(k) P. Khanra, P. Rajdev and A. Das, Angew. Chem., Int. Ed., 2024, e202400486 CAS;
(l) H. Itabashi, K. Tashiro, S. Koshikawa, S. Datta and S. Yagai, Chem. Commun., 2023, 59, 7375 RSC.
-
(a) A. Rajak and A. Das, Angew. Chem., Int. Ed., 2022, 61, e20211657 Search PubMed;
(b) A. Rajak and A. Das, Angew. Chem., Int. Ed., 2023, 62, e2023142 CrossRef.
-
(a) J. Bell, J. Blount, O. Briscoe and H. Freeman, Chem. Commun., 1968, 1656 RSC;
(b) S. P. Massie, Chem. Rev., 1954, 54, 797 CrossRef CAS;
(c) P. S. Gangadhar, G. Reddy, S. Prasanthkumar and L. Giribabu, Phys. Chem. Chem. Phys., 2021, 23, 14969 RSC.
- W. Zhou, Y. Wen, L. Ma, Y. Liu and X. Zhan, Macromolecules, 2012, 45, 4115 CrossRef CAS.
- S. Zimosz, A. Slodek, P. Gnida, A. Glinka, M. Ziółek, D. Zych, A. K. Pająk, M. Vasylieva and E. Schab-Balcerzak, J. Phys. Chem. C, 2022, 126, 8986 CrossRef CAS.
- J. K. Salunke, F. L. Wong, K. Feron, S. Manzhos, M. F. Lo, D. Shinde, A. Patil, C. S. Lee, V. A. L. Roy, P. S. Prakash and P. Wadgaonkar, J. Mater. Chem. C, 2016, 4, 1009 RSC.
-
(a) K. D. Thériault and T. C. Sutherland, Phys. Chem. Chem. Phys., 2014, 16, 12266 RSC;
(b) S. Thokala and S. P. Singh, ACS Omega, 2020, 5, 5608 CrossRef CAS.
- L. M. Sigmund, F. Ebner, C. Jöst, J. Spengler, N. Gönnheimer, D. Hartmann and L. Greb, Chem. – Eur. J., 2020, 26, 3152 CrossRef CAS.
-
(a) Y. Rout, A. Ekbote and R. Misra, J. Mater. Chem. C, 2021, 9, 7508 RSC;
(b) S. Revoju, A. Matuhina, L. Canil, H. Salonen, A. Hiltunen, A. Abate and P. Vivo, J. Mater. Chem. C, 2020, 8, 15486 RSC.
-
(a) C. Arivazhagan, S. Satapathy, A. Jana, P. Malakar, E. Prasad and S. Ghosh, Chem. – Eur. J., 2018, 24, 13213 CrossRef CAS;
(b) J. Gong, M. Yu, C. Wang, J. Tan, S. Wang, S. Zhao, Z. Zhao, A. Qin, B. Tang and X. Zhang, Chem. Commun., 2019, 55, 10768 RSC;
(c) S. Suganya, K. Debsharma, E. Ravindran, M. Mahato and E. Prasad, ACS Appl. Polym. Mater., 2020, 2, 1222 CrossRef CAS;
(d) N. V. Lakshmi, T. M. Babu and E. Prasad, Chem. Commun., 2016, 52, 617 RSC.
-
(a) T. Iwata and Y. Doi, Macromolecules, 1998, 31, 2461 CrossRef CAS;
(b) J. X. Zheng, H. Xiong, W. Y. Chen, K. Lee, R. M. V. Horn, R. P. Quirk, B. Lotz, E. L. Thomas, A.-C. Shi and S. Z. D. Cheng, Macromolecules, 2006, 39, 641 CrossRef CAS;
(c) J. Yang, T. Zhao, Y. Zhou, L. Liu, G. Li, E. Zhou and X. Chen, Macromolecules, 2007, 40, 2791 CrossRef CAS;
(d) M. Inam, G. Cambridge, A. P. Barry, Z. P. L. Laker, N. R. Wilson, R. T. Mathers, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2017, 8, 4223 RSC;
(e) X. He, Y. He, M. S. Hsiao, R. L. Harniman, S. Pearce, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2017, 139, 9221 CrossRef CAS;
(f) X. He, M. S. Hsiao, C. E. Boott, R. L. Harniman, A. Nazemi, X. Li, M. A. Winnik and I. Manners, Nat. Mater., 2017, 16, 481 CrossRef CAS;
(g) H. Qi, H. Zhou, Q. Tang, J. Y. Lee, Z. Fan, S. Kim, M. C. Staub, T. Zhou, S. Mei, Lin Han, D. J. Pochan, H. Cheng, W. Hu and C. Y. Li, Nat. Commun., 2018, 9, 3005 CrossRef;
(h) M. Inam, J. R. Jones, M. M. Pérez-Madrigal, M. C. Arno, A. P. Dove and R. K. O'Reilly, ACS Cent. Sci., 2018, 4, 63 CrossRef CAS;
(i) W. Yu, M. Inam, J. R. Jones, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2017, 8, 5504 RSC.
-
(a) B. A. G. Lamers, B. Van Genabeek, J. Hennissen, B. F. M. De Waal, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2019, 52, 1200 CrossRef CAS;
(b) Y. Kwon and K. T. Kim, Macromolecules, 2021, 54, 10487 CrossRef CAS.
-
(a) M. L. Di Lorenzo and R. Androsch, Polym. Int., 2019, 68, 320 CrossRef CAS;
(b) Z. Li, Y. Zhang, L. Wu, W. Yu, T. R. Wilks, A. P. Dove, H. Ding, R. K. O'Reilly, G. Chen and M. Jiang, ACS Macro Lett., 2019, 8, 596 CrossRef CAS;
(c) P. J. Hurst, A. M. Rakowski and J. P. Patterson, Nat. Commun., 2020, 11, 4690 CrossRef CAS;
(d) G. Virata and E. B. Gowd, Polym. Chem., 2022, 13, 838 RSC.
-
(a) B. Kalb and A. J. Pennings, Polymer, 1980, 21, 607 CrossRef CAS;
(b) S. J. Organ, A. Keller and H. H. Wills, J. Polym. Sci., Part B: Polym. Phys., 1986, 24, 2319 CrossRef CAS;
(c) T. Y. Zhang, X. S. Guo, Z. K. Zhang, J. T. Xu and Z. Q. Fan, Polymer, 2020, 208, 122979 CrossRef CAS;
(d) H. Abe, M. Harigaya, Y. Kikkawa, T. Tsuge and Y. Doi, Biomacromolecules, 2005, 6, 457 CrossRef CAS;
(e) H. Ni'mahab and E. M. Woo, CrystEngComm, 2014, 16, 4945 RSC;
(f) S. Nurkhamidah and E. M. Woo, J. Phys. Chem. B, 2011, 115, 13127 CrossRef CAS.
- C. Qiao, J. Zhao, S. Jiang, X. Ji, L. An and B. Jiang, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 1303 CrossRef CAS.
-
(a) B. Lotz, T. Miyoshi and S. Z. D. Cheng, Macromolecules, 2017, 50, 5995 CrossRef CAS;
(b) L. Jiang, T. Shen, P. Xu, X. Zhao, X. Li, W. Dong, P. Ma and M. Chen, e-Polymers, 2016, 16, 1 CAS;
(c) W. Xu, Y. Zheng and P. Pan, J. Polym. Sci., 2022, 60, 2136 CrossRef CAS;
(d) L. Liu, L. Zhu, Z. Chu and Z. Tong, Macromolecules, 2023, 56, 5984 CrossRef CAS.
-
(a) Y. Ge, Y. Wen, H. Liu, T. Lu, Y. Yu, X. Zhang, B. Li, S. T. Zhang, W. Li and B. Yang, J. Mater. Chem. C, 2020, 8, 11830 RSC;
(b) I. Yamazaki, N. Tamai and T. Yamazak, J. Phys. Chem., 1987, 91, 3572 CrossRef CAS;
(c) S. Karuppannan and J.-C. Chambron, Chem. – Asian J., 2011, 6, 964 CrossRef CAS.
-
(a) G. K. Bains, S. H. Kim, E. J. Sorin and V. Narayanaswami, Biochemistry, 2012, 51, 6207 CrossRef CAS;
(b) I. O. Aparin, G. V. Proskurin, A. V. Golovin, A. V. Ustinov, A. A. Formanovsky, T. S. Zatsepin and V. A. Korshun, J. Org. Chem., 2017, 82, 10015 CrossRef CAS.
-
(a) Q. Song, S. Goia, J. Yang, S. C. L. Hall, M. Staniforth, V. G. Stavros and S. Perrier, J. Am. Chem. Soc., 2021, 143, 382 CrossRef CAS;
(b) V. Kumar, B. Sk, S. Kundu and A. Patra, J. Mater. Chem. C, 2018, 6, 12086 RSC;
(c) A. Fujii, Y. Sekiguchi, H. Matsumura, T. Inoue, W.-S. Chung, S. Hirota and T. Matsuo, Bioconjugate Chem., 2015, 26, 537 CrossRef CAS.
-
(a) W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, Nat. Methods, 2019, 16, 815 CrossRef CAS;
(b) A. Rajak, C. K. Karan, P. Theato and A. Das, Polym. Chem., 2020, 11, 695 RSC;
(c) L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. Jame, Chem. Soc. Rev., 2020, 49, 5110 RSC.
- P. Rajdev, D. Basak and S. Ghosh, Macromolecules, 2015, 48, 3360 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*