
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructural catalyst: metallophthalocyanine and carbon nano-onion with enhanced visible-light photocatalytic activity towards organic pollutants†
Elzbieta
Regulska‡
 a,
Piotr
Olejnik‡
b,
Halyna
Zubyk
a,
Justyna
Czyrko-Horczak
a,
Manuel N.
Chaur
cd,
Monika
Tomczykowa
b,
Olena
Butsyk
e,
Krzysztof
Brzezinski
a,
Luis
Echegoyen
*f and
Marta E.
Plonska-Brzezinska
*b
a,
Piotr
Olejnik‡
b,
Halyna
Zubyk
a,
Justyna
Czyrko-Horczak
a,
Manuel N.
Chaur
cd,
Monika
Tomczykowa
b,
Olena
Butsyk
e,
Krzysztof
Brzezinski
a,
Luis
Echegoyen
*f and
Marta E.
Plonska-Brzezinska
*b
aFaculty of Chemistry, University of Bialystok, Ciolkowskiego 1K, 15-245 Bialystok, Poland
bFaculty of Pharmacy with the Division of Laboratory Medicine, Medical University of Bialystok, Mickiewicza 2A, 15-222 Bialystok, Poland. E-mail: marta.plonska-brzezinska@umb.edu.pl; Tel: +48 85 738 5687
cDepartamento de Química, Facultad de Ciencias Naturales y Exactas, Universidad del Valle, Cali, Colombia
dCentro de Excelencia en Nuevos Materiales (CENM), Universidad del Valle, Cali, Colombia
eInstitute of Physics of the Czech Academy of Sciences, Na Slovance 2, Prague 8, 182 21, Czech Republic
fDepartment of Chemistry, University of Texas at El Paso, 500 W. University Ave., El Paso, TX 79968, USA. E-mail: echegoyen@utep.edu
First published on 17th March 2020
Abstract
Metallophthalocyanine (MPc) and carbon nano-onion (CNO) derivatives were synthesized and characterized by using ultraviolet-visible spectroscopy, infrared and Raman spectroscopy, scanning electron microscopy with energy-dispersive X-ray spectroscopy and X-ray powder diffraction. The unmodified CNOs and MPc–CNO derivatives were used as photocatalysts for rhodamine B (RhB) degradation under visible-light irradiation. The photocatalytic studies revealed that the MPc–CNO nanostructural materials simultaneously exhibited a high absorption capacity and an excellent visible-light-driven photocatalytic activity towards RhB. These nanostructures possess great potential for use as active photocatalysts for organic pollutant degradation.
Introduction
Degradation of water pollutants through semiconductor mediated photocatalysis has attracted attention because it offers a promising pathway for environmental pollution problems.1–3 Various systems have been studied and employed as photocatalysts in the past few decades.4,5 Among these studied photocatalysts, carbon materials,6–8, organic derivatives or composites,9–13 metal oxides,14 inorganic nanoparticles and nanocrystals15,16 and hybrid materials combining two or more components with different chemical natures have been used.3,14,17,18 Often, phthalocyanines (Pcs), derivatives of the aromatic tetraazaporphyrins, also appear among these systems.19,20 Pcs are synthetic analogues of porphyrins, in which the nitrogen atoms in the aromatic polyene ring connect the four pyrrole groups instead of carbons.21 Pcs are planar aromatic macrocycles consisting of four isoindole units with delocalized electrons over the carbon and nitrogen atoms. Because of their structures, Pcs exhibit interesting electrical properties,22,23 strong absorption in the visible region,24–26 a good thermal and chemical stability,27 and can be applied to versatile organic reactions.28–31 The structure of Pcs enables them to be combined with macromolecular structures and/or metals, which broadens their applicability.32,33 The combination of Pcs with the metal atom (metallophthalocyanine, MPc) in the complex optimizes the Pc properties, mainly by increasing their photochemical,34,35 photocatalytic36 and electrochemical activity.32,37,38MPc complexes exhibit an extraordinary molecular stability and negligible toxicity.39 To date, MPcs and Pcs have been successfully incorporated as active components in semiconductor and electrochromic devices,40,41 organic field effect transistors,41,42 light-emitting devices,43 organic memristors,44 solar cells,45,46 light-driven micromotors,47 and sensors,48,49 as well as in photocatalytic applications,29,50–53 applications requiring antimicrobial and antioxidant activities,54 imaging and therapeutic applications,55etc. Within this context, Pc applications are restricted by the difficulties connected with the great tendency of the planar macrocycles to aggregate, which impairs the physico-chemical activity. The successful incorporation of Pcs or MPcs into larger macromolecular systems requires their self-organization and dispersibility to be controlled. These connections provide unique properties, such as increased surface areas, long lifetimes, facile recovery and easy separation of the catalyst. Among these systems, polymers56 and inorganic57,58 and organic nanoparticles59 have been studied. Synthetic efforts have also been devoted to implementing carbon nanostructures (CNs) into these macromolecular systems.39,60,61 In the case of these nanostructures, covalent and noncovalent modifications were applied.62–64 Although covalent modification first influences the electronic properties of CNs, noncovalent modification is relatively weak and can lead to the easy removal of the immobilized molecule from the surface, and hence, this type of modification is not applicable in many areas.
Fullerenes and carbon nanotubes (CNTs) are a type of CN that exhibit interesting photophysical properties,43,63,65–67 such as an excellent electron acceptor ability, a small reorganization energy and an ability to promote ultrafast charge separation together with very slow charge recombination.63 The first report regarding a covalently linking a Ni(II)Pc moiety to C60 through a Diels–Alder reaction was published in 1995.68 To date, a large variety of covalently and noncovalently modified fullerene and CNT acceptor units have been synthesized, and their photophysical properties were studied both in solution and in the solid phase.61,69,70
In this study, we used perfectly spherical CNs, called carbon nano-onions (CNOs). CNOs may have different sizes and shapes and are strongly connected to each other with their formation procedure.71–74 These structures, which are formed from nanodiamond particles (NDs), are the most common CN in this group, mainly because their high reactivity is connected with their perfectly spherical structures.72,75,76 CNOs derived from NDs are recognized as ‘zero-dimensional’ nanostructures due to their perfect spherical shape (6–8 graphitic shells and a diameter of 5–6 nm).77 Due to these properties, many covalent reactions can occur on CNOs to attach multiple groups to their surfaces. Among these covalent reactions are amidation,78 oxidation,79,80 1,3-dipolar cycloaddition,81,82 [2 + 1] cycloaddition,83 nucleophilic substitution,84 diazonium addition,85 reduction,84 fluorination86 and radical addition.87
Until now, attempts have been already made to use these CNs in photoactive devices. However, since then, no reports using CNOs as photocatalysts for the degradation of organic pollutants have been published, to the best of our knowledge. For example, CNOs were used as a hole collection layer in zinc-phthalocyanine-based organic photovoltaic devices.88,89 The photocurrent was increased by a factor of 5.5 compared to that of solar devices without CNOs.88 A device based on a perovskite crystalline film with oxidized CNOs incorporated into the hole transporting layer along with poly(3,4-ethylenedioxythiophene):polystyrene sulphonate (PEDOT:PSS) was prepared, resulting in a significant enhancement in the power conversion efficiency (PCE) from 11.07 to 15.26%.89 The larger CNO structures (30 nm in diameter) were also applied in dye sensitized solar cells and used as a counter electrode, but the PCE was comparable to that of a commonly used platinum electrode.90
We applied CNO as a platform for the assembly of MPcs. In this study, we covalently modified the outer CNO shells to form a chemically stable series of MPc-based CNOs. We tested these MPc-based CNOs as visible-light photocatalysts for the degradation of organic pollutants. For this study, a model system of rhodamine B (RhB) was used.
Experimental
Materials and methods
CNOs were obtained according to the method proposed by Kuznetsov,72 in which diamond nanoparticles (NDs, Carbodeon µDiamond Molto) with a diameter of 4.2 ± 0.5 nm and a ND content of ≥97 wt% were annealed. All the chemicals and solvents were commercially available and used without further purification: nanodiamond powder (µDiamond®Molto, Carbodeon); 1,2-dicyanobenzene (∼98%, Sigma-Aldrich); ammonia solution (25% pure p.a., Sigma-Aldrich); maleimide (∼99%, Sigma-Aldrich); tetrahydrofuran (pure p.a., Avantor); 2-propanol (pure p.a., Avantor); acetone (99.5% pure p.a., Avantor); 2-mercapto-4-methyl-5-thiazoleacetic acid (∼98%, Sigma-Aldrich); zinc chloride (≥99%, Sigma-Aldrich); nickel(II) chloride (∼98%, Sigma-Aldrich); copper(II) chloride (∼97%, Sigma-Aldrich); hydrochloric acid (35–38% pure p.a., Avantor); sulphuric acid (95% pure p.a., Avantor); sodium chloride (pure p.a., Avantor); aluminium powder (∼99.99%, Avantor); sodium (Sigma); 1-octanol (≥95%, Sigma-Aldrich); potassium permanganate (Avantor); dimethyl sulphoxide (DMSO, pure p.a., Avantor); and methanol (99.5% pure p.a., Avantor).Synthetic procedures
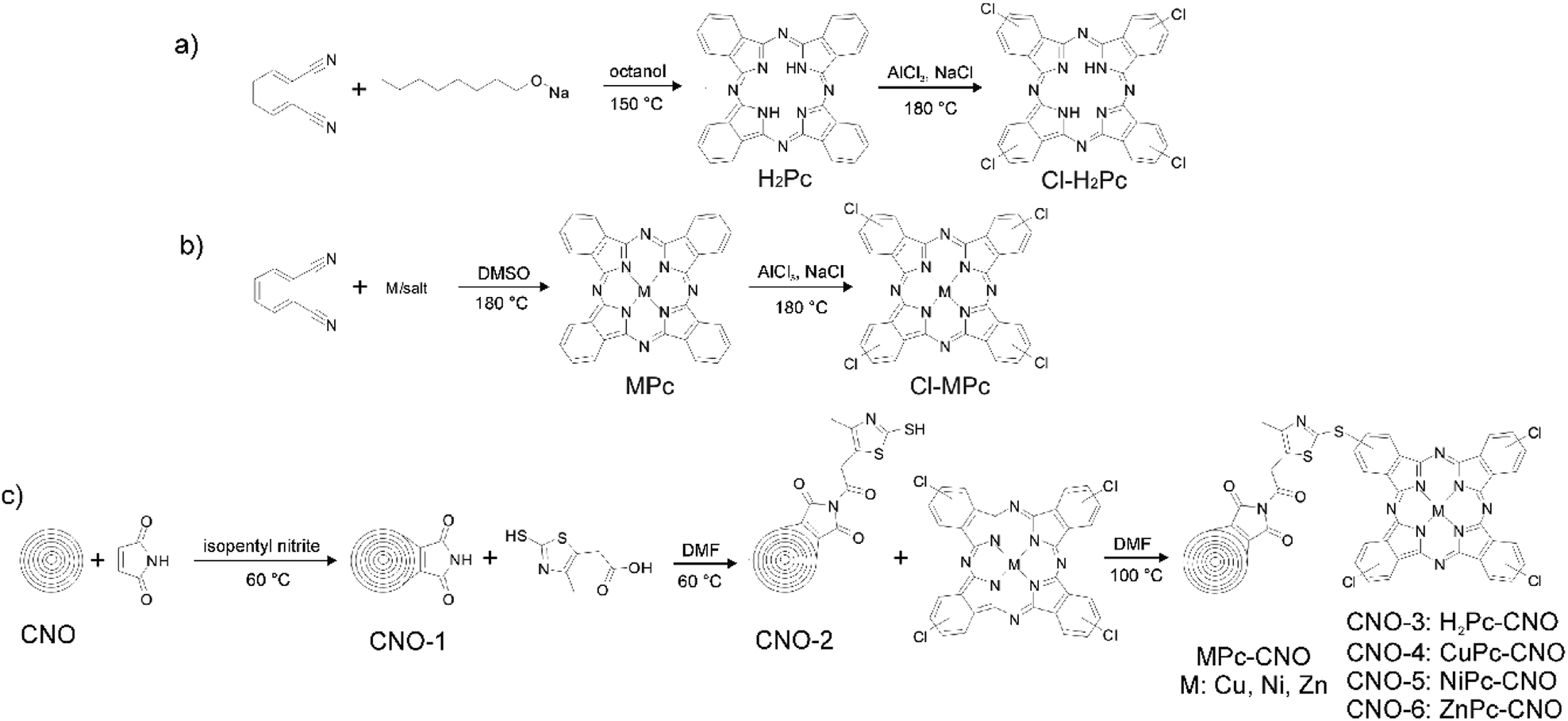 | ||
| Fig. 1 Schematic illustration of the whole synthesis procedure for (a) Pc, (b) MPc, and (c) Pc–CNO and MPc–CNO derivatives. | ||
Methods
The films were imaged by secondary electron scanning electron microscopy (SEM) using an FEI Tecnai S-3000N instrument (Tokyo, Japan). The accelerating voltage of the electron beam was 30 keV. The energy dispersive X-ray spectroscopy (EDX) analyses were performed with an ASPEX system fully integrated and automated with a scanning electron microscope (S-3000N). The 1H NMR spectra were recorded with an Agilent VNMRS system operated at 400 MHz, and CDCl3 was used as the solvent. The powder diffraction data were measured at 293 K using a SuperNova diffractometer (Agilent) with a CCD detector and a Cu-Kα radiation source, and the sample-to-detector distance was 148 mm. Prior to the experiment, all the samples were loaded into capillaries with a diameter of 0.5 mm.The room temperature Raman spectra obtained in the range between 3500 and 100 cm−1 were recorded with Renishaw Raman InVia microscope equipped with a high sensitivity ultra-low noise CCD detector. Radiation from an argon ion laser (514 nm) operated with an incident power of 1.15 mW was used as the excitation source. The Fourier transform infrared spectroscopy (FTIR) spectra were recorded in the range between 4000 and 200 cm−1 with NICOLET IN10 MX infrared microscope (Thermo Scientific) operated at room temperature under an N2 atmosphere. The microscope was operated mainly in the reflectance mode, and the MCT detector was cooled with liquid nitrogen. Additionally, the abovementioned MCT detector was utilized to map the MPc derivatives. The spectra were collected with a resolution of 4 cm−1, 256 scans were averaged to obtain a single spectrum and apodized with a triangular function, and a zero-filling factor of 1 was applied. The spectra were collected for a 100 µm (0.01 mm2 area) square region of the sample. All the spectra were corrected with conventional software to eliminate the variation of the analysed thickness with the wavelength.
The photocatalytic degradation experiments were investigated in a 50 mL glass cell. The reaction mixture consisted of 20 mL of the rhodamine B aqueous solution (12.5 mg L−1) and a photocatalyst (1.5 g L−1). Prior the photodegradation studies, adsorption experiments were carried out. The suspension of the model pollutant and the appropriate photocatalyst was kept in the dark while stirring for 1 hour to attain equilibrium. To monitor the progress of the degradation reaction during irradiation for 2 hours with simulated solar light (SUNTEST CPS+), samples were collected and centrifuged every 15 min, and the supernatant was analysed using UV-vis spectroscopy (HITACHI U-2800A).
Results and discussion
Synthesis of MPcs and MPc–CNOs
The synthesis of H2Pc was based on the most common procedure using phthalonitrile.91,92 The cyclotetramerization reaction of phthalonitrile proceeded in the presence of a reducing agent, sodium alkoxide in an alcoholic solution (Fig. 1a), that performed the function of a template during ring formation. Baumann et al. suggested that a nucleophilic attack by the alkoxide anion on the cyano group of phthalonitrile leads to form 1-alkoxy-3-iminoidoindolenine, and in the subsequent reduction and cyclization reactions, forms H2Pc.92 The analogous reaction was carried out in the presence of metal ions (Fig. 1b), which led to the formation of Pc moieties with a complexed metal ion at the centre of the ring (MPc). Compounds H2Pc and MPcs were further employed in the preparation of Cl–H2Pc and Cl–MPcs, respectively. Halogens were introduced at the periphery of the macrocyclic scaffold (Fig. 1a and b). Halogenated H2Pcs and MPcs were a starting point for the covalent modification of CNOs and the subsequent formation of the Pc–CNO and MPc–CNO derivatives.Simultaneously, the thiazole–maleimide functionalized CNOs (CNO-2) were synthesized using the two-step reaction presented in Fig. 1c, which is described in detail in the Experimental section. First, a Diels–Alder reaction was performed to incorporate maleimide into the CNOs.93 The sp2 carbon atoms of the CNOs serve as a dienophile and react with maleimide as a diene through a Diels–Alder reaction to form [4 + 2] cyclic adducts. The reaction results in maleimide-functionalized CNOs (CNO-1), which were applied as a platform for the further reaction with 2-mercapto-4-methyl-5-thiazoleacetic acid to synthesize the thiazole–maleimide CNO derivatives (CNO-2). Next, CNO-2 was used as a template for the synthesis of four different Pc–CNO derivatives, one metal-free phthalocyanine (H2Pc–CNO, assigned later as CNO-3) and three metal phthalocyanines containing either copper (CuPc–CNO, CNO-4), nickel (NiPc–CNO, CNO-5), or zinc (ZnPc–CNO, CNO-6).
Spectroscopic characteristics of the MPcs and MPc–CNOs
Fourier transform infrared spectroscopy (FTIR) and Raman spectroscopy were utilized as the main experimental techniques for the qualitative characterization of the metal-free and the MPc–CNO derivatives. The FTIR spectra of the CNOs and their MPc derivatives were recorded, as shown in Fig. 2. Additionally, the spectra of H2Pc and the MPcs are presented for comparison (Fig. 2.P(I)).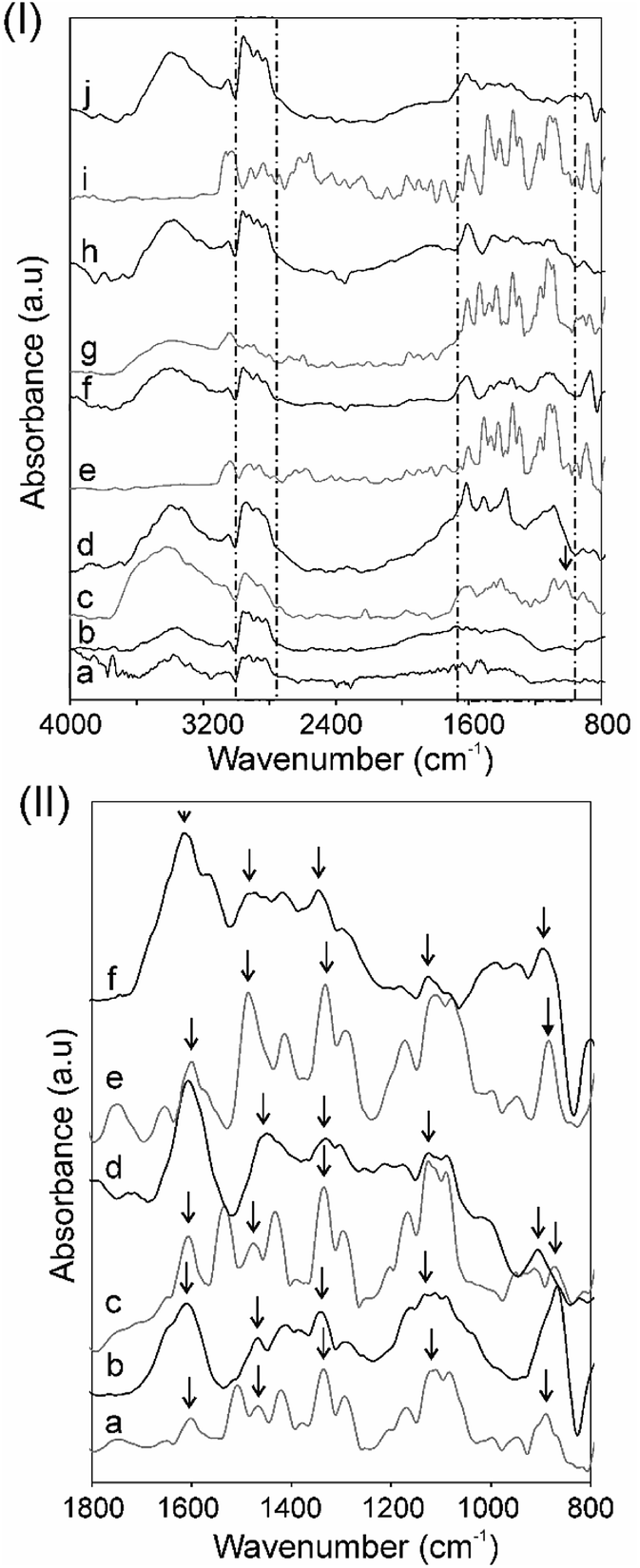 | ||
| Fig. 2 Panel (I) FT-IR spectra of (a) CNO-1, (b) CNO-2, (c) H2Pc, (d) CNO-3, (e) CuPc, (f) CNO-4, (g) NiPc, (h) CNO-5, (i) ZnPc, and (j) CNO-6. Panel (II) FT-IR spectra of (a) CuPc, (b) CNO-4, (c) NiPc, (d) CNO-5, (e) ZnPc, and (f) CNO-6. | ||
The broad, unshaped signals in the higher frequency range (3500–3100 cm−1 with a maximum ca. 3350 cm−1) correspond to free N–H stretching vibrations (Fig. 2.P(I)). The signals in the range between 3100–2900 cm−1 can be attributed to –C–H– asymmetric and symmetric stretching vibrations.21 Pcs have 18 π-conjugated electron aromatic ring systems, which are composed of four pyrrole units linked by four aza (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) groups at the α-carbon atoms of the pyrrole units.
C–) groups at the α-carbon atoms of the pyrrole units.
The infrared absorption band between ca. 1700 and 800 cm−1 is characteristic for metal-free and metal-containing Pcs. The peaks located at ca. 1610 cm−1 are correlated with the conjugated carbons (–CC–) and aza (–NC–) groups. The peaks located in the region 1330–880 cm−1 are correlated with the structure of the Pcs and/or a substituted metal (Fig. 2.P(II)).94 The three strong signals located in this region can be assigned to the pyrrole rings, –C–N– stretching and the –C–H– deformation vibrations in the ring. The band at 1010 cm−1 is attributed to the metal-free H2Pc (Fig. 2c.P(I)) and is used to check the purity of the MPc complexes (the N–H deformation modes).21,95,96 For the MPc derivatives, the peaks centred ca. 1540, 1490 and 1410 cm−1 are attributed to the –CN– and –CC– stretching vibrations of the phthalocyanine ring, and the peaks ca. 1090 and 880 cm−1 are attributed to the vibration of the MPc macrocycles: the isoindole deformation and aza vibration (Fig. 2.P(II)).95,97 In the lower wavenumbers range of the IR spectra from 1100 to 800 cm−1, a few more peaks were observed and can be assigned to planar –C–H– bending and out-of-plane deformations.94
The IR spectra analysis also showed some changes in the CNO structures after covalent attachment of Pc moieties. The spectra of the unsubstituted CNO (not shown) shows three broad signals in the range of 1540–1510, 1270–1240 and 1050–920 cm−1, which may be assigned to the innermost shell of the CNOs.79 There are some similarities between the CNO derivatives, the maleimide (CNO-1) and thiazole–maleimide functionalized CNOs (CNO-2), in this region (Fig. 2b.P(I) and 2c.P(I)). For these and the MPc–CNO derivatives, differences are visible in the higher wavenumber regions. The two broad bands in the range of 3430–3320 and 2960–2810 cm−1 can be attributed to C–H asymmetric and symmetric stretching vibrations and N–H stretching vibrations of the succinimide groups, respectively.98 These signals have higher intensities for the MPc–CNO derivatives in comparison to those of CNO-1 and CNO-2.
After the functionalization of the CNOs with the MPcs, the characteristics signals of phthalocyanines substituted with metals are also visible in the infrared spectra (1700–800 cm−1), which were discussed in detail. The peak located at ca. 1610 cm−1 correlated with the conjugated carbon atoms (–CC–) has a higher intensity for the MPc–CNO derivatives than for the MPcs (Fig. 2.P(II)). This observation successfully confirmed the incorporation of metallophthalocyanines into the multi-fullerene conjugated cages.
Fig. 3 shows the measurements performed using an infrared mapping method. The measurement maps show the optical distribution of the specific signals at a defined wavelength, with the signal intensity imaged using an appropriate colour. The red colour indicates the highest intensity signal for the whole spectrum. The images captured by an optical microscope reveal a fragment of the MPcs or MPc–CNO derivatives on the slide with dimensions of 700 × 800 µm. Fig. 3 illustrates the distribution profiles obtained for the specific spectra of the MPc and MPc–CNO derivatives and indicates the presence and uniform distribution of MPcs on the carbon nanoparticle surface. The arrangement profile obtained for the MPc based on the characteristic peaks (at 1410, 1330 or 1100 cm−1) shows the presence of MPcs across the entire experimental area (0.01 mm2 area).
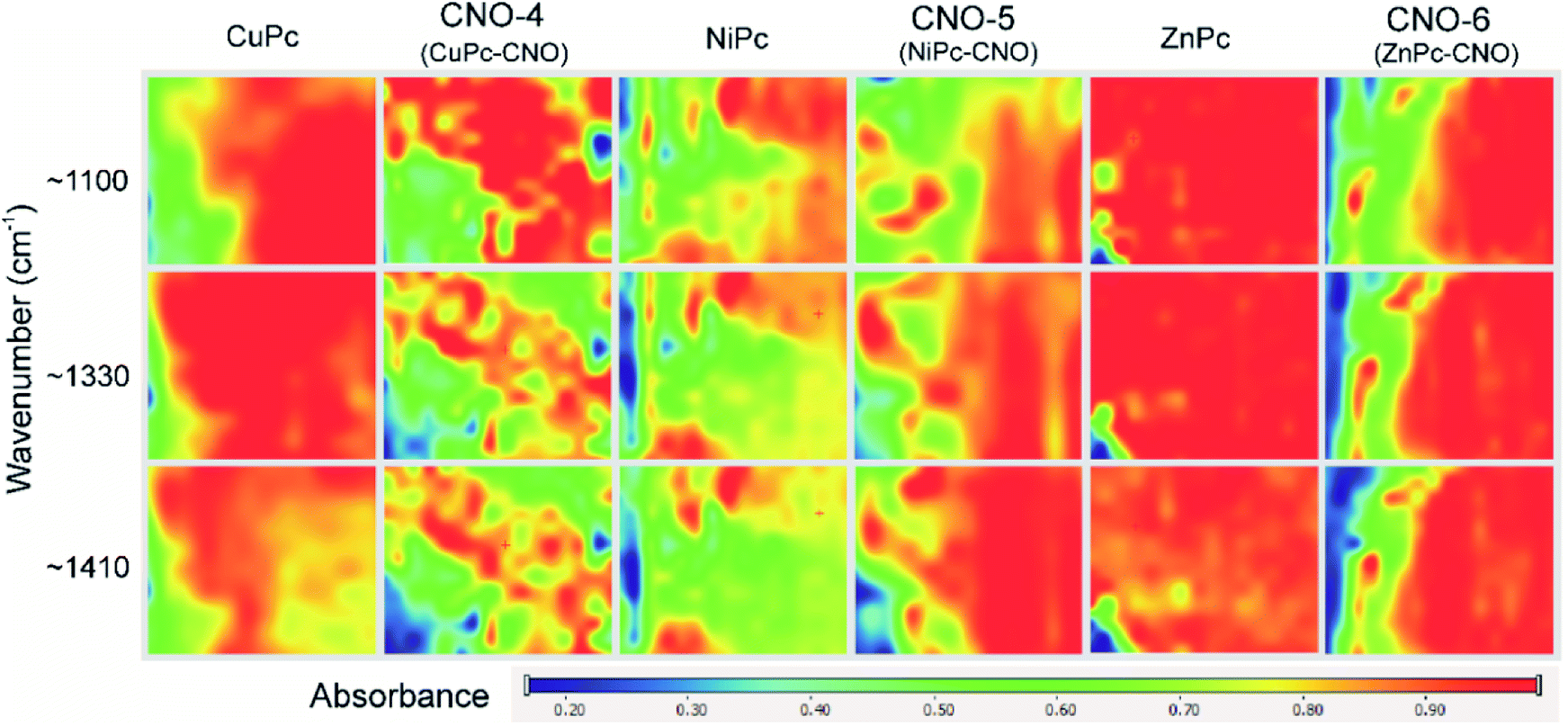 | ||
| Fig. 3 Infrared mapping set obtained for the MPc and MPc–CNO derivatives. | ||
Two characteristic areas of the CNs and Pc moieties are presented in the Raman spectra (Fig. SI1 and SI2†). The Raman spectra of carbonaceous materials are very sensitive to changes in the translational symmetry and the crystallite size.99 Fig. SI1 and SI2† show the Raman spectra of the pristine CNO, CNO-1, CNO-2, H2Pc, MPc, H2Pc–CNO and MPc–CNO samples. The spectra were obtained with an excitation wavelength of 514 nm using a He–Ne laser. The peak positions and intensities are summarized in Table 1.
| Sample | D (cm−1) | G (cm−1) | 2D (cm−1) |
|---|---|---|---|
| CNO | 1339 | 1576 | 2667 |
| CNO-1 | 1335 | 1576 | 2689 |
| CNO-2 | 1345 | 1577 | 2684 |
| CNO-3 | 1343 | 1585 | 2690 |
| CNO-4 | 1338 | 1585 | 2681 |
| CNO-5 | 1339 | 1597 | 2686 |
| CNO-6 | 1333 | 1582 | 2678 |
In general, the spectrum obtained for the pristine CNOs is composed of three characteristic peaks, which correspond to the contribution of the hexagonal mode characteristics of graphene or graphite.100 For most Raman spectroscopic analyses of carbon, two G and D bands are used to characterize CNs. The graphite line (G line at 1580(±5) cm−1) is assigned to the E2g species of the infinite crystals.101 The diamond band (D line at 1332(±5) cm−1), also frequently called a disorder band, is related to a zone-centre optical phonon with F2g symmetry.
It can be observed that the spectra obtained for the pristine CNOs also have these two intense bands: G at 1576 and D at 1340 cm−1 (Table 1).101 These bands are attributed to the first-order scattering band. From the spectrum of the pristine CNOs, the G band is downshifted from the position of planar graphite, which is correlated with the influence of bond bending on the spherical CNO shells.102 In addition to the strong G and D features observed in the Raman spectra of the CNOs, some second-order scattering bands at 2667 cm−1 were observed (Table 1) and detected at approximately twice the wavenumber of the D band.
The Raman spectra of the Pcs and their metal complexes have peaks in the region 1700–500 cm−1. The spectral range 1550–1350 cm−1 is characteristic of Pcs with different metal atoms and is unique for each Pc.103,104 In the Raman spectra of CuPc, we distinguish a band at 1524 cm−1 (B1g). This band is very important because it is sensitive to the size of the central metal ion. The band at 1524 cm−1 is considered as a ‘marker’ of crystal structure modification, and the shift in B1g shifted by 50 cm−1.104 The band at 1524 cm−1 observed for CuPc shifts to 1551 and 1504 cm−1 in the Raman spectra of NiPc and ZnPc, respectively (Fig. SI2†). This region also consists of overtones or combination bands corresponding to the –C–H– stretching modes in the 1550 to 1300 cm−1 range.21 Symmetric and antisymmetric –C–H– stretching modes are observed with similar frequencies for both non-metallated and metallated phthalocyanines.21
Additionally, in the 3200–2850 cm−1 region (Fig. SI2†), the –N–H– and –C–H– stretching vibrations observed for the unsubstituted Pc (H2Pc) are recorded at 3008, 2989, 2946, 2902 and 2899 cm−1. The absence of these bands in the MPc spectra may indicate the successful substitution of the H2Pc moieties with metals.
Ultraviolet-visible (UV-vis) absorption was used as an effective approach to characterize the optical properties of the CNO and MPc–CNO derivatives (Fig. 4). The UV-vis absorption spectra of the CNO derivatives show (Fig. 4a) broad absorption bands at 285 and 332 nm, which are characteristic of the π–π* electron transition in the polyaromatic systems of the curved graphite layers.105 The UV-vis absorption spectra of the MPcs showed a typical shape for Pcs, with B-band (in the region between 300 and 370 nm and centred at 320 and 335 nm) and Q-band (in the region between 550 and 750 nm and centred at 670 nm) with various intensities. The Zn–Pc and its CNO derivative have an additional shoulder centred at 385 nm (Fig. 4d). Three peaks and two shoulders were located in the absorption band in the UV region, which is preceded by the ultraviolet band of the Pc molecule.106 The analysis of the spectral data show differences between Zn–Pc (Fig. 4d), Cu–Pc (Fig. 4b) and Ni–Pc (Fig. 4c).
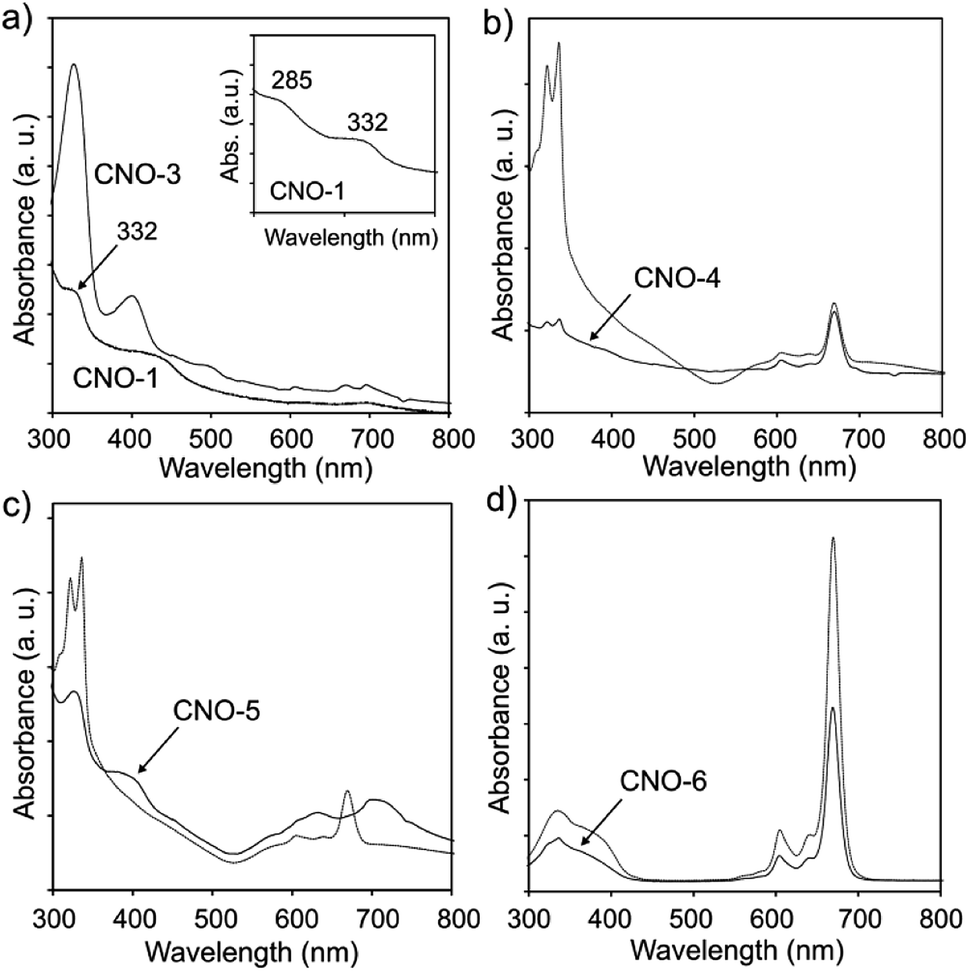 | ||
| Fig. 4 UV-vis spectra of (a) CNO-1 and CNO-3, (b) CuPc and CNO-4, (c) NiPc and CNO-5, and (d) ZnPc and CNO-6. | ||
The spectra recorded for Zn–Pc and its CNO derivative exhibit a high extinction coefficient and sharp Q bands, while the other two MPcs exhibit wider bands with lower intensities. This phenomena suggests strong aggregation interactions between the molecules. Additionally, the B-band in the spectra of Cu–Pc (Fig. 4b) is more intense than the Q-band, which also indicates significant aggregation.107 The MPc–CNO derivatives show the same trend and exhibit an obvious UV-vis light absorption ability. The morphology of the pristine metallated Pcs and their CNO derivatives were observed by scanning electron microscopy (SEM) (Fig. 5).
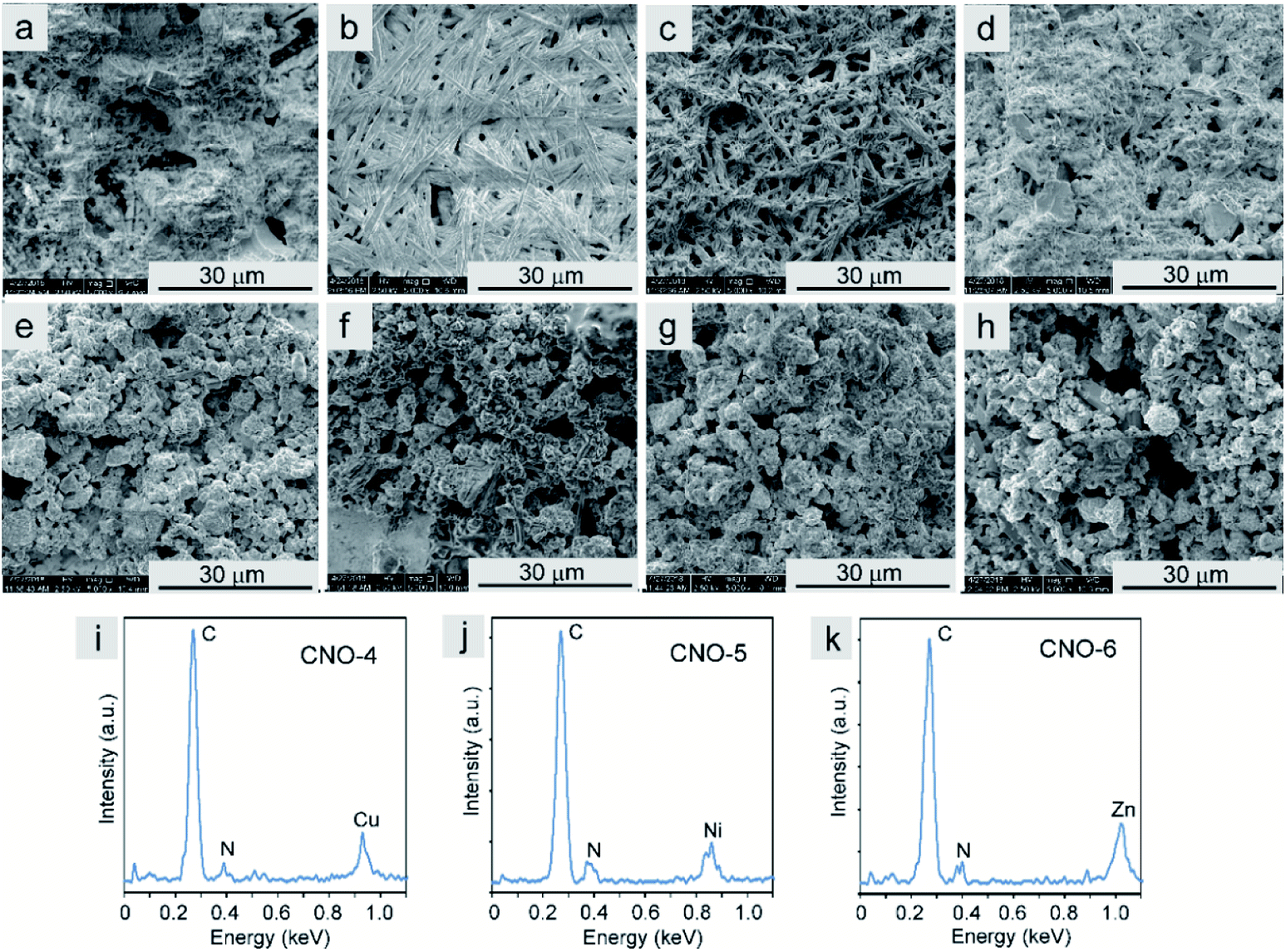 | ||
| Fig. 5 (a–h) SEM images of (a) H2Pc, (b) CuPc, (c) NiPc, (d) ZnPc, (e) CNO-3, (f) CNO-4, (g) CNO-5 and (h) CNO-6. (i–k) Energy dispersive X-ray spectroscopy analyses of (i) CNO-4, (j) CNO-5 and (k) CNO-6. | ||
The SEM images of an Au foil covered with the Pcs and the Pc–CNO derivatives are shown in Fig. 5a–d and e–h, respectively. The non-metallated Pcs and those with coordinated metal ions in the centre formed needle-like structures with different diameters and lengths. After covalent immobilization of these macrocycles on the CNO surface, the morphology of these systems radically changes to a morphology similar to that of carbon materials. The CNO-derivatives result in a homogeneous distribution of the Pcs on the CNO surfaces. The energy dispersive X-ray (EDX) spectra (Fig. 5i–k) of the CNO-4, CNO-5 and CNO-6 systems reveal that the derivatives mainly contain C, N, and the metals Cu, Ni or Zn. The EDX analysis confirmed the successful functionalization of the CNO surface with metallated Pcs.
XRD was performed to identify the phase composition and crystallinity of the synthesized Pc and the CNO–MPc derivatives. The XRD patterns of the pristine NDs (Fig. 6a), the pristine CNOs (Fig. 6b) and the functionalized CNOs (CNO-1, Fig. 6c) are presented in Fig. 6. The ND profile reveals the presence of a mixture of various phases. In the ND materials, the strongest reflection at 2θ = 43.8° corresponds to the (111) basal plane of the diamond structure, and the reflection at 2θ = 21.5° corresponds to a graphite-like amorphous phase.75,108 The conversion from NDs to CNOs was observed during the annealing process of the starting material (comparison of Fig. 6a and b).
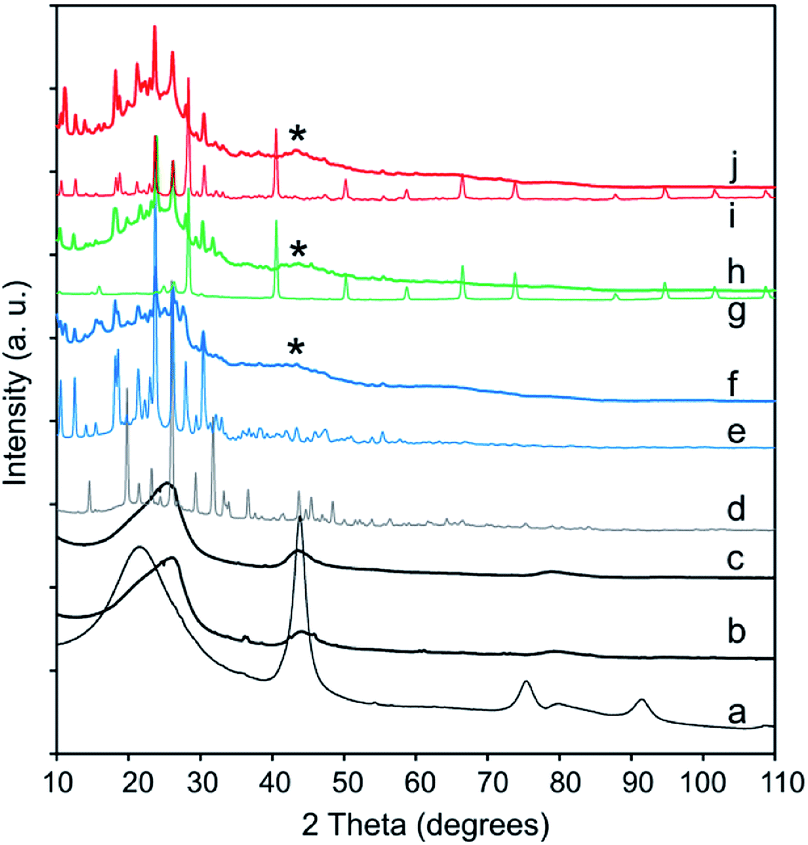 | ||
| Fig. 6 XRD patterns of (a) NDs, (b) CNOs, (c) CNO-1, (d) H2Pc, (e) CuPc, (f) CNO-4, (g) NiPc, (h) CNO-5, (i) ZnPc and (j) CNO-6. | ||
An asymmetric broad reflection in the range between 23–27° and a second one with a maximum at 43.0° were observed. They are attributed to the (002) and (100) planes of graphite, respectively (Fig. 6b and c).109 The broad signal centred approximately 23–27° suggests the contribution of some sp2-bonded carbons in the CNO samples, and can be assigned to graphitic carbon. This signal is relatively broad, has a weak intensity and is observed for every MPc–CNO derivative (Fig. 6f, h and i). For these derivatives, a reflection at approximately 43.0° is also observed (a broad reflection peaks marked with star in Fig. 6). Briefly, if H2 (Fig. SI3†) is substituted by the metal centre, then the (400) and (800) peaks at 2θ = 14.62 and 29.41°, respectively (marked in Fig. SI3†), have a lower intensity.110 All the profiles of the MPc–CNOs confirmed the presence of CNO and MPC components in the obtained materials.
Photocatalytic activity of MPc–CNOs
To test the photocatalytic activity of the MPc–CNOs for the degradation of organic pollutants, rhodamine B (RhB) was selected as a model compound representing a group of dye contaminants. The generally accepted mechanism for the photocatalytic degradation of organic molecules, such as RhB, is the formation of highly active species, which react with and degrade the targeted molecules to form smaller molecules that are less toxic, preferably water and carbon dioxide. Similar to other carbon nanostructures, e.g., fullerenes, CNOs can be used as semiconductors, as they demonstrate a photocatalytic activity. In the presented paper, we investigated the photocatalytic activity of the Pc-functionalized CNOs, which we compared with the behaviour of pristine CNOs and MPc, separately (Fig. 7). We found that both the unmetallated Pcs and all of the metallated Pcs demonstrated poor photocatalytic behaviours. The photodegradation efficiency (η) of RhB, defined as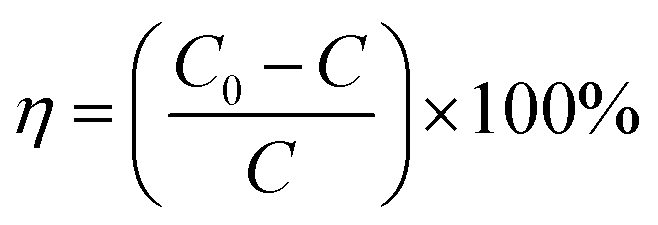 | (1) |
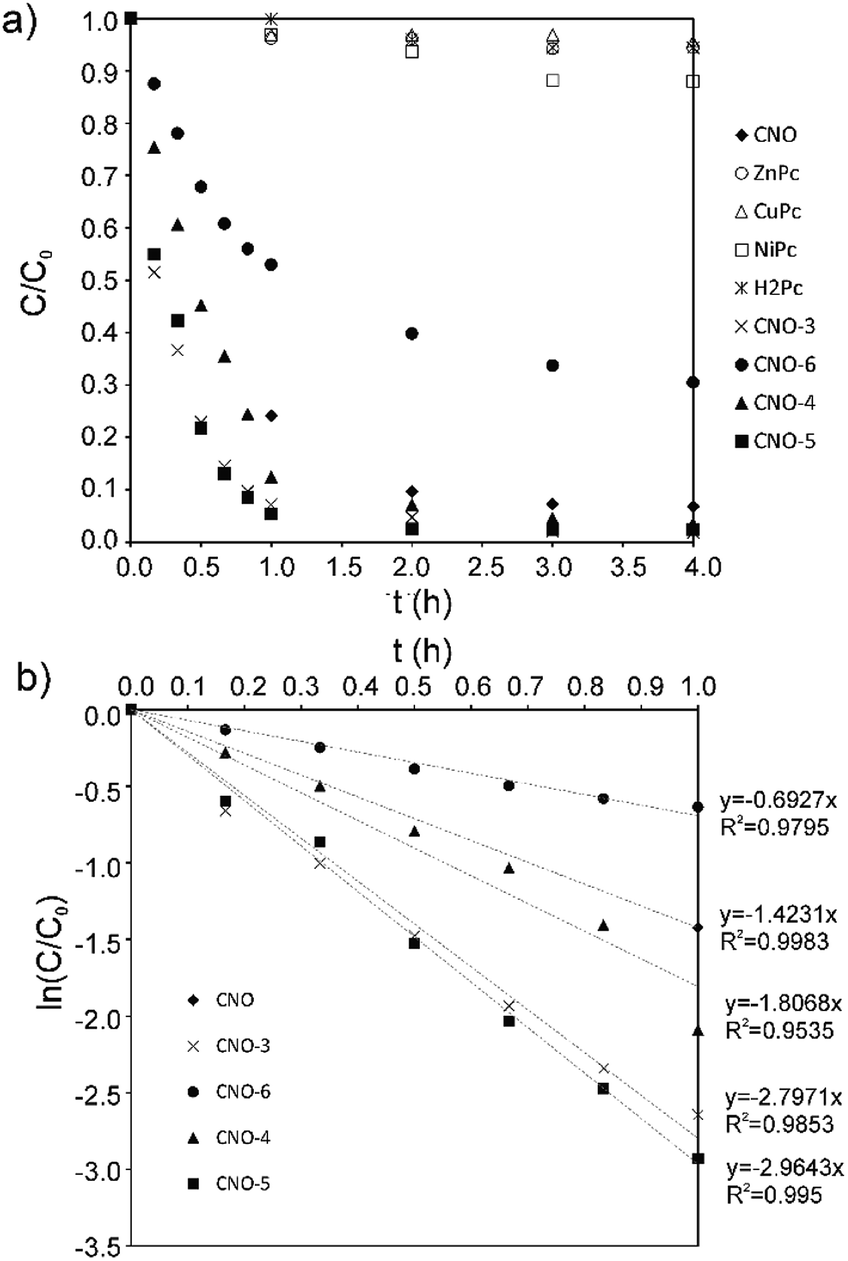 | ||
| Fig. 7 (a) The photocatalytic degradation profiles obtained for RhB with CNO, H2Pc, MPcs and MPc–CNOs. (b) The photocatalytic RhB degradation kinetics obtained with CNO and MPc–CNOs. | ||
| Parameter | CNO | CNO-3 | CNO-4 | CNO-5 | CNO-6 |
|---|---|---|---|---|---|
| η (%) | 93 | 98 | 97 | 98 | 70 |
| k (min−1) | 0.0235 | 0.0466 | 0.0301 | 0.0494 | 0.0115 |
Moreover, the photocatalytic reaction kinetics was investigated based on the pseudo-first order time-dependent relation expressed by the following equation:
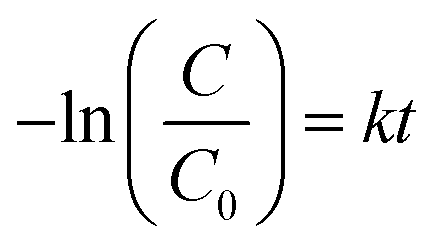 | (2) |
The calculated photocatalytic degradation reaction rate constant was the highest for CNO-5 (0.0494 min−1) and decreased in the following order: CNO-5 > CNO-3 > CNO-4 > CNO > CNO-6 (Table 2). There are two factors that contribute to the final photocatalytic activity of the applied material, namely, (i) the adsorption of the dye and (ii) photocatalytic degradation.
In fact, for CNO-6, which was functionalized with ZnPc to the higher extent, the smaller photocatalytic activity was observed. On the other hand, the final photodegradation efficiency depends also on the effective harvesting of the irradiation. In that respect the HOMO–LUMO gap is important. Indeed, the phthalocyanine-functionalized CNOs have lower HOMO–LUMO energy band gaps, and that enhances the photocatalytic activity. NiPc and CuPc have a smaller HOMO–LUMO band gap than ZnPc.112 The HOMO in CuPc is 3d metal-like, whereas in Ni and Zn phthalocyanines, the HOMO is localized on the Pc ring.112 However, the first ionization process removes an electron from the Pc a1u orbital in all cases. Moreover, also the formation of the stable and long-lived excited states plays a role. It was reported elsewhere that CNOs due to their multi-shell structure stabilize charge-transfer states.113 Therefore, merging MPcs with CNOs leads to the formation of the nanostructured catalyst that upon excitation generates the MPc˙+ and CNO˙− ephemeral species.113 The latter, in the aqueous environment contribute to the formation of hydroxyl and superoxide radicals, which are responsible for RhB degradation. Based on the observed results, we could conclude that the most advantageous effect of those factors was reached for CNO-5, CNO-3 and CNO-4.
Conclusions
Several Pc- and MPc-based derivatives possessing unique physico-chemical properties, including a photocatalytic activity towards organic pollutants, were synthesized. The absorption experiments showed that the functionalization of the CNO cages with MPc moieties results in the formation of photoactive systems. RhB was selected as a model compound representing a group of dye contaminants. The photodegradation efficiency of RhB did not reach more than 12% for the pristine MPcs. However, when the MPc–CNOs or pristine CNOs were applied as catalysts, the photodegradation efficiency significantly increased. The photocatalytic degradation efficiencies obtained using the MPc–CNOs and CNOs were as high as 98% or 93%, respectively. These pristine and MPc-based CNOs show a high photocatalytic activity in comparison with other Pc-functionalized CNs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science Center (NSC), Poland, for their generous support of this work (grant #2012/05/E/ST5/03800 to M. E. P.-B.). L. E. thanks the Robert A. Welch Foundation for an endowed chair, grant #AH-0033, and US NSF grants: PREM program (DMR-1205302) and CHE-1408865.References
- D. Wang, G. Ye, X. Wang and X. Wang, Adv. Mater., 2011, 23, 1122–1125 CrossRef CAS PubMed.
- Q. Liang, M. Zhang, C. Liu, S. Xu and Z. Li, Appl. Catal., A, 2016, 519, 107–115 CrossRef CAS.
- M. M. Mohamed, I. Ibrahim and T. M. Salama, Appl. Catal., A, 2016, 524, 182–191 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- S. Hu, Y. Ding, Q. Chang, J. Yang and K. Lin, Appl. Surf. Sci., 2015, 355, 774–777 CrossRef CAS.
- S. K. Movahed, Z. Piraman and M. Dabiri, J. Photochem. Photobiol., A, 2018, 351, 208–224 CrossRef CAS.
- H. L. Chen, C.-J. Li, C.-J. Peng, H.-J. Leu and W.-H. Hung, ACS Appl. Mater. Interfaces, 2017, 9, 327–334 CrossRef CAS PubMed.
- M. Zhang, Q. Liang, S. Xu and Z. Li, Optik, 2016, 127, 7993–8001 CrossRef CAS.
- S. Ma, Q. Li, Z. Cai, Z. Ye and Y. Zhou, Appl. Organomet. Chem., 2018, 32, e3966 CrossRef.
- Z.-G. Liu, J.-Y. Wan, Z. Yang, S.-Q. Wang and H.-X. Wang, Chem.–Asian J., 2016, 11, 1887–1891 CrossRef CAS PubMed.
- E. Regulska, D. M. Rivera‐Nazario, J. Karpinska, M. E. Plonska‐Brzezinska and L. Echegoyen, ChemistrySelect, 2017, 2, 2462–2470 CrossRef CAS.
- E. Regulska, D. Rivera-Nazario, J. Karpinska, M. Plonska-Brzezinska and L. Echegoyen, Molecules, 2019, 24, 1118 CrossRef CAS PubMed.
- G. Mele, R. Del Sole, G. Vasapollo, E. García-López, L. Palmisano and M. Schiavello, J. Catal., 2003, 217, 334–342 CrossRef CAS.
- X. Cui, Y. Wang, G. Jiang, Z. Zhao, C. Xu, A. Duan, J. Liu, Y. Wei and W. Bai, J. Mater. Chem. A, 2014, 2, 20939–20946 RSC.
- F. Huang, Z. Li, A. Yan, H. Zhao, H. Liang, Q. Gao and Y. Qiang, Sci. Rep., 2017, 7, 39973 CrossRef CAS PubMed.
- X. Liu, C. Chen, H. Ye, Y. Jia, Y. Wu, A. Jin, Y. Wang and X. Chen, Carbon, 2018, 131, 213–222 CrossRef CAS.
- A. Tiwari, N. V. Krishna, L. Giribabu and U. Pal, J. Phys. Chem. C, 2018, 122, 495–502 CrossRef CAS.
- Functional Phthalocyanine Molecular Materials, ed.J. Jiang, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 135 Search PubMed.
- H. Pan, C. Chen, K. Wang, W. Li and J. Jiang, Chem.–Eur. J., 2015, 21, 3168–3173 CrossRef CAS PubMed.
- C. Murray, N. Dozova, J. G. McCaffrey, S. FitzGerald, N. Shafizadeh and C. Crépin, Phys. Chem. Chem. Phys., 2010, 12, 10406 RSC.
- E. Ermiş, Y. Çimen, F. Dumludağ, A. R. Özkaya, B. Salih and Ö. Bekaroğlu, Polyhedron, 2013, 49, 129–137 CrossRef.
- S. Nakano, Y. Kage, H. Furuta, N. Kobayashi and S. Shimizu, Chem.–Eur. J., 2016, 22, 7706–7710 CrossRef CAS PubMed.
- C. Bozoğlu, M. Arıcı, A. L. Uğur, A. Erdoğmuş and A. Koca, Synth. Met., 2014, 190, 56–65 CrossRef.
- H. Abramczyk, B. Brożek-Płuska, K. Kurczewski, M. Kurczewska, I. Szymczyk, P. Krzyczmonik, T. Błaszczyk, H. Scholl and W. Czajkowski, J. Phys. Chem. A, 2006, 110, 8627–8636 CrossRef CAS PubMed.
- J. Blochwitz, M. Pfeiffer, T. Fritz and K. Leo, Appl. Phys. Lett., 1998, 73, 729–731 CrossRef CAS.
- İ. Değirmencioğlu, R. Bayrak, M. Er and K. Serbest, Polyhedron, 2011, 30, 1628–1636 CrossRef.
- L. Zhi, T. Gorelik, J. Wu, U. Kolb and K. Müllen, J. Am. Chem. Soc., 2005, 127, 12792–12793 CrossRef CAS PubMed.
- S. Z. Yildiz, S. Colak and M. Tuna, J. Mol. Liq., 2014, 195, 22–29 CrossRef CAS.
- C.-H. Lee, J. Guo, L. X. Chen and B. K. Mandal, J. Org. Chem., 2008, 73, 8219–8227 CrossRef CAS PubMed.
- S. Shimizu, S. Nakano, T. Hosoya and N. Kobayashi, Chem. Commun., 2011, 47, 316–318 RSC.
- E. Güzel, S. Güney and M. Kandaz, Dyes Pigm., 2015, 113, 416–425 CrossRef.
- T. Fukuda, J. Sato, N. Hashimoto and N. Kobayashi, Chem. Commun., 2014, 50, 14269–14272 RSC.
- S. Cogal, S. Erten-Ela, K. Ocakoglu and A. U. Oksuz, Dyes Pigm., 2015, 113, 474–480 CrossRef CAS.
- A. de la Escosura, M. V. Martínez-Díaz, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2006, 128, 4112–4118 CrossRef CAS PubMed.
- A. Wichmann, G. Schnurpfeil, J. Backenköhler, L. Kolke, V. A. Azov, D. Wöhrle, M. Bäumer and A. Wittstock, Tetrahedron, 2014, 70, 6127–6133 CrossRef CAS.
- H. Karaca, S. Sezer, Ş. Özalp-Yaman and C. Tanyeli, Polyhedron, 2014, 72, 147–156 CrossRef CAS.
- Z. Odabaş, E. B. Orman, M. Durmuş, F. Dumludağ, A. R. Özkaya and M. Bulut, Dyes Pigm., 2012, 95, 540–552 CrossRef.
- J. B. Brito, D. J. C. Gomes, V. D. Justina, A. M. F. Lima, C. A. Olivati, J. R. Silva and N. C. de Souza, J. Colloid Interface Sci., 2012, 367, 467–471 CrossRef CAS PubMed.
- M. E. Sánchez-Vergara, J. C. Alonso-Huitron, A. Rodriguez-Gómez and J. N. Reider-Burstin, Molecules, 2012, 17, 10000–10013 CrossRef PubMed.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- J. W. Perry, D. Alvarez, I. Choong, K. Mansour, S. R. Marder and K. J. Perry, Opt. Lett., 1994, 19, 625 CrossRef CAS PubMed.
- P. Zhu, P. Wang, W. Qiu, Y. Liu, C. Ye, G. Fang and Y. Song, Appl. Phys. Lett., 2001, 78, 1319 CrossRef CAS.
- Z. Lv, Q. Hu, Z. Xu, J. Wang, Z. Chen, Y. Wang, M. Chen, K. Zhou, Y. Zhou and S. Han, Adv. Electron. Mater., 2019, 5, 1800793 CrossRef.
- F. Li, J. Yuan, X. Ling, L. Huang, N. Rujisamphan, Y. Li, L. Chi and W. Ma, ACS Appl. Mater. Interfaces, 2018, 10, 42397–42405 CrossRef CAS PubMed.
- M. Urbani, G. de la Torre, M. K. Nazeeruddin and T. Torres, Chem. Soc. Rev., 2019, 48, 2738–2766 RSC.
- J. Tong, D. Wang, D. Wang, F. Xu, R. Duan, D. Zhang, J. Fan and B. Dong, Langmuir DOI:10.1021/acs.langmuir.9b02479.
- A. T. Bilgiçli, M. N. Yaraşir, M. Kandaz and A. Rıza Özkaya, Polyhedron, 2010, 29, 2498–2510 CrossRef.
- T. C. Canevari, T. M. Prado, F. H. Cincotto and S. A. S. Machado, Mater. Res. Bull., 2016, 76, 41–47 CrossRef CAS.
- X. Zhang, L. Yu, C. Zhuang, T. Peng, R. Li and X. Li, ACS Catal., 2014, 4, 162–170 CrossRef CAS.
- E. T. Saka and Y. Çağlar, Catal. Lett., 2017, 147, 1471–1477 CrossRef CAS.
- L. Fernández, V. I. Esteves, Â. Cunha, R. J. Schneider and J. P. C. Tomé, J. Porphyrins Phthalocyanines, 2016, 20, 150–166 CrossRef.
- X. Ma, M. Luo, L. Yan, N. Tang and J. Li, New J. Chem., 2019, 43, 9589–9595 RSC.
- D. Unluer, A. Aktas Kamiloglu, S. Direkel, E. Bektas, H. Kantekin and K. Sancak, J. Organomet. Chem., 2019, 900, 120936 CrossRef CAS.
- Y. Zhang and J. F. Lovell, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1420 Search PubMed.
- Y. Lei, R. Zhao, M. Xu, X. Zhao, X. Yang, H. Guo, J. Zhong and X. Liu, J. Mater. Sci.: Mater. Electron., 2012, 23, 921–927 CrossRef CAS.
- S. Tombe, E. Antunes and T. Nyokong, J. Mol. Catal. A: Chem., 2013, 371, 125–134 CrossRef CAS.
- A. Korkmaz and Y. Yilmaz, Main Group Chem., 2019, 18, 31–42 CAS.
- S. Hu, R. Tian, Y. Dong, J. Yang, J. Liu and S. Cao, RSC Adv., 2013, 3, 21447 RSC.
- V. A. Basiuk, N. Alzate-Carvajal, L. V. Henao-Holguín, E. V. Rybak-Akimova and E. V. Basiuk, Appl. Surf. Sci., 2016, 371, 16–27 CrossRef CAS.
- D. González-Rodríguez, T. Torres, M. Á. Herranz, L. Echegoyen, E. Carbonell and D. M. Guldi, Chem.–Eur. J., 2008, 14, 7670–7679 CrossRef PubMed.
- M. R. Axet, O. Dechy-Cabaret, J. Durand, M. Gouygou and P. Serp, Coord. Chem. Rev., 2016, 308, 236–345 CrossRef CAS.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed.
- G. Bottari, G. de la Torre and T. Torres, Acc. Chem. Res., 2015, 48, 900–910 CrossRef CAS PubMed.
- R. Koeppe, N. S. Sariciftci, P. A. Troshin and R. N. Lyubovskaya, Appl. Phys. Lett., 2005, 87, 244102 CrossRef.
- L. Moreira, J. Calbo, B. M. Illescas, J. Aragó, I. Nierengarten, B. Delavaux-Nicot, E. Ortí, N. Martín and J.-F. Nierengarten, Angew. Chem., Int. Ed., 2015, 54, 1255–1260 CrossRef CAS PubMed.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2015, 44, 6425–6433 RSC.
- G. Schick, K.-D. Kampe and A. Hirsch, J. Chem. Soc., Chem. Commun., 1995, 2023 RSC.
- R. Valencia, A. Rodríguez-Fortea, A. Clotet, C. de Graaf, M. N. Chaur, L. Echegoyen and J. M. Poblet, Chem.–Eur. J., 2009, 15, 10997–11009 CrossRef CAS PubMed.
- D. González-Rodríguez, C. G. Claessens, T. Torres, S. Liu, L. Echegoyen, N. Vila and S. Nonell, Chem.–Eur. J., 2005, 11, 3881–3893 CrossRef PubMed.
- M. Bystrzejewski, M. H. Rummeli, T. Gemming, H. Lange and A. Huczko, New Carbon Materials, 2010, 25, 1–8 CrossRef CAS.
- V. L. Kuznetsov, A. L. Chuvilin, Y. V. Butenko, I. Y. Mal'kov and V. M. Titov, Chem. Phys. Lett., 1994, 222, 343–348 CrossRef CAS.
- A. Kharlamov, M. Bondarenko, G. Kharlamova, N. Gubareni and V. Fomenko, Univers. J. Mater. Sci., 2013, 1(2), 78–86 Search PubMed.
- J.-C. Fan, H.-H. Sung, C.-R. Lin and M.-H. Lai, J. Mater. Chem., 2012, 22, 9794 RSC.
- S. Tomita, M. Fujii and S. Hayashi, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 245424 CrossRef.
- A. Palkar, F. Melin, C. M. Cardona, B. Elliott, A. K. Naskar, D. D. Edie, A. Kumbhar and L. Echegoyen, Chem.–Asian J., 2007, 2, 625–633 CrossRef CAS PubMed.
- A. Hirsch, Fullerenes and Related Structures, in Topics in Current Chemistry, 1999, vol. 199, pp. 1–65 Search PubMed.
- A. Palkar, A. Kumbhar, A. J. Athans and L. Echegoyen, Chem. Mater., 2008, 20, 1685–1687 CrossRef CAS.
- M. E. Plonska-Brzezinska, A. T. Dubis, A. Lapinski, A. Villalta-Cerdas and L. Echegoyen, ChemPhysChem, 2011, 12, 2659–2668 CrossRef CAS PubMed.
- M. E. Plonska-Brzezinska, A. Lapinski, A. Z. Wilczewska, A. T. Dubis, A. Villalta-Cerdas, K. Winkler and L. Echegoyen, Carbon, 2011, 49, 5079–5089 CrossRef CAS.
- V. Georgakilas, D. M. Guldi, R. Signorini, R. Bozio and M. Prato, J. Am. Chem. Soc., 2003, 125, 14268–14269 CrossRef CAS PubMed.
- C. T. Cioffi, A. Palkar, F. Melin, A. Kumbhar, L. Echegoyen, M. Melle-Franco, F. Zerbetto, G. M. A. Rahman, C. Ehli, V. Sgobba, D. M. Guldi and M. Prato, Chem.–Eur. J., 2009, 15, 4419–4427 CrossRef CAS PubMed.
- L. Zhou, C. Gao, D. Zhu, W. Xu, F. F. Chen, A. Palkar, L. Echegoyen and E. S.-W. Kong, Chem.–Eur. J., 2009, 15, 1389–1396 CrossRef CAS PubMed.
- A. Molina-Ontoria, M. N. Chaur, M. E. Plonska-Brzezinska and L. Echegoyen, Chem. Commun., 2013, 49, 2406 RSC.
- K. Flavin, M. N. Chaur, L. Echegoyen and S. Giordani, Org. Lett., 2010, 12, 840–843 CrossRef CAS PubMed.
- Y. Liu, R. L. Vander Wal and V. N. Khabashesku, Chem. Mater., 2007, 19, 778–786 CrossRef CAS.
- A. S. Rettenbacher, M. W. Perpall, L. Echegoyen, J. Hudson and D. W. Smith, Chem. Mater., 2007, 19, 1411–1417 CrossRef CAS.
- H. Mizuno, K. Nagano, S. Tomita, H. Yanagi and I. Hiromitsu, Thin Solid Films, 2018, 654, 69–76 CrossRef CAS.
- D. Zheng, G. Yang, Y. Zheng, P. Fan, R. Ji, J. Huang, W. Zhang and J. Yu, Electrochim. Acta, 2017, 247, 548–557 CrossRef CAS.
- I. Y. Y. Bu, Sol. Energy, 2014, 105, 236–242 CrossRef CAS.
- The Chemistry of Synthetic Dyes, ed. K. Venkatamaran, National Chemical Laboratory Pune, India, 1971, vol. 5 Search PubMed.
- F. Baumann, B. Bienert, G. Rösch, H. Vollmann and W. Wolf, Angew. Chem., 1956, 68, 133–150 CrossRef CAS.
- C.-M. Chang and Y.-L. Liu, Carbon, 2009, 47, 3041–3049 CrossRef CAS.
- R. Seoudi, G. S. El-Bahy and Z. A. El Sayed, J. Mol. Struct., 2005, 753, 119–126 CrossRef CAS.
- X.-X. Zhang, M. Bao, N. Pan, Y.-X. Zhang and J.-Z. Jiang, Chin. J. Chem., 2010, 22, 325–332 CrossRef.
- M. W. Wong, Chem. Phys. Lett., 1996, 256, 391–399 CrossRef CAS.
- Q. Li, H. Wang, Y. Li, Y. Li and Q. Duan, Dyes Pigm., 2018, 149, 261–267 CrossRef CAS.
- B. C. Smith, Infrared spectral interpretation: a systematic approach, CRC Press, Boca Raton, 1999 Search PubMed.
- R. J. Nemanich and S. A. Solin, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 392–401 CrossRef CAS.
- D. Codorniu Pujals, O. Arias de Fuentes, L. F. Desdín García, E. Cazzanelli and L. S. Caputi, Appl. Phys. A: Mater. Sci. Process., 2015, 120, 1339–1345 CrossRef CAS.
- E. D. Obraztsova, M. Fujii, S. Hayashi, V. L. Kuznetsov, Yu. V. Butenko and A. L. Chuvilin, Carbon, 1998, 36, 821–826 CrossRef CAS.
- D. Roy, M. Chhowalla, H. Wang, N. Sano, I. Alexandrou, T. W. Clyne and G. A. J. Amaratunga, Chem. Phys. Lett., 2003, 373, 52–56 CrossRef CAS.
- T. V. Basova, V. G. Kiselev, B.-E. Schuster, H. Peisert and T. Chassé, J. Raman Spectrosc., 2009, 40, 2080–2087 CrossRef CAS.
- D. R. Tackley, G. Dent and W. Ewen Smith, Phys. Chem. Chem. Phys., 2001, 3, 1419–1426 RSC.
- S. K. Sonkar, M. Ghosh, M. Roy, A. Begum and S. Sarkar, Mater. Express, 2012, 2, 105–114 CrossRef CAS.
- M. M. El-Nahass, K. F. Abd-El-Rahman and A. A. A. Darwish, Mater. Chem. Phys., 2005, 92, 185–189 CrossRef CAS.
- X. Wang, J. Zheng, K. Qiao, J. Qu and C. Cao, Appl. Surf. Sci., 2014, 297, 188–194 CrossRef CAS.
- S. Tomita, A. Burian, J. C. Dore, D. LeBolloch, M. Fujii and S. Hayashi, Carbon, 2002, 40, 1469–1474 CrossRef CAS.
- A. N. Popova, Coke Chem., 2017, 60, 361–365 CrossRef.
- G. Liu, T. Gredig and I. K. Schuller, EPL, 2008, 83, 56001 CrossRef.
- Y. Wan, Q. Liang, T. Cong, X. Wang, Y. Tao, M. Sun, Z. Li and S. Xu, RSC Adv., 2015, 5, 66286–66293 RSC.
- M.-S. Liao and S. Scheiner, J. Chem. Phys., 2001, 114, 9780–9791 CrossRef CAS.
- M. A. Hashmi and M. Lein, J. Phys. Chem. C, 2018, 122, 2422–2431 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00896f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |