
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a coumarin derivative of resorcin[4]arene with solvent-controlled chirality†
Anna Szafranieca and
Waldemar Iwanek *b
*b
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 60-614 Poznań, Poland
bFaculty of Chemical Technology and Engineering, UTP, University of Science and Technology, Seminaryjna 3, 85-326 Bydgoszcz, Poland
First published on 30th March 2020
Abstract
This paper presents the synthesis of a coumarin derivative of resorcin[4]arene (1) using a cascade thermolysis/Michael reaction. The influence of the hydrogen bonding system on the conformational rigidity and cyclochirality of the coumarin derivative of resorcin[4]arene was discussed; these properties depended on the proton-donor–acceptor properties of the solvent. Significant differences, which depended on the environment, in the coumarin derivative of resorcin[4]arene fluorescence were observed and discussed.
Introduction
Resorcin[4]arenes, despite the many years since their discovery, are still important components of supramolecular chemistry.1,2 Based on them, new functional supramolecules, including chiral ones, can be produced.3–5 They belong to the group of macrocyclic compounds, the structure and physicochemical properties of which can be modified in various ways. We can functionalize the “ortho” positions and the hydroxyl groups of the resorcin[4]arene platform, not only in terms of introducing additional moieties with the desired properties but also their specific number.6 This means that the skeleton of resorcin[4]arene can be used to synthesize new derivatives with diverse molecular architecture, chirality,7–11 and binding domains.12 Among other things, spatially developed structures such as cavitands,13–16 dimeric capsules,17–20 and hexamers21–24 can be constructed based on such skeletons, and these can be used to study the reactivity of chemical compounds in spatially limited or closed structures.25–28One possibility for modifying the structure of resorcin[4]arene was our use of the methoxy derivative of resorcin[4]arene for the thermal generation of the o-quinone methide derivative of resorcin[4]arene, followed by a cycloaddition reaction29–31 and Michael reaction with C, P – nucleophiles.32,33
Until now, the synthesis of cyclochiral derivatives of resorcin[4]arene was based on two approaches: (1) structural modifications of the skeleton of the resorcin[4]arene molecule via covalent bonds;4,5 (2) on the selection of appropriate substrates for the synthesis of cyclochiral resorcin[4]arene molecules.7,8,10,11 In this paper, we present a new concept for the synthesis of cyclochiral derivatives of resorcin[4]arene based on a hydrogen bond system. If we attach to the resorcin[4]arene platform groups with different proton-donor (DH)–acceptor (A) properties, as in Scheme 1, then due to the directivity of the hydrogen bonding system formed during the reaction, the product becomes cyclochiral. The hydrogen bonds formed during the reaction affect its stabilization and energy. The possibility of producing hydrogen bonding systems of the M and P type (Cahn–Ingold–Prelog rules) causes these products to occur in the form of a racemic mixture of enantiomers.
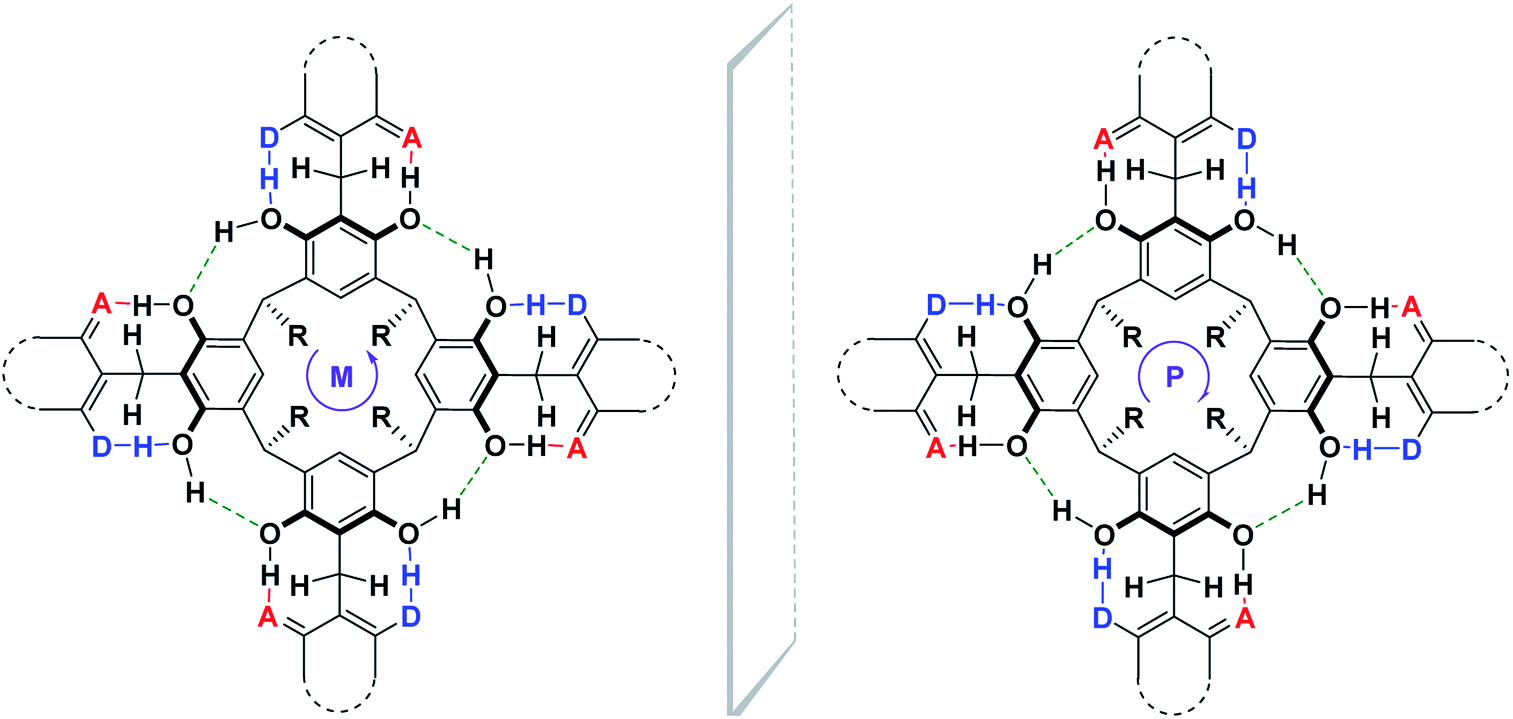 | ||
| Scheme 1 The general concept of cyclochiral (M, P) derivatives of resorcin[4]arene based on a hydrogen bond system. | ||
Solvent-mediated supramolecular conformational changes and chiroptical control of inherently chiral tertamethoxyresorcin[4]arenes were investigated.34 These studies concerned the basic skeleton of resorcin[4]arene. It was found, that in aprotic solvents the crown conformation (C4) is preferred, protic solvents favor the boat conformation (C2). Our work concerns changes stiffened via intramolecular hydrogen bond crown-out conformer of coumarin derivative of resorcin[4]arene under the influence of solvents with different proton-donor–acceptor properties.
We show the usefulness of the o-quinone methide intermediate of resorcin[4]arene in the Michael 1,4-addition reaction, opening up new possibilities for the synthesis of resorcin[4]arene derivatives with the interesting structural, chiral, receptor, and spectroscopic properties.
Results and discussion
Scheme 2 shows the cascade reaction of thermolysis/1,4-Michael addition of the methoxy derivative of resorcin[4]arene with 4-hydroxycoumarin in three solvents: dioxane, toluene, and acetic acid. This reaction gave coumarin derivative of resorcin[4]arene (1) with a yield of 70–85%, depending on the solvent used. The reaction took place most efficiently in toluene, leading after washing several times with a series of solvents to a spectrally pure product. Syntheses in amounts of several hundred milligrams were performed using an Anton-Paar Monowave 50 Reactor, conducting the reaction at 160 °C for 15 minutes. The synthesis procedure for compound (1) and its spectral characteristics are described in the experimental section. The resulting product was characterized by low solubility in organic solvents, which significantly limited the possibilities of testing its receptor properties.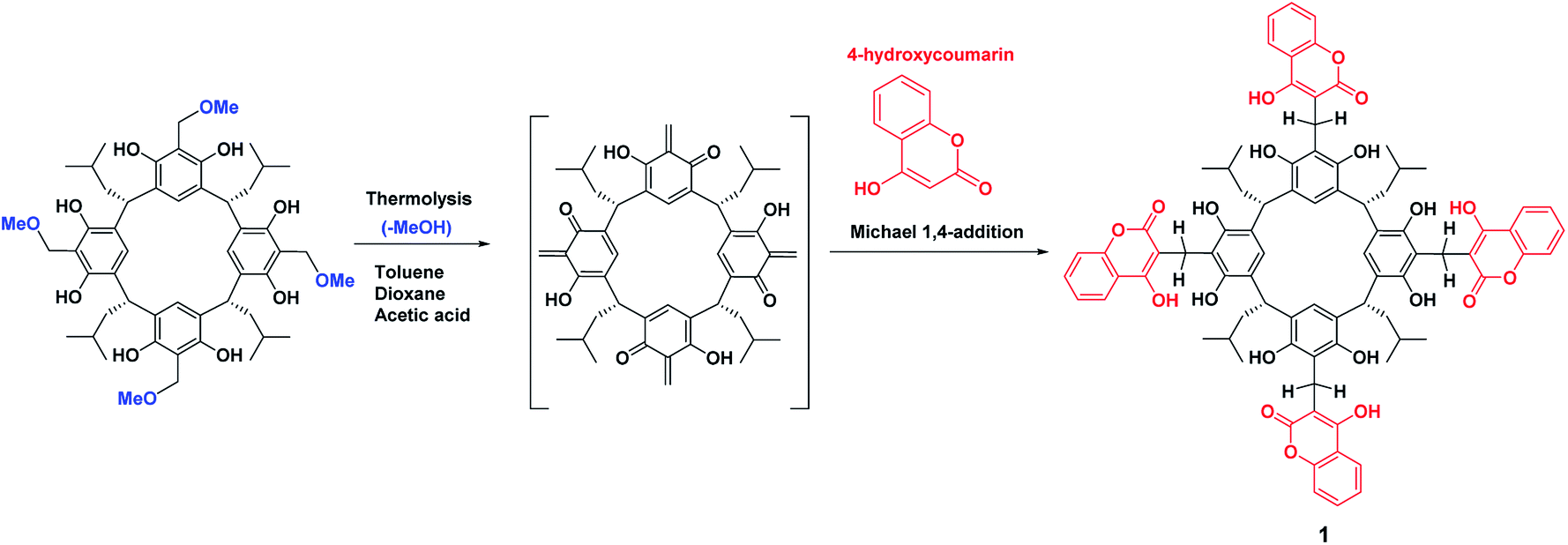 | ||
| Scheme 2 The sequence of the cascade reactions: thermolysis of methoxy derivative of resorcin[4]arene and the Michael 1,4-addition of generated o-quinone methide derivatives of resorcin[4]arene with 4-hydroxycoumarin. | ||
1H NMR spectra of compound (1) performed in chloroform and DMSO differed significantly. In chloroform, we observed spin–spin coupling of the diastereotopic protons of the methylene group –CH2 (g,g′) – Fig. 1. This indicated that the structure in this solvent gained stiffness by creating strong intramolecular hydrogen bonds. In addition, the methylene protons –CH2 (c,c′) in the lower rim of the compound (1) also become diastereotopic. It is possibly due to the generation of dissymmetry in the resorcin[4]arene skeleton.
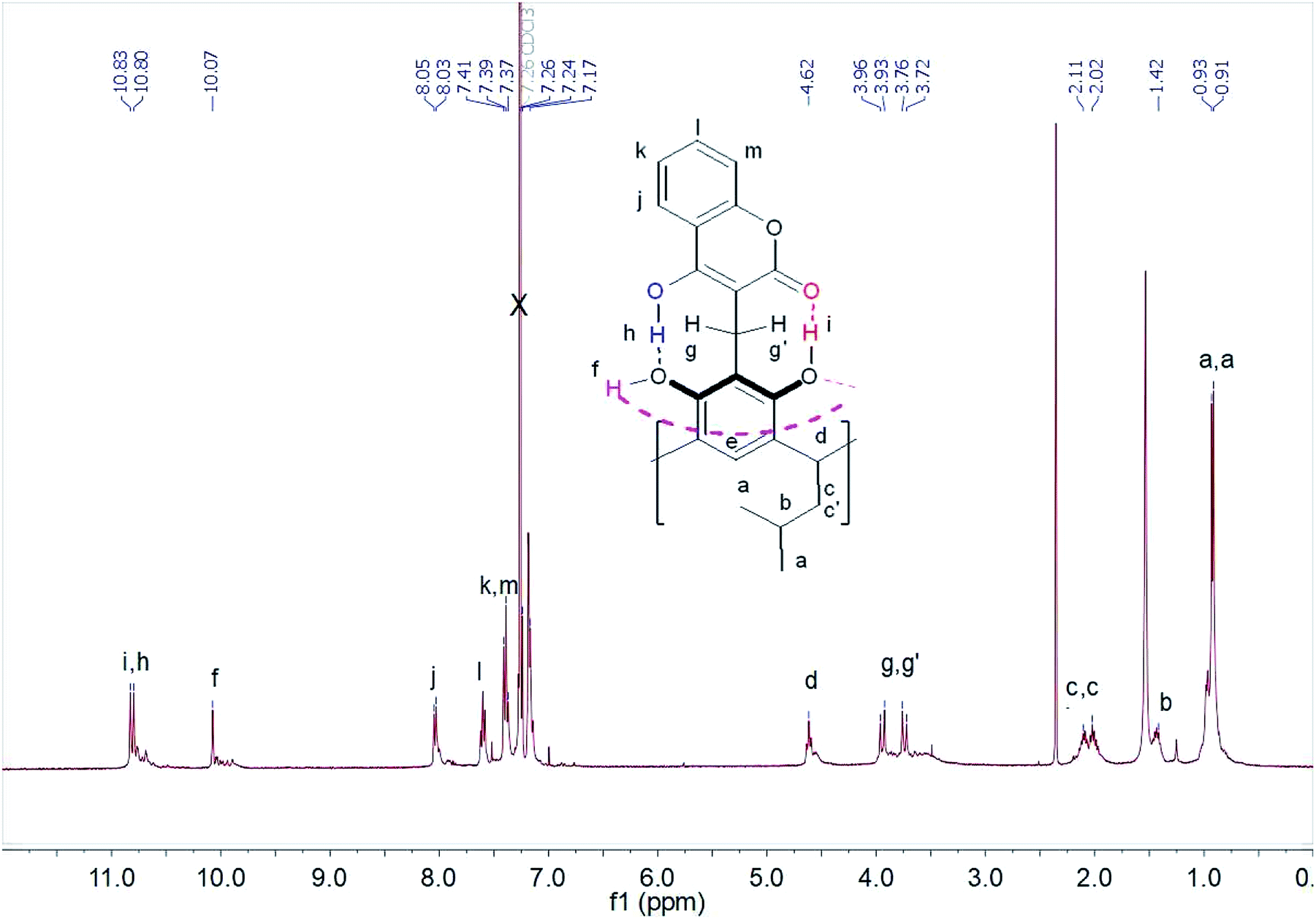 | ||
| Fig. 1 1H NMR spectrum of compound (1) in CDCl3 at 298 K. | ||
The protons participating in the formation of hydrogen bonds were in the range δ = 10–11 ppm. The proton at chemical shift δ = 10.83 ppm was assigned to the proton of the hydroxyl group OH(i) forming hydrogen bonds with the carbonyl group of the coumarin part of the resorcin[4]arene. However, the chemical shift δ = 10.80 ppm was assigned to the proton from the hydroxyl group OH(h) of the coumarin part of the resorcin[4]arene, which formed a hydrogen bond with the hydroxyl group OH(f) of the resorcin[4]arene ring. The proton signal of the hydroxyl group OH(f) at δ = 10.07 ppm was assigned to the hydroxyl group of the resorcin[4]arene, forming a hydrogen bond with oxygen from the hydroxyl group of the adjacent resorcinol unit in the resorcin[4]arene.
The assignment was made on the basis of ROESY spectra in CDCl3 of the mutual interactions of protons from hydroxyl groups (Fig. S8†) and their interactions with the diastereotopic protons of the methylene group (g,g′) – Fig. S9.† The proximity of aromatic protons of the coumarin parts of the compound (1) is not observed in the ROESY spectrum, which suggests the formation of an “out” conformer, with coumarin units located outside the cavity. In chloroform, an intramolecular hydrogen bonding system was formed, which stiffened the molecule of compound (1) and reduced the mobility of coumarin units.
The 1H-NMR spectrum in DMSO looked completely different. The diastereotopic protons of both methylene groups became singlets, while the protons of the hydrogen bonds gave a broad signal in the range of 3–5 ppm (Fig. 2).
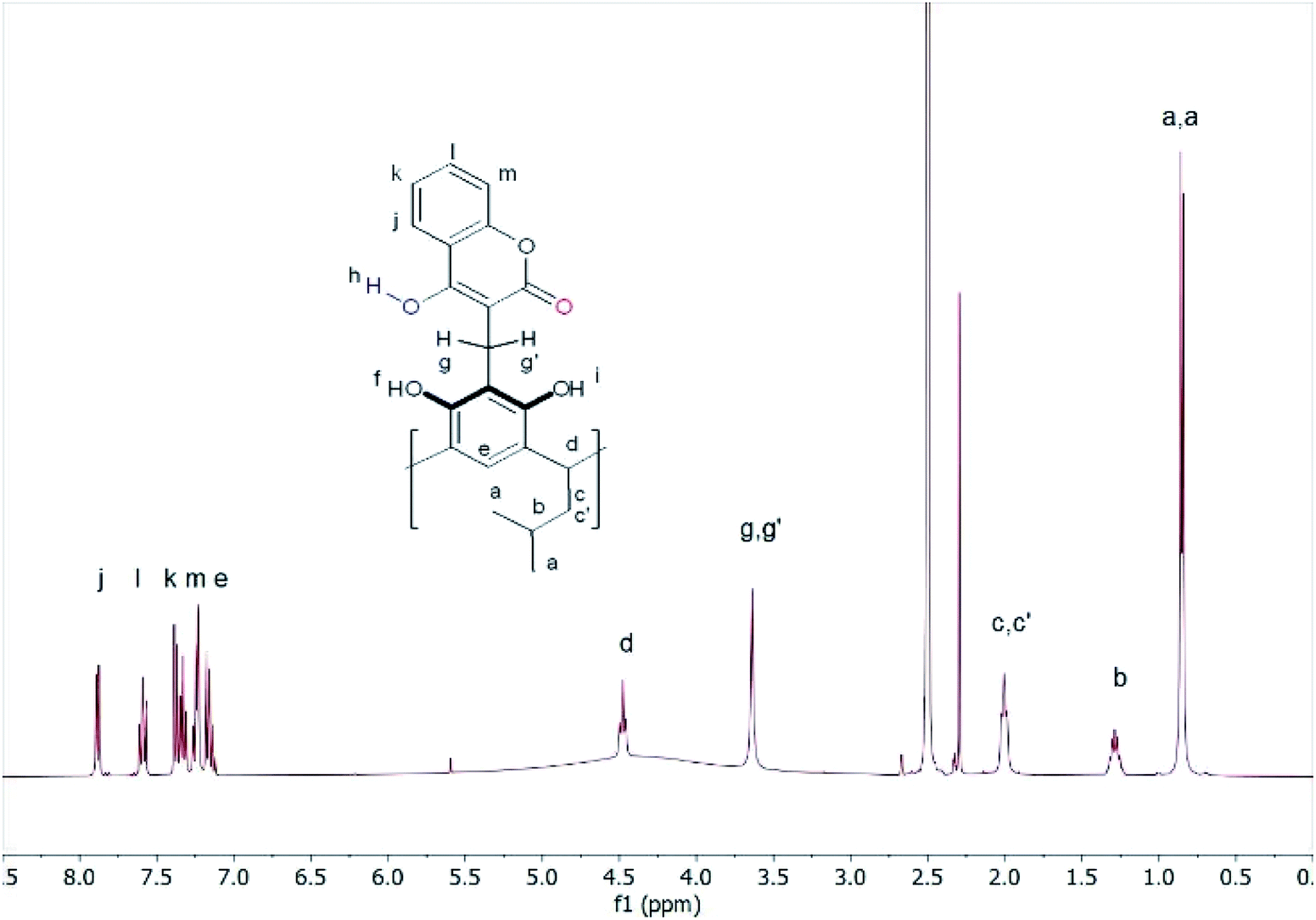 | ||
| Fig. 2 1H NMR spectrum of compound (1) in DMSO-d6 at 298 K. | ||
These changes demonstrated the high mobility of the attached coumarin fragments in DMSO and suggested that in this solvent, due to its high polarity and proton-acceptor nature, some of the intramolecular hydrogen bonds that maintain the rigidity of the molecule were broken. This allowed the high mobility of the coumarin units in the compound (1) in DMSO due to the formation of intermolecular hydrogen bonds and rapid proton exchange in DMSO. The addition of 2 drops of DMSO to the chloroform solution of compound (1) caused large changes in the 1H-NMR spectrum (Fig. S10†). The proton signals were significantly blurred, which indicated that the formation of the equilibrium system depending on the polarity and proton-acceptor.
NMR method is a very simple method of assessing the presence of an intramolecular hydrogen bond in a molecule.35 The difference in the chemical shifts of an OH or NH group in two solvents, Δδ = δ(DMSO) − δ(CDCl3), can be converted into the hydrogen bond acidity, A, of the group using the equation A = 0.0065 + 0.133Δδ. The NMR A value can be used as a quantitative assessment of intramolecular hydrogen bonding. Assuming the average value of 1H NMR chemical shift of hydroxyl groups OH in DMSO in the compound (1) as equal to δ = 4.55 ppm, descriptors A for OH group are respectively: A-OH(i) = 0.84, A-OH(h) = 0.83 and A-OH(f) = 0.74. These values indicate that in DMSO the molecule (1) does not form intramolecular hydrogen bonds affecting its stiffness.
Based on the 1D- and 2D-NMR spectra run in chloroform and DMSO, we postulated the formation of “out” conformer of compound (1), the dynamics of which were controlled by the solvent's proton-donor–acceptor properties and temperature. In chloroform, a solvent devoid of proton-donor–acceptor properties, an intramolecular hydrogen bond stiffened “out”-conformer of coumarin derivative of resorcin[4]arene was formed at room temperature (Scheme 3).
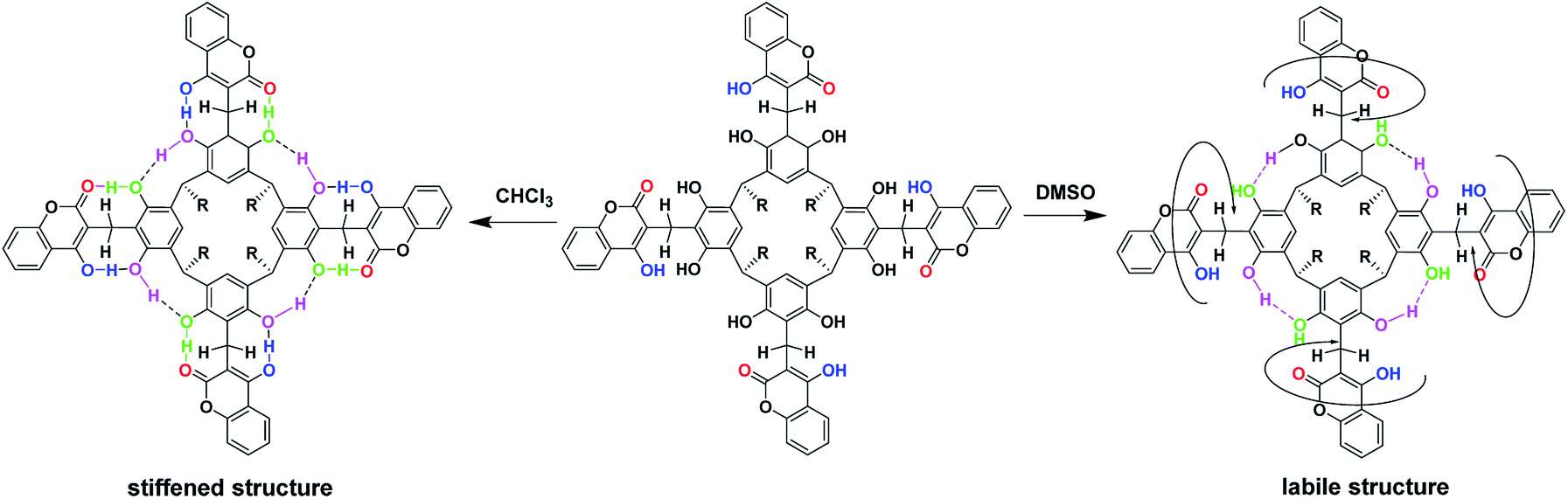 | ||
| Scheme 3 Conformational dynamics of compound (1) depending on the type of solvent. | ||
This stiffness, based on the intramolecular hydrogen bond system, determined the possibility of the formation of cyclochiral enantiomers of compound (1), as shown in Scheme 4. In turn, in DMSO, a solvent with strong proton-acceptor properties and high polarity, a labile conformer of compound (1) was formed, as a result of the breaking of some of the intramolecular hydrogen bonds in favor of the formation of intermolecular hydrogen bonds of compound (1) with DMSO.
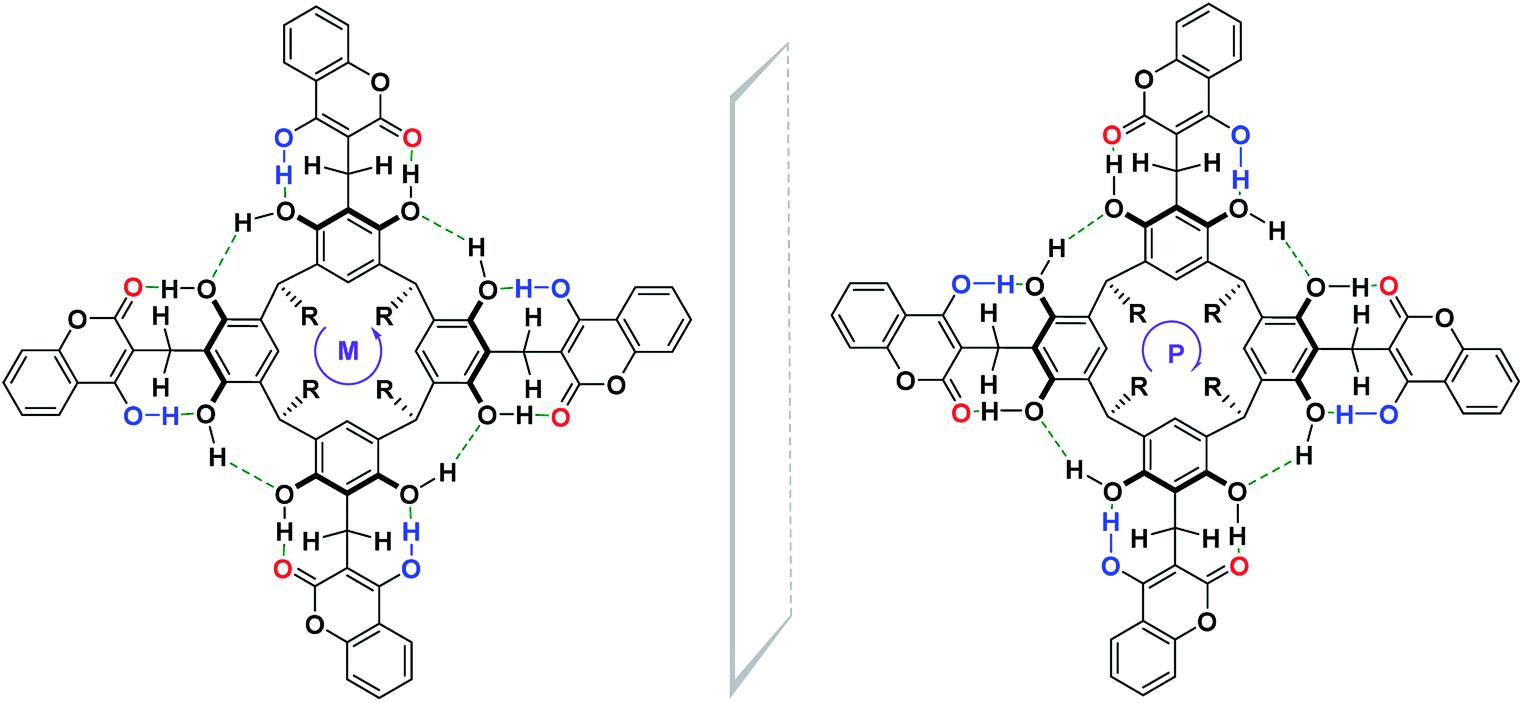 | ||
| Scheme 4 Structure of (M, P) enantiomers of cyclochiral coumarin derivative of resorcin[4]arene. | ||
Quantum-mechanical calculations of the kite conformer energy of compound (1) using the DFT/B3LYP/6-311(d,p) method for the gas phase, chloroform, and DMSO were respectively: gas-phase – 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 753.8259 (a.u.)/CHCl3 – 4753.8753 (a.u.)/DMSO – 4753.8995 (a.u.). The final calculated structure of “out” conformer of compound (1), regardless of the phase and type of solvent. In this conformation, the two opposite rings of the resorcin[4]arene move closer together, while the other two moves away. The calculated difference in the energy of compound (1) formation in chloroform and DMSO was 63.54 kJ mol−1 in favor of greater stability in DMSO. The calculated structures, taking into account hydrogen bonds and van der Waals clouds, are presented in Fig. 3. The structure of the molecule is shown from above and from the side (latter view for DMSO only) in a projection showing the cavity.
753.8259 (a.u.)/CHCl3 – 4753.8753 (a.u.)/DMSO – 4753.8995 (a.u.). The final calculated structure of “out” conformer of compound (1), regardless of the phase and type of solvent. In this conformation, the two opposite rings of the resorcin[4]arene move closer together, while the other two moves away. The calculated difference in the energy of compound (1) formation in chloroform and DMSO was 63.54 kJ mol−1 in favor of greater stability in DMSO. The calculated structures, taking into account hydrogen bonds and van der Waals clouds, are presented in Fig. 3. The structure of the molecule is shown from above and from the side (latter view for DMSO only) in a projection showing the cavity.
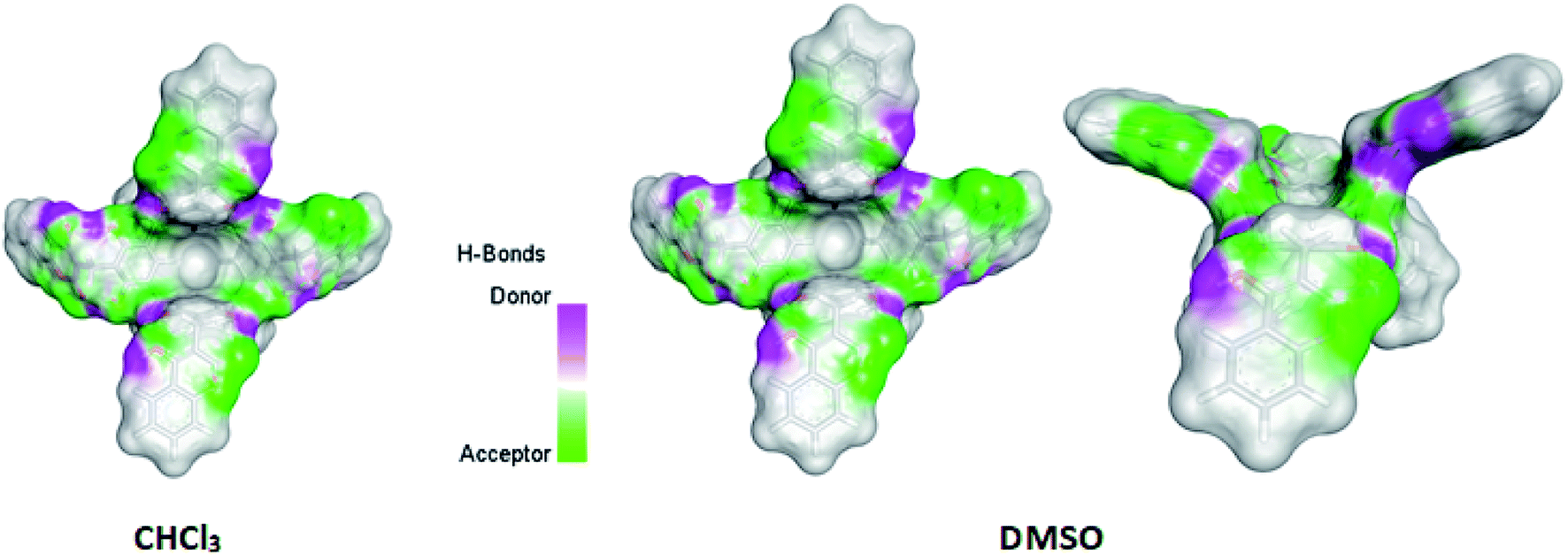 | ||
| Fig. 3 Calculated by DFT/B3LYP/6-311(d,p) structures of compound (1) in chloroform and DMSO. Side view of the molecule of compound (1) is also shown for DMSO. | ||
Because of their strong fluorescence, coumarin and its derivatives are often used as fluorescent markers in biological studies36 and as fluorescent chemosensor in cavitands.37 The inclusion of the coumarin units to the resorcin[4]arene platform should enhance the fluorescence of compound (1) relative to pure 4-hydroxycoumarin. Measurements of absorption and fluorescence spectra in chloroform and DMSO, as well as comparatively to 4-hydroxycoumarin, showed significant differences in fluorescence for compound (1), depending on the type of solvent.
The UV-Vis spectrum of compound (1) differs little from the spectrum of 4-hydroxycoumarin in chloroform, maintaining a similar structure. The UV-Vis spectrum of compound (1) is more widened, less structured and slightly bathochromic in relation to 4-hydroxycoumarin in DMSO (Fig. S17†). The fluorescence spectra of compound (1) in the tested solvents are clearly different. As mentioned, increased intensity of fluorescence would be expected for compound (1). However, fluorescence spectrum measurements show the opposite trend. Fluorescence of compound (1) in chloroform is very strongly quenched (Fig. 4). The fluorescence spectrum is heavily noisy, despite the fact that concentrations of compound (1) in chloroform and DMSO were equal and amounted to 2.64 × 10−6 M. Most likely, the reason for this fact is the intramolecular, non-radiative dissipation of absorbed energy through the formation of intramolecular hydrogen bonds in the compound (1).
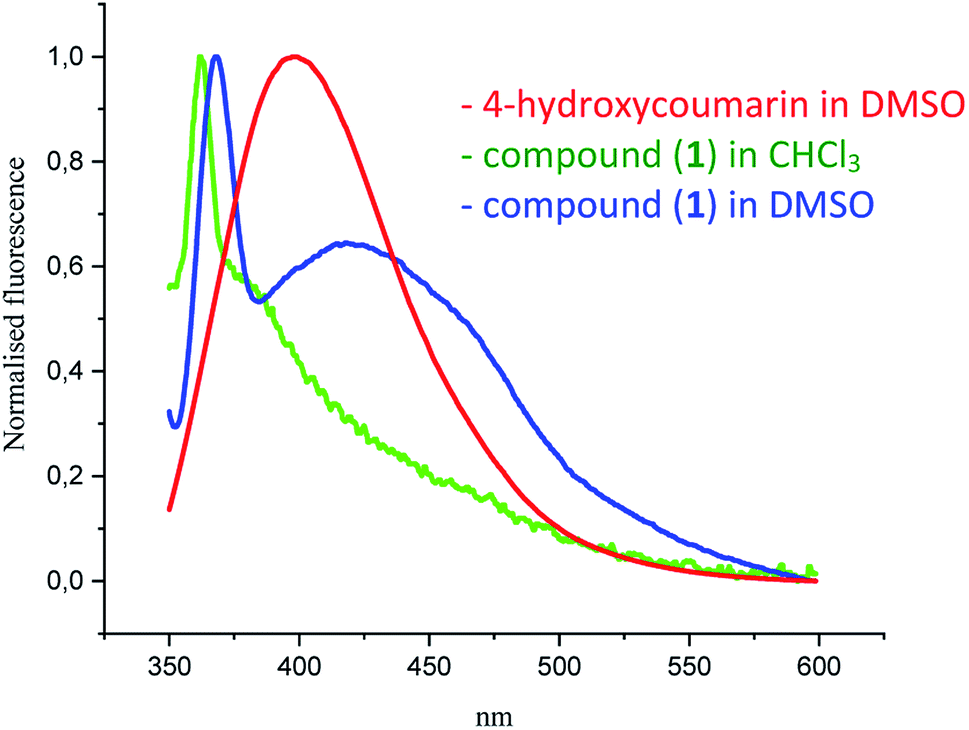 | ||
| Fig. 4 Normalized fluorescence spectra of compound (1) in DMSO and chloroform (2.64 × 10−6 M, λexc = 320 nm) and 4-hydroxycoumarin (7.03 × 10−7 M, λexc = 320 nm) in DMSO. | ||
The intensity of fluorescence of compound (1) in DMSO was clearly higher than in chloroform and shifted by 25 nm in relation to 4-hydroxycoumarin. This fact can be explained by the lack of hydrogen bonds between coumarin units and the resorcin[4]arene skeleton in DMSO and its greater polarity. This results in fewer intramolecular channels for the non-radiative dissipation of absorbed energy, which causes an increasing intensity of fluorescence of compound (1) in DMSO.
Depending on the directivity of the hydrogen bonds formed, compound (1) may appear in the form of a racemic mixture, with an equimolar amount of both enantiomers, as shown in Scheme 4.
Due to the intramolecular hydrogen bonding system giving rigidity to compound (1) in chloroform, the influence of a chiral auxiliary on the directivity of the hydrogen bonding system being formed and on the determination of its stereochemistry was investigated. For this purpose, chiral compounds of various proton-donor–acceptor natures, i.e. D-(+)-camphorsulfonic acid (proton donor) and S-(−)-phenylethylamine (proton acceptor) were used. The measurements were carried out in an NMR cuvette, adding 4 equivalents of the appropriate chiral auxiliaries in CDCl3 to a carefully weighed amount of compound (1). As a result of the addition of the chiral auxiliary, a precipitate formed from the chloroform solution, which significantly reduced the quality of the obtained spectra. Fig. S11 and S12† show the 1H NMR spectrum of compound (1) with D-(+)-camphorsulfonic acid in CDCl3. Under chiral conditions, the diastereotopic methylene protons (g,g′) signal present in the form of doublets should increase twice. We observed this for the example of the methylene proton (g′), whose signal becomes a double doublet in the presence of the chiral factor D-(+)-camphorsulfonic acid.
No changes in the 1H NMR spectrum of compound (1) in CDCl3 were observed directly after the addition of S-(−)-phenylethylamine as a proton acceptor. The spectrum measured in the presence of the amine again after 24, 48 and 72 hours indicated that the rigid conformation changed to a labile conformation (Fig. S13†). The reason was the formation of intermolecular hydrogen bonds of compound (1) with amine molecules at the expense of intramolecular hydrogen bonds maintaining the rigidity of the molecule.
Experimental section
NMR spectra were measured using 400 MHz spectrometers. Mass spectra were recorded with the electrospray (ESI) technique and a TOF analyzer. The reaction was carried out using a Monowave 50 reactor (Anton Paar). UV-Vis spectra were measured using a Shimadzu UV-1900. Fluorescence spectra were measured using a Hitachi F-7100 fluorescence spectrophotometer. Reagents and solvents were obtained from Sigma-Aldrich, Fluka, and Merck and were used without purification. Methoxy derivative of resorcin[4]arene was synthesized according to literature.38Compound (1)
The methoxy derivative of resorcin[4]arene (0.112 mmol, 100 mg), 4-hydroxy-coumarin (0.448 mmol, 72.6 mg, 4 equivalents), and toluene (acetic acid, dioxane) (5 ml) were stirred at 160 °C for 15 min in a Monowave 50 reactor. After cooling to room temperature, the precipitate was filtered, washed slowly with toluene, chloroform, acetone, and methanol, and then dried (135 mg, 85% yield in toluene, 77% yield in acetic acid, 81% yield in dioxane). White crystals, mp > 300 °C (decomposition). 1H NMR (400 MHz, DMSO-d6) δ = 7.88 (dd, J1 = 8.1 Hz, J2 = 1.5 Hz, 4H, j), 7.60–7.56 (m, 4H, l), 7.40–7.30 (m, 8H, k,m), 7.24 (s, 4H, e), 4.48 (t, J = 7.7 Hz, 4H, d), 3.65 (s, 8H, g,g′), 2.01 (t, J = 7.0 Hz, 8H, c,c′), 1.36–1.23 (m, 4H, b), 0.86 (d, J = 6.60 Hz, 24H, a) ppm. 13C NMR (100 MHz, DMSO-d6) δ = 166.13 165.75, 152.01, 149.39, 131.53, 124.56, 123.82, 123.61, 122.50, 118.08, 116.13, 114.16, 102.02, 79.15, 42.62, 25.74, 22.70, 19.92 ppm. HRMS ESI m/z for C84H80O20 [M–H]− calcd 1407.5165, found 1407.5154.Conclusion
We present an efficient and fast synthesis of new coumarin derivatives of resorcin[4]arene. The 1H NMR spectrum of this compound varies greatly depending on the solvent. Based on 1D- and 2D-NMR spectra and quantum-mechanical calculations, an “out” conformation was proposed for this derivative. The calculated values of descriptor A for hydroxyl groups OH indicate the formation of very weak intramolecular hydrogen bonds. However, in chloroform, a solvent devoid of proton-donor–acceptor properties, the formation of 12 weak intramolecular hydrogen bonds stiffens molecule 1 so that it becomes cyclochiral. The cyclochirality of this molecule has been attempted to be demonstrated using chiral auxiliaries with different proton-donor–acceptor properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr R. Dobosz for quantum-mechanical calculations and indebted to CI TASK Gdańsk, for the supply of computer time and providing programs.References
- D. J. Cram and J. M. Cram, Container Molecules and Their Guests, Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, The Royal Society of Chemistry, Cambridge, 1994 Search PubMed.
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Resorcinarenes, Tetrahedron, 1996, 52, 2663 CrossRef CAS.
- W. Iwanek and A. Wzorek, Introduction to the chirality of resorcinarenes, Mini-Rev. Org. Chem., 2009, 6, 398 CrossRef CAS.
- M. Wierzbicki, H. Jędrzejewska and A. Szumna, Chiral Calixarenes, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, Waltham, MA, 2014 Search PubMed.
- A. Szumna, Inherently chiral concave molecules-from synthesis to applications, Chem. Soc. Rev., 2010, 39, 4274 RSC.
- A. Wzorek, J. Mattay and W. Iwanek, Diastereoselective synthesis of boron-derivatives of resorcinarenes from amino alcohols and triethylborane, Tetrahedron: Asymmetry, 2007, 7, 815 CrossRef.
- C. Agena, Ch. Wolff and J. Mattay, First synthesis, isolation and complete characterization of both enantiomers of inherently chiral resorc[4]arenes by monofunctionalization, Eur. J. Org. Chem., 2001, 2977 CrossRef CAS.
- J. L. McIldowie, M. Mocerino, B. W. Skelton and A. H. White, Facile Lewis acid catalyzed synthesis of C4 symmetric resorcinarenes, Org. Lett., 2000, 2, 3869 CrossRef PubMed.
- W. Iwanek, R. Fröhlich, P. Schwab and V. Schurig, The synthesis and crystallographic structures of novel bora-oxazino-oxazolidine derivatives of resorcarene, Chem. Commun., 2002, 2516 RSC.
- S. Wiegmann and J. Mattay, Inherently chiral resorcin[4]arenes: a new concept for improving the functionality, Org. Lett., 2011, 13, 3226 CrossRef CAS PubMed.
- S. Wiegmann, B. Neumann, H.-G. Stammler and J. Mattay, Inherently chiral cyano-substituted resorcin[4]arene: a promising starting point for further functionalization, Eur. J. Org. Chem., 2012, 3955 CrossRef CAS.
- Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M.-X. Wang, Springer International Publishing, Switzerland, 2016 Search PubMed.
- C. L. D. Gibb and B. C. Gibb, Well-defined, organic nanoenvironments in water: the hydrophobic effect drives a capsular assembly, J. Am. Chem. Soc., 2004, 126, 11408 CrossRef CAS PubMed.
- D. A. Leigh, P. Linnane, R. G. Pritchard and G. Jackson, Unusual host–guest π-arene ⋯ H bonding in a ‘hooded’ cavitand: the first solid-state structure of a calix[4]resorcinarene with underivatised hydroxy groups, J. Chem. Soc., Chem. Commun., 1994, 4, 389 RSC.
- E. S. Barrett, J. L. Irwin, A. J. Edwards and M. S. Sherburn, Superbowl container molecules, J. Am. Chem. Soc., 2004, 126, 16747 CrossRef CAS PubMed.
- I. Pochorovski, M. O. Ebert, J.-P. Gisselbrecht, C. Boudon, W. B. Schweizer and F. Diederich, Redox-switchable resorcin[4]arene cavitands: molecular grippers, J. Am. Chem. Soc., 2012, 134, 14702 CrossRef CAS PubMed.
- J. Rebek Jr, Hydrogen-bonded capsules: molecular behavior in small spaces, World Scientific Publishing, Singapore, 2016 Search PubMed.
- K. Kobayashi and M. Yamanaka, Self-assembled capsules based on tetrafunctionalized calix[4] resorcinarene cavitands, Chem. Soc. Rev., 2015, 44, 449 RSC.
- K. Wang, J. H. Jordan and B. C. Gibb, Molecular protection of fatty acid methyl esters within a supramolecular capsule, Chem. Commun., 2019, 55, 11695 RSC.
- P. Jacopozzi and E. Dalcanale, Metal-Induced Self-Assembly of Cavitand-Based Cage Molecules, Angew. Chem., Int. Ed. Engl., 1997, 36, 613 CrossRef CAS.
- T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, Ch. Näther and J. Mattay, Self-Assembly of 2,8,14,20-Tetraisobutyl-5,11,17,23-tetrahydroxyresorc[4]arene, Eur. J. Org. Chem., 1999, 9, 2257 CrossRef.
- L. R. MacGillivray and J. L. Atwood, A chiral spherical molecular assembly held together by 60 hydrogen bonds, Nature, 1997, 389, 469 CrossRef CAS.
- P. Jin, S. J. Dalgarno and J. L. Atwood, Mixed metal-organic nanocapsules, Coord. Chem. Rev., 2010, 254, 1760 CrossRef CAS.
- L. Avram and J. Cohen, Diffusion NMR of molecular cages and capsules, Chem. Soc. Rev., 2015, 44, 586 RSC.
- Q. Zhang, L. Catti and K. Tiefenbacher, Catalysis inside the Hexameric Resorcinarene Capsule, Acc. Chem. Res., 2018, 51, 2107 CrossRef CAS PubMed.
- C. Gaeta, C. Talotta, M. De Rosa, P. La Manna, A. Soriente and P. Neri, The Hexameric Resorcinarene Capsule at Work: Supramolecular Catalysis in Confined Spaces, Chem.–Eur. J., 2019, 25, 4899 CrossRef CAS PubMed.
- A. Pappalardo, R. Puglisi and G. T. Sfrazzetto, Catalysis inside Supramolecular Capsules: Recent Developments, Catalysts, 2019, 9, 630 CrossRef CAS.
- S. Gambaro, P. La Manna, M. De Rosa, A. Soriente, C. Talotta, C. Gaeta and P. Neri, The Hexameric resorcinarene capsule as a Brønsted acid catalyst for the synthesis of bis(heteroaryl)methanes in a nanoconfined space, Front. Chem., 2019, 7, 687 CrossRef PubMed.
- W. Iwanek, K. Stefańska, A. Szumna and M. Wierzbicki, Inherently chiral heterocyclic resorcinarenes using a Diels–Alder reaction, RSC Adv., 2016, 6, 13027 RSC.
- K. Stefańska, H. Jędrzejewska, M. Wierzbicki, A. Szumna and W. Iwanek, The inverse demand oxa-Diels-Alder reaction of resorcinarenes: An experimental and theoretical analysis of regioselectivity and diastereoselectivity, J. Org. Chem., 2016, 81, 6018 CrossRef PubMed.
- K. Stefańska, A. Szafraniec, M. Szymański, M. Wierzbicki, A. Szumna and W. Iwanek, Chiral chromane[4]arenes synthesised by cycloaddition reactions of o-quinomethine resorcin[4]arenes, New J. Chem., 2019, 43, 2687 RSC.
- W. Iwanek, K. Stefańska, A. Szumna and M. Wierzbicki, Synthesis of resorcinarene phosphonium salts and the effect of counteranion on their structure, Tetrahedron, 2016, 72, 142 CrossRef CAS.
- W. Iwanek, K. Stefańska, A. Szumna and M. Wierzbicki, Solvent-free synthesis and structure of 2-naphthol derivatives of resorcinarenes, Tetrahedron, 2015, 71, 2222 CrossRef CAS.
- Ch. Schiel, G. A. Hembury, V. V. Borovkov, M. Klaes, C. Agena, T. Wada, S. Grimme, Y. Inoue and J. Mattay, New Insights into the Geometry of Resorc[4]arenes: Solvent-Mediated Supramolecular Conformational and Chiroptical Control, J. Org. Chem., 2006, 71, 976 CrossRef CAS PubMed; D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangili, J. S. Kim and W. Lin, Correction to coumarin-based small-molecule fluorescent chemosensors, Chem. Rev., 2019, 119, 10403 CrossRef PubMed.
- M. H. Abraham, R. J. Abraham, W. E. Acree, E. Aliev, A. J. Leo and W. L Whaley, An NMR method for the quantitative assessment of intramolecular hydrogen bonding; application to physicochemical, environmental, and biochemical properties, J. Org. Chem., 2014, 79, 11075 CrossRef CAS PubMed.
- C. S. Francisco, T. C. Valim, A. C. Neto and V. Lacerda, Coumarin-based hybrids as fluorescent probes for highly selective chemosensing and biological target imaging, Aldrichimica Acta, 2019, 52, 51 Search PubMed.
- Y. J. Jang, B. S. Moon, M. S. Park, B. G. Kang, J. Y. Kwon, J. Sung, J. Hong, Y. L. Yoon, K. D. Leeb and J. Yoona, New cavitand derivatives bearing four coumarin groups as fluorescent chemosensors for Cu2+ and recognition of dicarboxylates utilizing Cu2+ complex, Tetrahedron Lett., 2006, 47, 2707 CrossRef CAS.
- M. Urbaniak and W. Iwanek, Synthesis of alkoxymethyl derivatives of resorcinarene via the Mannich reaction catalyzed with iminodiacetic acid, Tetrahedron, 2006, 62, 1508 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00368a |
| This journal is © The Royal Society of Chemistry 2020 |