
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of nanostructured hybrid materials: twin polymerization of urethane-based twin prepolymers†
D. Uhlig a,
S. Spangea,
A. Seiferta,
K. Nagela,
S. Andersb,
L. Krollb,
R. Stollc,
F. Thielbeerd,
P. Müllerd and
K. Schreiter*a
a,
S. Spangea,
A. Seiferta,
K. Nagela,
S. Andersb,
L. Krollb,
R. Stollc,
F. Thielbeerd,
P. Müllerd and
K. Schreiter*a
aDepartment of Polymer Chemistry, Chemnitz University of Technology, Faculty of Natural Science, D-09107 Chemnitz, Germany. E-mail: katja.schreiter@chemie.tu-chemnitz.de
bDepartment of Lightweight Structures and Polymer Technology, Chemnitz University of Technology, Faculty of Mechanical Engineering, D-09107 Chemnitz, Germany
cBASF Polyurethanes GmbH, Elastogranstraße 60, 49448 Lemfoerde, Germany
dBASF SE, Carl-Bosch Straße 38, 60756 Ludwigshafen, Germany
First published on 12th September 2018
Abstract
Organic–inorganic hybrid materials with urethane functionalities were obtained by simultaneous twin polymerization of twin prepolymers in combination with the ideal twin monomer 2,2′-spirobi[4H-1,3,2-benzodioxasiline]. The twin prepolymers consist of a urethane-based prepolymer with reactive terminal groups which can react during the twin polymerization process. Nanostructured hybrid materials with integrated dialkylsiloxane crosslinked urethane structures, phenolic resin and SiO2 are obtained in a one pot process. The effects of the polymerization temperature as well as those of various catalysts and reagent ratios on the polymerization behavior were investigated. The molecular structures of the obtained materials were determined by 13C- and 29Si-{1H}-CP-MAS NMR spectroscopies. HAADF-STEM-measurements were performed to prove the distribution of silicon in the hybrid material.
Introduction
Nanostructured materials are one of the emerging areas of materials science and have attracted much attention because of their possible use in areas such as microelectronics, sensors, photovoltaics, electro-optical instruments, energy storage, conversion and separation processes as well as in catalysis.1–9 In recent years a variety of methods for the preparation of organic–inorganic hybrid materials (HMs) have been developed which deliver defined nanostructures with domain sizes of 2 to 100 nm. In particular silica-based organic–inorganic HMs have been investigated since they combine the distinctive properties of organic polymers (e.g. malleability, good impact resistance, flexibility) and silica (e.g. high mechanical strength, transparency, good chemical and thermal stability) as well as some special or new types of properties based on their specific microstructure.9The sol–gel procedure and template-assisted polymerization are the most frequently used methods for preparing silica-based organic–inorganic HMs both on the micro scale and at the molecular level.10,11 However, most established synthetic methods require subsequent processing steps.
In recent years the twin polymerization (TP) turned out to be an elegant tool to prepare nanostructured HMs with domain sizes from 0.5 up to 10 nm whereby two different polymer phases result from only one single source monomer.12–16 For instance, the polymerization of the “ideal” twin monomer 2,2′spirobi-[4H-1,3,2-benzodioxasiline] (1) leads to a HM consisting of phenolic resin and silica (Scheme 1a).13,14 Different types of so-called twin monomers (TMs) are suitable for the TP.16–24 Depending on their molecular structure, two of these monomers can be polymerized simultaneously to generate ternary organic–inorganic HMs (see Scheme 1b).17,25,26
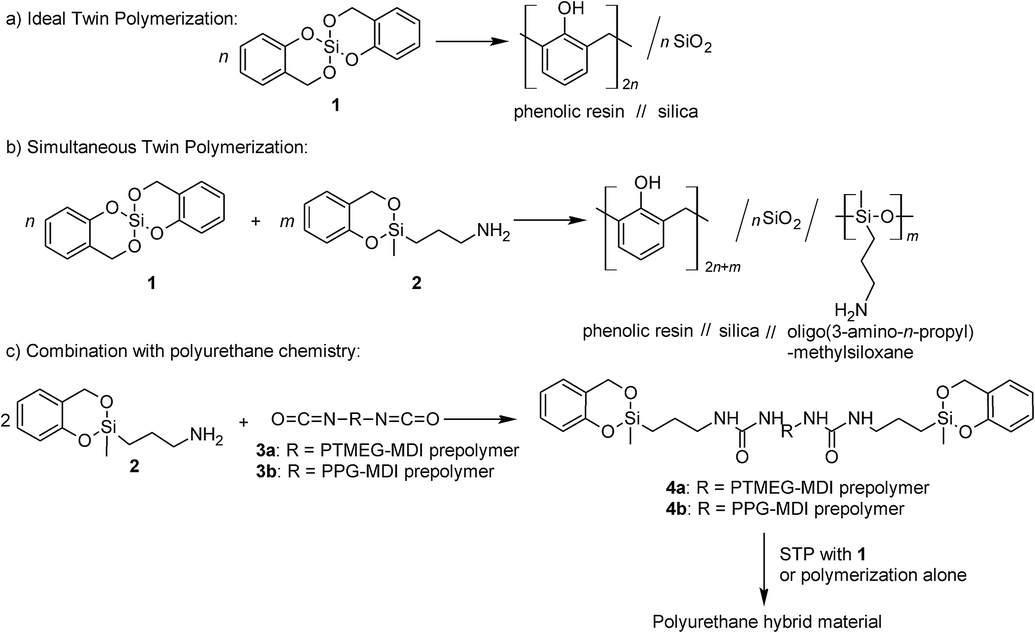 | ||
| Scheme 1 (a) The “ideal” twin polymerization (TP) of 1 to generate a hybrid material consisting of phenolic resin and silica. (b) The introduction of (oligo)-dialkylsiloxane into phenolic resin/silica hybrid materials is possible by simultaneous TP (STP) of 1 with 2. (c) End-capping of a polyurethane prepolymer with 2 and the subsequent STP results in hybrid materials consisting of phenolic resin, silica and dialkylsiloxane-crosslinked PU. | ||
Polyurethanes (PUs) are outstanding materials due to their versatility and ability to generate tailor-made properties. The diversity had already been pointed out by O. Bayer in the very first article about PUs.27 These materials fulfil the varied requirements of modern applications as form-memory polymers, adhesives, foams, fibers, thermoplastic elastomers and coatings.28–30 As mentioned before, fabrication of nanocomposites or HMs consisting of silica species enables the improvement of material properties like tensile strength or flame retardancy.9,31 By combining the versatile world of PU chemistry with that of nanostructured HMs accessible by TP, such multicomponent HMs can become accessible. The objective of this work is to present a new concept for combining PU chemistry with that of organic–inorganic HM chemistry.
We describe here the first synthesis of nanostructured phenolic resin/urethane/SiO2 HMs by simultaneous twin polymerization (STP) using a novel type of urethane-based twin prepolymer and 1. A urethane-based twin prepolymer is a macromolecule or an oligomer molecule, which can be incorporated into the TP by means of its reactive groups. The synthesis of such twin prepolymers is a subtask of this work. They can be synthesized by use of an amino functionalized TM with isocyanate terminated prepolymer (Scheme 1c).
In the course of the (simultaneous) TP process various scenarios are possible for the formation of the molecular structure of the resulting polymer networks. The inorganic polymer (A)n can be obtained by reaction with reactive parts from the same or another polymer strand [Z]r or by a combination of both of these groups (Scheme 2).
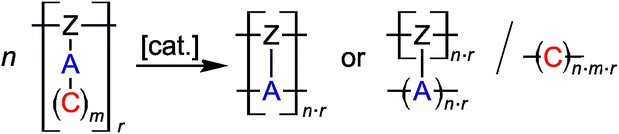 | ||
| Scheme 2 Twin polymerization of a twin prepolymer.14 | ||
Advantageously, the TP process of TM species with benzodioxasiline increments as reactive organic moiety can be induced thermally or by the use of acids or bases as catalyst.13,14,16,18 In this work, the polymerizations of the considered twin prepolymer systems will be thermally induced and triggered by the use of diazabicyclo[2.2.2]octane (DABCO), or diazabicyclo[5.4.0]undec-7-ene (DBU) as base catalyst, respectively. The polymerization process is carried out in the melt, which is advantageous for possible applications as an adhesion promoter.
Experimental
Materials and methods
Diphenylmethane-4,4′-diisocyanate (MDI), polytetramethylene ether glycole (PTMEG, average molar weight 1000 g mol−1), polypropylene ether glycole (PPG, average molar weight 1000 g mol−1) and PTMEG-MDI-prepolymer (1500 g mol−1) was provided by the BASF SE.Salicylic alcohol (ABCR), 3-aminopropyl-dimethoxymethylsilane (TCI), tetra-N-butylammonium fluoride (1 M solution; ABCR), diazabicyclo[2.2.2]octane (DABCO) (Sigma Aldrich), diazabicyclo[5.4.0]-undec-7-ene (DBU) (fluorochem) were used as received.
All solvents were dried with appropriate drying agents and freshly distilled before usage.
Liquid state 1H NMR (250.1 MHz), 13C NMR (62.9 MHz) and 29Si NMR (49.7 MHz) spectra were recorded with a Bruker DRX 250 NMR spectrometer. The residual signal of the solvent CDCl3 was used as an internal standard relative to tetramethylsilane (TMS; δ = 0 ppm). The 29Si NMR spectra are referenced to TMS (δ = 0 ppm).
Solid state NMR spectra were collected at 9.4 T on a Bruker Avance 400 spectrometer equipped with double tuned probes capable of MAS (magic angle spinning). 13C-{1H}-CP-MAS NMR spectra were measured at 100.6 MHz in 3.2 mm standard zirconium oxide rotors (BRUKER) spinning at 15 kHz. Cross polarization with a contact time of 3 ms was used to enhance the sensitivity. The recycle delay was 6 s. Spectra were referenced externally to tetramethylsilane (TMS) as well as to adamantane as secondary standard (38.48 ppm for 13C). 29Si-{1H}-CP-MAS NMR spectroscopy was performed at 79.5 MHz using 3.2 mm rotors spinning at 12 kHz. The contact time was 3 ms and the recycle delay 6 s. Shifts were referenced externally to tetramethylsilane (0 ppm) with the secondary standard being tetrakis(trimethylsilyl)silane (−9.5 ppm). All spectra were collected with 1H decoupling using a TPPM pulse sequence.
The high annular dark field (HAADF) scanning transmission electron microscopy (STEM) images were recorded with a Technai G2-F-20ST with HAADF-STEM-detector. The samples were prepared by ultramicrotomy.
Electron microscopy images were taken by using an instrument from type Nova NanoSEM 200 of FEI Company after sputtering with platinum.
The DSC measurements were performed by a DSC 1 (Mettler Toledo) and a DSC 204 F1 Phoenix (Netzsch) in a 40 μL aluminum pan and a N2-flow of 50 mL min−1 in a temperature range of 30–300 °C or 30–350 °C with a heating rate of 10 K min−1. The highest and the lowest temperature was held for 3 minutes, respectively.
TGA measurements were performed by a Thermogravimetric Analyzer 7. The samples were measured under air atmosphere, with a heating rate of 10 K min−1 from 30–900 °C.
The TG/MS measurements were performed by a TG 209 F1 Iris (Netzsch) coupled to a Aëolos mass spectrometer (Bruker). The samples were prepared in a 85 μL aluminum pan and measured under He-flow of 25 mL min−1 in a temperature range of 25–350 °C with a heating rate of 10 K min−1. Before the start of the measurement, the sample room with sample was subjected twice to an evacuation and rinsing cycle with helium.
Size exclusion chromatography (SEC) was performed with a PL-GPC 50 plus from Polymer Laboratories equipped with a PL-AS RT auto sampler and a PC-RI detector. THF was used as eluent with a flow rate of 1.0 mL min−1 at 40 °C. The column was a PLgel MIXED-D.
The ATR-FTIR-spectra were recorded with a FTS 165 spectrometer (BioRad). A Golden Gate-single reflection-ATR-stage (L.O.T. Oriel GmbH) was used. The samples were pressed against the detection system with a sapphire stamp (5 bar).
Elemental analysis was determined with a Vario El analyser (Elementar analysensysteme GmbH).
Synthetic procedures
Polymerization
Detailed information are summarized in the ESI. The monomer amounts, extractable contents as well as the found silicon contents are listed in Tables 2/3.
Extraction
The obtained materials were cut into small pieces with a scalpel. About 1 g of the material pieces were placed in an extraction thimble and Soxhlet-extracted with 200 mL dichloromethane for 48 h. The extracted material was dried in a vacuum oven at 40 °C until weight constant. The solvent was removed under reduced pressure to give the concentrated extract.Results and discussion
The reactive prepolymers 4a and 4b were obtained by end-capping of the PU prepolymers 3a or 3b with 2-(3-aminopropyl)-2-methyl-4H-1,3,2-benzodioxasiline (2), respectively.In accordance to the classification of TM species and polymerization processes, a so-called twin prepolymer was generated (Scheme 3).14
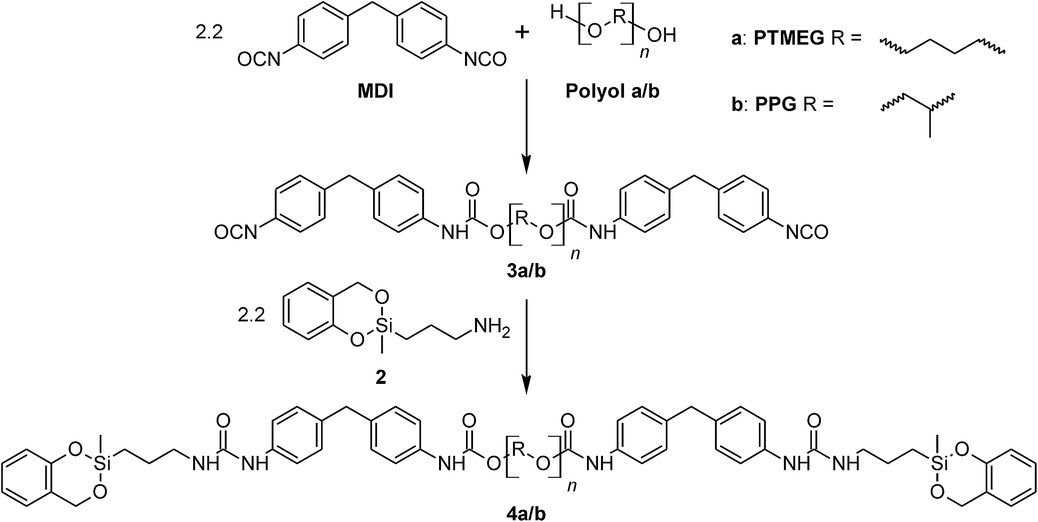 | ||
| Scheme 3 General procedure for preparation of the twin prepolymer 4a and 4b. | ||
A representative 1H NMR spectrum of 4a is shown in Fig. 1. The disappearance of the NH2 signal and the down field shift of the neighboring CH2 group of 2 (signal 5) prove the successful reaction of the amino functionality with isocyanate.
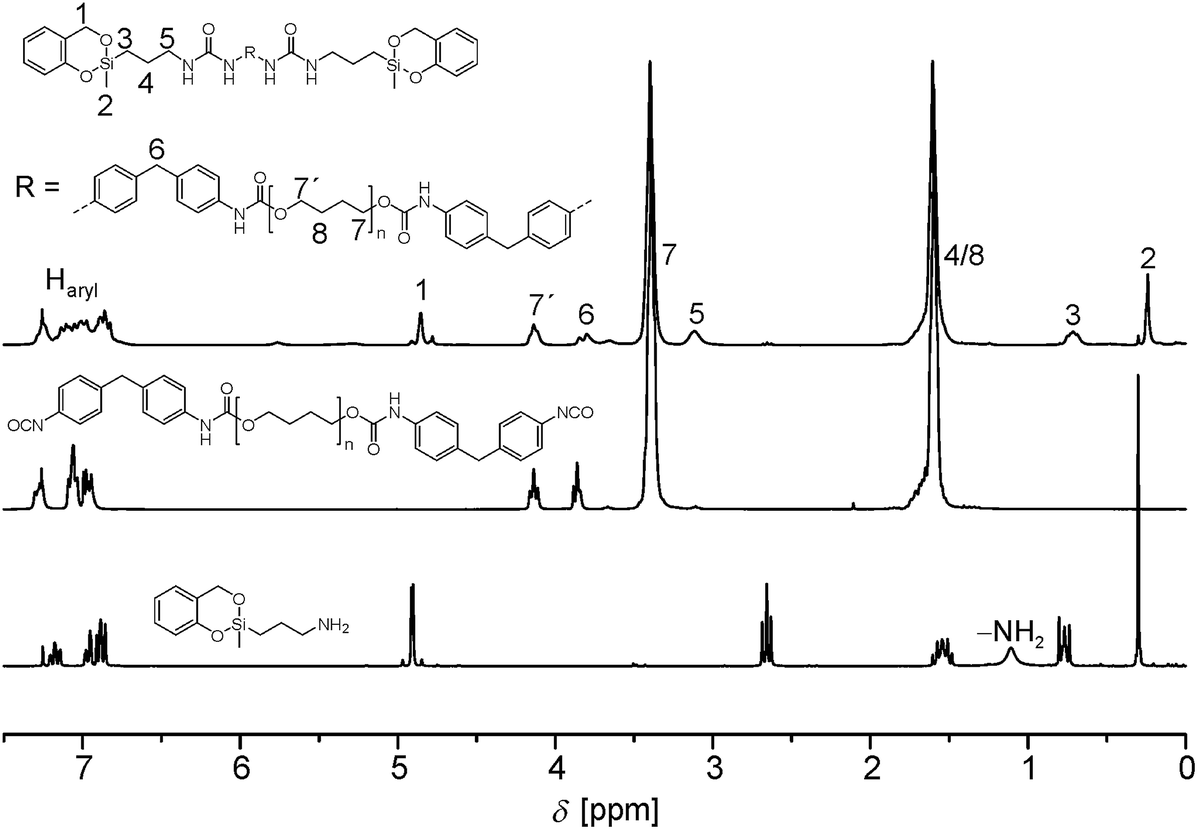 | ||
| Fig. 1 1H NMR spectrum of 4a (top) in combination with the spectra of the corresponding educts 2 (bottom) and 3a (middle, all spectra measured in CDCl3). | ||
Due to the required excess of diisocyanate used during the preparation of the prepolymer, low molecular by-products can be detected by GPC measurement (Fig. S4/S5 and Tables S1/S2†). Those by-products have to be taken into account for the subsequent polymerization and its interpretation.
Thermal initiation – preliminary investigation
Polymerization ability of the twin prepolymers 4a and 4b were studied using differential scanning calorimetry (DSC). Various reaction scenarios are apparent from the temperature profiles, showing the reaction of a twin prepolymer alone, in the presence of a second TM or with a potential catalyst (Fig. 2). Homogenous monomer mixtures were prepared by melting all components at 100 °C prior to the DSC measurements.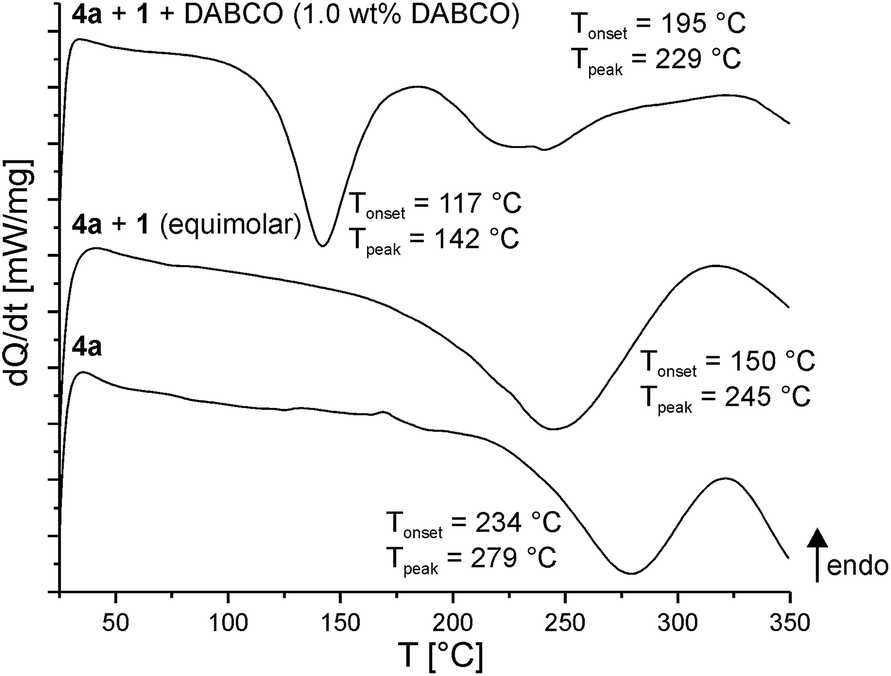 | ||
| Fig. 2 DSC curves of the twin prepolymer 4a alone, with equimolar amounts of 1 and an equimolar mixture of 4a and 1 with 1.0 wt% of base catalyst (DABCO); heating rate 10 K min−1. | ||
Upon heating the twin prepolymer 4a, an exothermic peak at 279 °C (Tonset = 234 °C, Fig. 2 bottom) is observed. The addition of 1 reduces the trigger temperature as indicated by the exothermic peak to 245 °C (Tonset = 150 °C, Fig. 2 middle). If DABCO is added as a catalyst to that mixture a further reduction of the polymerization trigger temperature (Fig. 2 top) is measured. Two exothermic peaks (Tonset1 = 117 °C, Tpeak1 = 142 °C; Tonset2 = 195 °C, Tpeak2 = 229 °C) are obtained, the first one indicating its catalytic activity.
The addition of lactic acid as catalyst to the mixture of 4a and 1 already leads to polymerization at a temperature of approx. 80 °C. However, a prerequisite for a polymerization in the melt of 4a and 1 is a mixing of the components at approx. 100 °C. Otherwise, inhomogeneous products are created due to the high viscosity of the reaction mixture. Therefore, acid catalysis was not preferred in this work, although this problem can be solved by using a solvent but that is not useful for the objective of this work.
4b alone shows an exothermic peak at 293 °C. The addition of 1 and DABCO results in similar trends to lower temperatures as observed for 4a (Table 1 and Fig. S12†).
| Tonset,exo (Tpeak,exo) [°C] | ||
|---|---|---|
| 4a | 4b | |
| Pure | 234 (279) | 245 (293) |
| +1 | 150 (245) | 191 (237) |
| +1 + DABCO | 117 (142) | 134 (148) |
| 195 (229) | ||
These preliminary results were confirmed by reproducing these polymerizations on a multi-gram scale and subsequent analysis of the products. Monolithic HMs were prepared by thermal polymerization, with the addition of either DABCO or DBU as catalysts.
Thermally induced (simultaneous) twin polymerization
The prepolymers 4a or 4b were polymerized simultaneously with 1 at 170 °C, 190 °C, 210 °C as well as 230 °C for 2 h using a stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 2). The yellow colored materials obtained are readily foamed and elastic. According to the concept of STP, the resulting organic–inorganic HM should consist of phenolic resin, SiO2 and oligodialkylsiloxane (ODAS) with integrated urethane structural units which also have a cross-linking function. The SiO2 and ODAS groups can form Si–O–Si bonds and thus form a class II hybrid structure among the phenolic resin/SiO2/ODAS hybrid compounds according to the classification of Sanchez32 (Scheme 4). This is verified by 29Si and 13C solid state NMR spectroscopy of the HMs (Fig. S6†).
:1 (Table 2). The yellow colored materials obtained are readily foamed and elastic. According to the concept of STP, the resulting organic–inorganic HM should consist of phenolic resin, SiO2 and oligodialkylsiloxane (ODAS) with integrated urethane structural units which also have a cross-linking function. The SiO2 and ODAS groups can form Si–O–Si bonds and thus form a class II hybrid structure among the phenolic resin/SiO2/ODAS hybrid compounds according to the classification of Sanchez32 (Scheme 4). This is verified by 29Si and 13C solid state NMR spectroscopy of the HMs (Fig. S6†).
| Sample | Molar ratio [mol%] n4a/b:n1 |
Temp. [°C] | Extr. cont. [wt%] | Silica cont. [wt%] |
|---|---|---|---|---|
| HM5a_170 | 50:50 |
170 | 47 | 10.7 |
| HM5a_190 | 190 | 47 | 11.1 | |
| HM5a_210 | 210 | 35 | 9.2 | |
| HM5a_230 | 230 | 36 | 10.5 | |
| HM5b_170 | 50:50 |
170 | 84 | — |
| HM5b_190 | 190 | 49 | 11.1 | |
| HM5b_210 | 210 | 33 | 11.5 | |
| HM5b_230 | 230 | 45 | 9.2 |
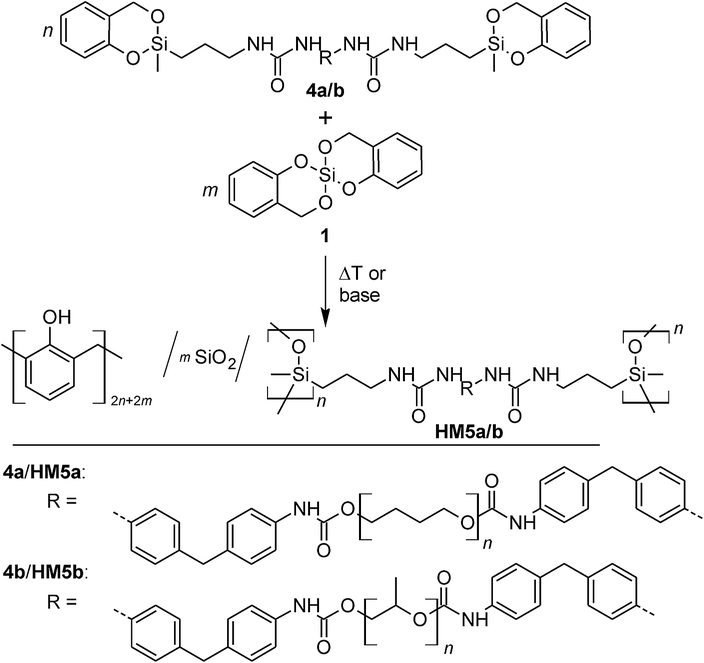 | ||
| Scheme 4 Reaction scheme for the STP of 4a/b with 1. | ||
Each of the 29Si-{1H}-CP-MAS NMR spectrum of HMs show the expected Q and D signals resulting from monomer 1 and the corresponding twin prepolymer, respectively. The Q4 signals measured at −110 ppm are the most intense signals in the silica range (= Q) of the 29Si NMR spectrum in each case. The cross polarization technique used to enhance the sensitivity overestimates Q2 (−90 ppm) and Q3 (−100 ppm) over Q4 because these are silicon species with hydrogen in close proximity resulting in a more efficient polarization transfer from 1H to 29Si. The high intensity of the Q4 signals in comparison to Q2 and Q3 found in the products clearly shows that the SiO2 network must nearly be fully condensed.
The D1, D(Q) and D2 signals observed in the range from −10 to −23 ppm correspond to bifunctional (D) silicon structures originating from 4a, or 4b. D(Q) signals are also found at about −16 to −17 ppm. These are characteristic for the STP products indicating a reduced flexibility of the oligodialkylsiloxane (ODAS) part and thus the binding of ODAS to the SiO2 network. According to the classification of STP processes,25 this is therefore a real simultaneous twin copolymerization. The observation of a D1 (one siloxane bond, Si–O–Si–) signal at ≈−7 ppm probably corresponds to end groups of ODAS. No signals from the monomer reactants were detected in the solid state 29Si NMR spectra.
The formation of phenolic resin, ODAS and urethane structures can be proven by 13C-{1H}-CP-MAS NMR spectroscopy. The o,o′- and o,p′-bonded phenolic resin structures can be detected by the signals in the aromatic region (signals 3 and 5, Fig. S6†). The signal for the bridging methylene group (signal 4) is overlaid with the intense signal of the CH2 groups of the ether fragment (signal 15). The ODAS structure gives a signal for the methyl group at 0 ppm. The signals for the carbon atoms of the alkyl chains of ODAS are observed in the expected region of 18–70 ppm, whereby the most intense carbon signals at 26 and 70 ppm are due to the methylene groups of the ether fragments (signals 14 and 15). Furthermore, the presence of signals at 42 ppm (signal 11), between 110–140 ppm (signal 12) and 155 ppm (signal 10 and 13), shows that the urethane fragment is bonded to HM after the polymerization and subsequent extraction.
Soxhlet-extraction of the raw HM with dichloromethane (DCM) gave extractable fractions, which were identified as prepolymer and low molecular weight products such as salicylic alcohol (Fig. S7 and Table S7†). Depending on the polymer backbone chosen, the quantity of the extractable fraction was very different. Using 4a gives an extractable portion of 47 wt% whereas with 4b the extractable portion increases to 84 wt%. These extraction experiments together with the results of the DSC (Fig. S12†) show that for the system 4b:1 a polymerization temperature of 170 °C is too low (Table 2).
Thus, the HMs of the monomers 1 and 4a or 4b were polymerized at higher temperatures (190 °C/210 °C/230 °C) to increase the conversion.
Increasing the polymerization temperature to 210 °C results in a reduction of the extractable proportions of the resulting HMs to ≈35 wt% for 4a and ≈33 wt% for 4b (T = 210 °C, n4a/b:n1 = 1:1) (Table 2). Subsequent DSC measurements do not show any subsequent reactions during the thermal treatment of the materials (Fig. S13†). The large amount of extractable portions can, on the one hand, be attributed to this sterically demanding prepolymer, on the other hand because the polymerization process is a bulk polymerization, i.e. the increasing viscosity during polymerization is a limiting factor for complete conversion. At a temperature of 230°C highly foamed partly dark-colored HMs were obtained. TG-MS measurements show that during the polymerization process CO2 and traces of water are liberated (Fig. S17†). Urethane and urea increments are, after biuret and allophanate substructures, the most thermally labile compounds in a PU formulation.33–35 In order to reduce the thermal load on the samples and thus avoid dissociation of the urethane bonds, only the twin prepolymer 4a was selected for further experiments involving variation of the monomer ratios at a polymerization temperature of 170 °C. Varying the monomer ratio of 4a:1 gives no clear trend with regard to the amount of extractable material (Table 3). NMR spectras of the extract showed again prepolymer residues and low molecular weight products such as salicylic alcohol (Fig. S8 and Table S8†).
| Sample | Monomer ratio n4a:n1 [mol%] |
Extr. cont. [wt%] | Silica cont. [wt%] (TGA) | Silica cont. [wt%] (calc.) |
|---|---|---|---|---|
| HM5a_05 | 5:95 |
21 | 20.9 | 17.6 |
| HM5a_15 | 15:85 |
33 | 18.7 | 13.0 |
| HM5a_50 | 50:50 |
47 | 10.7 | 8.1 |
| HM5a_85 | 85:15 |
25 | 6.1 | 6.5 |
| HM5a_100 | 100:0 |
14 | 5.7 | 6.2 |
Solid-state 13C and 29Si NMR spectroscopy proves the formation of the phenolic resin, silica and ODAS (Fig. 3). The different monomer ratios of 4a:1 are reflected in the NMR spectra and the signal intensities of phenolic resin/ODAS varies correspondingly. An increasing content of the monomer 4a leads to formation of more ODAS structures and thus to more intense D signals, as are shown in the 29Si NMR spectra (Fig. 3). A lower monomer 4a ratio gives a strong signal at −17 ppm, which represents D(Q) species. As expected, HM5a_100 shows only D and no Q signals because of the absence of 1 as reactant.
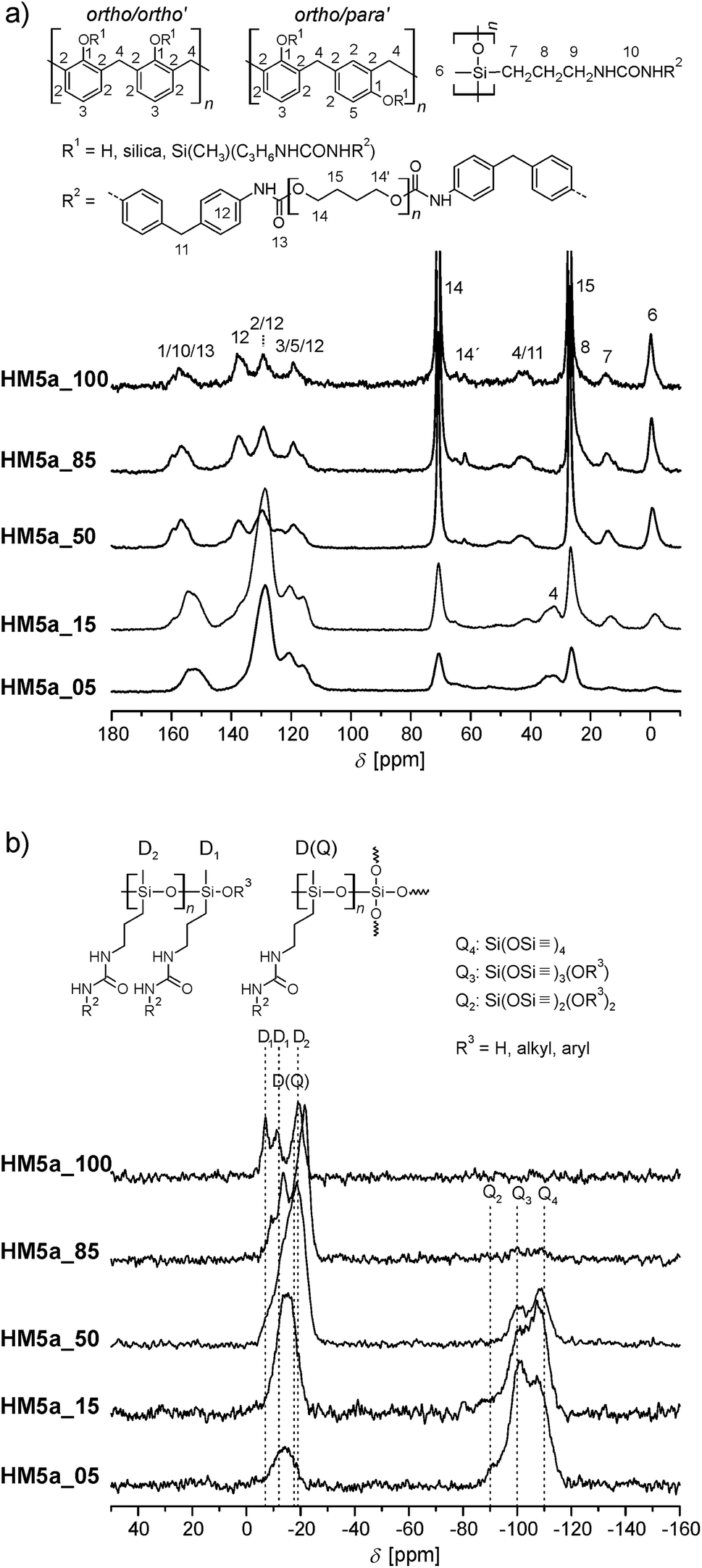 | ||
| Fig. 3 Solid-state-NMR spectra of selected samples with different molar ratios of the monomers 4a and 1. (a) 13C-{1H}-CP-MAS@15 kHz (number of scans: 5a_100 14190; 5a_85 39776; 5a_50 14077; 5a_15 47345; 5a_05 15476) (b) 29Si-{1H}-CP-MAS@12 kHz (number of scans: 5a_100 14184; 5a_85 45596; 5a_50 62126; 5a_15 15012; 5a_05 14716). | ||
The signals of the phenolic carbon and the urethane carbon atom have quite similar chemical shifts within 13C solid state NMR spectra and cannot be distinguished due to broad line widths. The 13C NMR signals in the chemical shift range of 110 to 140 ppm can be assigned to aromatic carbons. The phenolic resin as well as the used aromatic isocyanate contains such structures. Thus, it cannot be determined with certainty whether phenolic resin is formed in the homo polymerization of prepolymer 4a (n4a:n1 = 100:0). In order to determine whether the twin prepolymer participates in the polymerization reaction under formation of phenolic resin a third twin prepolymer 4c was synthesized using the aliphatic diisocyanate IPDI (Scheme 5; ESI†). In the absence of aromatic carbon atoms in the isocyanate component it is possible to determine unequivocally whether phenolic resin is being formed during the homo-polymerization of the twin prepolymer by 13C NMR spectroscopy. The spectra of 4c (in CDCl3) and that of its corresponding homo-polymer HM5c_100 are shown in Fig. 4. After the polymerization (170 °C, 2 h) and extraction of the HM, characteristic signals in the range 110–133 ppm are seen, demonstrating the formation of phenolic resin even in the absence of co-monomer 1.
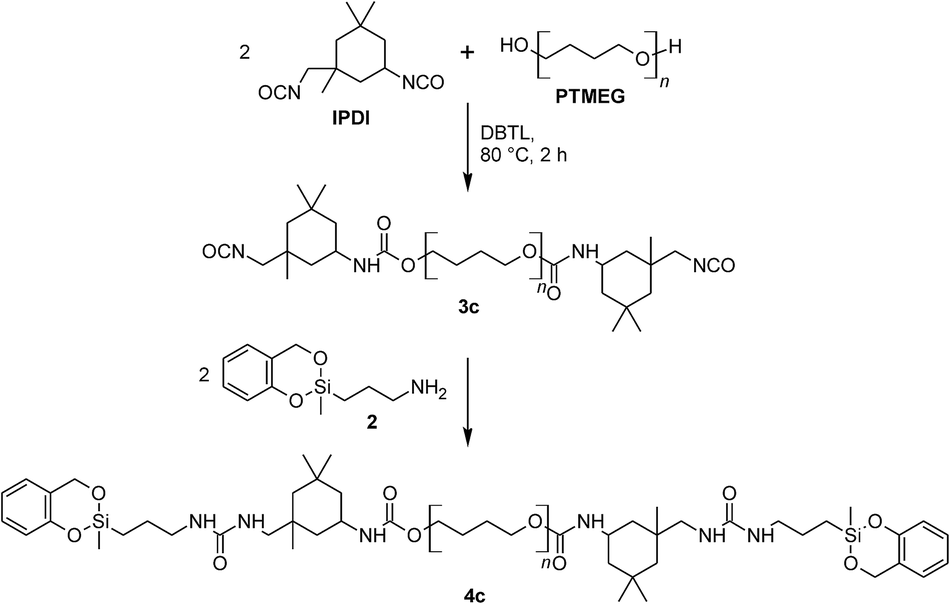 | ||
| Scheme 5 Preparation of the aliphatic prepolymer 3c with DBTL-catalyst and the end-capping with 2 to obtain 4c. | ||
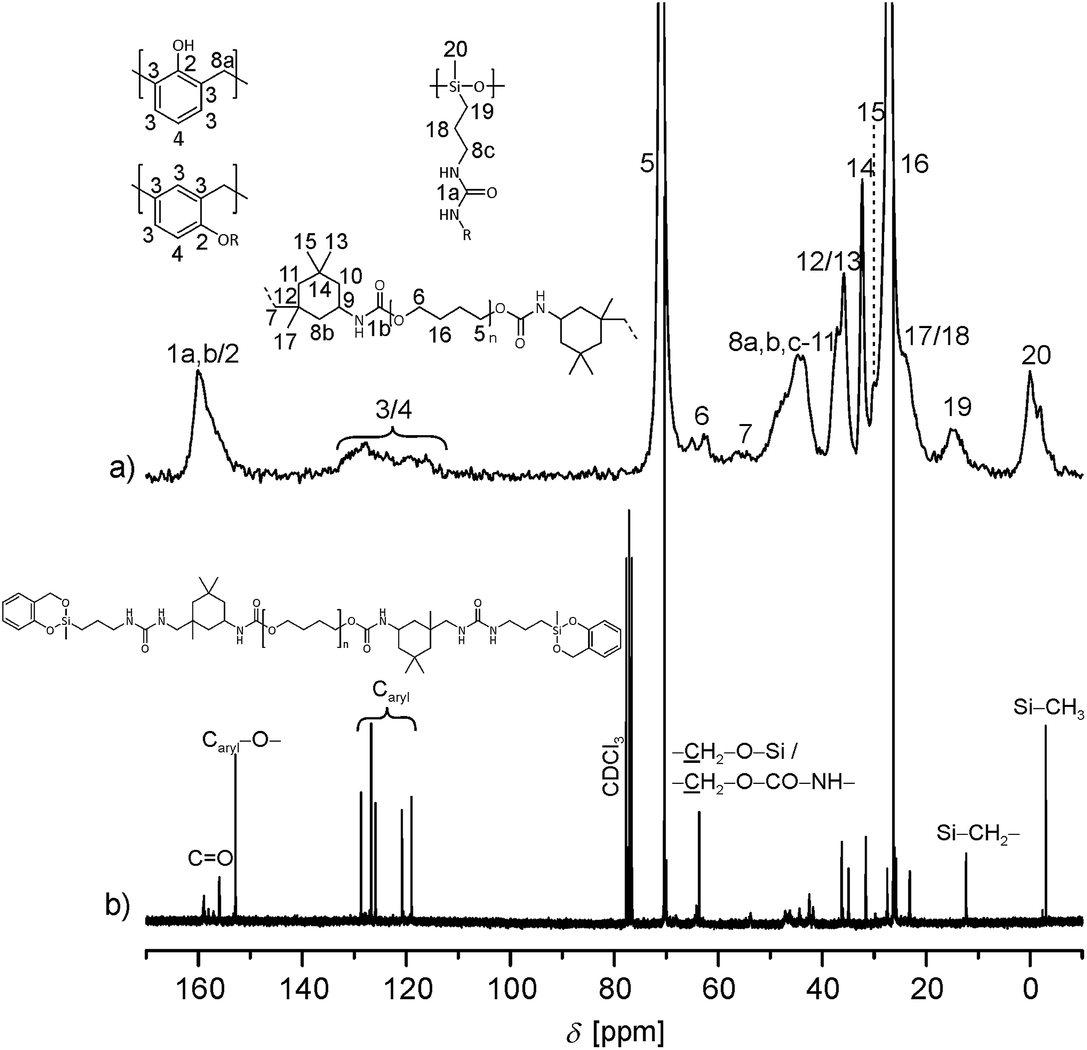 | ||
| Fig. 4 (a) 13C-{1H}-CP-MAS spectrum of the extracted polymer HM5c_100 prepared by the thermally induced polymerization of 4c. (b) Liquid-state NMR of 4c (in CDCl3). | ||
As shown in Fig. 3(a), the characteristic 13C signals of the urethane bond and the phenolic resin cannot be identified separately from each other (signals 1, 9 and 12 in Fig. 3). 29Si-{1H}-CP-MAS NMR spectroscopy is used to show the successful polymerization of the dialkyl siloxane increments of the TM end groups (Fig. 3).
In order to test whether the urea increment is present after the polymerization, the HM was investigated by ATR-FT-IR spectroscopy. The materials HM5a-100/85/50/15/5 as well as 4a show, in comparison with prepolymer 3a, an additional band at 1642 cm−1 assigned to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ourea group. In prepolymer 3a this band is, as expected, not detectable (Fig. S18†).
Ourea group. In prepolymer 3a this band is, as expected, not detectable (Fig. S18†).
All HM were investigated by thermogravimetric analysis (TGA). The samples were heated to 900 °C under air atmosphere with a heating rate of at 10 K min−1 and held for 10 minutes at this temperature in order to guarantee complete removal of the organic content. The quantity of the residue reflects the amount of inorganic material introduced in the HM by variation of the monomers used in each experiment. The thermogravimetric analysis (TGA) of the organic–inorganic HMs shows a slight loss in weight of 4.8 wt% (HM5a_15) to 9.6 wt% (HM5a_85) between 30 and 300 °C.
Base catalyzed simultaneous twin polymerization
Thermally initiated STP of 4a and 1 fabricates to a phenolic resin/SiO2/ODAS nanocomposite in a single step. However, the TP can also be catalyzed by acids or bases. The addition of acid (e.g. lactic acid) to a mixture of 4a and 1 reduces the polymerization temperatures to 80 °C. Unfortunately, the high viscosity results in poor mixing and lead to heterogeneous products. In comparison, the base-catalyzed polymerization of 4a and 1 requires activation temperatures higher than 120 °C as concluded from DSC measurements (see Fig. 2). Thus, the necessary polymerization temperature lies between those of the thermally induced and the acid-catalyzed polymerizations. 1,4-diazabicyclo[2.2.2]octane (DABCO) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were used as base catalysts. A selection of the experiments performed is shown in Table 4.| Sample | Catalyst (base) | Conc. (base) [mol%] | T [°C] | Extr. cont. [wt%] | Silica cont. [wt%] |
|---|---|---|---|---|---|
| 4a and 1 | |||||
| HM5a_base_1 | DABCO | 2.3 | 120 | 18 | 8.3 |
| HM5a_base_2 | 9.0 | 120 | 17 | 9.6 | |
| HM5a_base_3 | 140 | 19 | 9.6 | ||
| HM5a_base_4 | DBU | 6.0 | 150 | 18 | 9.0 |
|
|||||
| 4b and 1 | |||||
| HM5b_base_1 | DABCO | 6.0 | 140 | 15 | 8.7 |
The base-catalyzed STP results in clearly lower extractable portions compared with the thermally induced TP despite the lower reaction temperatures. The soluble extracts show the same composition as for the HM produced by thermal initiation without base as catalyst (Fig. S11†).
High angle annular dark field scanning transmission electron microscopy (HAADF-STEM) investigations were carried out to achieve information on the sizes of the phase domains of the HMs obtained. This technique shows an elemental contrast as well as the contrast attributed to the thickness and the density of the sample. With increasing atomic weight, a higher signal intensity is observed. Thus, the silicon-rich domains of the HM are brighter than the organic polymer. The HAADF-STEM pictures of selected samples with different monomer compositions of 4a and 1 show bright SiO2 clusters with a size of about 2 nm. However, the monomer compositions chosen do not have a significant effect on the domain size (Fig. 5). Si-rich domains are clearly smaller than those found in comparable phenolic resin-silicon dioxide composite materials which were synthesized via sol–gel process.36,37
 | ||
| Fig. 5 HAADF-STEM-images of hybrid materials with different monomer ratios of 4a and 1. | ||
Fig. 6 shows HAADF-STEM pictures of HMs produced from thermally induced and base catalyzed (DABCO) STP of 4a and 1 in equimolar ratio. The silicon distribution within HM5a_base_2 is slightly more homogeneous compared with HM5a_50. STPs of both pre-polymers, 4a and 4b, respectively, in combination with 1 result in HMs which show a quite similar structure in the high resolution TEM pictures: bright SiO2 clusters with a size of approximately 2 nm (HM5a_50 polymerized at 170 °C, HM5b_190 polymerized at 190 °C, Fig. 6). The monomer ratios chosen have no significant effect on the size of the phase domains.
 | ||
| Fig. 6 HAADF-STEM images of the HM prepared by the STP of 4a and 1 with the use of a base catalyst (HM5a_base_2, DABCO, 9.0 mol%/1 wt%, 120 °C, 2 h), the HM prepared by thermally induced STP of 4a and 1 (HM5a_50, equimolar, 170 °C, 2 h) and the HM prepared by thermally induced STP of 4b and 1 (HM5b_190, equimolar, 190 °C, 2 h). | ||
Conclusions
Novel twin prepolymers 4a–c were synthesized and their polymerization ability to produce nanostructured HMs was demonstrated. It was shown that the choice of polymer backbone in the twin prepolymers has an effect on the necessary trigger temperature of the TP. Organic–inorganic HMs are accessible by means of the synergistic combination of twin prepolymers 4a/b with 2,2′-spirobi[4H-1,3,2-benzo-dioxasiline] (1) using STP. The interpenetrating networks resulting from three different polymers result in nanostructured phase domains of approximately 2–4 nm in size. The different networks are formed on the same time scale. Therefore the phase separation is very small and very similar to the already known hybrid materials from twin polymerization. This represents a completely new procedure for incorporating various types of silicon and phenolic resin into nanoscale urethane formulations. The careful choice of monomer composition and trigger process for polymerization procedure is key for the material properties of the HM produced. A correlation of the molecular structure of the organic–inorganic HM with urethane increments with their macroscopic properties is important for the construction of nanocomposites with specific properties required for industrial applications, e.g. construction of strongly adhering, flexible interlayers in lightweight structures, which is the aim of future work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the BASF SE for particular financial support, the providing of chemicals as well as the measurement of the HAADF-STEM-images. This work was also supported by the Deutsche Forschungsgemeindschaft (SCHR 1469/1-1 and the Federal Cluster of Excellence EXC 1075 “Technology Fusion for Multifunctional Lightweight Structures”). Furthermore, we acknowledge Prof. Hietschold and Prof. Tegenkamp for the opportunity of measuring electron microscopy images.Notes and references
- M. Christopher Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- M. Antonietti, Sci. Bull., 2016, 61, 1662–1664 CrossRef.
- A.-H. Lu, W.-C. Li, G.-P. Hao, B. Spliethoff, H.-J. Bongard, B. B. Schaack and F. Schüth, Angew. Chem., Int. Ed., 2010, 49, 1615–1618 CrossRef PubMed.
- Y. Yamauchi, N. Suzuki, L. Radhakrishnan and L. Wang, Chem. Rec., 2009, 9, 321–339 CrossRef PubMed.
- M. Antonietti, Nat. Mater., 2003, 2, 9 CrossRef PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef PubMed.
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef.
- H. S. Thiam, W. R. W. Daud, S. K. Kamarudin, A. B. Mohammad, A. A. H. Kadhum, K. S. Loh and E. H. Majlan, Int. J. Hydrogen Energy, 2011, 36, 3187–3205 CrossRef.
- Y. Chen, S. Zhou, H. Yang and L. Wu, J. Appl. Polym. Sci., 2005, 95, 1032–1039 CrossRef.
- S. Karataş, Z. Hoşgör, N. Kayaman-Apohan and A. Güngör, Prog. Org. Coat., 2009, 65, 49–55 CrossRef.
- S. Karataş, Z. Hoşgör, N.-K. Apohan and A. Güngör, J. Polym. Res., 2010, 17, 247–254 CrossRef.
- S. Spange, P. Kempe, A. Seifert, A. A. Auer, P. Ecorchard, H. Lang, M. Falke, M. Hietschold, A. Pohlers, W. Hoyer, G. Cox, E. Kockrick and S. Kaskel, Angew. Chem., Int. Ed., 2009, 48, 8254–8258 CrossRef PubMed.
- P. Kempe, T. Löschner, A. A. Auer, A. Seifert, G. Cox and S. Spange, Chem. – Eur. J., 2014, 20, 8040–8053 CrossRef PubMed.
- T. Ebert, A. Seifert and S. Spange, Macromol. Rapid Commun., 2015, 36, 1623–1639 CrossRef PubMed.
- P. Kitschke, A. A. Auer, T. Löschner, A. Seifert, S. Spange, T. Rüffer, H. Lang and M. Mehring, ChemPlusChem, 2014, 79, 1009–1023 CrossRef.
- S. Spange and S. Grund, Adv. Mater., 2009, 21, 2111–2116 CrossRef.
- M. Göring, A. Seifert, K. Schreiter, P. Müller and S. Spange, Chem. Commun., 2014, 50, 9753–9756 RSC.
- A. Mehner, A. Pohlers, W. Hoyer, G. Cox and S. Spange, Macromol. Chem. Phys., 2013, 214, 1000–1010 CrossRef.
- S. Oschatz, A. Lange, S. Csihony, G. Cox, O. Gronwald, A. Seifert and S. Spange, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2312–2320 CrossRef.
- C. Leonhardt, A. Seifert, S. Csihony, H. Sommer and M. Mehring, RSC Adv., 2016, 6, 3091–3098 RSC.
- L. Kaßner, K. Nagel, R.-E. Grützner, M. Korb, T. Rüffer, H. Lang and S. Spange, Polym. Chem., 2015, 6, 6297–6304 RSC.
- P. Kitschke, M. Walter, T. Rüffer, A. Seifert, F. Speck, T. Seyller, S. Spange, H. Lang, A. A. Auer, M. V. Kovalenko and M. Mehring, J. Mater. Chem. A, 2016, 4, 2705–2719 RSC.
- F. Böttger-Hiller, A. Mehner, S. Anders, L. Kroll, G. Cox, F. Simon and S. Spange, Chem. Commun., 2012, 48, 10568–10570 RSC.
- A. Mehner, T. Rüffer, H. Lang, A. Pohlers, W. Hoyer and S. Spange, Adv. Mater., 2008, 20, 4113–4117 CrossRef.
- T. Löschner, A. Mehner, S. Grund, A. Seifert, A. Pohlers, A. Lange, G. Cox, H.-J. Hähnle and S. Spange, Angew. Chem., 2012, 124, 3312–3315 CrossRef.
- J. Weißhuhn, T. Mark, M. Martin, P. Müller, A. Seifert and S. Spange, Polym. Chem., 2016, 7, 5060–5068 RSC.
- O. Bayer, Angew. Chem., 1947, 59, 257–272 CrossRef.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef PubMed.
- N. Adam, G. Avar, H. Blankenheim, W. Friederichs, M. Giersig, E. Weigand, M. Halfmann, F.-W. Wittbecker, D.-R. Larimer, U. Maier, S. Meyer-Ahrens, K.-L. Noble and H.-G. Wussow, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- U. Meier-Westhues, Polyurethane – Lacke, Kleb- und Dichtstoffe, Vincentz Network GmbH & Co. KG, Hannover, 2007 Search PubMed.
- H. Zou, S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893–3957 CrossRef PubMed.
- C. Sanchez and F. Ribot, New J. Chem., 1994, 18, 1007–1047 Search PubMed.
- M. Ravey and E. M. Pearce, J. Appl. Polym. Sci., 1997, 63, 47–74 CrossRef.
- F. E. Rogers and T. J. Ohlemiller, J. Macromol. Sci., Chem., 1981, 15, 169–185 CrossRef.
- S. V. Levchik and E. D. Weil, Polym. Int., 2004, 53, 1585–1610 CrossRef.
- H. Sardon, L. Irusta, M. J. Fernández-Berridi, M. Lansalot and E. Bourgeat-Lami, Polymer, 2010, 51, 5051–5057 CrossRef.
- Y. Zhu and D.-X. Sun, J. Appl. Polym. Sci., 2004, 92, 2013–2016 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05310c |
| This journal is © The Royal Society of Chemistry 2018 |