
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction mechanisms in swelling clays under ionizing radiation: influence of the water amount and of the nature of the clay mineral†
M. Lainéa,
E. Balanb,
T. Allardb,
E. Paineau c,
P. Jeunessed,
M. Mostafavid,
J.-L. Robertb and
S. Le Caër*a
c,
P. Jeunessed,
M. Mostafavid,
J.-L. Robertb and
S. Le Caër*a
aLIONS, NIMBE, CEA, CNRS, Université Paris Saclay, CEA Saclay, F-91191 Gif-sur-Yvette Cedex, France. E-mail: sophie.le-caer@cea.fr
bIMPMC, Sorbonne Universities, UPMC, CNRS UMR-7590, MNHN, IRD, 4 Place Jussieu, F-75252 Paris Cedex 05, France
cLaboratoire de Physique des Solides, CNRS, Univ. Paris-Sud, Université Paris-Saclay, F-91405 Orsay Cedex, France
dLaboratoire de Chimie Physique, CNRS/Université Paris-Sud, Bâtiment 349, F-91405 Orsay, France
First published on 3rd January 2017
Abstract
We have studied the H2 production under ionizing radiation of water confined in synthetic saponite and montmorillonite as a function of the relative humidity. The H2 radiolytic yields in the dry systems are very similar to that measured in a non-swelling clay mineral. They are 2–3 times higher with one water layer in the interlayer space, evidencing very efficient energy transfers and efficient recombination reactions due to a high confinement. With two water layers, the H2 yields decrease as compared to the previous case, but remain higher than in bulk water, proving that recombination reactions of hydrogen atoms are less efficient. Electron paramagnetic resonance measurements evidence that reactivity changes significantly with the number of water layers. Saponite and montmorillonite give similar results, showing that reactivity is driven by the amount of water and that the details of the clay structure play a less important role. Lastly, the behavior of natural vs. synthetic swelling clays is discussed. The presence of impurities, even in small quantities, significantly alters energy transfers and has a positive implication for the geological nuclear waste management.
Introduction
Currently, the nuclear industry represents one of the best carbon footprint energy sources although the nuclear waste management remains a major issue. In particular, High Activity (HA) and Long Life-Medium Activity (LL-MA) nuclear wastes have to be isolated from our environment during several thousands of years until they reach the natural radioactivity. To tackle this issue, one of the alternatives chosen by several countries is deep geological disposal.1,2 In France, HA and LL-MA nuclear wastes would be buried at several hundred meters below the surface in a clay stratum. The corresponding clay minerals, mainly smectites, present good swelling and retention properties making them materials of choice for the long term disposal of nuclear wastes. However, these swelling clays contain both hydroxyl groups and water molecules and are prone to release H2 when they are submitted to ionizing radiation.3–9 This specific reactivity can have a significant impact on the safety of nuclear waste disposal.Clay minerals, and more generally the phyllosilicates, are layered structures. Each layer can be decomposed in geometrically distinct sheets. One of these sheets is composed of TO4 tetrahedral (T) sites usually occupied by Si4+, Al3+ or Fe3+ cations interconnected by three vertices, which combine themselves into pseudo-hexagons (TO4)6. This tetrahedral sheet is always bonded to an octahedral sheet formed of octahedral (O) sites, usually occupied by Al3+, Fe3+, Mg2+ or Fe2+ cations. Clay minerals are called dioctahedral if 2/3 of octahedral sites are filled by trivalent cations (such as montmorillonite) or trioctahedral if all octahedral sites are filled by divalent cations (such as saponite). When the basic layer consists in one octahedral sheet sandwiched by two tetrahedral sheets, the clay mineral is referred to as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or TOT clay (Fig. 1). Because of the frequent substitution of cations in tetrahedral and/or octahedral sites, the layers can display an excess negative charge which is compensated by the insertion of cations in the interlayer space (Fig. 1). These cations can be surrounded by water molecules depending on the relative humidity of the medium. When water molecules penetrate between the layers, they force them apart, leading to a stepwise swelling. Consequently, 0, 1, 2 layer(s) of water molecules are usually found in the interlayer space around the cations,10 with basal spacings around 10, 12 and 15 Å, respectively. As the TOT layer has a thickness of 9 Å (Fig. 1), this implies that the interlayer space is a highly 2D confined environment.
:1 or TOT clay (Fig. 1). Because of the frequent substitution of cations in tetrahedral and/or octahedral sites, the layers can display an excess negative charge which is compensated by the insertion of cations in the interlayer space (Fig. 1). These cations can be surrounded by water molecules depending on the relative humidity of the medium. When water molecules penetrate between the layers, they force them apart, leading to a stepwise swelling. Consequently, 0, 1, 2 layer(s) of water molecules are usually found in the interlayer space around the cations,10 with basal spacings around 10, 12 and 15 Å, respectively. As the TOT layer has a thickness of 9 Å (Fig. 1), this implies that the interlayer space is a highly 2D confined environment.
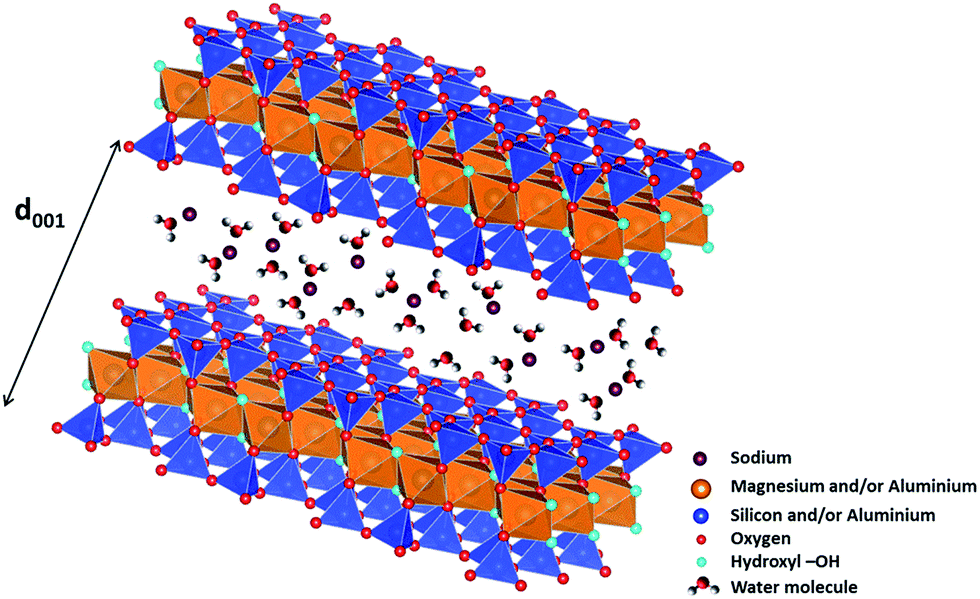 | ||
| Fig. 1 Structure of 2/1 phyllosilicates (TOT), two TO4 (T = Si4+ and/or Al3+) tetrahedral sheets on each side of an octahedral sheet occupied by magnesium and/or aluminium cations, forming 2D layers (figure made using the VESTA software).11 Each TOT layer has a thickness of roughly 9 Å. Compensating cations (here sodium) are present in interlayer space and are surrounded by water molecules. The d001 value obtained from X-ray diffraction experiments enables determining the thickness of the interlayer space, and hence, the number of water layers in it. | ||
Despite its environmental significance, only few studies discuss the behavior under radiation of those complex and confining systems.5,12–17 Indeed, energy transfer phenomena should be correctly considered as well as pathways of reactive species within the interlayer space of clay minerals that is highly confined, and in which specific effects may be expected. In a previous study,5 we have focused our attention on natural montmorillonites of Camp-Berteau (Marocco). The purpose of the present work is to understand in details how the nature of the clay mineral and the water content influence the H2 production under ionizing radiation. In this framework, synthetic montmorillonite and saponite (TOT clay minerals) were chosen as representative models of swelling clays that will help understanding the processes at stake. Their chemical formulae are respectively Na0.5(Al1.5Mg0.5)Si4O10(OH)2 for montmorillonite and Na0.5Mg3(Si3.5Al0.5)O10(OH)2 for saponite. The difference between these minerals is thus related to the location of the excess negative charge of the TOT layers, i.e. in the octahedral or the tetrahedral sheet, respectively. The determination of H2 production yields as a function of the relative humidity together with EPR measurements provides a picture of H2 reaction mechanisms. Pulse radiolysis experiments performed at the picosecond time scale on natural films, together with previous results obtained with natural montmorillonites5 will help understanding the reactivity differences between natural and synthetic samples. The implications for the nuclear waste management will then be discussed.
Experimental
Sample conditioning, irradiation and gas chromatography
Synthetic montmorillonite and saponite were synthesized using high purity chemicals (99.9%) by hydrothermal method from gels of appropriate compositions prepared according to conventional gelling method.18–20The samples were first dehydrated at 100 °C for 24 hours in order to remove any water remaining from synthesis and then placed in different desiccators at known relative humidity (RH) until equilibrium is reached: 3% (using silica gel), 11% (LiCl saturated solution), 43% (K2CO3 saturated solution), 74% (NaNO3 saturated solution) and 97% (K2SO4 saturated solution). Sample weight stability attests that equilibrium is reached. The driest possible state without damaging the clay structure (0%) was obtained by heating the samples directly in the irradiation ampoules (see below) under primary vacuum at 110 °C for 48 hours. The samples were controlled by a thorough characterization; infrared spectroscopy (IR), thermogravimetric analysis (TGA) and X-ray diffraction (XRD) are given in ESI SI-1, SI-2 and SI-3,† respectively. The total water content of each sample was determined from TGA measurements (see below).
The conditioning procedure for irradiation experiments and gas chromatography analysis was previously described.21 Briefly, samples were placed in a Pyrex glass ampoule and then irradiated using a Titan Beta, Inc. linear accelerator (LINAC)22 with 10 MeV electron pulses of 10 ns duration at a repetition rate of 2 Hz to avoid macroscopic heating. The dose per pulse (30 ± 2 Gy per pulse, with 1 Gy = 1 J kg−1) was determined using the Fricke dosimetry.23 H2 formed under irradiation was measured by gas chromatography (μGC-R3000 SRA Instrument) using ultra-high purity argon as a carrier gas. Samples were also characterized after irradiation to ensure that irradiation conditions have no effect on their water content (see SI-4†).
Thermogravimetric analysis (TGA)
Thermogravimetric Analysis (TGA) were performed with a Mettler-Toledo TGA/DSC 1 analyzer. Samples were placed in 70 μL alumina crucibles and heated to 900 °C at a heat flow of 10 °C min−1 under a dinitrogen flux of 50 mL min−1. Data were analyzed with the STARe software. Differential Thermal Analysis (DTA) was also obtained by plotting the derivate function of TGA data in order to identify the different weight loss domains: roughly from 40 to 110 °C and from 110 to 200 °C (SI-2†).Electron paramagnetic resonance (EPR)
EPR experiments were carried out at low temperature (90 to 240 K) at a microwave power of 1 mW thanks to a Bruker EMX plus spectrometer in X-band bridge (9.1–9.6 GHz) equipped with a cold nitrogen flux device. The sample conditioning and subsequent irradiation was performed according to the method previously described.21 Our previous study evidenced that 15 kGy was an appropriate dose to perform the EPR measurements. Therefore all the spectra presented below were recorded at this dose. Data were analyzed with the Win EPR software and modeled spectra were obtained using the ZFSFIT code which is based on a numerical diagonalization of the spin Hamiltonian.24Picosecond pulse radiolysis experiments
Pulse radiolysis experiments (transient absorption spectroscopy measurements) were carried out at the ELYSE facility.25–27 The overall time resolution of the set-up is 10 ps. It consists of a pump–probe experiment where reactive species are generated directly in the samples by an electron beam and then detected by UV-Visible spectroscopy. Investigated samples were prepared in the form of self-supporting films with a thickness optimized (about 500 μm) so as to be optically transparent but to allow signal detection. This could not be achieved with synthetic samples due to the opacity of the films obtained. To overcome this issue, two natural clay minerals were used instead: a montmorillonite from Arizona Na0.555(Al1.375Mg0.535Fe0.0853+)(Si3.975Al0.025)O10(OH)2 (ref. 28) and a beidellite from Idaho Na0.474(Al1.812Mg0.09Fe0.1123+)(Si3.574Al0.426)O10(OH)2.29 These natural samples have been chosen because they display a contrasted location of the excess negative charge, i.e. mainly octahedral for the montmorillonite and mainly tetrahedral for the beidellite, as for the synthetic samples. They also contain different amounts of structural iron Fe3+. The films were prepared by successive deposition from aqueous suspensions.28 They were then placed in front of the electron beam in a controlled relative humidity chamber, designed for that purpose, and equipped with pellicle uncoated windows allowing the passage of the laser beam. Two significantly different relative humidities were studied: 11% and 97%. Due to the very low absorbance of the species detected, the signal was monitored at 782 nm only. At this wavelength, only the electron, which has a high molar extinction coefficient, is expected to give a signal.30–33 Unfortunately, due to the low detected absorbances, it was then impossible to record the spectrum of the electron.Results and discussion
Water amount and dihydrogen production measurements
TGA measurements of synthetic montmorillonite (Fig. 2a) and saponite (Fig. 2b) enable us to monitor the weight loss during the heating of the sample and, hence, to determine the water “type” together with the corresponding weight percentage (%) as a function of RH (Fig. 2c and d). Bonded Water (BW) corresponds to water molecules strongly bonded to the cation and to the surface of the sheets. Free Water (FW) corresponds to water molecules more weakly bonded. The sum represents the total water content. Roughly, free water weight loss occurs between 40 and 110 °C while bonded water weight loss occurs between 110 and 200 °C. Fig. 2c and d show that the bonded water content is almost constant, within the error bars, with RH in both systems, whereas free water increases with relative humidity, as expected. In samples at 0% RH, only BW is present. FW is detected at 3% RH for montmorillonite and saponite.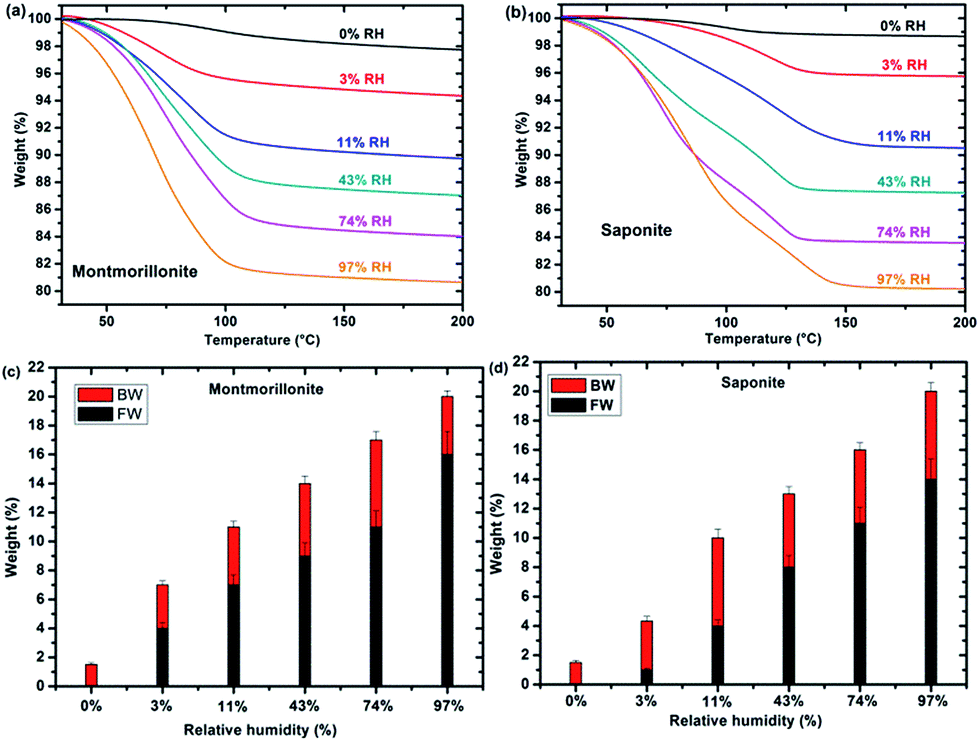 | ||
| Fig. 2 TGA from 30 to 200 °C of synthetic montmorillonite (a) and saponite (b) according to relative humidity (upper part). Free Water (FW, in black), Bonded Water (BW, in red) and Total Water Content (TWC) (in weight percent) are represented for synthetic montmorillonite (c) and saponite (d) (lower part). | ||
Under irradiation, dihydrogen is produced. The cumulated H2 production evolves linearly with the dose, for the whole sets of samples and relative humidity conditions (see SI-5†). The corresponding H2 radiolytic yields, expressed in mol J−1 (Fig. 3a) were obtained from the slope of the lines.
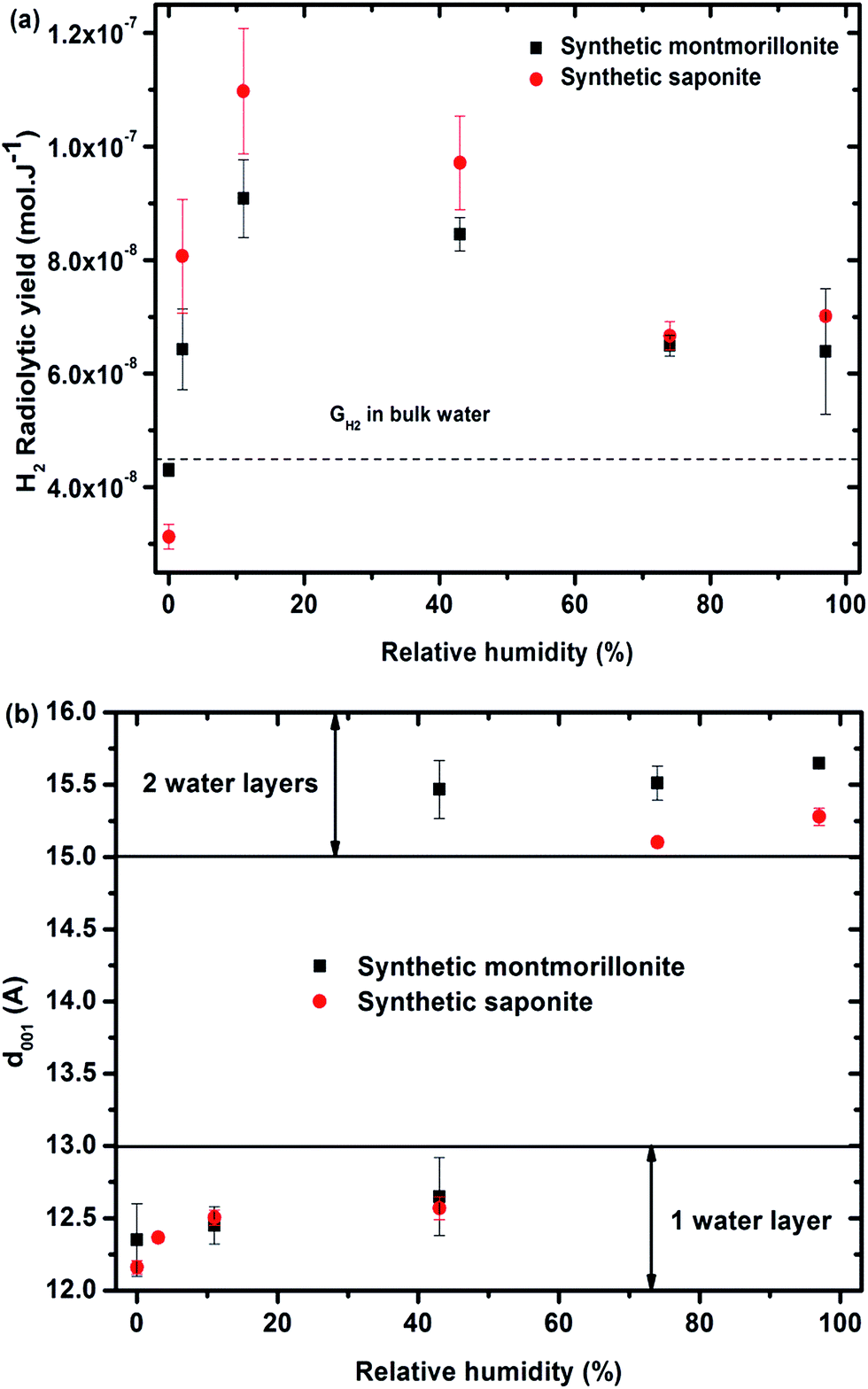 | ||
Fig. 3 (a) H2 radiolytic yields as a function of the relative humidity for synthetic montmorillonite (■) and saponite ( ). The value obtained in liquid bulk water is given as a comparison (4.5 × 10−8 mol J−1) to unravel the specific behavior of confined water. (b) d001 distance as a function of the relative humidity obtained for synthetic montmorillonite (■) and saponite ( ). The value obtained in liquid bulk water is given as a comparison (4.5 × 10−8 mol J−1) to unravel the specific behavior of confined water. (b) d001 distance as a function of the relative humidity obtained for synthetic montmorillonite (■) and saponite ( ). No value for montmorillonite at 3% RH is given here, as the assignment of the peaks was unlikely in this case. For the montmorillonite at 43% RH, two peaks were obtained that account for the simultaneous presence of 1 and 2 water layer(s) in the interlayer space (see SI-3†). ). No value for montmorillonite at 3% RH is given here, as the assignment of the peaks was unlikely in this case. For the montmorillonite at 43% RH, two peaks were obtained that account for the simultaneous presence of 1 and 2 water layer(s) in the interlayer space (see SI-3†). | ||
The evolution of the H2 yield with RH is similar for montmorillonite and saponite, indicating that the excess charge location in the clay structure only plays a minor role on the mechanisms of H2 production in synthetic samples. In parallel, the number of water layers in the interlayer space (Fig. 3b) was determined from XRD d001 values (see SI-3†). A direct comparison of both trends suggests that the amount of inter-layer water molecules strongly affects the H2 production. For the driest state (0%), the H2 radiolytic yield measured for both samples ((4.3 ± 0.1) × 10−8 and (3.1 ± 0.2) × 10−8 mol J−1 for montmorillonite and saponite, respectively) is lower than the one measured in bulk water (4.5 × 10−8 mol J−1), but consistent with the value recently obtained in non-swelling clay minerals ((2.9 ± 0.3) × 10−8 mol J−1 in talc).21 At 0% RH, the higher d001 value measured in the case of montmorillonite as compared to saponite may account for the higher H2 yield determined at this RH (Fig. 3b). Noteworthy, if one water layer is always present, even in the driest state, TGA measurements evidence that only bonded water (and not free water) is found in both samples at 0% RH (Fig. 2c and d).
From 0% to 11% RH, the H2 production yield strongly increases and becomes even higher than the value obtained in bulk water. The d001 is still consistent with one water layer in the interlayer space but the number of water molecules increases with RH as attested by ATR-FTIR measurements (SI-1†). A full energy transfer occurs from the irradiated layers to the interlayer space.
Compared with bulk water, the dissociation of the O–H bond could be facilitated by interaction with inter-layer sodium cations, which increases the O–H bond polarization.34 Moreover, the molecules being highly confined into the interlayer space (about 2.5 Å), the recombination of reactive species and formation of H2 molecules could be favored.
At higher RH, from 11% to 74%, the H2 radiolytic yield is first stable (11–43% RH) and then decreases slightly, but still remains higher than in bulk water.
In this RH range, more water molecules penetrate in the interlayer space (≈5 Å) where globally two water layers are observed. The confinement effect becomes less pronounced and the recombination of hydrogen atoms is less efficient. Adsorbed water molecules are also present. Above 74% RH, the H2 radiolytic yield remains constant. The water molecules being organized in two water layers in the interlayer space, a fraction of water molecules exhibits a bulk-like behavior34–37 and direct radiolysis of water also takes place. Indeed, the water mass percentage is about 20% in the most hydrated samples (Fig. 2c and d).
Electron paramagnetic resonance experiments to identify the radical defects created upon irradiation
Only paramagnetic defects are detected by EPR in irradiated samples. Contrary to our previous results on talc,21 these defects are not stable and only observed at low temperatures. The spectra obtained at 90 K for both clays with various water contents look very similar (Fig. 4 for montmorillonite; the spectra obtained for saponite are given in SI-6†). This is an additional evidence that the reaction mechanisms under ionizing radiation are very similar, whatever the charge location in the layer of the swelling clay. Thus, only the spectra of montmorillonite will be discussed in further details afterwards (see SI-6† for saponite results). At 0% RH (i.e., the driest system, Fig. 4a), radical H˙ atoms (i.e., electron trapped on a proton) are detected. Their signal consists of a doublet centered at geff = 2.026 with a hyperfine splitting of 50.3 mT.21,27 Note that the actual g-value of this centre differs from the effective one because of the relatively high value of the hyperfine interaction (with respect to Zeeman splitting). The g-value (∼2.002) obtained at a second order perturbation level38 is similar to previously reported values for H˙ atoms in other irradiated clay minerals.5,21,38 A superhyperfine structure is observed, due to the proximity of nonzero nuclear spin atoms such as 25Mg.21,39 The central part of the spectrum displays an overlap of spectra related to six distinct paramagnetic centers, labelled from (1) to (6) (Fig. 4d, Table 1). A tentative identification of these centers can be done from measured EPR parameters (g-tensor principal values), based on literature data.21,40–49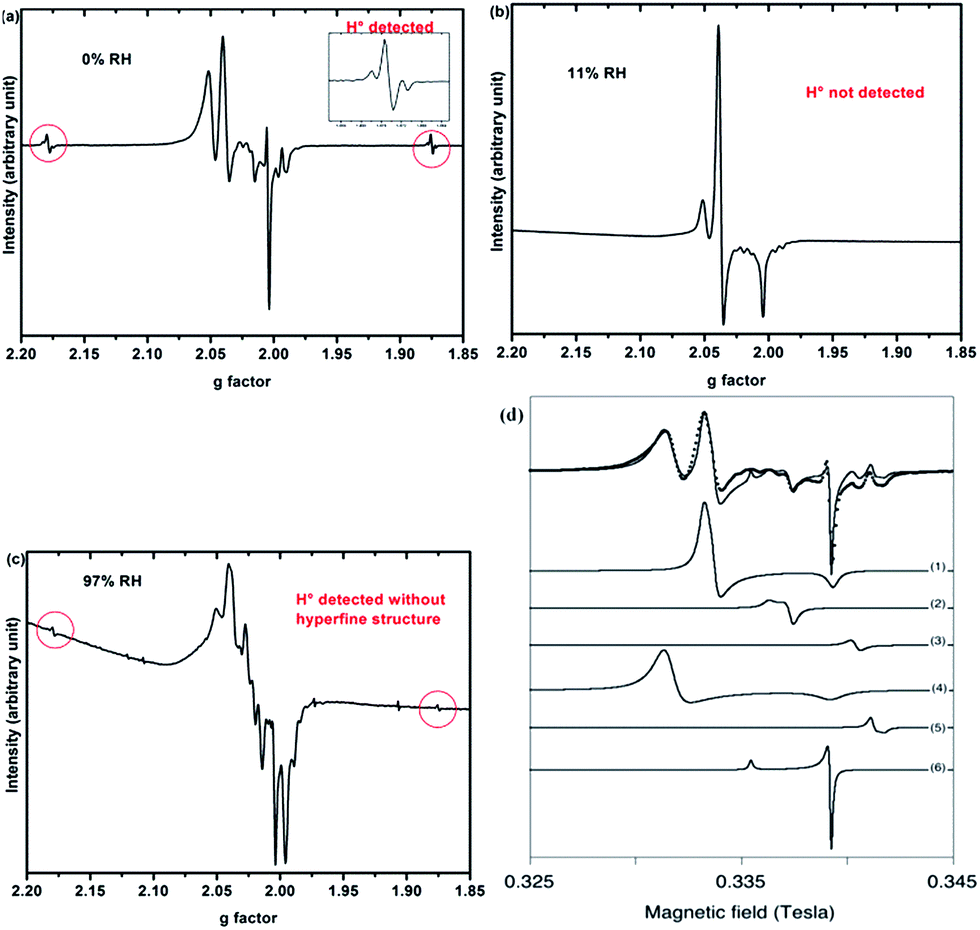 | ||
| Fig. 4 EPR spectra of synthetic montmorillonite at (a) 0% RH, (b) 11% RH and (c) 97% RH irradiated at 15 kGy. The g-values are effective ones. The spectra are recorded at 90 K. (d) Experimental data (black dots) of montmorillonite 0% with the corresponding fit (black line) together with the six components of this fit. Corresponding g values are summarized in Table 1. | ||
| Center | (1) | (2) | (3) | (4) | (5) | (6) | |
|---|---|---|---|---|---|---|---|
| g | ⊥ | 2.0365 | 2.0145 | 1.9965 | 2.0495 | 1.9920 | 2.0035 |
| ∥ | 2.0028 | 2.0210 | 2.0035 | 1.9885 | 2.0260 | ||
| Attribution | –O˙− | Unknown | Trapped e− | –O˙− | Trapped e− | Unknown | |
The centers (1) and (4) are attributed to “–MgO−” ion-radicals in different environments arising from the lysis of the MgO–H bond, as already observed in MgO samples43–49 and in synthetic talc.21 It corresponds to a hole located on an oxygen atom not bonded to silicon and linked to magnesium atoms. Using formal ionic charges, the defects detected by EPR are more explicitly described as –(Mg2+)n(O˙−) where n is the number of Mg atoms coordinated to the oxygen.50 Except in the description of reaction mechanisms, this defect will be referred to as –O˙− hereafter. The isotropic signal at g = 1.9965 (center (3)) was also observed in synthetic talc (g = 1.996)21 and is attributed to a trapped electron according to its g value lower than ge = 2.0023 for the free electron. Similarly, the center (5) displays a g value lower than the one of the free electron, indicating a trapped electron in a non-isotropic configuration. The remaining last two centers (2) and (6) are present in both irradiated montmorillonite and saponite (Fig. 4a and SI-6†). Although not fully identified, they share some similarity with O hole centers located on the Si–O bond of other clay samples such as kaolinite or natural montmorillonite.51 Unfortunately, under our experimental conditions and due to the instability of defects at room temperature, we were not able to perform angular variation experiments. Therefore, a more detailed assignment of the defects was not possible.
When the relative humidity increases, the EPR spectrum changes drastically (Fig. 4b). At 11% RH, hydrogen atoms are no longer detected and the signal of the defects is clearly modified. Centers (1) and (4) (corresponding to –O˙−) and (6) are still present; whereas the three other defects are hardly detected. According to those results, the –O˙− ion-radicals are still formed but not in the same proportion as at 0% RH. At 97% RH, with two water layers in the interlayer space (Fig. 4c), H˙ radicals are detected again but without any superhyperfine structure, suggesting that these H˙ radicals are not in the same environment as those detected in Fig. 4a. The lack of super-hyperfine structure leads us to hypothesize that they are located farther from Mg atoms, potentially in the interlayer space or in the ditrigonal cavity.
The six centers described in Fig. 4a are still present but their spectrum is superimposed to broad signal centered around g = 2.025. The width of this new signal likely originates from magnetic interactions between the centers, suggesting that it is related to some clustering of the defects.
The EPR spectra thus evidence that reaction mechanisms are very sensitive to the water content of the samples. For example, –O˙− centers (1) and (4) have their respective intensities that greatly change in proportion with RH. Globally, the intensity of the EPR spectra, normalized to the sample mass, decreases when RH increases. Fig. 5 shows the evolution of the intensity of the EPR signal with RH for both samples. Spectra are normalized with respect to the same sample mass. Relative humidity has an important impact on the nature and the amount of the created defects. The decrease of the amount of defects with RH highlights the protective effect of water on the formation of defects, as already observed in the case of nanoporous silica.8
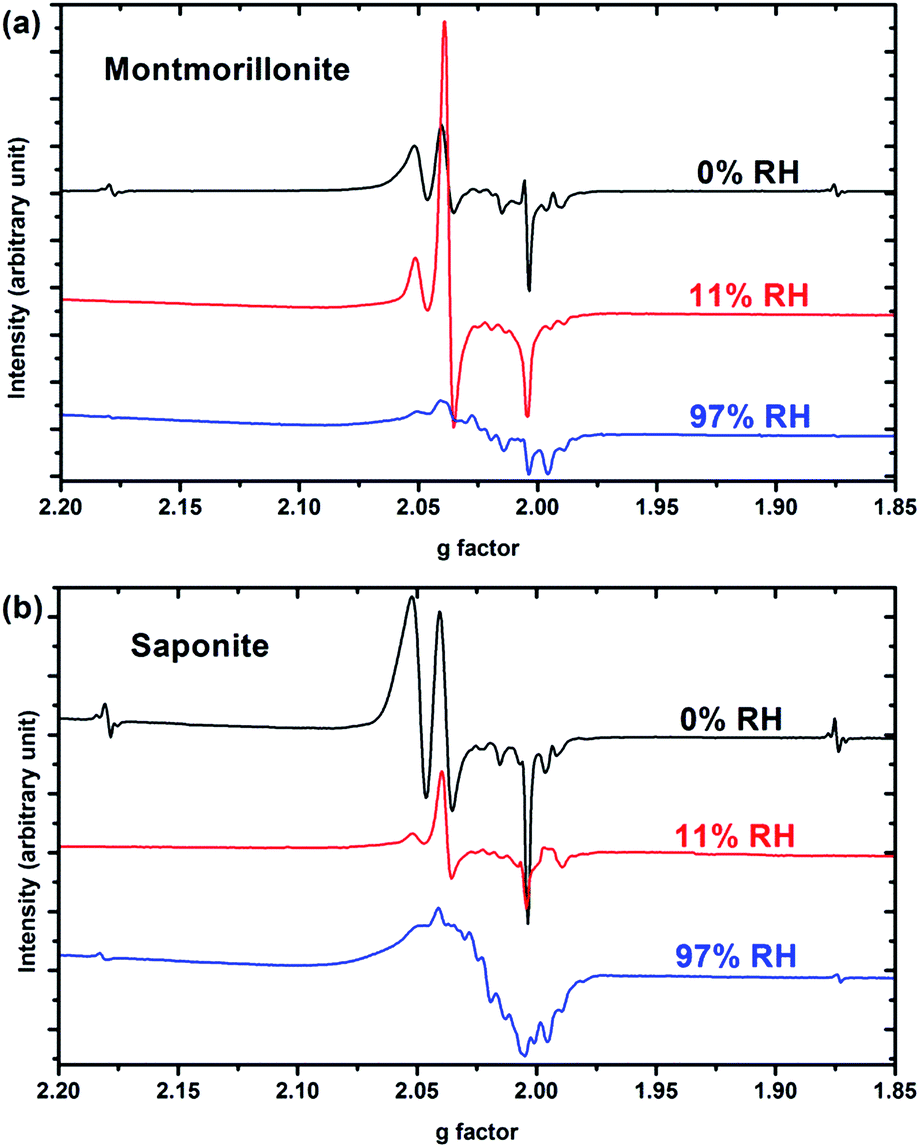 | ||
| Fig. 5 EPR spectrum of (a) synthetic montmorillonite and (b) synthetic saponite at 0%, 11% and 97% RH irradiated at 15 kGy. The spectra are recorded at 90 K. Spectra are normalized with respect to the same sample mass and reported as a function of effective g-values. | ||
Picosecond pulse radiolysis experiments
A more thorough understanding of the first stages of reactions issued from the ionizing radiation–matter interaction can be obtained from pulse radiolysis experiments. As stated in the experimental section, the production of films that are thick enough but remain optically transparent is a prerequisite for that kind of experiments and has been achieved only for natural swelling clay samples (montmorillonite and beidellite) with similar difference in charge location as the natural ones. This allows us to compare the short time reactivity in the two natural samples, which still display a different charge location in the structural layers. As the observed signal is very weak and, in order to increase the signal/noise ratio, it was chosen to work at 782 nm, which is the fundamental wavelength of the laser. Moreover, this wavelength range (700–800 nm) is known to be characteristic of the signature of the solvated electron in water.52,53 The kinetics, depicted in Fig. 6, is thus attributed to the decay of the electron. Fig. 6a presents the evolution of normalized absorbance at 782 nm for both montmorillonite from Arizona and beidellite at two different relative humidities whereas Fig. 6b highlights the low absorbance value obtained for these systems. For each sample, relative humidity has no significant influence on the electron decay: the signals are very similar and the characteristic time constants are the same, whatever RH. This suggests that the detected electrons are formed and trapped within the layer.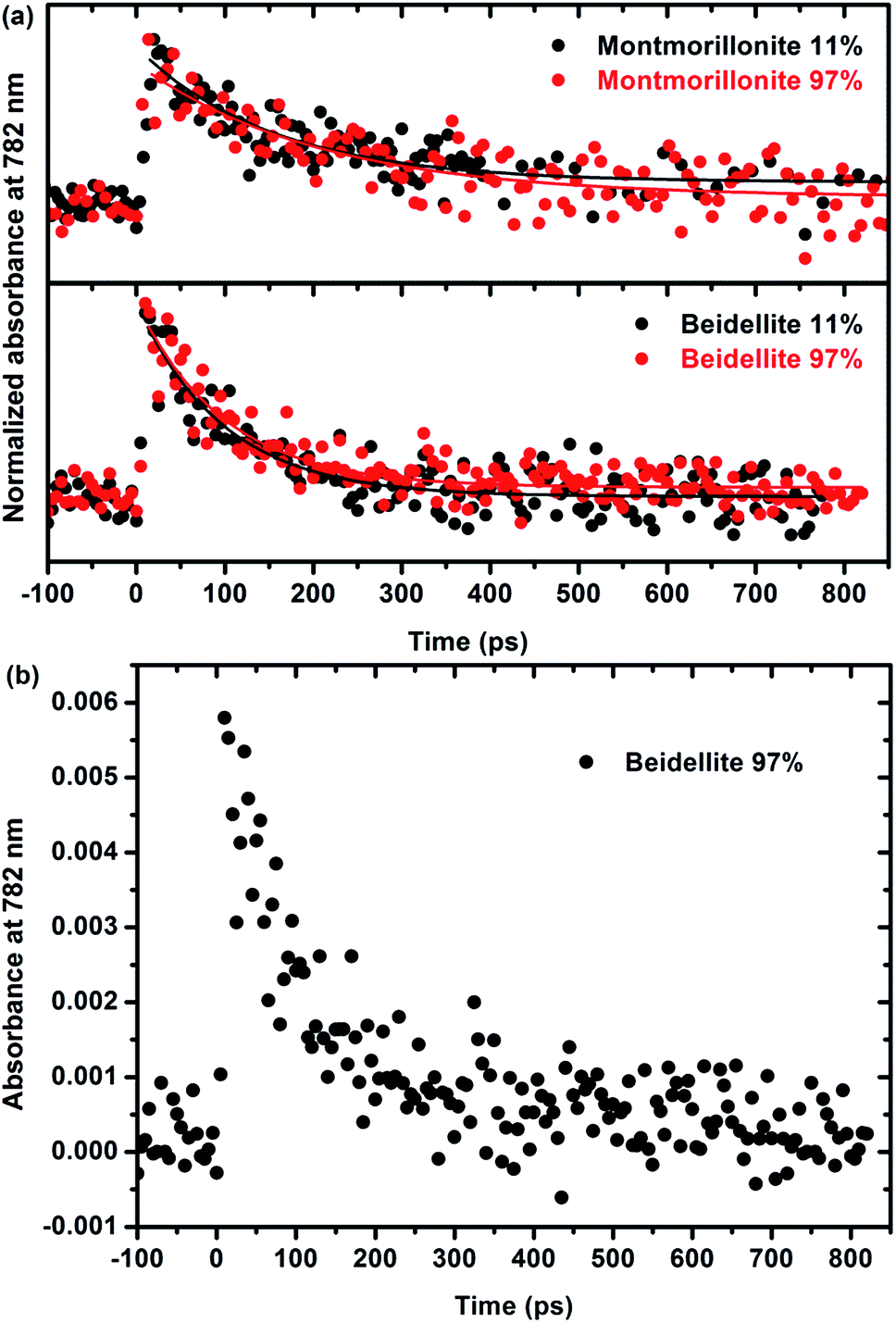 | ||
| Fig. 6 (a) Normalized absorbance at 782 nm of the electron decay and corresponding fits for natural swelling clays: Arizona montmorillonite (upper part) and beidellite (lower part) at two relative humidities. The corresponding time constants are 180 ± 30 ps for the montmorillonite from Arizona and 100 ± 10 ps for beidellite. The fits do not depend on RH. (b) Absorbance at 782 nm of the electron decay in natural beidellite at 97% relative humidity showing the very low values of the measured absorbance (0.006 at the maximum). | ||
Moreover, electrons decay very quickly (a few hundreds of ps), as compared to a few microseconds in bulk water.54,55 They are quickly trapped in the material, likely by impurities such as Fe3+. This interpretation is consistent with the lack of relation between the water content and the kinetics. It is also consistent with the shorter time (100 ± 10 ps) measured in beidellite as compared to montmorillonite from Arizona (180 ± 30 ps). Beidellite indeed displays a higher amount of Fe3+ (about 32% more) than montmorillonite. Noteworthy, the reduction of Fe3+ is difficult to observe by EPR. Nevertheless, the behavior of the two samples remains relatively close. As stated above, the location of the charge in the clay structure does not seem to have a major influence on its reactivity neither at short time (ultra-fast spectroscopy with natural samples) nor at long time (radiolytic yields and EPR experiments for synthetic samples). The reactivity appears to be mainly controlled by transition element impurities in the natural samples and by the water amount in the synthetic samples. The nature of the swelling clay itself plays a less important role.
Discussion
Synthetic montmorillonite and saponite exhibit the same evolution of their dihydrogen radiolytic yield (GH2) with RH (Fig. 3). All our experimental data evidence that, for the synthetic samples, the H2 radiolytic yields are mainly controlled by the amount and the nature of water, and not by the composition of the sheets.The hydration state of swelling clay minerals can be rationalized thanks to literature data.36 At very low relative humidity (from 0.1 to 2%), water molecules are adsorbed at the edge or at the surface of sheets and they hydrate cations at the external surfaces of sheets. These first water molecules are strongly bonded. For one water layer, cations and water molecules are usually located in the interlayer midplane.36,56,57 They interact with the cations and are not prone to form inter-molecular hydrogen bonds at low RH.58 The corresponding concentration of the compensating cation is very high (more than 10 mol L−1). Nevertheless, the sodium cations are not reactive towards the electron. The introduction of additional water molecules will then enable the formation of a H-bond network in the inter-layer space. They exhibit infrared spectra similar to those of bulk water with a maximum around 3400 cm−1 for the O–H stretching band.59 At the highest RH (>70%), another type of water molecule occurs in the intergranular porosity,36 i.e. the space between the clay particles. They exhibit a “bulk water” behavior.
At 0% relative humidity, one water layer is present (Fig. 3b) but it consists of “bonded water” as proven by TGA in Fig. 2c and d. This leads to a radiolytic yield lower than the one in bulk water but consistent with values obtained in synthetic talc21 (Mg3Si4O10(OH)2), which is a TOT clay mineral without water molecules in its interlayer space ((2.9 ± 0.3) × 10−8 mol J−1). These bonded water molecules together with structural –OH groups lead to H2 yield similar to that of talc which possesses only structural hydroxyl groups and possibly some water molecules adsorbed on the border of sheets.
Upon irradiation, electron/hole pairs (e−/h+)5,17 are generated:
 | (R1) |
Electrons can be trapped in the structure as proven by EPR spectroscopy (Fig. 4a):
 | (R2) |
As in talc,21 a source of hydrogen is dissociative electronic attachment on –MgOH, leading to the formation of –MgO− that is not detectable by EPR (R3). Moreover, the hole leads to the formation of the –O˙− defect and of H+ (R4) that can lead to the formation of the hydrogen atom after reduction (R5). Hydrogen atoms dimerize then into H2 (R6).
| e− + –MgOH → –MgO− + H˙ | (R3) |
| –(Mg2+)(O2−)(H+) + h+ → –(Mg2+)(O˙−) + H+ | (R4) |
| e− + H+ → H˙ | (R5) |
| H˙ + H˙ → H2 | (R6) |
Noteworthy and contrary to talc, the presence of the water molecules in the interlayer space destabilizes significantly the defects detected by EPR. This may be attributed to a fluctuation of the local electric field. Indeed, in talc, the defects are stable at room temperature. Even the hydrogen atoms are stable at 300 K for few days.21 In the present case, the defects are detected in the temperature range 90–240 K but are no longer present at 300 K.
When RH increases, the contribution of water molecules to the H2 yield, in addition to the one of –OH groups and bonded water molecules, is revealed.
From 3 to 11% RH, the water layer in the interlayer space exhibits a very specific behavior. Indeed, the O–H bond is weakened by electronic impoverishment. So, during irradiation, this confined water having only a few degrees of freedom is easily broken by efficient energy transfer (i.e. electron transfer here) mechanisms from the layers to the interlayer space (as no transition element impurities are present). Electron and hole can migrate in the interlayer space where the first is hydrated whereas the other can react with confined water:
| e− + H2O → eaq− | (R7) |
| h+ + H2O → H+ + HO˙ | (R8) |
Solvated electron and hydrogen radical formed in the clay structure can react to form dihydrogen (R6):
| eaq− + H+ → H˙ + H2O | (R9) |
| eaq− + eaq− → H2 + 2HO− | (R10) |
| eaq− + H˙ → H2 + HO− | (R11) |
All these reactions are favored by the confined geometry (2.5 Å) in one direction. This change in reaction mechanisms is reflected by the change in EPR spectra when increasing RH (Fig. 4a and b). The presence of free water (Fig. 2) destabilizes the hydrogen atom, even it is formed within the layers, and it is no longer detected (Fig. 4b). Noteworthy, trapped electrons are not present at 11% RH (Fig. 4b). This proves that they have been efficiently transferred to the interlayer space. Water has indeed an impact on the relative amount of the defects created in the layers, such as –O˙−.
When RH increases from 43 to 97%, a second water layer is present in the interlayer space. The environment is less confined (interlayer spacing about 5 Å), making those previous mechanisms less efficient. Moreover, the radiolysis of adsorbed water molecules will also take place. Hence, dihydrogen radiolytic yields decrease to a constant value comparable to the bulk water value. Again, EPR spectra evidence a change in the reaction mechanisms (Fig. 4c). Even if the presence of a broad paramagnetic component complicates the interpretation, hydrogen atoms are clearly detected. They may be formed in the interlayer space, and no longer in the phyllosilicate layers. The detection of these hydrogen atoms indicates a stabilizing role of the clay compared to what is observed in other materials. Nevertheless, they are only detected at low temperatures, contrary to the case of talc,21 which has no water molecules.
In synthetic swelling clay minerals the reactivity is thus controlled by the water amount, as the energy transfer from the materials to the interlayer space is total. It is not the case in natural swelling clay minerals where the presence of impurities as Fe3+ decreases markedly the efficiency of the energy transfer by trapping electrons. This is evidenced in Fig. 6, where the trapping of electrons takes place in hundreds of ps after the electron pulse, depending on the Fe3+ content, whereas in bulk water, the lifetime of the electron is a few microseconds. Indeed, as measured in natural montmorillonites from Camp-Berteau5 where Fe3+ is present, the H2 radiolytic yield is always smaller than the value in water. Thus, the efficient trapping of electrons by trivalent iron ions prevents the dihydrogen formation, as Fe3+ is known to be a good scavenger of the pre-solvated electron:
| Fe3+ + e− → Fe2+ | (R12) |
Concerning the geological disposal, the use of a natural swelling clay, that has good water retention properties to avoid the leak of nuclear waste, with a high Fe3+ content seems to be recommended to avoid dihydrogen production. Nevertheless, the reduction of structural trivalent iron in smectite by (bio)chemical reactions is known to increase layer charge and cation exchange capacity, albeit with decreasing swelling capacity owing to some collapse of layers, particularly in the case of K+ fixation.60 If such results are confirmed in the case of the radiolytical reduction of iron (electron displacements inside the layer), then the corresponding evolution of properties will have to be taken into account in the safety assessment of nuclear waste disposals. In addition, as the H2 radiolytic yield increases with RH in natural samples,5 the production of H2 will be minimized if the RH can be kept as low as possible.
Conclusions
We have studied here the behaviour under ionizing radiation of two prototype synthetic swelling clay minerals (montmorillonite and saponite). The evolution of the radiolytic yield is strongly dependent on the relative humidity: it increases from a value comparable to the one obtained in synthetic anhydrous clay minerals21 to a maximum value (more than twice the value in bulk water) and then decreases but remains slightly higher than the value in bulk water, due to the lowering of the confinement effect. The high values measured highlight the great efficiency of the energy transfer from the sheets to the interlayer space and the strong recombination of reactive species (hydrogen atoms) within the highly confined interlayer space. Moreover, the H2 yields measured are very similar in both clay minerals: energy transfer is very efficient and the reactivity is driven by the water amount, not by the location of the excess negative charge.This picture is quite different in natural swelling clay minerals. In this case, we have shown through picosecond pulse radiolysis that electrons are quickly (100–200 ps) trapped by transition element impurities inside the layers. In natural systems, the reactivity is driven by the amount of the impurities that are able to trap the electrons.
Concerning the radiolytic production of H2, the best choice for swelling clay minerals as engineered barrier for deep geological disposal of nuclear wastes is natural clay minerals, with a significant content in Fe3+ that will trap electrons and prevents their transfer to the interlayer space. In this case, the RH has to be as low as possible to minimize the H2 production under ionizing radiation.
Acknowledgements
This work was supported by a grant from Région Ile-de-France in the framework of DIM Oxymore. The authors thank Dr D. Simeone from LRC CARMEN of CEA Saclay for his help in XRD experiments.Notes and references
- B. Grambow, Elements, 2016, 12, 239–245 CrossRef.
- W. Miller, R. Alexander, N. Chapman, J. McKinley and J. A. T. Smellie, Geological Disposal of Radioactive Wastes and Natural Analogues, Elsevier, 9th edn, 2000 Search PubMed.
- N. Brodie-Linder, S. Le Caer, M. S. Alam, J. P. Renault and C. Alba-Simionesco, Phys. Chem. Chem. Phys., 2010, 12, 14188–14195 RSC.
- F. Brunet, T. Charpentier, S. Le Caer and J. P. Renault, Solid State Nucl. Magn. Reson., 2008, 33, 1–11 CrossRef CAS PubMed.
- C. Fourdrin, H. Aarrachi, C. Latrille, S. Esnouf, F. Bergaya and S. Le Caër, Environ. Sci. Technol., 2013, 47, 9530–9537 CrossRef CAS PubMed.
- J. A. LaVerne, J. Phys. Chem. B, 2005, 109, 5395–5397 CrossRef CAS PubMed.
- S. Le Caer, Water, 2011, 3, 235–253 CrossRef CAS.
- S. Le Caer, P. Rotureau, F. Brunet, T. Charpentier, G. Blain, J. P. Renault and J. C. Mialocq, ChemPhysChem, 2005, 6, 2585–2596 CrossRef CAS PubMed.
- P. Rotureau, J. P. Renault, B. Lebeau, J. Patarin and J. C. Mialocq, ChemPhysChem, 2005, 6, 1316–1323 CrossRef CAS PubMed.
- E. Ferrage, B. Lanson, B. A. Sakharov and V. A. Drits, Am. Mineral., 2005, 90, 1358 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- P. Bouniol and E. Bjergbakke, J. Nucl. Mater., 2008, 372, 1–15 CrossRef CAS.
- R. R. Brey, R. Rodriguez, J. F. Harmon and P. Winston, Waste Manag., 2001, 21, 581–588 CrossRef CAS PubMed.
- D. Gournis, A. E. Mantaka-Marketou, M. A. Karakassides and D. Petridis, Phys. Chem. Miner., 2000, 27, 514–521 CrossRef CAS.
- M. Holmboe, K. K. Norrfors, M. Jonsson and S. Wold, Radiat. Phys. Chem., 2011, 80, 1371–1377 CrossRef CAS.
- M. Holmboe, S. Wold, M. Jonsson and S. Garcia-Garcia, Appl. Clay Sci., 2009, 43, 86–90 CrossRef CAS.
- J. K. Thomas, Chem. Rev., 2005, 105, 1683–1734 CrossRef CAS PubMed.
- D. L. Hamilton and C. M. B. Henderson, Mineral. Mag., 1968, 36, 832–838 CAS.
- L. Bergaoui, J.-F. Lambert, R. Franck, H. Suquet and J.-L. Robert, J. Chem. Soc., Faraday Trans., 1995, 91, 2229–2239 RSC.
- M. S. Karmous, H. Ben Rhaiem, J. L. Robert, B. Lanson and A. Ben Haj Amara, Appl. Clay Sci., 2009, 46, 43–50 CrossRef CAS.
- M. Lainé, T. Allard, E. Balan, F. Martin, H. J. Von Bardeleben, J.-L. Robert and S. L. Caër, J. Phys. Chem. C, 2016, 120, 2087–2095 Search PubMed.
- J. C. Mialocq, B. Hickel, G. Baldacchino and M. Juillard, J. Chim. Phys. Phys.-Chim. Biol., 1999, 96, 35–43 CrossRef CAS.
- H. Fricke and J. E. Hart, in Radiation Dosimetry, ed. F. H. Attix and W. C. Roesch, Academic press, New York and London, 2nd edn, 1966, vol. 2, pp. 167–232 Search PubMed.
- G. Morin and D. Bonnin, J. Magn. Reson., 1999, 136, 176–199 CrossRef CAS PubMed.
- J. Belloni, H. Monard, F. Gobert, J. P. Larbre, A. Demarque, V. De Waele, I. Lampre, J. L. Marignier, M. Mostafavi, J. C. Bourdon, M. Bernard, H. Borie, T. Garvey, B. Jacquemard, B. Leblond, P. Lepercq, M. Omeich, M. Roch, J. Rodier and R. Roux, Nucl. Instrum. Methods Phys. Res., Sect. A, 2005, 539, 527–539 CrossRef CAS.
- J. L. Marignier, V. de Waele, H. Monard, F. Gobert, J. P. Larbre, A. Demarque, M. Mostafavi and J. Belloni, Radiat. Phys. Chem., 2006, 75, 1024–1033 CrossRef CAS.
- J. Ma, P. Archirel, P. Pernot, U. Schmidhammer, S. Le Caër and M. Mostafavi, J. Phys. Chem. B, 2016, 120, 773–784 CrossRef CAS PubMed.
- E. Paineau, I. Bihannic, C. Baravian, A.-M. Philippe, P. Davidson, P. Levitz, S. S. Funari, C. Rochas and L. J. Michot, Langmuir, 2011, 27, 5562–5573 CrossRef CAS PubMed.
- V. Robin, E. Tertre, D. Beaufort, O. Regnault, P. Sardini and M. Descostes, Appl. Geochem., 2015, 59, 74–84 CrossRef CAS.
- J. P. Keene, Radiat. Res., 1964, 22, 1–13 CrossRef CAS.
- R. Laenen, T. Roth and A. Laubereau, Phys. Rev. Lett., 2000, 85, 50–53 CrossRef CAS PubMed.
- J. P. Jay-Gerin and C. Ferradini, J. Chim. Phys. Phys.-Chim. Biol., 1994, 91, 173–187 CAS.
- E. Keszei and J.-P. Jay-Gerin, J. Chim. Phys. Phys.-Chim. Biol., 1989, 33, 183–184 CAS.
- W. Xu, C. T. Johnston, P. Parker and S. F. Agnew, Clays Clay Miner., 2000, 48, 120–131 CAS.
- J. L. Bishop, C. M. Pieters and J. O. Edwards, Clays Clay Miner., 1994, 42, 702–716 CAS.
- E. Rinnert, PhD thesis, Université Henri Poincaré, Nancy 1, France, 2004.
- G. Sposito and R. Prost, Chem. Rev., 1982, 82, 553–573 CrossRef CAS.
- K. Toriyama, A. Lund and M. Okazaki, Phys. Chem. Chem. Phys., 2000, 2, 4697–4701 RSC.
- V. I. Feldman, F. F. Sukhov and A. Y. Orlov, J. Chem. Phys., 2008, 128, 214511 CrossRef PubMed.
- T. Allard and G. Calas, Appl. Clay Sci., 2009, 43, 143–149 CrossRef CAS.
- T. Allard, P. Ildefonse, L. P. Del Villar, S. Sorieul, M. Pelayo, B. Boizot, E. Balan and G. Calas, Eur. J. Mineral., 2003, 15, 629–640 CrossRef CAS.
- B. R. Angel, J. P. E. Jones and P. L. Hall, Clay Miner., 1974, 10, 247–270 CAS.
- L. I. Barsova, T. K. Yurik and V. I. Spitsyn, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 879–883 CrossRef.
- C. Di Valentin, D. Ricci, G. Pacchioni, M. Chiesa, M. C. Paganini and E. Giamello, Surf. Sci., 2002, 521, 104–116 CrossRef CAS.
- O. Diwald, M. Sterrer, E. Knözinger, P. V. Sushko and A. L. Shluger, J. Chem. Phys., 2002, 116, 1707–1712 CrossRef CAS.
- S. A. Dolgov, T. Kärner, A. Lushchik, A. Maaroos, S. Nakonechnyi and E. Shablonin, Phys. Solid State, 2011, 53, 1244–1252 CrossRef CAS.
- T. Ito, M. Watanabe, K. Kogo and T. Tokuda, Z. Phys. Chem., 1981, 124, 83 CrossRef CAS.
- M. Sterrer, O. Diwald, E. Knözinger, P. V. Sushko and A. L. Shluger, J. Phys. Chem. B, 2002, 106, 12478–12482 CrossRef CAS.
- A. I. Volkov, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 815–829 RSC.
- G. Pacchioni and H. Freund, Chem. Rev., 2013, 113, 4035–4072 CrossRef CAS PubMed.
- T. Allard, E. Balan, G. Calas, C. Fourdrin, E. Morichon and S. Sorieul, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 277, 112–120 CrossRef CAS.
- Y. Kimura, J. C. Alfano, P. K. Walhout and P. F. Barbara, J. Phys. Chem., 1994, 98, 3450–3458 CrossRef CAS.
- F. Torche and J.-L. Marignier, J. Phys. Chem. B, 2016, 120, 7201–7206 CrossRef CAS PubMed.
- J. Bonin, I. Lampre and M. Mostafavi, Radiat. Phys. Chem., 2005, 74, 288–296 CrossRef CAS.
- J. W. T. Spinks and R. J. Woods, An Introduction to Radiation Chemistry, John Wiley and Sons, Inc., New York, Toronto, 3rd edn, 1990 Search PubMed.
- E. Ferrage, B. Lanson, L. J. Michot and J.-L. Robert, J. Phys. Chem. C, 2010, 114, 4515–4526 CAS.
- E. Ferrage, B. A. Sakharov, L. J. Michot, A. Delville, A. Bauer, B. Lanson, S. Grangeon, G. Frapper, M. Jimenez-Ruiz and G. J. Cuello, J. Phys. Chem. C, 2011, 115, 1867–1881 CAS.
- C. T. Johnston, G. Sposito and C. Erickson, Clays Clay Miner., 1992, 40, 722–730 CAS.
- S. Le Caer, S. Pin, S. Esnouf, Q. Raffy, J. P. Renault, J. B. Brubach, G. Creff and P. Roy, Phys. Chem. Chem. Phys., 2011, 13, 17658–17666 RSC.
- J. W. Stucki, Fundamentals. Developments in clay science, in Handbook of Clay Science, ed. F. Bergaya and G. Lagaly, Elsevier, 2nd edn, 2013, vol. A Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24861f |
| This journal is © The Royal Society of Chemistry 2017 |