
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical oxidative C–H/S–H cross-coupling between enamines and thiophenols with H2 evolution†
Dandan
Li
a,
Shuaibing
Li
a,
Chong
Peng
a,
Lijun
Lu
b,
Shengchun
Wang
b,
Pan
Wang
b,
Yi-Hung
Chen
b,
Hengjiang
Cong
b and
Aiwen
Lei
 *b
*b
aSchool of Chemistry and Chemical Engineering, Xuchang University, Xuchang 461000, Henan, P. R. China
bCollege of Chemistry and Molecular Sciences, Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, Hubei, P. R. China. E-mail: aiwenlei@whu.edu.cn
First published on 18th January 2019
Abstract
Electrochemical oxidative C–H/S–H cross-coupling has been developed to construct the C–S bond in a highly straightforward and efficient manner. Various enamines and (hetero)aryl thiols could be transformed smoothly under undivided electrolytic cell conditions. Moreover, this electrosynthesis strategy not only avoided the use of chemical oxidants and transition metal catalysts, but also exhibited excellent atom economy.
Introduction
Vinyl sulfides are important structural units which are widely found in natural products and bioactive molecules.1 Continuous endeavors have been focused on developing efficient methods for their synthesis.2 One of the most straightforward strategies is the direct cross-coupling of olefins and thiols/thiophenols. However, thiols/thiophenols are more prone towards the addition to olefins.3 In addition, thiols/thiophenols are easily over-oxidized in the presence of oxidants.3c–g,4 Due to the existence of these problems, it has become difficult to obtain the vinyl sulfides through the C–H/S–H cross-coupling.Over the past few decades, radical–radical cross-coupling reactions have been recognized as a powerful and efficient protocol for the construction of carbon–carbon and carbon–heteroatom bonds.5 This strategy might provide a solution for the synthesis of vinyl sulfides by the cross-coupling of an alkene radical and a thiyl radical. It has been reported that an enamine can generate the enamine radical cation under oxidation and/or transition-metal catalyst conditions.6 And thiophenol can produce the thiyl radical which might couple with the enamine radical cation to furnish the vinyl sulfide. However, stoichiometric amounts of chemical oxidants and/or metal catalysts are often required in these transformations.7 Therefore, considering the development of green chemistry and sustainable chemistry, it is necessary to develop a more effective and greener method to construct the C–S bond.
Electrosynthesis is considered to be a versatile and environmentally friendly synthetic strategy and has received considerable attention.8 In this process, the use of external oxidants could be efficiently avoided. Thiols/thiophenols (RSH) can generate a thiyl radical at the anode.9 And the generated thiyl radical undergoes a rapid dimerization to form a disulfide, which could be reduced at the cathode to furnish a thiyl radical.9b,c The reaction is reversible and an equilibrium exists between the thiyl radical and disulfide.9c For this reason, the sulfur radical can be considered as a persistent radical. On the other hand, an enamine might be oxidized to form a radical cation by losing one electron at the anode. According to the persistent radical effect,10 we speculate that this radical-cation intermediate, serving as a transient radical, could couple with the thiyl radical. Herein, a new radical–radical cross coupling between enamines and thiophenols toward C–S bond formation is described in this work (Scheme 1).
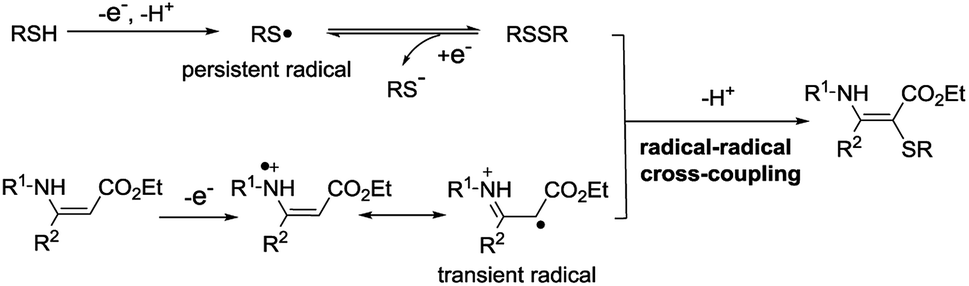 | ||
| Scheme 1 Selective radical–radical cross-coupling between enamines and thiols. | ||
Results and discussion
With the hypothesis mentioned above, initial studies were focused on (Z)-ethyl 3-(allylamino)-3-phenylacrylate (1a) and 4-chlorothiophenol (2a) as model substrates to test the reaction conditions. By using nBu4NBF4 as electrolyte under a 3 mA constant current, 2a can react selectively with the double bond of the enamine to give the product 3aa in 89% yield (Table 1, entry 1). We did not observe the product of the reaction of thiophenol with the allyl double bond. Further optimization was carried out by changing the supporting electrolyte. However, lithium perchlorate showed decreased reaction efficiency (entry 2). Only a trace amount of the coupling product was isolated using nBu4NPF6 as the electrolyte (entry 3). We also examined the effect of the electrode material. A platinum plate cathode showed a similar reactivity to the iron plate cathode (entry 4), while carbon cloth or a nickel plate cathode was less effective than the iron plate cathode (entries 5 and 6). Moreover, the effect of solvent was also explored. DMSO afforded the desired product in a lower yield (entry 7). DMF and MeOH were not suitable for this transformation (entries 8 and 9). These results indicated that CH3CN was the most effective for this reaction. Increasing the constant current led to a reduced yield (entry 10). When the reaction was carried out in open air, the yield of 3aa was sharply reduced to 24% (entry 11). As expected, the reaction failed to provide any desired product without electricity under atmospheric conditions (entry 12).|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Standard conditions: C (cloth) anode, Fe cathode, constant current = 3 mA, 1a (0.20 mmol), 2a (0.4 mmol), nBu4NBF4 (0.1 mmol), CH3CN (10.0 mL), room temperature, N2, 5 h. b Isolated yield, n.d. = not detected. | ||
| 1 | None | 89 |
| 2 | LiClO4 instead of nBu4NBF4 | 70 |
| 3 | n Bu4NPF6 instead of nBu4NBF4 | Trace |
| 4 | Pt plate cathode | 81 |
| 5 | Ni plate cathode | 71 |
| 6 | Carbon cloth cathode | 71 |
| 7 | DMSO instead of CH3CN | 69 |
| 8 | DMF instead of CH3CN | n.d. |
| 9 | MeOH instead of CH3CN | n.d. |
| 10 | 5 mA instead of 3 mA, 3 h | 74 |
| 11 | Under air | 24 |
| 12 | No electricity, under air | n.d. |
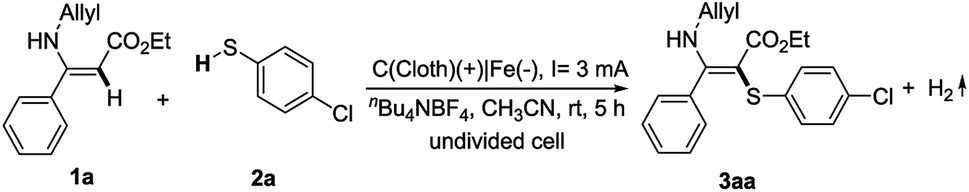
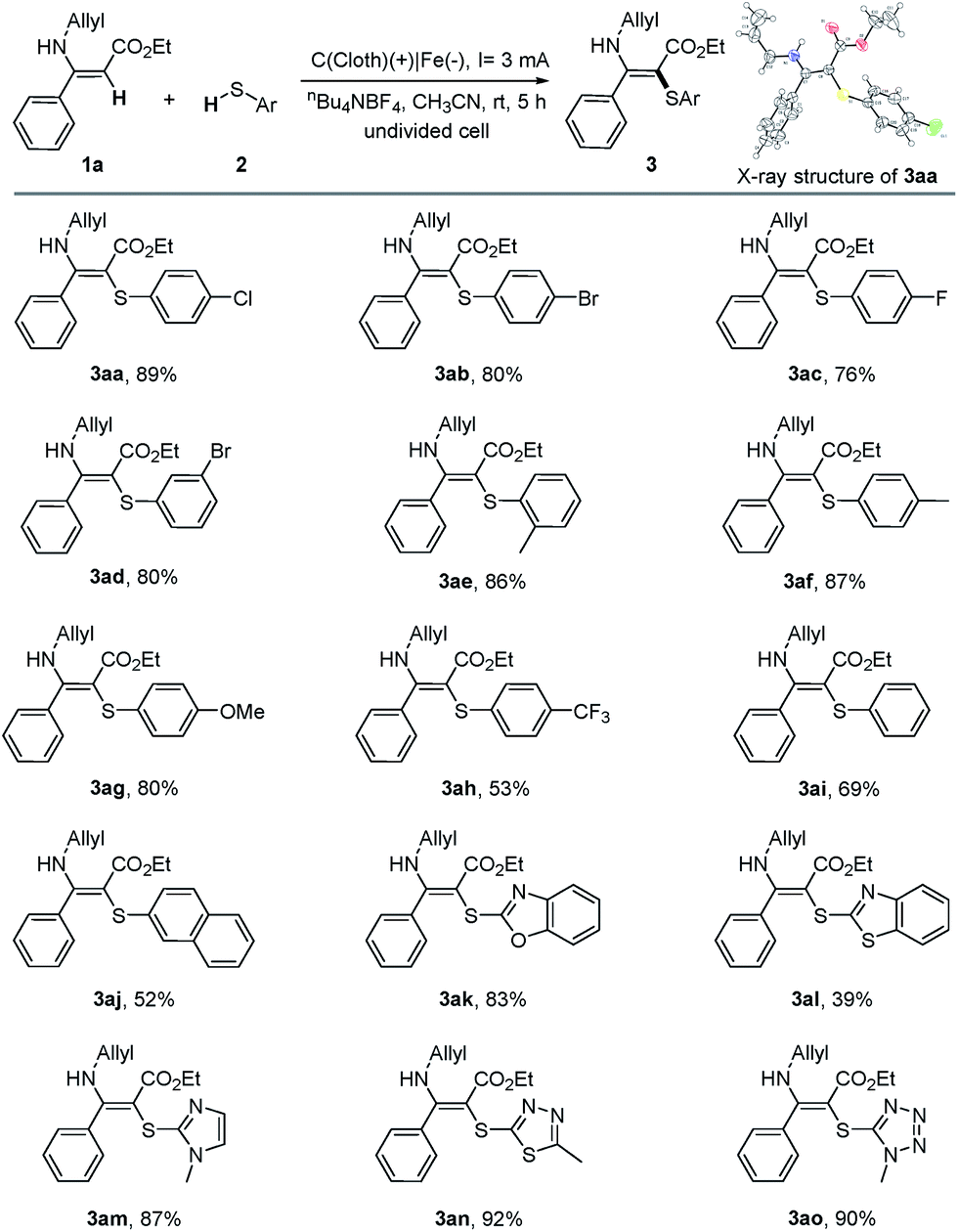
With the optimal conditions in hand, we then turned to investigate the scope and the limitations of the electrochemical oxidative cross-coupling reaction. First, a range of thiophenols bearing various substitution patterns were applied as substrates in this C–S bond construction reaction, giving the corresponding vinyl sulfides in moderate to good yields (Table 2). Thiophenols containing halogen atoms such as Cl, Br, and F reacted smoothly and led to the corresponding products (3aa–3ad) in 76–89% yields. Similarly, thiophenols substituted with an electron-neutral substituent (Me) or the strong electron donating groups (OMe) also showed good reactivity in this transformation (3ae–3ag). It should be noted that the substrate bearing the electron-withdrawing p-CF3 group could also afford the target product, although in a relatively low yield (3ah).
| a Standard conditions: C (cloth) anode, Fe cathode, constant current = 3 mA, 1a (0.20 mmol), 2a (0.4 mmol), nBu4NBF4 (0.1 mmol), CH3CN (10.0 mL), room temperature, N2, 5 h. |
|---|
|
Benzenethiol could afford 69% yield (3ai) under the standard conditions while 2-naphthalenethiol gave 52% yield (3aj). Furthermore, the reaction could be successfully extended to heteroaryl thiols, furnishing the corresponding products in good to excellent yields (3ak, 3am–3ao). 2-Mercaptobenzothiazole gave 3al in a relatively low yield owing to the poor solubility in acetonitrile. We also attempted mercaptans as substrates; however, either no or a trace amount of products were detected.
Encouraged by the above results, we next explored the scope of enamines (Table 3). Enamines 1b–i bearing electron-donating or electron-withdrawing groups on the aromatic ring were tolerated under the electrooxidation conditions, affording the desired products in high yields (3ba–3ia). The substrates containing naphthalene and thiophene groups were also suitable for this transformation (3ja and 3ka). Moreover, we also attempted (Z)-ethyl 3-(methylamino)but-2-enoate, (Z)-ethyl 3-(methylamino)-3-phenylacrylate, and (Z)-ethyl 3-phenyl-3-(phenylamino)acrylate, all of them could successfully convert to the desired products with a good reaction efficiency (3la–3na).
| a Standard conditions: C (cloth) anode, Fe cathode, constant current = 3 mA, 1a (0.20 mmol), 2a (0.4 mmol), nBu4NBF4 (0.1 mmol), CH3CN (10.0 mL), room temperature, N2, 5 h. Isolated yields are shown. |
|---|

|
With the established electrochemical method, the gram-scale reaction was performed on a 3 mmol scale. The reaction of (Z)-ethyl 3-(allylamino)-3-phenylacrylate (1a) and 4-chlorothiophenol (2a) proceeded smoothly and the desired 3aa was obtained in 66% yield (0.74 g, Scheme 2). This result indicates that this electrochemical cross-coupling reaction has great potential in practical synthesis.
 | ||
| Scheme 2 Gram scale synthesis. | ||
In order to gain insights into the reaction mechanism, several control experiments were conducted (Scheme 3). 4-Chlorothiophenol (2a) underwent dimerization to afford bis-(4-chlorophenyl) disulfide 4a, which was obtained in 83% yield under the standard reaction conditions (Scheme 3a). Then, when replacing 4-chlorothiophenol (1a) with bis-(4-chlorophenyl) disulfide (4a) under the standard conditions, this reaction could still give the desired C–S formation product in 90% yield. The reaction failed to provide any target product without electricity (Scheme 3c). These results indicated that disulfides could be transformed into the corresponding thiyl radicals under the electrocatalytic conditions and might be the key intermediate for the transformation.
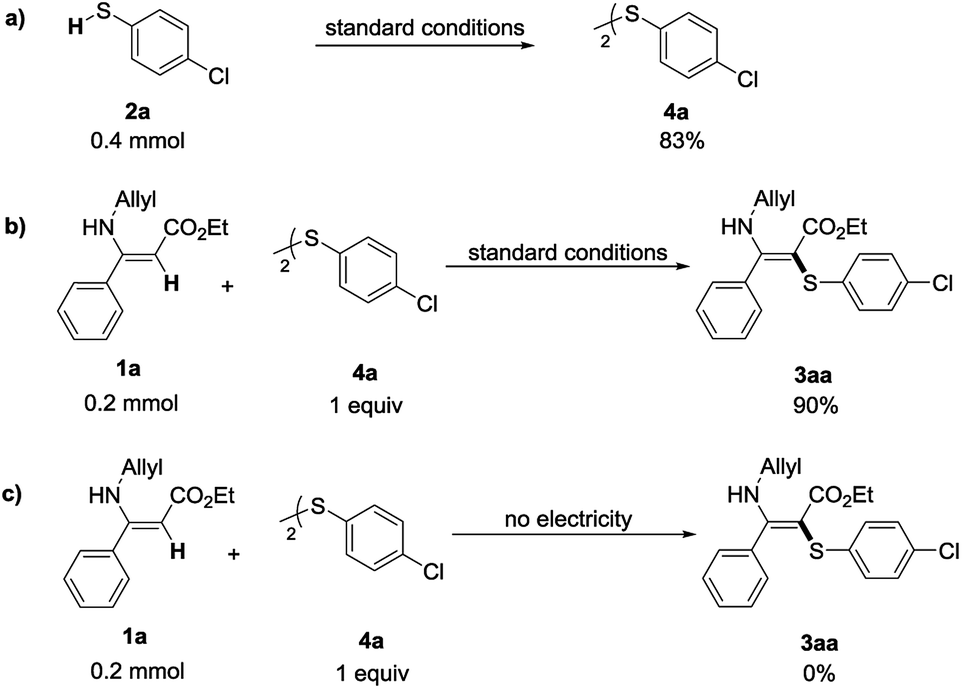 | ||
| Scheme 3 Control experiments. | ||
Additionally, EPR experiments (Fig. 1) showed the presence of C-centred radicals and S-centred radicals which were trapped by 5,5-dimethyl-1-pyrroline N-oxide (C-centred radicals: g = 2.0068, AH = 14.5 G, AN = 21.3 G and S-centred radicals: g = 2.007, AH = 13.1 G, AN = 12.2 G) under standard conditions (details showed in the ESI†). In the next step, cyclic voltammetry (CV) experiments were carried out to study the redox potential of the substrates (Fig. S1–S3†). An obvious oxidation peak of 1a in acetonitrile was observed at 1.35 V vs. Ag/AgCl. The oxidation peak of 2a could also be observed at about 1.44 V. At the same time, there was an oxidation peak at 1.38 V for the solution of 1a and 2a. We proposed that both substrates could be oxidized under the electrolytic conditions.
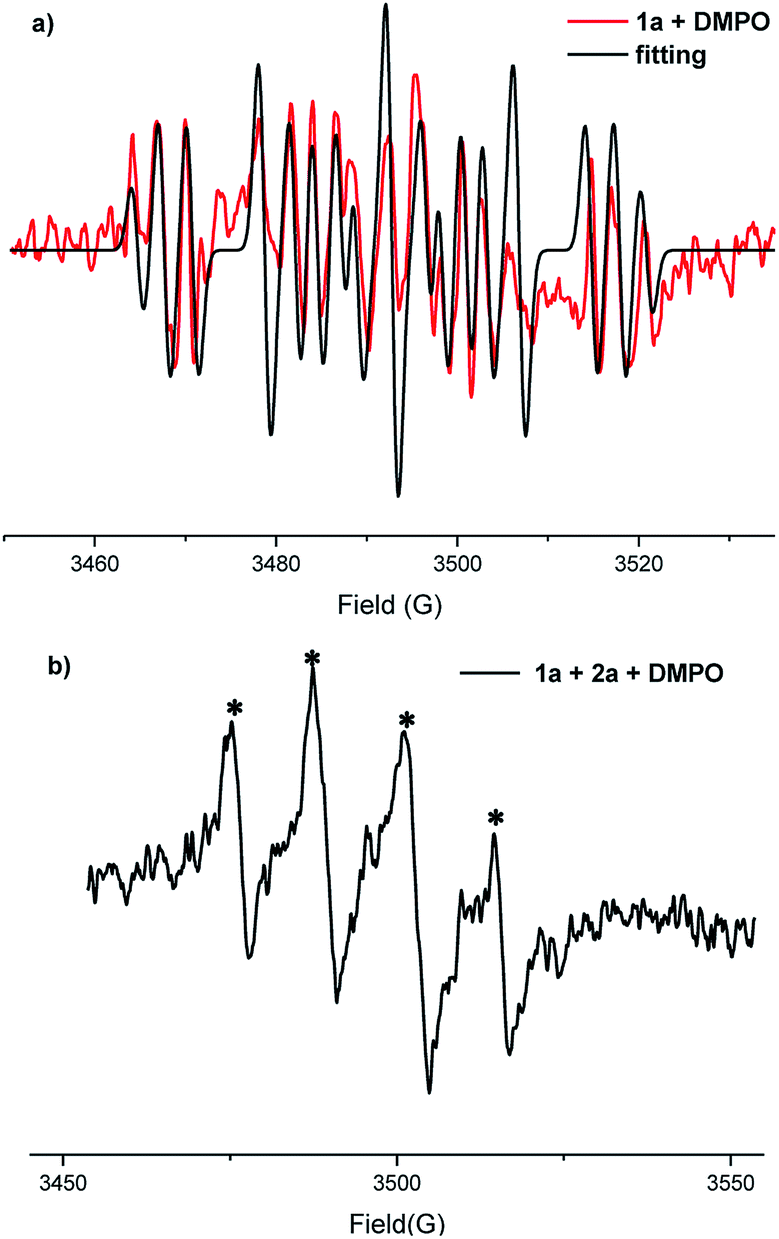 | ||
| Fig. 1 (a) EPR measurements of a solution in CH3CN of nBu4NBF4 and 1a in the presence of DMPO under constant current conditions for 8 min. (b) EPR measurements of 1a and 2a under the same reaction conditions for 5 min. | ||
Based on the above experimental results and literature reports, a plausible mechanism for the C–S formation process is outlined in Scheme 4. Thiophenol is converted to the sulfur radical by anodic oxidation which rapidly dimerizes to generate the disulfide. The disulfide gets one electron at the cathode to afford the disulfide radical anion. Subsequently, this radical anion cleaves to give the thiyl radical and thiolate anion. The thiolate anion can be converted to the thiyl radical at the anode. Meanwhile, an enamine can also be oxidized by the anode to give a radical intermediate A which could isomerize to the corresponding imine radical B. The imine radical intermediate B can directly couple with the sulfur radical to afford intermediate C. Finally, C would isomerize to afford the target product. At the same time, concomitant cathodic reduction of a proton leads to hydrogen evolution. It should be noted that a mechanism involving the homolytic substitution pathway11 could not be completely excluded.
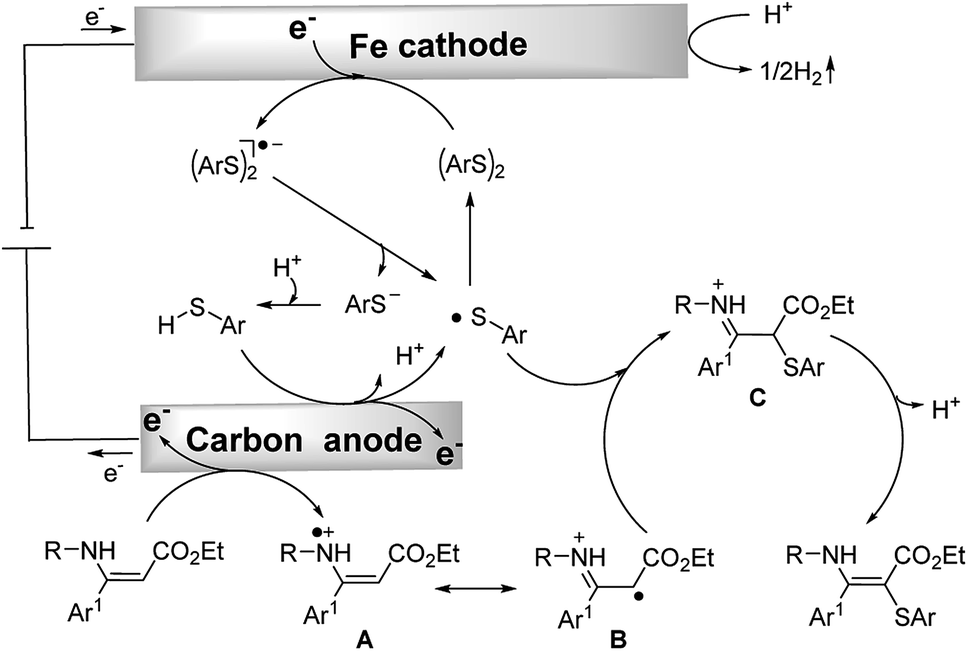 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, an efficient and highly selective C–H/S–H cross-coupling reaction between enamines and thiophenols has been demonstrated for the first time. Various vinyl sulfides could be obtained in good to excellent yields under electrochemical oxidative conditions. This synthetic method obviated the usage of external oxidants and exhibited good functional group tolerance and higher atom economy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21390402, 21520102003), the Hubei Province Natural Science Foundation of China (2017CFA010), the Fundamental Research Funds for the Central Universities and the Program of Introducing Talents of Discipline to Universities of China (111 Program) and the Key Project of Education Department of Henan Province (No. 17A150050).Notes and references
- (a) M. Ceruti, G. Balliano, F. Rocco, P. Milla, S. Arpicco, L. Cattel and F. Viola, Lipids, 2001, 36, 629 CrossRef CAS PubMed; (b) P. Johannesson, G. Lindeberg, A. Johansson, G. V. Nikiforovich, A. Gogoll, B. Synnergren, M. Le Greves, F. Nyberg, A. Karlen and A. Hallberg, J. Med. Chem., 2002, 45, 1767 CrossRef CAS; (c) J. S. Lazo, K. Nemoto, K. E. Pestell, K. Cooley, E. C. Southwick, D. A. Mitchell, W. Furey, R. Gussio, D. W. Zaharevitz, B. Joo and P. Wipf, Mol. Pharmacol., 2002, 61, 720 CrossRef CAS; (d) H. S. Sader, D. M. Johnson and R. N. Jones, Antimicrob. Agents Chemother., 2004, 48, 53 CrossRef CAS PubMed; (e) A. Lavecchia, S. Cosconati, V. Limongelli and E. Novellino, ChemMedChem, 2006, 1, 540 CrossRef CAS PubMed; (f) D. Pappo and Y. Kashman, Org. Lett., 2006, 8, 1177 CrossRef CAS PubMed; (g) S. Pasquini, C. Mugnaini, C. Tintori, M. Botta, A. Trejos, R. K. Arvela, M. Larhed, M. Witvrouw, M. Michiels, F. Christ, Z. Debyser and F. Corelli, J. Med. Chem., 2008, 51, 5125 CrossRef CAS PubMed; (h) F. R. Ianiski, M. M. Bassaco, A. G. Vogt, A. S. Reis, M. P. Pinz, G. T. Voss, R. L. de Oliveira, C. C. Silveira, E. A. Wilhelm and C. Luchese, Med. Chem. Res., 2018, 27, 46 CrossRef CAS.
- (a) S. Blechert, Nachr. Chem., Tech. Lab., 1979, 27, 634 CrossRef CAS; (b) N. V. Zyk, E. K. Beloglazkina, M. A. Belova and N. S. Dubinina, Russ. Chem. Rev., 2003, 72, 769 CrossRef CAS; (c) C. G. Bates, P. Saejueng, M. Q. Doherty and D. Venkataraman, Org. Lett., 2004, 6, 5005 CrossRef CAS PubMed; (d) S.-R. Guo and Y.-Q. Yuan, Synth. Commun., 2008, 38, 2722 CrossRef CAS; (e) M. S. Kabir, M. L. Van Linn, A. Monte and J. M. Cook, Org. Lett., 2008, 10, 3363 CrossRef CAS PubMed; (f) S. Ranjit, Z. Duan, P. Zhang and X. Liu, Org. Lett., 2010, 12, 4134 CrossRef CAS PubMed; (g) H.-L. Kao and C.-F. Lee, Org. Lett., 2011, 13, 5204 CrossRef CAS PubMed; (h) J. Feng, G. Lu, M. Lv and C. Cai, Asian J. Org. Chem., 2014, 3, 77 CrossRef CAS; (i) E. S. Degtyareva, J. V. Burykina, A. N. Fakhrutdinov, E. G. Gordeev, V. N. Khrustalev and V. P. Ananikov, ACS Catal., 2015, 5, 7208 CrossRef CAS; (j) H.-Y. Tu, B.-L. Hu, C.-L. Deng and X.-G. Zhang, Chem. Commun., 2015, 51, 15558 RSC.
- (a) E. L. Tyson, M. S. Ament and T. P. Yoon, J. Org. Chem., 2013, 78, 2046 CrossRef CAS PubMed; (b) S.-F. Zhou, X. Pan, Z.-H. Zhou, A. Shoberu and J.-P. Zou, J. Org. Chem., 2015, 80, 3682 CrossRef CAS PubMed; (c) H. Wang, Q. Lu, C. Qian, C. Liu, W. Liu, K. Chen and A. Lei, Angew. Chem., Int. Ed., 2016, 55, 1094 CrossRef CAS PubMed; (d) H. Cui, W. Wei, D. Yang, Y. Zhang, H. Zhao, L. Wang and H. Wang, Green Chem., 2017, 19, 3520 RSC; (e) D. Limnios and C. G. Kokotos, Adv. Synth. Catal., 2017, 359, 323 CrossRef CAS; (f) Y. Wang, W. Jiang and C. Huo, J. Org. Chem., 2017, 82, 10628 CrossRef CAS PubMed; (g) Y. Zhang, Z. R. Wong, X. Wu, S. J. L. Lauw, X. Huang, R. D. Webster and Y. R. Chi, Chem. Commun., 2017, 53, 184 RSC; (h) K. Choudhuri, A. Mandal and P. Mal, Chem. Commun., 2018, 54, 3759 RSC.
- L. Wang, H. Yue, D. Yang, H. Cui, M. Zhu, J. Wang, W. Wei and H. Wang, J. Org. Chem., 2017, 82, 6857 CrossRef CAS PubMed.
- (a) The Molecular World: Mechanism and Synthesis, ed. P. Taylor, Royal Society of Chemistry, 2002 Search PubMed; (b) L. Zhou, S. Tang, X. Qi, C. Lin, K. Liu, C. Liu, Y. Lan and A. Lei, Org. Lett., 2014, 16, 3404 CrossRef CAS PubMed; (c) J. L. Jeffrey, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 8404 CrossRef CAS PubMed; (d) Z. Huang, D. Zhang, X. Qi, Z. Yan, M. Wang, H. Yan and A. Lei, Org. Lett., 2016, 18, 2351 CrossRef CAS PubMed; (e) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed; (f) S. Wang, S. Tang and A. Lei, Sci. Bull., 2018, 63, 1006 CrossRef CAS; (g) C. Zheng, F. Lu, H. Lu, J. Xin, Y. Deng, D. Yang, S. Wang, Z. Huang, M. Gao and A. Lei, Chem. Commun., 2018, 54, 5574 RSC.
- (a) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2005, 127, 3672 CrossRef CAS PubMed; (b) J. Ke, Y. Tang, H. Yi, Y. Li, Y. Cheng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 6604 CrossRef CAS PubMed; (c) K. Wu, Z. Huang, X. Qi, Y. Li, G. Zhang, C. Liu, H. Yi, L. Meng, E. E. Bunel, J. T. Miller, C.-W. Pao, J.-F. Lee, Y. Lan and A. Lei, Sci. Adv., 2015, 1, e1500656/1 CAS; (d) M. Meciarova, P. Tisovsky and R. Sebesta, New J. Chem., 2016, 40, 4855 RSC.
- (a) Y. Jiang, G. Liang, C. Zhang and T.-P. Loh, Eur. J. Org. Chem., 2016, 3326, DOI:10.1002/ejoc.201600588; (b) J.-P. Wan, S. Zhong, L. Xie, X. Cao, Y. Liu and L. Wei, Org. Lett., 2016, 18, 584 CrossRef CAS PubMed; (c) Y. Siddaraju and K. R. Prabhu, J. Org. Chem., 2017, 82, 3084 CrossRef CAS PubMed; (d) P. Ghosh, A. K. Nandi, G. Chhetri and S. Das, J. Org. Chem., 2018, 83, 12411 CrossRef CAS PubMed; (e) B. Hu, P. Zhou, K. Rao, J. Yang, L. Li, S. Yan and F. Yu, Tetrahedron Lett., 2018, 59, 1438 CrossRef CAS.
- (a) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605 RSC; (b) J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS PubMed; (c) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC; (d) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77 CrossRef CAS PubMed; (e) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (f) J. I. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS PubMed; (g) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS PubMed; (h) M. D. Karkas, Chem. Soc. Rev., 2018, 47, 5786 RSC; (i) K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375 RSC; (j) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS; (k) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175 CrossRef CAS; (l) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27 CrossRef CAS.
- (a) P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009 CrossRef CAS PubMed; (b) P. Huang, P. Wang, S. Tang, Z. Fu and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 8115 CrossRef CAS PubMed; (c) Y. Yuan, Y. Chen, S. Tang, Z. Huang, A. Lei and A. Lei, Sci. Adv., 2018, 4, eaat5312 CrossRef PubMed.
- H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef CAS PubMed.
- A. L. J. Beckwith and S. A. M. Duggan, J. Chem. Soc., Perkin Trans. 2, 1994, 1509 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1868011. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05143g |
| This journal is © The Royal Society of Chemistry 2019 |