
Highly selective organo-gallium hydroxamate mediated inhibition of antibiotic resistant Klebsiella pneumoniae†
Rebekah N.
Duffin
 ,
Jenisi T. A.
Kelderman
,
Megan E.
Herdman
and
Philip C.
Andrews
*
,
Jenisi T. A.
Kelderman
,
Megan E.
Herdman
and
Philip C.
Andrews
*
School of Chemistry, Monash University, Clayton, Melbourne, VIC 3800, Australia. E-mail: phil.andrews@monash.edu
First published on 11th November 2024
Abstract
Five complexes of gallium derived from hydroxamic acids have been synthesised, characterised, and their anti-bacterial activity and mammalian cell toxicity established. These are three metal–organic complexes; [Ga(BPHA)3] 1, [Ga(BHA-H)3] 2, [Ga(SHA-H2)(SHA-H)3] 3, and two heteroleptic organometallic complexes [GaMe2(BPHA)] 4, and [GaMe(BHA-H)2] 5, along with the iron complex [Fe(BPHA)3] 6 (BPHA-H = N-benzoyl-N-phenylhydroxamic acid, BHA-H2 = phenylhydroxamic acid, and SHA-H3 = salicylhydroxamic acid). Solid-state structures of 1, 4–6 were identified by single-crystal X-ray crystallography. Complexes 1 and 6 adopt an octahedral geometry at the metal centre, while the organometallic complexes 4 and 5 adopt, respectively, tetrahedral and trigonal bipyramidal geometry. The solution and solid-state chemistry of complex 3 was found to differ: the solution state is composed of an equilibrium mixture of the bis-complexed hydroximate–hydroxamate species and the homoleptic tris-hydroxamate species, with the solid state preferring the bis-complexed hydroximate–hydroxamate composition. The methyl gallium complexes 4 and 5 differed in their preferred composition with 4 forming the expected dimethyl hydroxamate complex while 5 stabilises as the methyl bis-hydroxamate complex. Complexes were tested for the anti-microbial activity against a series of Gram-positive and Gram-negative bacteria, with an emphasis on the Gram-negative Klebsiella pneumoniae. While the metal–organic complexes 1, 2, 3 and 6 showed little to no activity towards either the bacteria or mammalian cells, the alkyl gallium complexes 4 and 5 were found to have exceptional activity toward K. pneumoniae in RPMI-HS media with MIC values of 78 nM. Interestingly, [GaMe2(OH)] also showed significant activity with an MIC of 156 nM.
Introduction
Klebsiella pneumoniae is a major opportunistic nosocomial pathogen of the Enterobacteriaceae family.1 It is one of the ESKAPE bacteria, classified as nosocomial pathogens that exhibit multidrug resistance and virulence, with the remaining five encompassing Enterococcus faecium, Staphylococcus aureus, Pseudomonas aeruginosa, Acinetobacter baumannii and Enterobacter spp.2 Typically K. pneumoniae is found in the flora of the mouth, skin and intestines. However, it often inhabits medical devices and high-touch surfaces, creating substantial infection risk for patients with compromised and weakened immune systems.3 Biofilm formation on medical devices poses a major threat due to unintentional infection, especially in catheter patients.4 For two main reasons K. pneumoniae infections tend to be chronic: biofilm formation provides a protective barrier in vivo toward host immune responses and antibiotics, and common nosocomial strains tend to display multi-drug resistance due to the extended-spectrum β-lactamases (ESBL) or carbapenem exposure, causing difficulties in drug selection for treatment.5–7K. pneumoniae, like many pathogens, have evolved sophisticated ways in which to either evade host immunity or inhibit it. For example: a thick capsule at the cell surface enables evasion of host phagocytosis by immune cells (e.g., macrophages, neutrophils) in hypervirulent strains.8,9 Furthermore, the evolution of bacterial efflux pumps to actively export antibiotics has led to the rapid development of antibiotic resistance. These efflux pumps are also able to remove host derived antimicrobial agents, such as antimicrobial peptides.10
Like most pathogens K. pneumoniae requires iron from exogenous sources. There are at least 12 distinctive transport systems for iron in K. pneumoniae found in four major categories: Ferrous transporters (FeO), ABC transporters, hemophore-based uptake, and siderophore uptake systems. Only the siderophore based systems and two of the ABC transporters are required for full virulency.11 Siderophore mediated uptake is the major acquisition pathway for K. pneumoniae, utilising this high iron affinity transporter to obtain iron in the iron-poor environments of their mammalian hosts. Scarcity of iron tends to cause an increase in the production of these siderophores in an attempt to maintain a high degree of pathogenicity and homeostasis for further growth and infection. Hypervirulent strains also secrete a higher concentration of these siderophores in comparison to classical strains.12–14 These siderophores generally contain carboxylate, catecholate, and hydroxamate functional groups as main metal chelation sites.
Gallium is known to mimic iron in vivo. Ga(III) binds to and is transported by similar proteins, including transferrin, lactoferrin and serum albumin. The main uptake pathway for Ga into cells has been shown to be mediated by transferrin receptors (TfR) but the very different redox chemistry, which requires reduction of Fe(III) to Fe(II) for TfR, indicates that the uptake processes are not exactly the same and that different non-TfR pathways are possible.15
As discussed, K. pneumoniae will up-regulate production of the siderophores aerobactin and enterobactin in iron-poor environments.15,16 There is therefore the potential to exploit this nutritional requirement using gallium as an iron pathway inhibitor. The chemical similarities between Ga(III) and Fe(III) make it exceptionally difficult for bacteria to distinguish between the two, ultimately leading to uptake of gallium via siderophores and inhibition of redox-active pathways. This mechanistic pathway, the “Trojan Horse effect” is therefore the target of a variety of potential gallium-based therapeutics.17,18 Deferoxamine is a polyfunctional siderophore mimic which is used therapeutically to treat iron overload in the body, primarily using hydroxamate chelation. Though there have been investigations into the antibacterial activity of Ga(III) deferoxamine complexes they were found to exhibit similar or higher toxicity than the parent Ga(III) salt, suggesting broad antimicrobial properties.19 Beyond this there is only limited research into the structure and bio-activity of more simple gallium hydroxamate complexes.
Recently, we have shown that complexation with Ga(III) can reactivate previously inactive quinolone antibiotics in low iron environments. Importantly, the most active complexes had the organogallium [GaMe2] moiety at their core. The complexes exhibited highly potent activity toward four antibiotic resistant strains of K. pneumoniae in RPMI media supplemented with 10% human AB serum,20 much greater that their homoleptic [GaL3] analogues. Similarly, a class of dimethyl gallium quinolinolates were also observed to be exceptional inhibitors of K. pneumoniae in reduced iron media,21 and were again much superior to their tris-quinolinolate analogues. This hints at the uniqueness of the heteroleptic organogallium complexes compared with the traditional homoleptic salts and metal–organic complexes.
As such, our efforts have focussed in on the differences in the homoleptic and heteroleptic gallium complexes and the effect of lipophilicity, hydrolytic stability, protein exchange kinetics, and solubility in their biological activity.20 This current study draws on this, and the role of hydroxamate chelation, in exploring the synthesis, characterisation and antibacterial activity of a range of gallium hydroxamate complexes.
Three structurally diverse hydroxamic acids; N-benzoyl-N-phenylhydroxamic acid (BPHA-H), phenylhydroxamic acid (BHA-H2), and salicylhydroxamic acid (SHA-H3), shown in Fig. 1, were used in the synthesis of three metal–organic gallium complexes and one iron complex [Ga(BPHA)3] 1 [Ga(BHA-H)3] 2, and [Ga(SHA-H2)(SHA-H)] 3, [Fe(BPHA)3] 6, and two heteroleptic organometallic complexes, [GaMe2(BPHA)] 4 and [GaMe(BHA-H)2] 5. These are shown in Fig. 2.
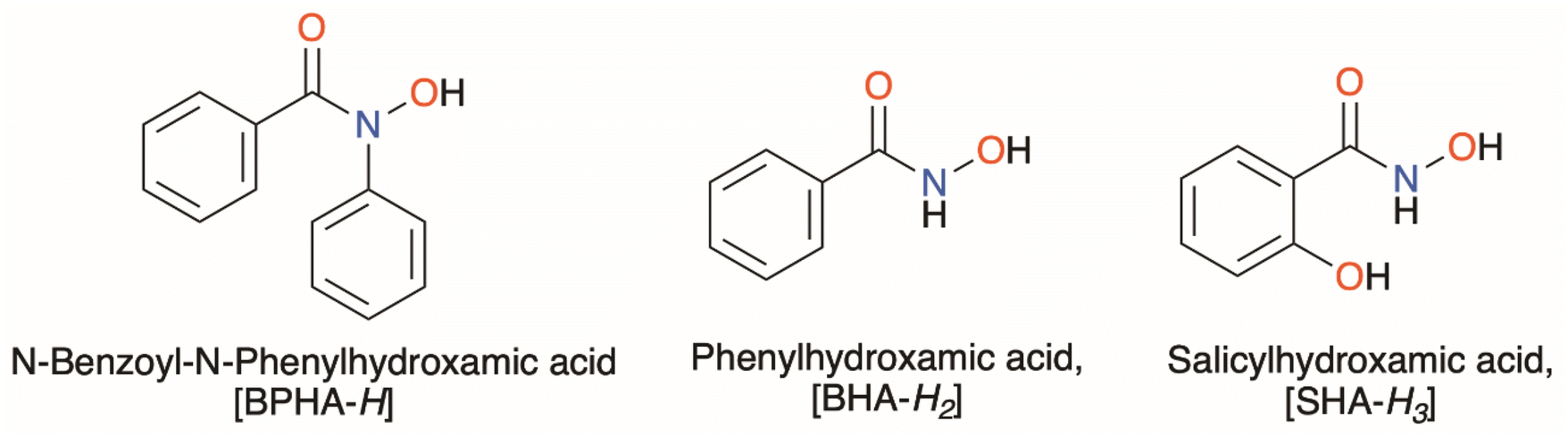 | ||
| Fig. 1 The three hydroxamic acids used in this study: BPHA-H = N-benzoyl-N-phenylhydroxamic acid, BHA-H2 = phenylhydroxamic acid, and SHA-H3 = salicylhydroxamic acid. | ||
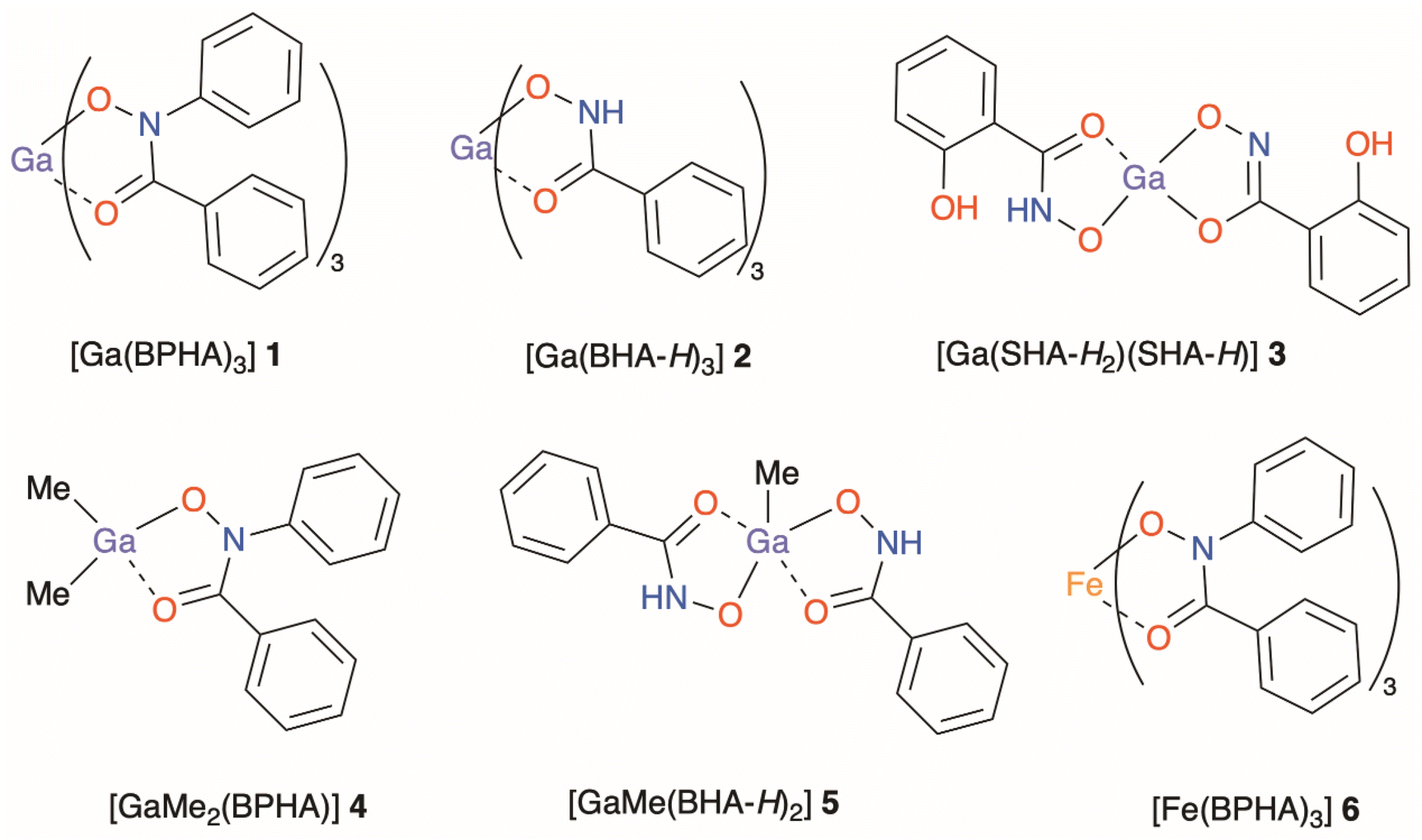 | ||
| Fig. 2 Complexes synthesised in this study: [Ga(BPHA)3] 1, [Ga(BHA-H)3] 2, and [Ga(SHA-H2)(SHA-H)] 3, [GaMe2(BPHA)] 4, [GaMe(BHA-H)2] 5 and [Fe(BPHA)3] 6. | ||
In addition to K. pneumoniae, the compounds were also analysed for their antibiotic activity against a range of Gram-positive and Gram-negative bacteria including methicillin resistant S. aureus (MRSA), vancomycin resistant Enterococcus (VRE), E. coli, P. aeruginosa and A. baumannii. Cytotoxicity toward mammalian control cells was also assessed and the selectivity index of the complexes calculated.
Results and discussion
Synthesis and characterisation
Gallium complexes [Ga(BPHA)3] 1, [Ga(BHA-H)3] 2, and [Ga(SHA-H2)(SHA-H)] 3, were synthesised (Scheme 1) through the solid addition of the gallium nitrate to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol:water solution of the hydroxamic acid heated to 60 °C. NaHCO3 was then added to bring the solution pH up to ca. 7 at which point the metal complex precipitated. The products, as white to off-white powders, were collected via gravity filtration and washed with copious amounts of water before air drying.
:1 ethanol:water solution of the hydroxamic acid heated to 60 °C. NaHCO3 was then added to bring the solution pH up to ca. 7 at which point the metal complex precipitated. The products, as white to off-white powders, were collected via gravity filtration and washed with copious amounts of water before air drying.
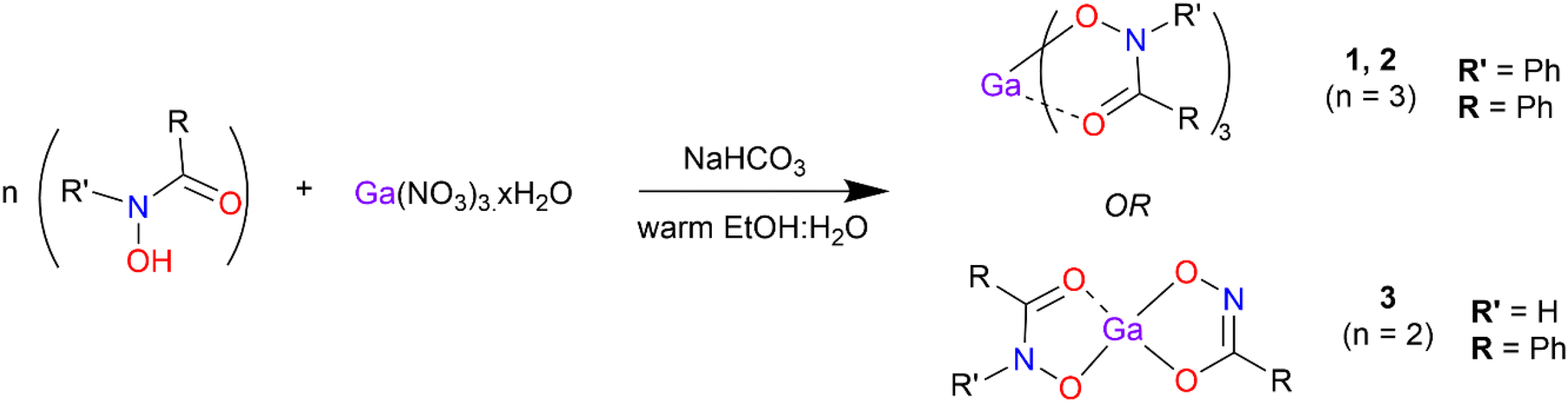 | ||
| Scheme 1 Synthetic pathway for synthesis of the gallium hydroxamate/hydroximate complexes 1 and 2 and 3. | ||
The organometallic heteroleptic complexes [GaMe2(BPHA)] 4 and [GaMe(BHA-H)2] 5 were synthesised through the direct addition of neat trimethyl gallium to a suspension of the pre-dried hydroxamic acid under inert conditions in dry hexane at 0 °C (Scheme 2). Despite multiple attempts to synthesise and isolate the dimethyl gallium benzohydroxamate complex, [GaMe2(BHA-H)], the reaction would rapidly proceed to the formation of the mono-methyl bis-hydroxamate complex 5, [GaMe(BHA-H)2]. To maximise the yield of 5, the required 2:1 reaction stoichiometry was used. For complex 4, the opposite was observed wherein attempted formation and isolation of a bis-hydroxamate complex, [GaMe(BPHA)2], would invariably result in isolation of the dimethyl mono-hydroxamate complex 4. Attempts to synthesise an organogallium complex derived from SHA-H3 consistently resulted in the formation of multiple products and a pure isolable complex could not be obtained. Since the biological assays require complex purity, this reaction was not studied further at this time.
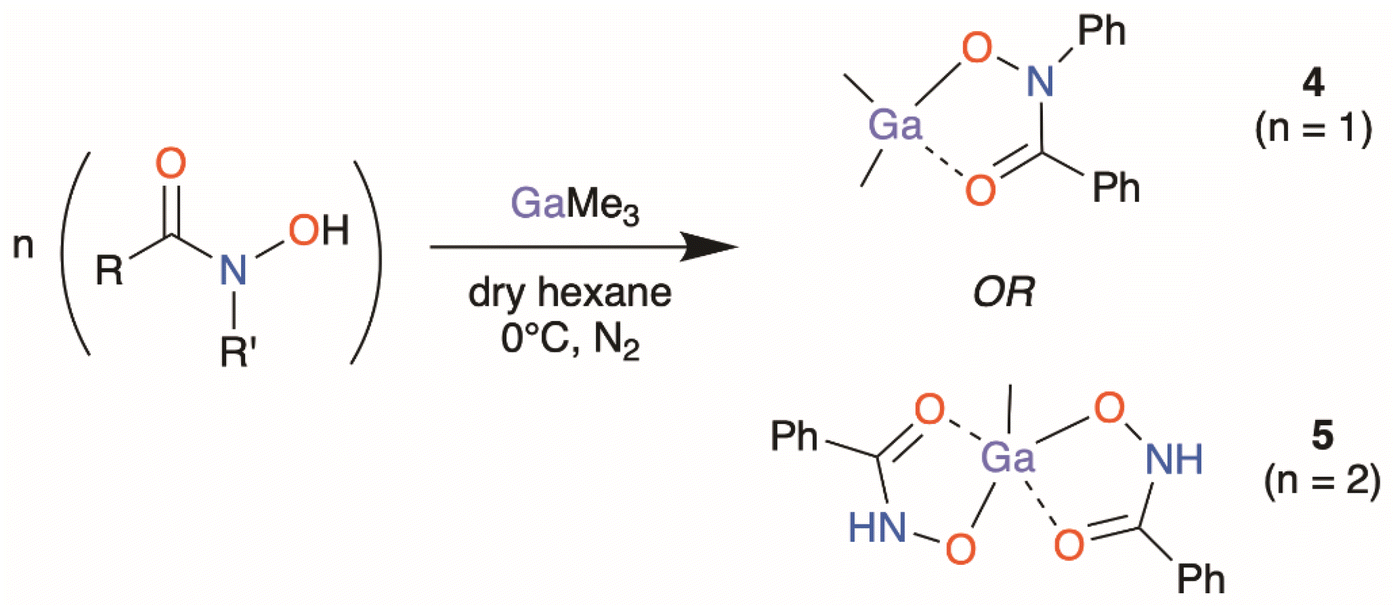 | ||
| Scheme 2 Synthetic pathway for the acquisition of the organometallic gallium complexes 4 and 5. | ||
These gallium complexes were all characterised through 1H and 13C NMR spectroscopy, FT-IR spectroscopy, ESI-mass spectrometry, elemental analysis, melting point analysis, and where applicable single crystal X-ray diffraction.
During the attempted syntheses of these complexes, several crystalline products were obtained in low yield which were indicative of the impact of solvent, ligand redistribution and hydrolysis on the reaction mixtures and products. These were; the heteroleptic methanolysis product [Ga(BPHA)2(OMe)] 7, the tetra-nuclear oxido cluster [Ga4Me7(BPHA)O4] 8, the mono-methyl complex [GaMe(BPHA)2] 9, and [{GaMe2(BHA-H)}{Ga2Me4O}], 10. A summary of the conditions under which these complexes were obtained, their X-ray data, and any further analysis is provided in the ESI.†
Understanding that there is always the potential for exchange of gallium for iron in biological media (and also in vivo) the iron complex [Fe(BPHA)3] 6, being the analogue of 1, was synthesised from the reaction of FeCl3 with BPHA-H (Scheme 3). This was able to be crystallised and the solid-state structure determined. It was further characterised by ESI-MS, FT-IR, and elemental analysis.
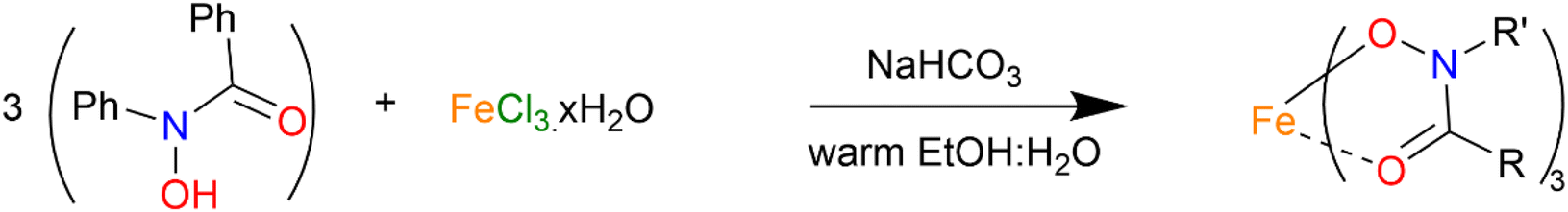 | ||
| Scheme 3 Synthetic pathway for the formation of complex [Fe(BPHA)3] 6. | ||
X-ray crystallography
Single crystals of complexes 1, 4, 5 and 6 were obtained through slow evaporation in solvent. The additional solid-state structures for 7, 8 and 10 are given in the ESI (Fig. S12–S15†). The tris-hydroxamato complexes [Ga(BPHA)3] 1 and [Fe(BPHA)3] 6 crystallise with high symmetry, and with a distorted octahedral coordination environment, Fig. 3. A detailed summary of bond lengths and angles for the solid-state structures of these complexes are given in in the ESI (Table S1†). Due to the high symmetry of 1, and axial rotation of the ligands, the carbon and nitrogen atoms in the ‘O–C–N–O’ backbone of BPHA are superimposed and so were modelled over the two positions. The degree of distortion away from an ideal octahedron was found to be greater for iron than gallium, with the iron complex exhibiting an angle of 98.17(14) through the O(1A)–Fe–O(1B) bond.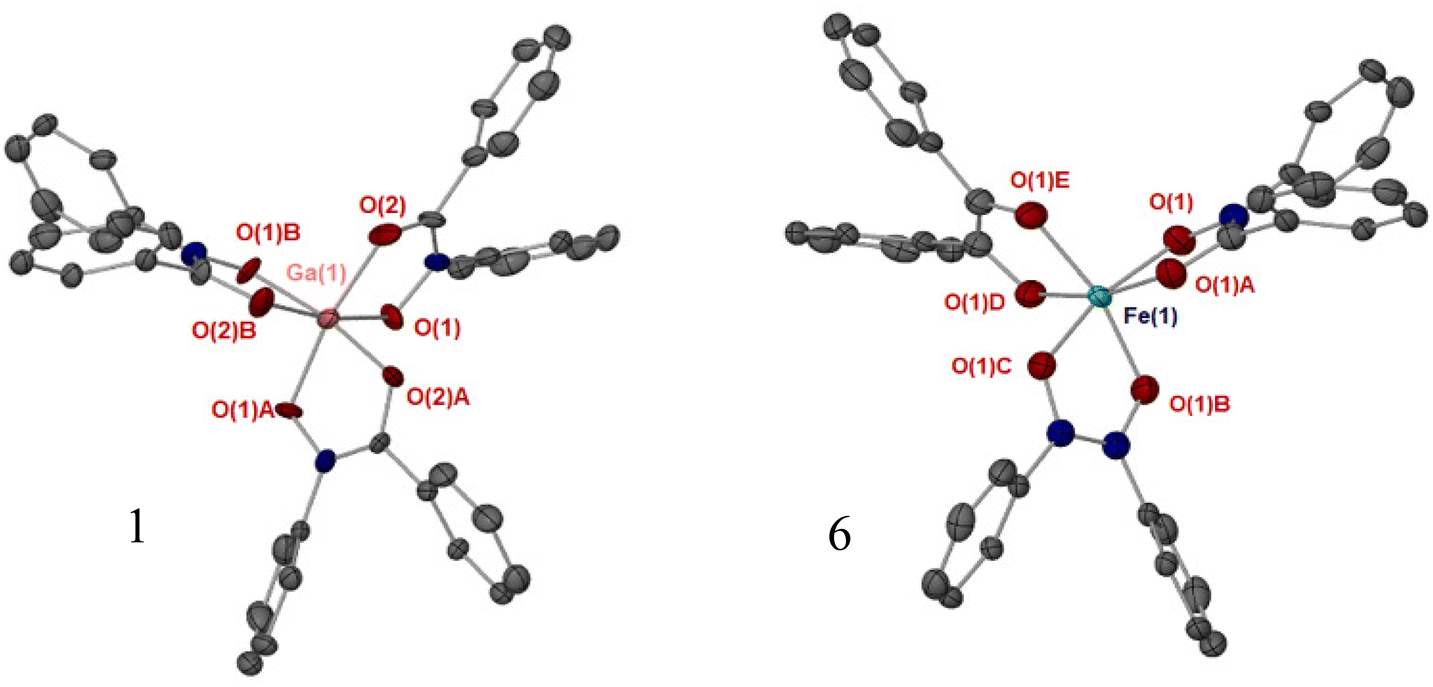 | ||
| Fig. 3 Solid-state structures of complexes [Ga(BPHA)3] 1 and [Fe(BPHA)3] 6. Thermal ellipsoids at 50% probability, hydrogen atoms have been omitted for clarity. Symmetry operators: 1, (1 − X + Y, 1 − X, Z):(1 − Y, X − Y, Z); 6 (Y, X, ½ − Z):(−Y, X − Y, Z):(−X, −X + Y, ½ − Z):(−X + Y, −X, Z):(X − Y, −Y, ½ − Z). Selected bond length (Å) 1: Ga(1)–O(1), 1.972(2), O(1)–N(1), 1.389(12), O(1)–C(2), 1.257(28). 6: Fe(1)–O(1), 2.001(3), O(2)–C(1): 1.322(5) Angles (°) G1: O(1)–Ga(1)–O(2), 91.64(8), O(1)–Ga(1)–O(1B), 169.6(1). 6: O(1)–Fe(1)–O(2), 92.45(12), O(1)–Fe(1)–O(1B), 166.1(2). | ||
Crystals of complexes 4 and 5 were obtained through slow evaporation in toluene. The coordination environments and geometries for complexes [GaMe2(BPHA)] 4 and [GaMe2(BPHA)] 5 were found to be similar to the those we recently reported for di-methyl and mono-methyl gallium quinolinolato complexes. As shown in Fig. 4, complex 4 is four coordinate with a typical tetrahedral geometry, while 5 is five coordinate and trigonal bipyramidal.22
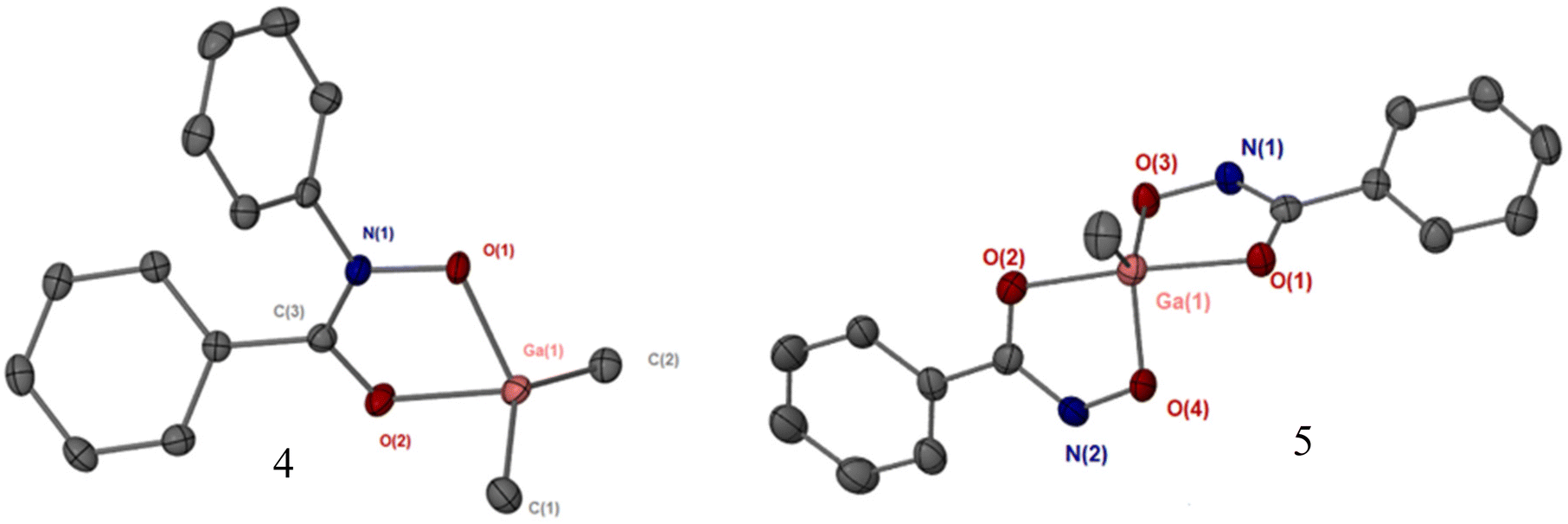 | ||
| Fig. 4 Crystal structures of complex 4, [GaMe2(BPHA)] and complex 5, [GaMe(BHA-H)2] (note: interstitial toluene molecule in 5 excluded for clarity). Thermal ellipsoids at 50%, hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): 4, [GaMe2(BPHA)]: Ga(1)–O(1), 1.958(1), Ga(1)–O(1′), 2.544(1), Ga(1)–O(2), 2.062(1), Ga(1)–C(1), 1.949(2), Ga(1)–C(2), 1.951(2). O(1)–Ga(1)–O(1′) 68.86(5), O(1)–Ga(1)–O(2) 79.47(5), O(2)–Ga(1)–O(1′), 147.6(1), C(1)–Ga(1)–O(1), 113.4(1), C(2)–Ga(1)–O(1) 110.0(1). 5, [GaMe(BHA-H)2]: Ga(1)–O(1), 2.040(2), Ga(1)–O(2), 2.067(2), Ga(1)–O(3), 1.906(2), Ga(1)–O(4), 1.902(2), Ga(1)–C(1), 1.935(2). O(1)–Ga(1)–O(2), 157.8(1), O(1)–Ga(1)–O(3) 82.18(6), O(1)–Ga(1)–O(4), 89.28(7), O(3)–Ga(1)–O(4), 108.2(1), C(1)–Ga(1)–O(1), 101.8(1), C(1)–Ga(1)–O(3). | ||
As evidenced by the bond lengths, and illustrated in Fig. 2, the mono-anionic hydroxamate ligands in complexes 1, 4, 5 and 6 all show a similar bonding {O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(–R)–N–O−} pattern for the metal chelating moiety.
C(–R)–N–O−} pattern for the metal chelating moiety.
FT-IR data correlates well with the hydroxamate ligand conformation observed in complexes 1, 4, 5 and 6 (ESI†). In all complexes, the typical broad O–H stretch at ca. ∼3100 cm−1 found in the spectrum of each of the hydroxamic acids is absent in the spectrum of each of the corresponding complexes. Supporting this, Table 1 provides the carbonyl absorbances and amide absorbances for each of the gallium complexes and the subsequent shift to lower wavenumbers of the corresponding symmetric and asymmetric stretches in the metal complexes, evidence of mono-anion formation, metal complexation, and chelation.
| Complex | IR absorbance (cm−1) | ||||
|---|---|---|---|---|---|
| COsym |
COasym |
N–H | CN |
C–H | |
| 1 [Ga(BPHA)3] | 1582 | 1517 | 3050 | — | — |
| 2 [Ga(BHA-H)3] | 1598 | 1517 | 3356 | — | — |
| 3 [Ga(SHA-H2)(SHA-H)3] | 1574 | 1516 | — | 1601 | — |
| 4 [GaMe2(BPHA)] | 1586 | 1535 | 3060 | — | 2961 |
| 5 [GaMe(BHA-H)2] | 1601 | 1534 | 3194 | — | 3007 |
For complex 2, complexation of the hydroxamate ligand was identified by the significant shift in the carbonyl stretches from 1622 cm−1 to 1598 cm−1 similar to that observed for 1. Supporting this, a weak N–H stretch is observed at 3356 cm−1 which is blue-shifted compared to the strong N–H stretching signal 3286 cm−1 for the hydroxamic acid.
In contrast, the FT-IR spectrum of 3 coupled with elemental analysis data indicates that the solid-state structure of this complex is heteroleptic, composed of a hydroxamate (SHA-H2) and hydroximate (SHA-H) ligand chelating into the Ga centre, [Ga(SHA-H2)(SHA-H)]. Thermogravimetric analysis (TGA) suggests the presence of one moiety of water per compound (ESI Fig. S2†). The absence of the N–H stretch at 3285 cm−1 suggested deprotonation of the imine functional group. Furthermore, the additional large hydroxyl signal of the phenol further masks the N–H absorbance. The strong CN band at 1601 cm−1, absent in the FTIR spectra for other five complexes, alludes to the presence of the hydroximate ligand.
For complexes 1 and 2 the hydroxamate form was observed in the NMR spectra (see ESI†). The predominant form in the solid state for complexes 2 and 3 was confirmed by elemental analysis to be the hydroxamate–hydroximate bis-hydroxamate substitution, with solution state data highlighting a shift to a tris-substituted hydroxamate state (Fig. S1†). Thermogravimetric analysis (TGA) revealed that complexes 2 and 3 were hygroscopic with a loss of 3.8567% and 3.0727% mass respectively at 100 °C, equivalent to one H2O molecule for one molecule of 2 and one H2O for two 3 molecules, as confirmed via elemental microanalysis (Fig. S2 and S4†). Complex 1 was observed to be the tris-hydroxamate in both the solution and solid-states, supported by the X-ray data. For the alkyl analogues 4, and 5 only the hydroxamate form was observed as confirmed by XRD analysis, showing the carbonyl oxygen providing a dative interaction and stabilising the complex through chelation of the ligand. The absence of the hydroxyl proton of the free acids in the NMR spectrum provided further evidence of deprotonation and metal-chelation. Shifts were observed in the aromatic regions of the free acids versus the complexes. An example spectrum of complex 1, 4 and free BPHAH can be found in the ESI.†
Changes in complex composition in the solid-state versus solution state was also able to be confirmed with ESI-MS. For 3, both ([Ga(SHA-H2)(SHA-H)] + Na+, 395.0)+ and ([Ga(SHA-H2)3] + Na+, 548.0)+ were observed.
Biological testing
| Complex | Aqueous solubility (μg mL−1) |
|---|---|
| 1 [Ga(BPHA)3] | 4.90 |
| 2 [Ga(BHA-H)3] | 41.8 |
| 3 [Ga(SHA-H2)(SHA-H)3] | 33.6 |
| 4 [GaMe2(BPHA)] | 0.989 |
| 5 [GaMe(BHA-H)2] | 0.851 |
| Complex | IC50 (μM) |
|---|---|
| 1 [Ga(BPHA)3] | 67.3 |
| 2 [Ga(BHA-H)3] | >100 |
| 3 [Ga(SHA-H2)(SHA-H)3] | >100 |
| 4 [GaMe2(BPHA)] | >100 |
| 5 [GaMe(BHA-H)2] | >100 |
| 6 [Fe(BPHA)3] | >100 |
| Complex | MIC (μM) | ||||||
|---|---|---|---|---|---|---|---|
| PA | AB | KP | KP1074 | EC | VRE | MRSA | |
| 1 [Ga(BPHA)3] | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 |
| 2 [Ga(BHA-H)3] | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 |
| 3 [Ga(SHA-H2)(SHA-H)3] | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 | ≥100 |
| 4 [GaMe2(BPHA)] | 100 | 100 | 100 | 100 | ≥100 | ≥100 | ≥100 |
| 5 [GaMe(BHA-H)2] | 100 | 100 | 100 | 100 | ≥100 | ≥100 | ≥100 |
| 6 [Fe(BPHA)3] | — | — | ≥100 | ≥100 | — | — | — |
| Complex | MIC (μM) | ||
|---|---|---|---|
| PA | AB | KP1074 | |
| 1 [Ga(BPHA)3] | ≥100 | ≥100 | ≥100 |
| 2 [Ga(BHA-H)3] | ≥100 | ≥100 | ≥100 |
| 3 [Ga(SHA-H2)(SHA-H)3] | ≥100 | ≥100 | ≥100 |
| 4 [GaMe2(BPHA)] | 100 | 100 | 0.078 |
| 5 [GaMe(BHA-H)2] | 100 | 100 | 0.078 |
| 6 [Fe(BPHA)3] | — | — | ≥100 |
| [GaMe2(OH)] | — | — | 0.156 |
As we have observed before, the controls for the reference strain of K. pneumoniae did not show significant growth in RPMI-HS, and hence no MIC was obtainable, and we believe is most likely due to the presence of antimicrobial peptides found in human serum.20,21 Furthermore, the presence of the MagA gene in MDR K. pneumoniae, which up-regulates the production of hypermucoviscosity, is said to demonstrate an extraordinarily high resistance to the antimicrobial properties of complement-human serum.28
With the increased activity toward KP1074 obtained, controls were then assessed to determine whether the complex itself exudes the activity. The free acids BHA-H2 and BPHA-H were analysed for their inhibitory activity at the same concentration as the complexes. The iron complex [Fe(BPHA)3] 6 and the potential hydrolysis product [GaMe2OH] were also analysed at the same concentration gradient. Unsurprisingly, the tris-hydroxamato Fe complex and the free acids exhibited no activity in RPMI-HS, however [GaMe2OH] exhibited activity comparable to complexes 4 and 5 with an MIC of 156.0 nM.
While this is significant, 4 and 5 still have activity two-fold higher indicating that the hydroxamate ligands are important in the overall antibacterial activity but that the ‘GaMe2’ and ‘GaMe’ moieties are critical in their effect on MDR K. pneumoniae (see ESI† for example inhibition images).
GaM has been shown to have a high bioavailability due to the prolonged absorption into the blood stream, allowing uptake of the Ga3+ ions by transferrin as opposed to rapid formation of gallate [Ga(OH)4]−.30,31 It is possible that complexes 1 and 2 are rapidly hydrolysed in aqueous media, leading to gallate formation, possibly rapidly exchange with iron (discussed further), and/or undergo rapid ligand exchange with transferrin or lactoferrin, thereby preventing uptake by the bacteria that rely less on methods of host-peptide iron sequestration. In contrast, the alkyl complexes may provide a greater degree of hydrolytic stability given the relatively inert nature of the remaining Ga–Me bonds. A study by Coggin et al. in the early 90s detailed the hydrolytic stability of the Ga–C bond in four-coordinate gallium complexes and determined with in vivo rat studies that protein adducts of [GaMe2]+ are either kinetically labile or not formed.32 Recently, we determined that complexation may play a role in potential iron exchange processes in iron-depleted media. Our analysis of both organo gallium quinolonates and quinolinolates showcased a lower degree of protein association in RPMI-HS than their homoleptic analgoues.20,21 Therefore, we investigated whether this property would translate to the siderophore mimicking hydroxamates. To allow for direct comparison in this experiment, only hydroxamate bearing complexes derived from BHA-H2 and BPHA-H were explored, thus excluding complex 3. However, because of its significant activity [GaMe2(OH)] was examined. Triplicate gallium analysis of both the digested extracted protein pellet phase and aqueous supernatant by ICP-MS supported the hypothesis that the metal–organic complexes 1 and 2 are rapidly hydrolysed and/or exchanged with iron into mammalian iron sequestering proteins, whereas the increased hydrolytic stability of the alkyl gallium complexes 4, 5 and [GaMe2(OH)] does not allow free access to Ga3+ ions in solution, leading to decreased uptake by transferrin and/or lactoferrin (Table 6 and Fig. S11†). Additionally, this uptake is supported further by the similar levels of Ga observed for [GaMe2(OH)] (Table 6). As previously mentioned, in iron poor conditions K. pneumoniae will up-regulate the production of siderophores in order to maximise its ability to scavenge iron.12 These combined factors may provide the basis for the increased activity of complexes 4 and 5 and [GaMe2(OH)] toward K. pneumoniae in RPMI-HS.
| Complex | Supernatant initial (%) | Pellet initial (%) | Supernatant 24 h (%) | Pellet 24 h (%) |
|---|---|---|---|---|
| 1 [Ga(BPHA)3] | 19.9 + 7.27 | 80.1 + 12.6 | 16.2 + 2.04 | 83.8 + 8.37 |
| 2 [Ga(BHA-H)3] | 16.4 + 8.95 | 83.6 + 5.75 | 19.0 + 7.10 | 81.0 + 18.0 |
| 4 [GaMe2(BPHA)] | 72.4 + 1.95 | 27.6 + 4.69 | 20.1 + 6.44 | 79.9 + 18.2 |
| 5 [GaMe(BHA-H)2] | 83.5 + 8.95 | 16.5 + 0.945 | 23.3 + 4.60 | 76.7 + 19.3 |
| [GaMe2(OH)] | 78.2 + 4.21 | 21.8 + 1.22 | 36.0 + 2.94 | 64.0 + 12.04 |
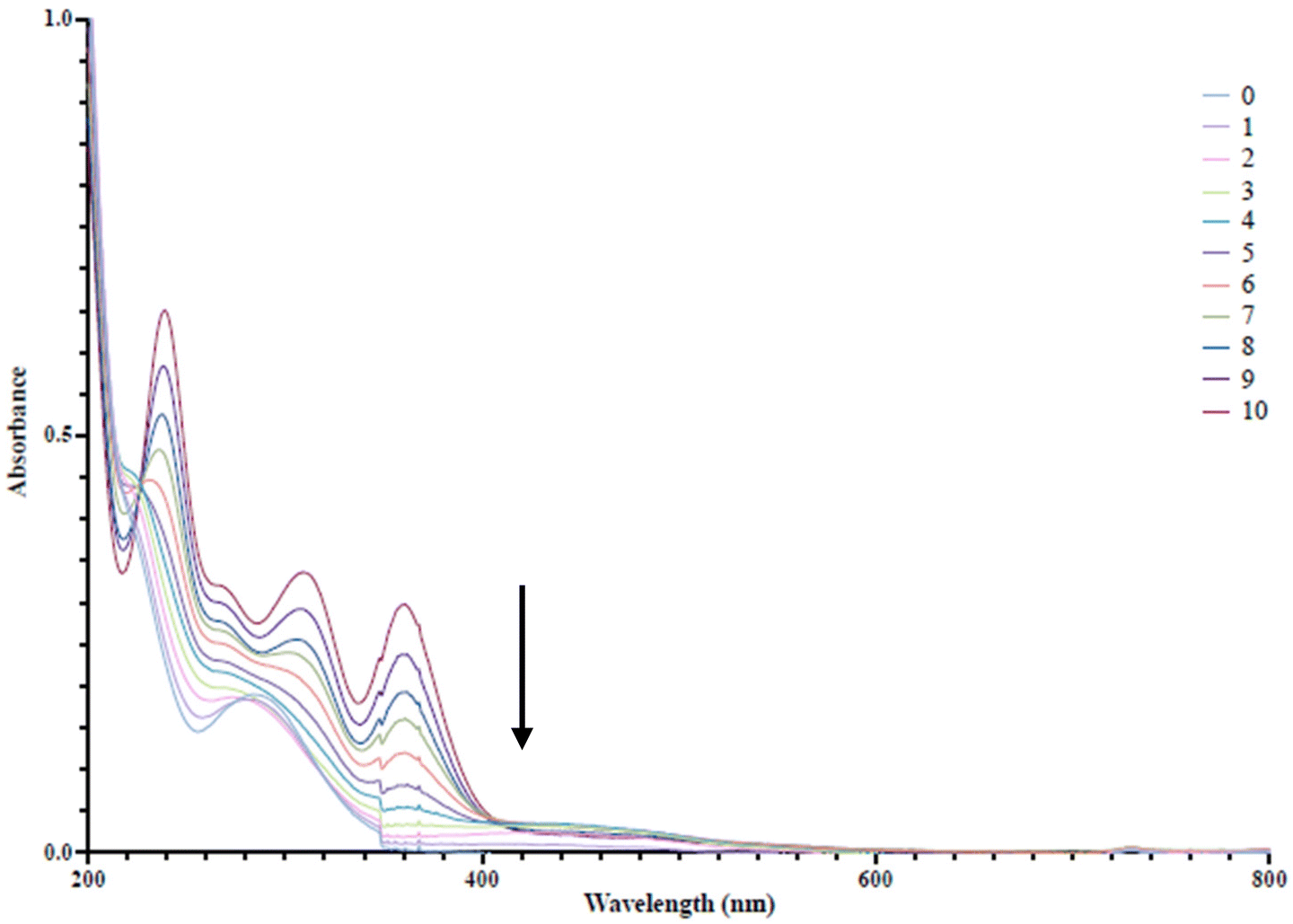 | ||
| Fig. 5 UV-visible spectra of aliquots taken after 1 mL addition of 10 μM FeCl3 used to determine the ‘end-point’ of titration. | ||
Conclusions
Five gallium complexes; three metal–organic [Ga(BPHA)3] 1, [Ga(BHA-H)3] 2, [Ga(SHA-H2)(SHA-H)] 3, and two heteroleptic organometallic [GaMe2(BPHA)] 4, [GaMe(BHA-H)2] 5, and the iron complex [Fe(BPHA)3] 6, were synthesised and fully characterised. Complex 3 was found to adopt a mixed ligand hydroximate–hydroxamate composition [Ga(SHA-H2)(SHA-H)] in the solid state which in solution exists in an equilibrium mixture with the homoleptic tris-hydroxamato complex [Ga(SHA-H)3]. The complexes were found to be stable to air and moisture in the solid-state over a period of three months. Single crystal XRD studies confirmed that 1 and 6 adopted analogous O′O chelating six coordinate environments at the metal centres, though the Fe complex shows greater distortion from ideal octahedral coordination than the Ga complex. Complex 4 presented as four-coordinate tetrahedral while 5 presented as trigonal bipyramidal with a 5-coordinate centre.These complexes were tested for their anti-microbial activity and mammalian cell viability. Only 1 exhibited any degree of mammalian cell inhibition, with an IC50 value of 67.3 μM. The other four complexes were found to be non-toxic towards L929 mouse fibroblast cells at 100 μM, the highest concentration tested.
The anti-bacterial activity of the complexes is dependent on the media in which the assays were conducted. In the iron supplement LB media, no activity was observed towards either of the Gram-positive strains of MRSA or VRE. However, some activity was seen towards the three Gram-negative strains of P. aeruginosa, A. baumannii, and K. pneumoniae, with all showing MIC values of 100 μM.
Analysis in the low iron, and more physiologically relevant, RPMI-HS media showed a drastic increase in the antibacterial activity of complexes 4 and 5 toward K. pneumoniae, with MIC values as low as 78.0 nM. No change in activity was observed for the metal–organic complexes 1 and 2. This radical difference in activity may be due in part to the increased hydrolytic stability of the methyl gallium (Ga–C) bonds and the propensity for the metal–organic gallium complexes to readily exchange with iron, as demonstrated by both the protein uptake analysis and UV-Vis titration study.
With nanomolar in vitro activity, the methyl gallium hydroxamate complexes 4 and 5 provide for a new and promising class of gallium-based therapeutics for challenging K. pneumoniae infections and biofilms.
Experimental section
General
Reagents were purchased from Sigma Aldrich, Merck, Otsuka Pharmaceuticals and Thermo Fisher without the need for further purification. Solvents were purchased from Merck and where required dried using an MBRUAN SPS-800 solvent purification system and stored over 4 Å molecular sieves. All reaction requiring inert conditions were performed under an atmosphere of nitrogen, using oven dried glassware and utilising a Schlenk manifold and technique. 1H NMR and 13C NMR were recorded on a Bruker Ultrashield Plus 600 DRX600 spectrophotometer (600 MHz), chemical shifts were referenced to the appropriate solvent, DMSO-d6. Variable temperature NMR was recorded on a Bruker Avance DRX400 spectrophotometer (400 MHz) Melting point analysis was conducted in open end capillary tubes, on a digital Stuart Scientific melting point apparatus SMP10. Infrared Spectra were recorded on an Agilent Technologies Cary 630 FTIR spectrometer, using a range of 4000–500 cm−1. Mass spectrometry (ESI) was obtained utilising a Micromass Platform QMS spectrometer, with an electrospray source and a cone voltage of 35 eV. Elemental analysis was obtained from the School of Chemistry, Monash University. Solubility of complexes in H2O was determined using inductively coupled plasma mass spectrometry (ICP-MS) on a PerkinElmer NexION 2000 ICP-MS instrument. An internal standard of 20 ppb Ag/As in 2–3% HNO3 was used and 11-point calibration curves of gallium and indium were generated. Samples were prepared by adding compound 100 μL of H2O, vortexing for 30 s to ensure the point of saturation has been reached. These samples were left to incubate overnight at room temperature before being centrifuged at 3000 rpm for 5 min and a 10 μL aliquot of solution was diluted to 10 mL using 2–3% HNO3. All volumes were determined by weight and the dilution factor was used to calculate the final solubility value. Ultraviolet–visible spectra were recorded on an Agilent Technologies Cary 100 UV-visible spectrometer in the range of 200 to 800 nm. Absorbance readings were conducted in 10 mm innovative Lab Supply ES Quartz cuvettes using acetonitrile as a solvent. Blank measurements were conducted using acetonitrile. Thermogravimetric analysis (TGA) was carried out using a Mettler Toledo TGA/DSC 1 Star System with heating up to 400 °C.X-ray crystallography
Crystallographic data was collected on an OXFORD XtaLAB Synergy, Dualflex, HyPix diffractometer equipped with an OXFORD Cryosystems 700 Cryostream and cooled to 173(2) K. Each compound was solved and refined using SHELXL-2014/7 utilising the graphical interface X-Seed or Olex2.37,38 Unless otherwise indicated, all non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms were placed in calculated positions using a riding model with C–H = 0.95–0.98 Å and Uiso(H) = xUiso(C), x = 1.2 or 1.5 unless otherwise indicated.Biological testing
Human serum (H4522), Fetal Bovine Serum (FBS), Dulbecco's modified Eagle's medium, penicillin–streptomycin solution (pen–strep), and GlutaMAX™ Supplement was purchased from Gibco/Thermo-Fisher. RPMI-1640 medium was purchased from the Biomedicine Discovery Institute, Monash University. Mouse fibroblasts; NCTC clone 929 [L cell, L-929, derivative of Strain L] (ATCC CCL-1).General procedures
Caution: GaMe3 is a pyrophoric liquid and can ignite on contact with air and moisture. It should always be handled carefully under inert atmosphere conditions with appropriate glassware and equipment.1H NMR (600 MHz, DMSO-d6) δ = 5.28 (1H, s OH), −0.52 (6H, s, Hmethyl). Elemental analysis; calcd (%): C 20.6, H 6.04, found: C 20.8, H 6.03.
:50 ethanol:water before gentle heating to 60 °C. To this, 0.5–1 mmol of the metal nitrate was added to the mixture as a solid. The solution was stirred as NaHCO3 was added dropwise till a pH of approximately 7–8. Visible effervescence of carbon dioxide was observed as well as precipitation as the pH approached neutral. The white to off-white powders were collected via gravity filtration and washed with copious water before air drying.
:50 mixture of ethanol and water and heated gently to ∼60 °C before the addition of 0.5 mmol (0.209 g) of solid [Ga(NO3)3·9H2O]. NaHCO3 was added dropwise until pH of approximately 7–8. Effervescence of CO2 was observed. The resultant white precipitate was gravity filtered and washed 3 × 15 mL of H2O yielding a white solid.
Yield 0.295 g (83%), m.p. 238–242 °C, 1H NMR (600 MHz, DMSO-d6) δ = 7.44–7.29 (30H, m, Har). 13C NMR (151 MHz, DMSO-d6) δ = 162.5 (CO), 140.7 (Cquaternary), 130.9 (Car), 130.3 (Car), 129.2 (Car), 128.8 (Car), 128.7 (Car), 128.4 (Car), 126.1 (Car). IR (cm−1): 3053 w v(C–Har), 1583 w, 1522 s v(CO), 1462 m, 1436 m, 1161 s, 760 s, 718 m, 698 s, 669 s. Elemental analysis; calcd (%): C 66.31, H 4.28, N 5.95, found: C 66.16, H 4.21, N 5.82. High-resolution ESI-MS+m/z: ([GaC39H30N3O6 + Na]+) calcd: 728.1282, found: 728.1267. CCDC: 2150403.†
:50 mixture of ethanol and water and heated gently to ∼60 °C before the addition of 1.0 mmol (0.420 g) of solid [Ga(NO3)3·9H2O]. NaHCO3 was added dropwise until pH of approximately 7–8. Effervescence of CO2 was observed. The resultant white precipitate was gravity filtered and washed 3 × 15 mL of H2O yielding a white solid.
Solid state: 82.5% yield (0.28 g, 0.82 mmol) of 2. m.p. 250 °C (decomp). FT-IR (cm−1): 3371 w v(N–H), 3194 bw, 3060 w v(C–Har), 2826 w, 1599 m v(CN), 1566 m v(CO), 1523 m, 1483 m, 1444 w, 1363 m, 1150 m, 1048 m, 1027 m, 920 s, 781 m, 690 s. Elemental analysis; calcd (%) [2 + H2O]: C 46.84, H 3.65, N 7.80, found: C 46.66, H 3.60, N 7.77.
Solution state: 1H NMR (600 MHz, DMSO-d6) δ = 12.79 (3H, bs, NH), 7.80 (6H, d, Ho, J = 44 Hz), 7.56 (3H, t, Hp, J = 24 Hz), 7.54 (6H, t, Hm, J = 28 Hz). 13C NMR (151 MHz, DMSO-d6) δ = 160.1 (CO), 130.6 (Cquaternary), 130.70 (Cquaternary), 128.0 (Co), 127.2 (Co) 126.0 (Cm) 124.5 (Cm). High resolution ESI-MS+m/z: ([GaC14H11N2O4 + Na]+) calcd: 362.9867, found: 362.9842; ([GaC21H18N3O6 + Na]+) calcd: 500.0343, found: 500.0310.
:50 mixture of ethanol and water and heated gently to ∼60 °C before the addition of 1.0 mmol (0.420 g) of solid [Ga(NO3)3·9H2O]. NaHCO3 was added dropwise until pH of approximately 7–8. Effervescence of CO2 was observed. The resultant pink-white precipitate was gravity filtered and washed 3 × 15 mL of H2O.
Solid state: Yield 0.27 g (72.7%), m.p. 245 °C (decomp), FT-IR (cm−1): 3065 bw (O–Hph), 1599 s v(CN), 1569 m v(CO), 1512 s, 1475 s, 1364 m, 1304 m, 1247 s, 1147 s, 1096 s, 1033 s, 923 s, 744 s. Elemental analysis; theoretical (%) (3 + ½H2O): C 44.02, H 3.17, N 7.33. Found: C 44.20, H 3.32, N 7.38. Note: 0.5 equiv. of H2O observed in TGA trace.
Solution state: [Ga(SHA-H2)(SHA-H)] + [Ga(SHA-H2)3]: 1H NMR (600 MHz, DMSO-d6) δ = 12.62 (0.12H, d, Him), 11.72 (4H, bs, NH/OH), 10.20 (0.05H, bs, Him), 9.33 (0.75H, bs, OHph), 7.73 (1H, dd, hydroximate Ho), 7.67 (1H, dd, hydroxamate Ho), 7.37 (1H, td, hydroxamate Hp), 7.32 (1H, td, hydroximate Hp), 6.90 (4H, m, hydroximate & hydroxamate Hm). 13C NMR (151 MHz, DMSO-d6) δ = 166.2 (CO), 159.6 (CN), 159.4 (hydroxamate Cquaternary), 155.8 (hydroximate Cquaternary), 133.4 (hydroxamate Cphenol), 132.3 (hydroximate Cphenol), 128.5 (hydroximate Co), 127.0 (hydroxamate Co), 119.3 (hydroximate Cm), 118.7 (hydroxamate Cm), 117.3 (hydroxamate Cm), 116.3 (hydroximate Cm), 114.2 (hydroximate Cp), 114.0 (hydroxamate Cp). High resolution ESI-MS+m/z: ([GaC14H11N2O6 + Na]+) calcd: 394.9765, found: 394.9747; ([GaC21H18N3O9 + Na]+) calcd: 548.0191, found: 548.0166.
O). FT-IR (cm−1): 3060 b, 2962 b, 2910 v(C–H), 1586 s (CO), 1534 s (C–N), 1439 s, 1187 m, 1151 s, 1075 m, 1037 m, 939 s, 914 s, 770 s, 693 s. High resolution ESI-MS+m/z: [Ga(CH3)2(C6H5CON(C6H5)O)3 + H] calcd: 312.0513. found: 312.0546. Elemental analysis; calcd (%): C 57.62, H 5.15, N 4.52, found: C 57.74, H 5.17, N 4.49. CCDC: 2380238.†
O). FT-IR (cm−1): 3193 b, 2848 b (C–H), 2722 b (N–H), 1601 s (CO), 1535 s (C–N), 1485 m, 1446 w, 1363 w, 1315 w, 1143 s, 1074 s, 1027 m, 923 s, 789 m, 688 s. High resolution ESI-MS+m/z: [GaCH3(C6H5CONHO)2 + H] calcd: 357.0361. Found: 357.0358. Elemental analysis (5 + 0.33 toluene, observed in NMR) calcd (%): C 53.72, H 4.49, N 6.88. Found: C 53.69, H 4.59, N 7.23. CCDC: 2380239.†
O), 1463 m, 1432 s, 1160 m, 1047 m, 1021 m, 938 s, 761 s, 720 m, 693 s, 668 s. Elemental analysis calcd (%): C 67.64, H 4.37, N 6.07, found: C 67.51, H 4.34, N 6.13. High-resolution ESI-MS+m/z: ([FeC39H30N3O6 + H]+) calcd: 693.1556, found: 693.1557; ([FeC39H30N3O6 + Na]+) calcd: 715.1376, found: 715.1375. CCDC: 2150396.†
Author contributions
Professor Phil Andrews: primary investigator and supervisor, writing, editing and revision of manuscript. Dr Rebekah Duffin: data collection, data analysis, research-based activities, writing, editing and revision of manuscript. Equal contribution of authorship. Miss Jenisi Kelderman: data collection, data analysis, research-based activities, writing. Equal contribution of authorship. Dr Megan Herdman: data analysis, research-based activities.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for complexes 1–10 have been deposited at the Cambridge Crystallographic Data Centre (CCDC). The relevant CCDC number can be found for each complex in the Experimental sections (incl ESI†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Australian Research Council (DP220103632) and Monash University for financial support. We would also like to thank Dr Craig Forsyth (Monash) for assistance with X-ray crystallography.References
-
M. Wu and X. Li, Mol. Med. Microbiol, Elsevier, 2015, 10, 1547–1564 Search PubMed
.
- M. S. Mulani, E. E. Kamble, S. N. Kumkar, M. S. Tawre and K. R. Pardesi, Front. Microbiol., 2019, 10, 539 CrossRef
- R. Podschun and U. Ullmann, Clin. Microbiol. Rev., 1998, 11, 589–603 CrossRef CAS
- C. Schroll, K. B. Barken, K. A. Krogfelt and C. Struve, BMC Microbiol., 2010, 10, 1–10 CrossRef
- J. Jagnow and S. Clegg, Microbiology, 2003, 149, 2397–2405 CrossRef CAS
- L. S. Munoz-Price, L. Poirel, R. A. Bonomo, M. J. Schwaber, G. L. Daikos, M. Cormican, G. Cornaglia, J. Garau, M. Gniadkowski and M. K. Hayden, Lancet Infect. Dis., 2013, 13, 785–796 CrossRef
- K. Kuo, Y. Shen and K. Hwang, J. Microbiol. Immunol. Infect., 2007, 40, 248–254 Search PubMed
- G. Cortés, N. Borrell, B. de Astorza, C. Gómez, J. Sauleda and S. Albertí, Infect. Immun., 2002, 70, 2583–2590 CrossRef
- H. Sahly, R. Podschun, T. A. Oelschlaeger, M. Greiwe, H. Parolis, D. Hasty, J. R. Kekow, U. Ullmann, I. Ofek and S. Sela, Infect. Immun., 2000, 68, 6744–6749 CrossRef CAS
- E. Padilla, E. Llobet, A. Doménech-Sánchez, L. Martínez-Martínez, J. A. Bengoechea and S. Albertí, Antimicrob. Agents Chemother., 2010, 54, 177–183 CrossRef CAS PubMed
- B. Li, Y. Zhao, C. Liu, Z. Chen and D. Zhou, Future Microbiol., 2014, 9, 1071–1081 CrossRef CAS PubMed
- T. A. Russo, A. S. Shon, J. M. Beanan, R. Olson, U. MacDonald, A. O. Pomakov and M. P. Visitacion, PLoS One, 2011, 6, e26734 CrossRef CAS PubMed
- M. A. Bachman, S. Lenio, L. Schmidt, J. E. Oyler and J. N. Weiser, mBio, 2012, 3, e00224–e00211 CrossRef CAS PubMed
- M. A. Bachman, J. E. Oyler, S. H. Burns, M. Caza, F. Lépine, C. M. Dozois and J. N. Weiser, Infect. Immun., 2011, 79, 3309–3316 CrossRef CAS
- D. C. Bailey, E. J. Drake, T. D. Grant and A. M. Gulick, Biochemistry, 2016, 55, 3559–3570 CrossRef CAS PubMed
- M. A. Bachman, V. L. Miller and J. N. Weiser, PLoS Pathog., 2009, 5, e1000622 CrossRef
- C. R. Chitambar, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2044–2053 CrossRef CAS
- A. B. Kelson, M. Carnevali and V. Truong-Le, Curr. Opin. Pharmacol., 2013, 13, 707–716 CrossRef CAS PubMed
- J. A. A. Huayhuaz, H. A. Vitorino, O. S. Campos, S. H. P. Serrano, T. M. Kaneko and B. P. Espósito, J. Trace Elem. Med. Biol., 2017, 41, 16–22 CrossRef CAS
- T. Sultana, R. N. Duffin, V. L. Blair and P. C. Andrews, Chem. Commun., 2023, 59, 11093–11096 RSC
- R. N. Duffin and P. C. Andrews, J. Inorg. Biochem., 2023, 249, 112371 CrossRef CAS PubMed
- R. N. Duffin, V. L. Blair, L. Kedzierski and P. C. Andrews, Eur. J. Med. Chem., 2020, 186, 111895 CrossRef CAS PubMed
- M. J. McKeage, S. J. Berners-Price, P. Galettis, R. J. Bowen, W. Brouwer, L. Ding, L. Zhuang and B. C. Baguley, Cancer Chemother. Pharmacol., 2000, 46, 343–350 CrossRef CAS
- T. Riss and R. Moravec, Assay Drug Dev. Technol., 2004, 2, 51–62 CrossRef CAS PubMed
- R. S. Go and A. A. Adjei, J. Clin. Oncol., 1999, 17, 409–422 CrossRef CAS PubMed
- Y. He, Q. Zhu, M. Chen, Q. Huang, W. Wang, Q. Li, Y. Huang and W. Di, Oncotarget, 2016, 7, 70803–70821 CrossRef
- Y. T. Wang, Y. Fang, M. Zhao, M. X. Li, Y. M. Ji and Q. X. Han, MedChemComm, 2017, 8, 2125–2132 RSC
- C. Y. Effah, T. Sun, S. Liu and Y. Wu, Ann. Clin. Microbiol. Antimicrob., 2020, 19, 1–9 CrossRef CAS PubMed
- S. Hijazi, D. Visaggio, M. Pirolo, E. Frangipani, L. Bernstein and P. Visca, Front. Cell. Infect. Microbiol., 2018, 8, 316 CrossRef PubMed
- C. R. Chitambar, D. P. Purpi, J. Woodliff, M. Yang and J. P. Wereley, J. Pharmacol. Exp. Ther., 2007, 322, 1228–1236 CrossRef CAS PubMed
- L. R. Bernstein, T. Tanner, C. Godfrey and B. Noll, Met.-Based Drugs, 2000, 7, 33–47 CrossRef CAS
- D. K. Coggin, C. J. Mathias and M. A. Green, Nucl. Med. Biol., 1994, 21, 283–285 CrossRef CAS
- C. R. Chitambar and Z. Zivkovic, Cancer Res., 1987, 47, 3929–3934 CAS
- W. R. Harris and L. Messori, Coord. Chem. Rev., 2002, 228, 237–262 CrossRef CAS
- W. R. Harris and V. L. Pecoraro, Biochemistry, 1983, 22, 292–299 CrossRef CAS
- É. A. Enyedy, O. Dömötör, K. Bali, A. Hetényi, T. Tuccinardi and B. K. Keppler, J. Biol. Inorg. Chem., 2015, 20, 77–88 CrossRef PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112–122 CrossRef CAS
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- R. N. Duffin, V. L. Blair, L. Kedzierski and P. C. Andrews, Dalton Trans., 2018, 47, 971–980 RSC
- R. N. Duffin, V. L. Blair, L. Kedzierski and P. C. Andrews, J. Inorg. Biochem., 2018, 189, 151–162 CrossRef CAS
- R. N. Duffin, V. L. Blair, L. Kedzierski and P. C. Andrews, J. Inorg. Biochem., 2020, 203, 110932 CrossRef CAS
- R. N. Duffin, V. L. Blair, L. Kedzierski and P. C. Andrews, J. Inorg. Biochem., 2021, 219, 111385 CrossRef CAS PubMed
- A. Pathak, V. L. Blair, R. L. Ferrero, L. Kedzierski and P. C. Andrews, J. Inorg. Biochem., 2017, 177, 266–275 CrossRef CAS PubMed
- M. Werrett, M. Herdman, R. Brammananth, U. Garusinghe, W. Batchelor, P. Crellin, R. Coppel and P. C. Andrews, Chem. – Eur. J., 2018, 24, 12938–12949 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2150403, 2150396, 2380238, 2380239, 2150393, 2330100, 2330099 and 2330101. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02440k |
| This journal is © The Royal Society of Chemistry 2025 |