
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphonopeptides containing free phosphonic groups: recent advances†
Paweł Kafarski
 *
*
Department of Bioorganic Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-305 Wrocław, Poland. E-mail: pawel.kafarski@pwr.edu.pl
First published on 9th July 2020
Abstract
Phosphonopeptides are mimetics of peptides in which phosphonic acid or related (phosphinic, phosphonous etc.) group replaces either carboxylic acid group present at C-terminus, is located in the peptidyl side chain, or phosphonamidate or phosphinic acid mimics peptide bond. Acting as inhibitors of key enzymes related to variable pathological states they display interesting and useful physiologic activities with potential applications in medicine and agriculture. Since the synthesis and biological properties of peptides containing C-terminal diaryl phosphonates and those with phosphonic fragment replacing peptide bond were comprehensively reviewed, this review concentrate on peptides holding free, unsubstituted phosphonic acid moiety. There are two groups of such mimetics: (i) peptides in which aminophosphonic acid is located at C-terminus of the peptide chain with most of them (including antibiotics isolated from bacteria and fungi) exhibiting antimicrobial activity; (ii) non-hydrolysable analogues of phosphonoamino acids, which are useful tools to study physiologic effects of phosphorylations.
 Paweł Kafarski | Paweł Kafarski was born in 1949. He studied chemistry at Wrocław University of Science and Technology where his scientific adventure started with M. Sc. Thesis, completed in 1971, followed by doctoral thesis (1977) both under the supervision of Prof. Przemysław Mastalerz. Prof. Mastalerz subsequently supervised his scientific career for many years. In his laboratory Paweł Kafarski worked on the synthesis of organophosphorus compounds and their potential biological activities. In 1976/1977 he interrupted his PhD studies and spent nine months at Marquette University at Milwaukee working in the laboratory of Prof. Sheldon E. Cremer on the synthesis of phosphetanes. In 1989 he spent six months in the laboratory of Prof. Henri-Jean Cristau at Ecole Nationale Superieure de Chimie at Montpellier elaborating the procedure for the synthesis of phosphonopeptides containing P–N bond in their structures. Scientific activity of Paweł Kafarski was concentrated on elaboration of synthetic procedures suitable to produce phosphonate inhibitors (most likely in enantiomerically pure forms) of physiologically important enzymes, to mention only: aminopeptidases (targets for anti-cancer and anti-malarial drugs), cathepsin C (potential anti-tumor agents), glutamine synthetase (target for ant-tuberculosis agents), urease (antibacterials for treatment of stomach ulcer and stone formation in urinary tract) or L-phenylalanine ammonia lyase (potential herbicides). The design of ligands for these targets relied on knowledge of molecular mechanisms of the catalyzed reactions and on three-dimensional structures of the chosen proteins. He coauthored over 350 papers, which are well cited in the literature (over 6500 independent citations). He promoted 39 PhDs. |
A Introduction
Phosphonopeptides can be broadly defined as mimetics of peptides in which carboxylic acid or peptide bond are replaced by phosphonic or related (phosphinic, phosphonous) moiety at any position of the peptide chain.1 Also mimetics of phosphopeptides with phosphate moiety being superseded by phosphonic acid may be included into that category.On structural basis, phosphonopeptides could be divided into: (i) peptides, in which carboxylic acid group is replaced by phosphonic one (ii) peptides, in which phosphonic or phosphinic acid unit is mimicking peptide bond; and (iii) peptides in which phosphonic moiety replaces phosphate fragment in O-phosphonopeptides. Typical examples of the first class are: synthetic antibacterial agent alafosfalin,2 phosphonic acid analogue of alanylalanine (compound 1) and natural antibiotic bialaphos,3,4 a mimic of glutamylalanylalanine (compound 2), both shown in Fig. 1. Diaryl (especially diphenyl) esters of such phosphonopeptides may be also classified to this group, however, will not be discussed in this review because synthesis and biological activity of these peptidyl diaryl phosphonates has been reviewed in detail.5–8 Compound 3,9 shown in Fig. 1 may serve as an example of this group. It acts as inhibitor of X-linked inhibitor of apoptosis protein thus being a potential anticancer agent. A potent inhibitor of several aminopeptidases – an analogue of homophenylalanyl-homophenylalanine (compound 4,10 Fig. 1) represents the second group, whereas hexapeptide containing O-phosphonotyrosine mimic (compound 5)11 represents third group of phosphonopeptides. The latter one is one of the first effective inhibitors of protein-tyrosine phosphatase 1B, and thus became promising lead compound for treatment of type 2 diabetes. Also peptides containing phosphonate or phosphinate moiety in place of peptide bond will not be considered here since the synthesis and physiologic activities of these compounds are comprehensively reviewed.9,12–16
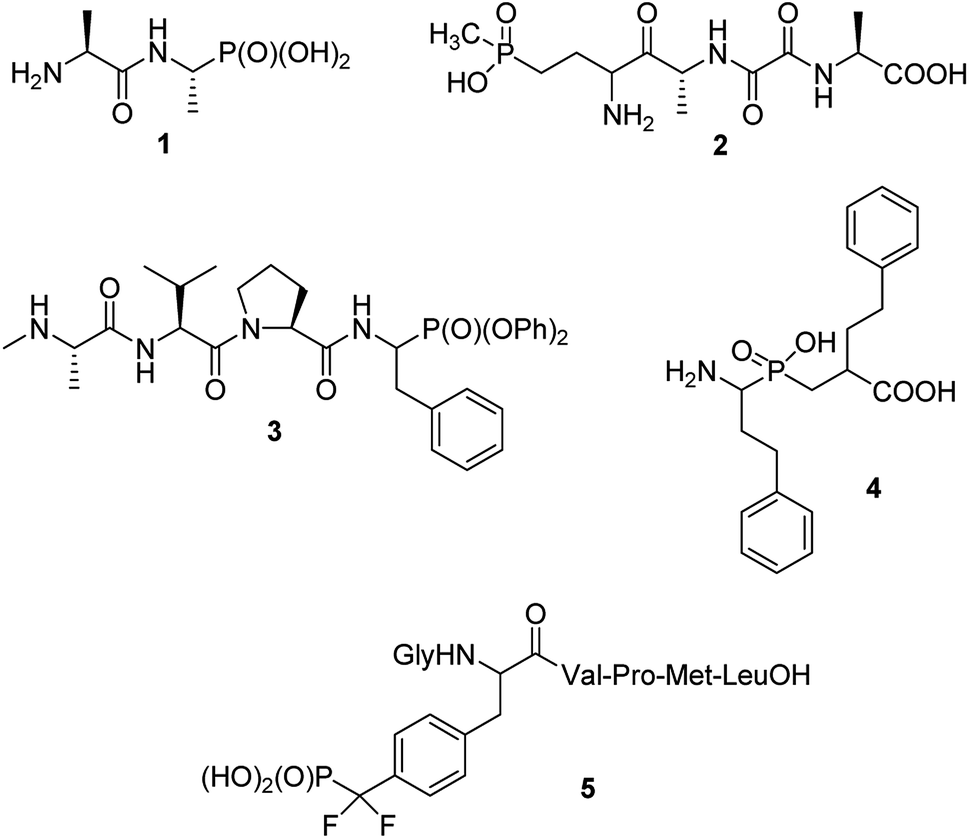 | ||
| Fig. 1 Representative examples of structurally different phosphonopeptides. | ||
In this paper recent advances of chemistry and biology of phosphonopeptides holding free phosphonate or phosphinate groups will be discussed in some detail.
B Peptides containing C-terminal aminophosphonic acids
Synthesis of peptides containing C-terminal phosphonic acid analogues of amino acids was intensively studied at the end of XX century. This was stimulated by promising activity and determination of molecular mode of action of alafosfalin (1) and bialaphos (2). Peptidyl parts of these molecules function as targeting unit. Thus, the peptides are efficiently transported through bacterial (or fungal) membranes and after hydrolysis release aminophosphonic acids, which exert their toxic action by inhibiting vital enzymes. In the case of alafosfalin it is an analogue of alanine, a potent inhibitor of alanine racemase17 and in the case of bialaphos it is phosphinothricin (compound 6), an inhibitor of glutamine synthetase and potent herbicide (Fig. 2).18 This mechanism of action was found for majority of the phosphonopeptides isolated from bacteria and fungi (Fig. 3), namely: phosalacine (compound 7, releases phosphinothricin),19 rhizocticins and plumbemycins (compounds 8, release inhibitor of threonine synthase),20 and dehydrophos (compound 9).21 The action of the latter one has quite unusual since phosphonic analogue of dehydroalaninate formed after cleavage of peptide bond is spontaneously converted into methyl acetylphosphonate (an analogue of pyruvic acid), which is strongly antibacterial by acting most likely as antimetabolite of pyruvate.22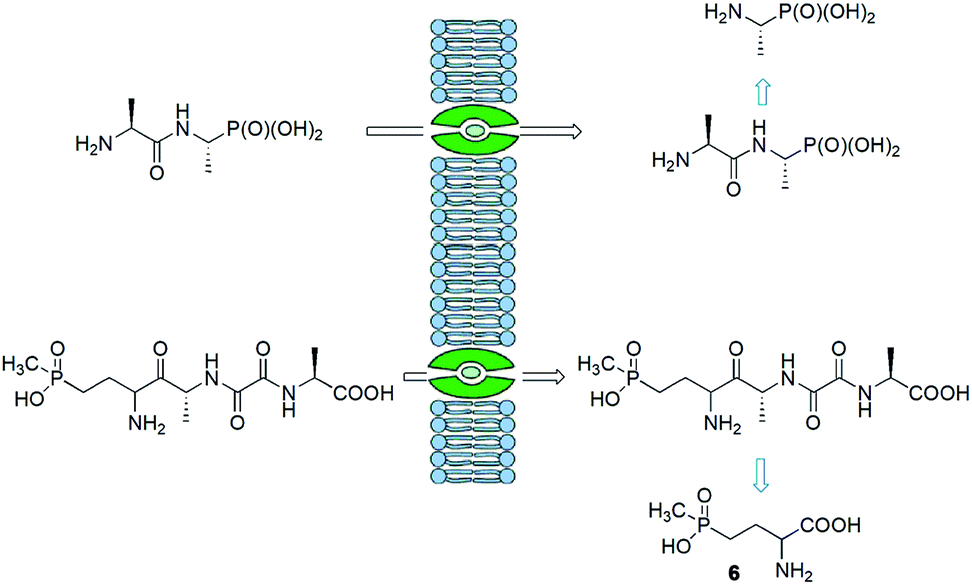 | ||
| Fig. 2 Mechanism of action of alafosfalin and bialaphos. | ||
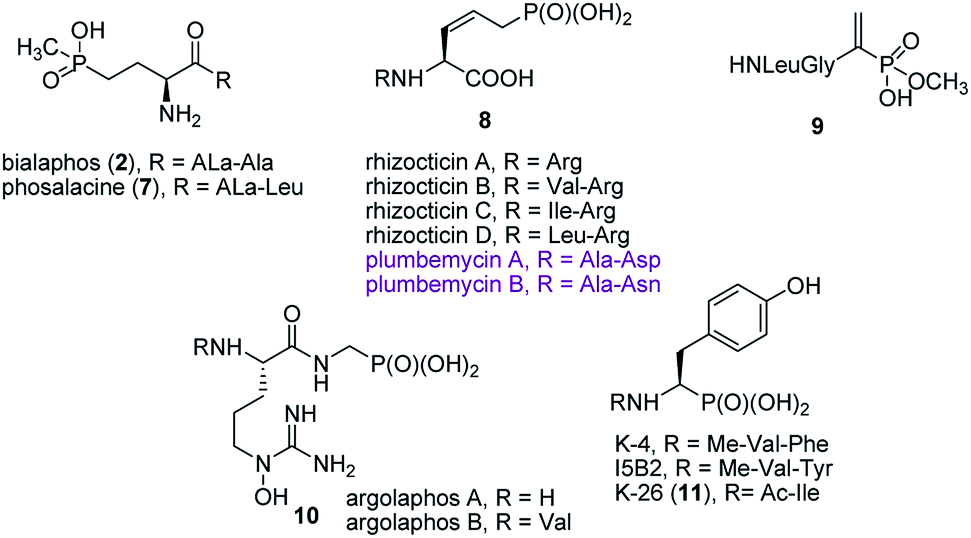 | ||
| Fig. 3 Naturally occurring phosphonopeptide antibiotics. | ||
The peptidyl structure of these antibiotics cause that they have limited utility in human medicine. It is because peptides are readily hydrolysed in human body fluids and thus obtained aminophosphonic acids are not able to cross bacterial cell barrier and exert antibiotic action.
Published in 2015 work of Metcalf and van der Donk brought a significant breakthrough in search for phosphonate antibiotics. By clever combination of the mining of genome of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 of actinomycetes and selective labeling of phosphonate metabolites they rediscovered a huge number of old phosphonates and found out 19 new compounds including two phosphonopeptides – argolaphases (compounds 10, Fig. 3).23
000 of actinomycetes and selective labeling of phosphonate metabolites they rediscovered a huge number of old phosphonates and found out 19 new compounds including two phosphonopeptides – argolaphases (compounds 10, Fig. 3).23
A separate group of naturally occurring phosphonic peptidomimetics is the K-26 (compound 11)24 and its analogues (Fig. 3), a family of bacterial secondary metabolites, tripeptides terminated by an unusual phosphonate analog of tyrosine. They are produced by three different actinomycetales and act as potent inhibitors of human angiotensin-I converting enzyme selectively targeting the eukaryotic family of the enzyme.25
Intensive search for synthetic bacterial phosphonopeptides was stimulated by the premise that phosphonic acid moiety, similarly to sulfonic one, may mimic carboxylic function. Consequently, aminophosphonic acids should act as false substrates for enzymes involved in amino acid metabolism and thus function as their inhibitors. Intensive studies on their inhibitory action towards various enzymes have indicated that this concept, in majority of cases, is invalid. Anyway, this assumption resulted in elaboration of effective procedures for preparation of this class of phosphonopeptides.26
Consequently, in last decade only singular papers devoted to the synthesis of phosphonopeptides, which used dialkyl aminophosphonates as substrates and applied classic peptide bond formation methods, have been published. They concern (see Fig. 4): synthesis of analogue of enkephalin (compound 12),27 analgesic activity of which has not been described, N-substituted analogues of 1-(N-glycylamino)-1-1-methylethylphosphonic acids (compound 13 as representative example),28 peptides of trifluoromethylated β-aminophosphonates (compounds 14)29 and phosphonic acid analogue of glutamic acid acylated with various enantiomers of prolyl-proline (for example, compound 15).30 The latter peptides found an application in organic synthesis as effective bifunctional organocatalysts for enantioselective 1,4-Michael addition of aldehydes to nitroalkenes.31
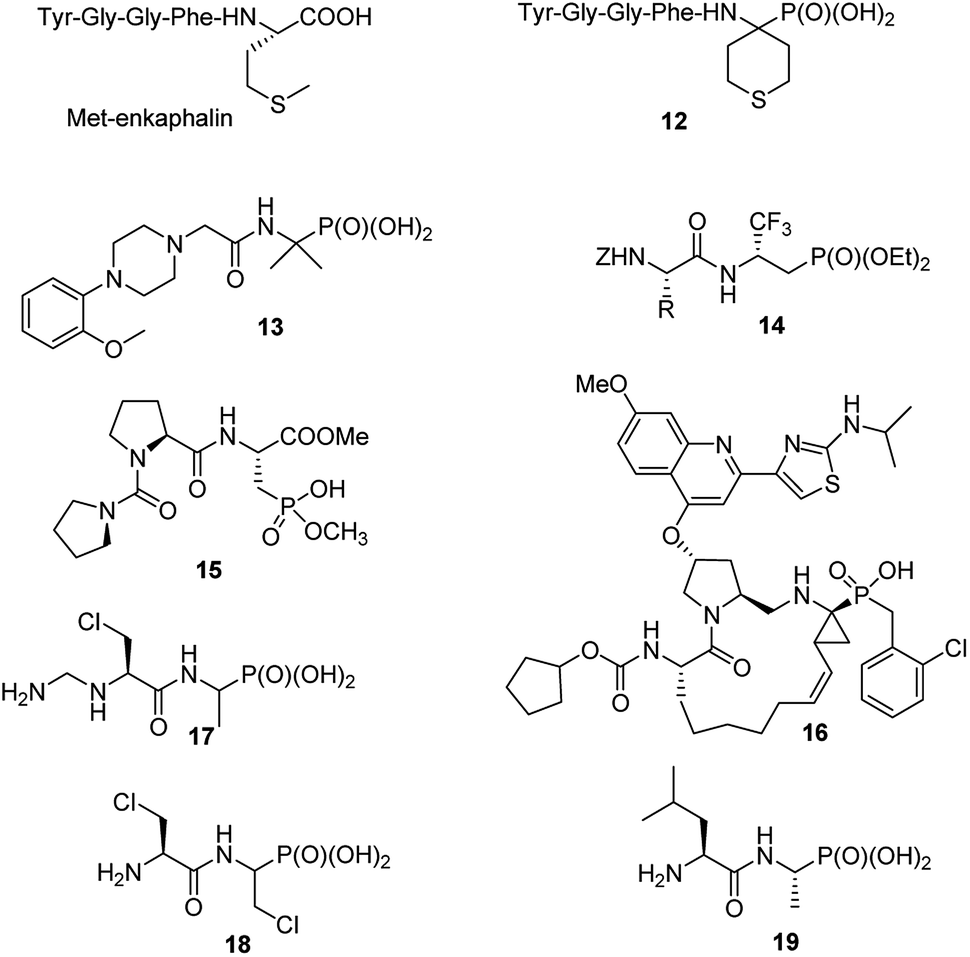 | ||
| Fig. 4 Structurally variable peptides containing C-terminal aminophosphonic acids. | ||
Also classic liquid-phase peptide synthesis was applied for the preparation of a series of phosphonopeptides of quite complex structures being potent inhibitors of hepatitis C virus NS3 protease, with compound 16 (Fig. 4) showing the highest affinity to the enzyme. This compound also significantly inhibited proliferation of the virus in cell base assay.32
The resistance of bacteria to antibacterial agents is rising to dangerously high levels in all parts of the world. Simultaneously limited number of effective new antibiotics has been developed in the last few decades. One of the solutions here is to reinvestigate of old or abandoned drugs for reuse in medicine or for serving as lead compounds for the design of novel antibacterials.
Recently alafosfalin and its analogues, containing additionally second antibacterial agent - β-chloroalanine in the peptide chain, were synthesized by standard mixed carboxylic–carbonic anhydride method and were studied as new weapons against bacterial infections (for representative example see compounds 17 and 18 in Fig. 4).33,34 Since the introduction of nonproteinogenic amino acids into peptide chain usually inhibits or slower its hydrolysis these compounds have a chance to be more stable in body fluids.
C-terminal domain of the dehydrophos (compound 9) biosynthetic enzyme DphH catalyzes the condensation of Leu-tRNA with phosphonic analogue of alanine, yielding dipeptide 19 (Fig. 4) – phosphonic acid analogue of leucylalanine. This enzyme is highly tolerant to structural variations in both aminoacyl-tRNAs and aminophosphonic acids and was used for the preparation of series phosphonodipeptides of interesting antibacterial properties.35
Quite original, approach was used to the synthesis of natural phosphono peptide – dehydrophos (Fig. 5).36 Reaction started from readily available aminomethylenebisphosphonic acid, which was N-protected with carbobenzoxyl (Z) moiety and then esterified by action of triethyl orthoformate. Removal of Z protection by hydrogenolysis followed by acylation with Boc-L-leucine resulted in peptide 20. This peptide was used as substrate for Horner–Wadsworth–Emmons reaction (a version of popular Wittig reaction), which gave dipeptide 21 containing C-terminal phosphonic analogue of dehydroalanine. Hydrolysis of its methyl group by action of strong base (Dabco®) followed by acidification with Dowex® is accompanied by rearrangement and decomposition of the desired dipeptide. Standard removal of Boc from compound 20 with trifluoroacetic acid followed by acylation with Fmoc-Gly an then Horner–Wadsworth–Emmons reaction resulted in peptide 22, which gave clean hydrolysis of one methyl group and after deprotection of Fmoc with Amberlite® afforded dehydrophos in nearly quantitative yield.
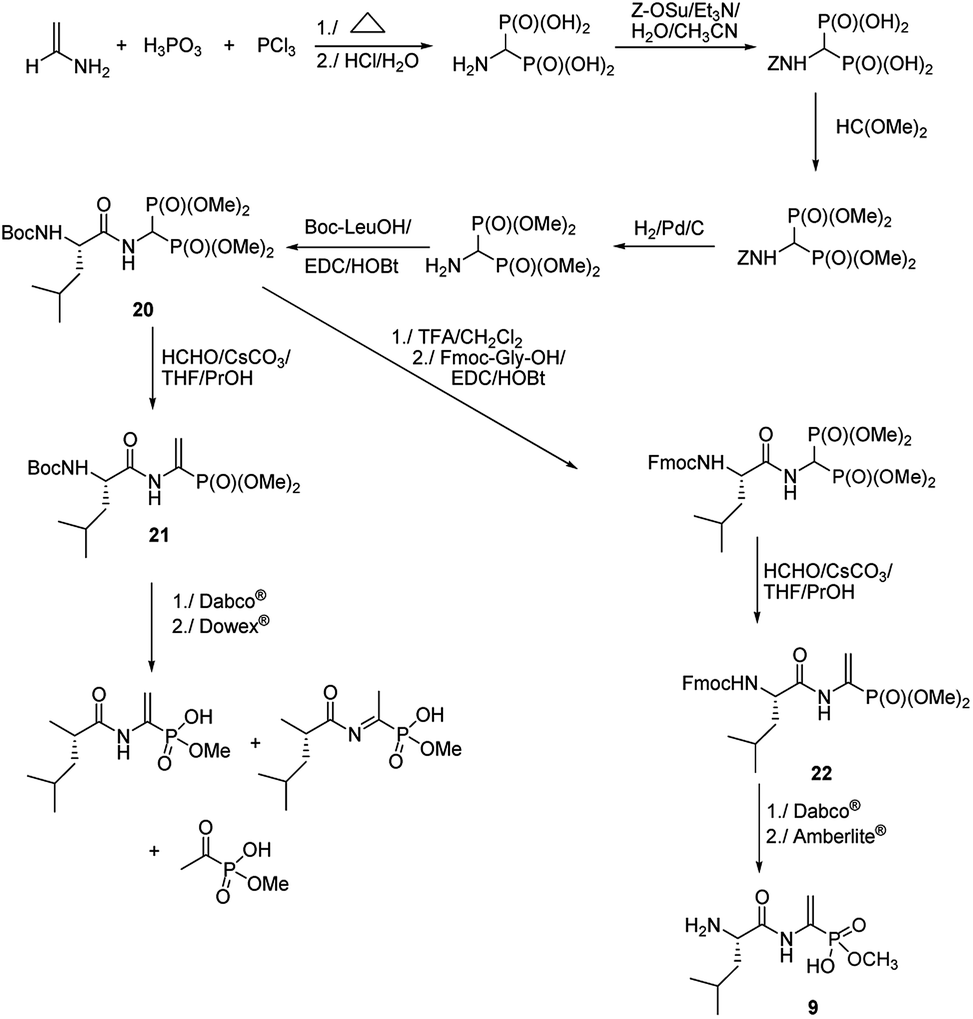 | ||
| Fig. 5 Synthesis of dehydrophos. | ||
The limited number of papers dealing with application of classic methods of phosphonopeptide synthesis is not surprising if considering that they require accessibility to appropriate esters of aminophosphonic acids with free unblocked amino moiety, which have to be synthesized by independent procedures and are not readily available.37,38
Therefore, more efforts have been devoted to the elaboration of non-classical methods for preparation of peptides containing C-terminal aminophosphonic acids. One of the most obvious solutions seems to be the introduction of phosphonate moiety at final steps of peptidomimetic synthesis. Thus, Mannich-type reaction of N-protected amides of amino acids with aldehydes and phosphorus trichloride (classical procedure for the preparation of aminophosphonic acids) or aryldichlorophosphine afforded nearly equimolar mixtures of diastereomers of the desired phosphonopeptides 23 with good yields (Fig. 6).39–41 Similar reaction of N-protected taurine amides, aldehydes and aryldichlorophosphines was used to prepare sulphonophosphinodipeptides (compounds 24, Fig. 6).41–43
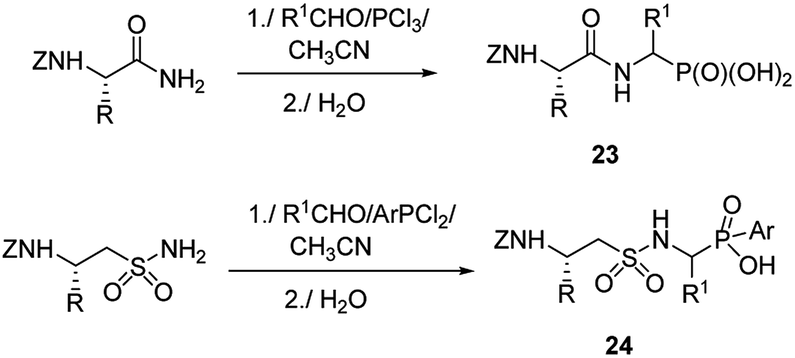 | ||
| Fig. 6 Synthesis of phosphonopeptides by Mannich-like approach. | ||
Another approach is to use peptidylamides as substrates is their reaction with α-isocyanatophosphonate, which afforded phosphono peptides with thiourea fragment replacing peptide bond (Fig. 7).
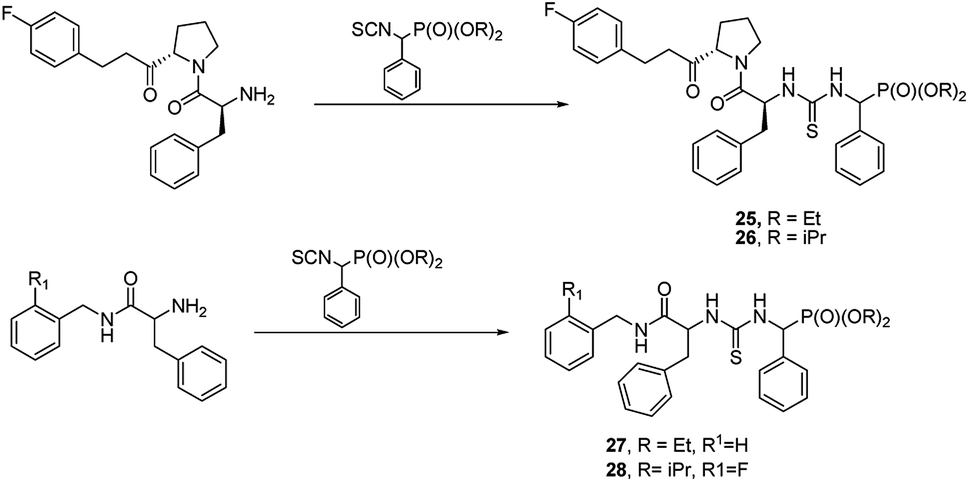 | ||
| Fig. 7 Thiourea containing phosphonopeptides. | ||
These peptides exhibited promising antiproliferative activity against several cancer cell lines (BCG-823, A-549, PC-3, Bcap-37 and BCG-823) with compounds 25, 26, 27 and 28 being the most active.44,45
An addition of diethyl or dimethyl phosphites to aldehydes derived from S-phenylalanyl-S-alanine resulted in diastereomeric mixtures of dipeptidyl 2-amino-1-hydroxyphosphonates (Fig. 8). The formation of compounds of (SSS) configuration was privileged (72–92% de).46 The obtained phosphonopeptides appeared to be promising inhibitors of cathepsins, with compound 29 (Fig. 9) being the most effective low-molecular weight inhibitor of cathepsin K described so far (Ki = 0.32 nM).
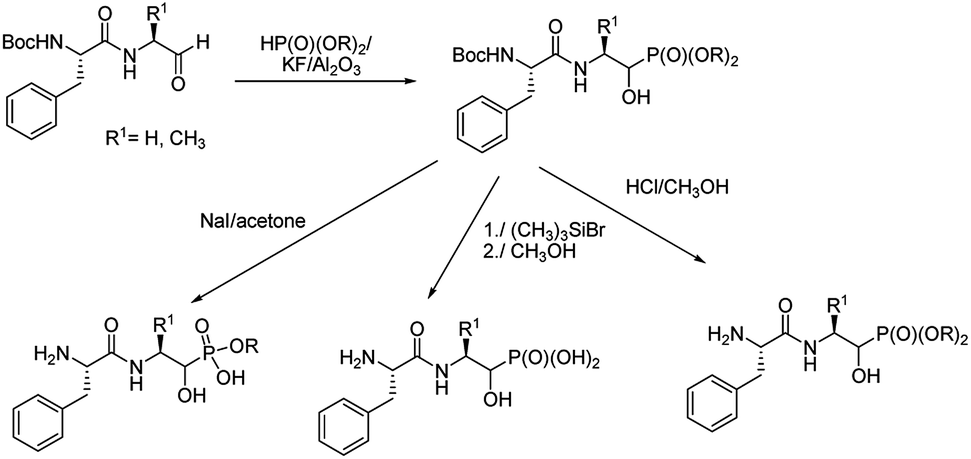 | ||
| Fig. 8 Synthesis of peptides containing 1-hydroxy-2-aminoalkylphosphponates. | ||
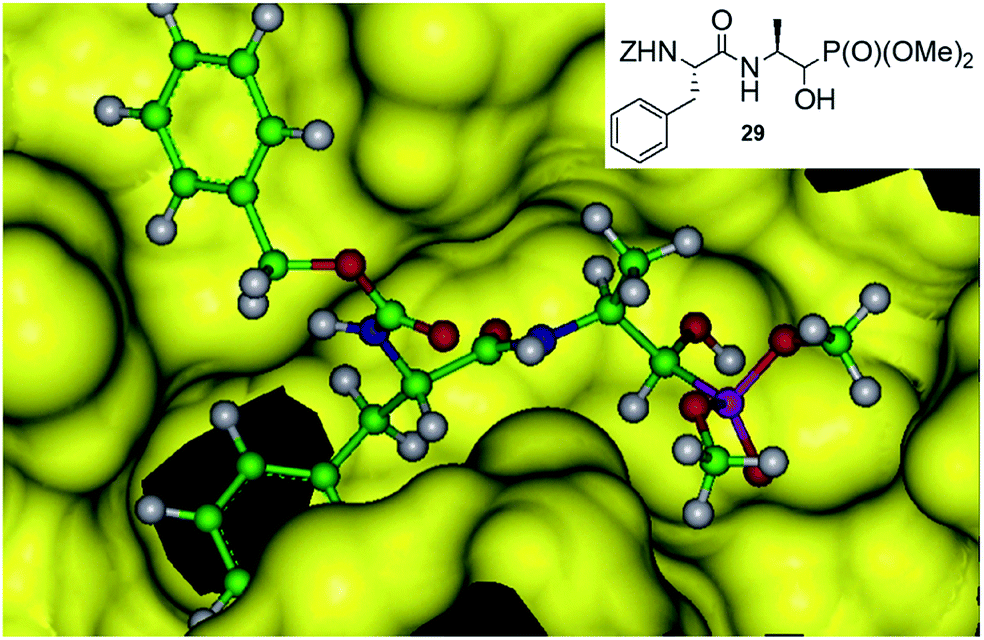 | ||
| Fig. 9 Mode of binding of peptide 28 by cathepsin K. | ||
Molecular modeling of binding of the most effective inhibitors towards cathepsins C and K suggest that hydroxyphosphonate fragment (but not phosphonic group as expected) acts as transition state analogue in this case (Fig. 9).46 However, the possibility that hydroxyphosphonates decompose upon binding with the enzyme and release aldehyde, which is a real inhibitor, cannot be also ruled out.
An interesting example of synthesis of phosphonopeptides using aminophosphonic acids as substrates is an application of one-pot microvawe-assisted multicomponent Ugi reaction. It provided a series of dipeptides 30 containing α, β and γ aminophosphonates as C-terminal ones (Fig. 10). A high level of structural diversity may be obtained by this approach providing a platform for the synthesis of novel peptidomimetics.
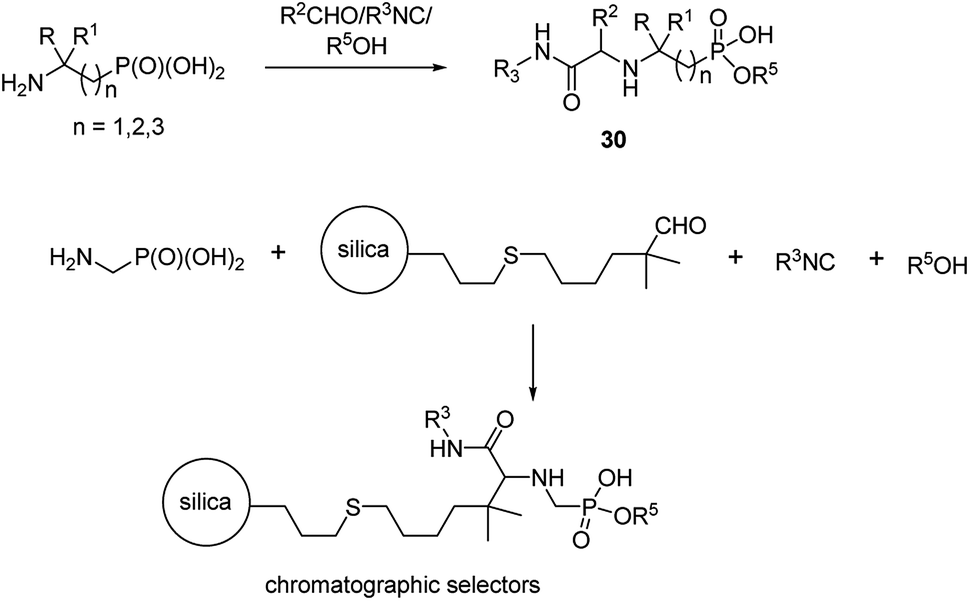 | ||
| Fig. 10 Phosphonopeptides designed as phases for chromatography. | ||
This reaction was also used to generate zwitterionic, chromatographic selectors (Fig. 10), which might be considered as new reversed-phase/ion-exchangers with interesting properties.47,48
Functionalization of the aminophosphonate of already existing peptides also seems to be an attractive approach for synthesis of more complex structures. An example of such an approach is a simple conversion of N-(1,2,2,2-tetrachloroethyl)amides (compounds 31, Fig. 11), which were obtained from N-phtalylamino acid amides, into 1-acylamino-2,2,2-trichloroethylphosphonates (compounds 32, Fig. 11). This is a good starting point to obtain diethyl 5-amino-1,3-oxazol-4-ylphosphonates (compounds 33 and 34). Availability of the latter ones opened an original route to structurally variable phosphonopeptides 35 and 36.49–53
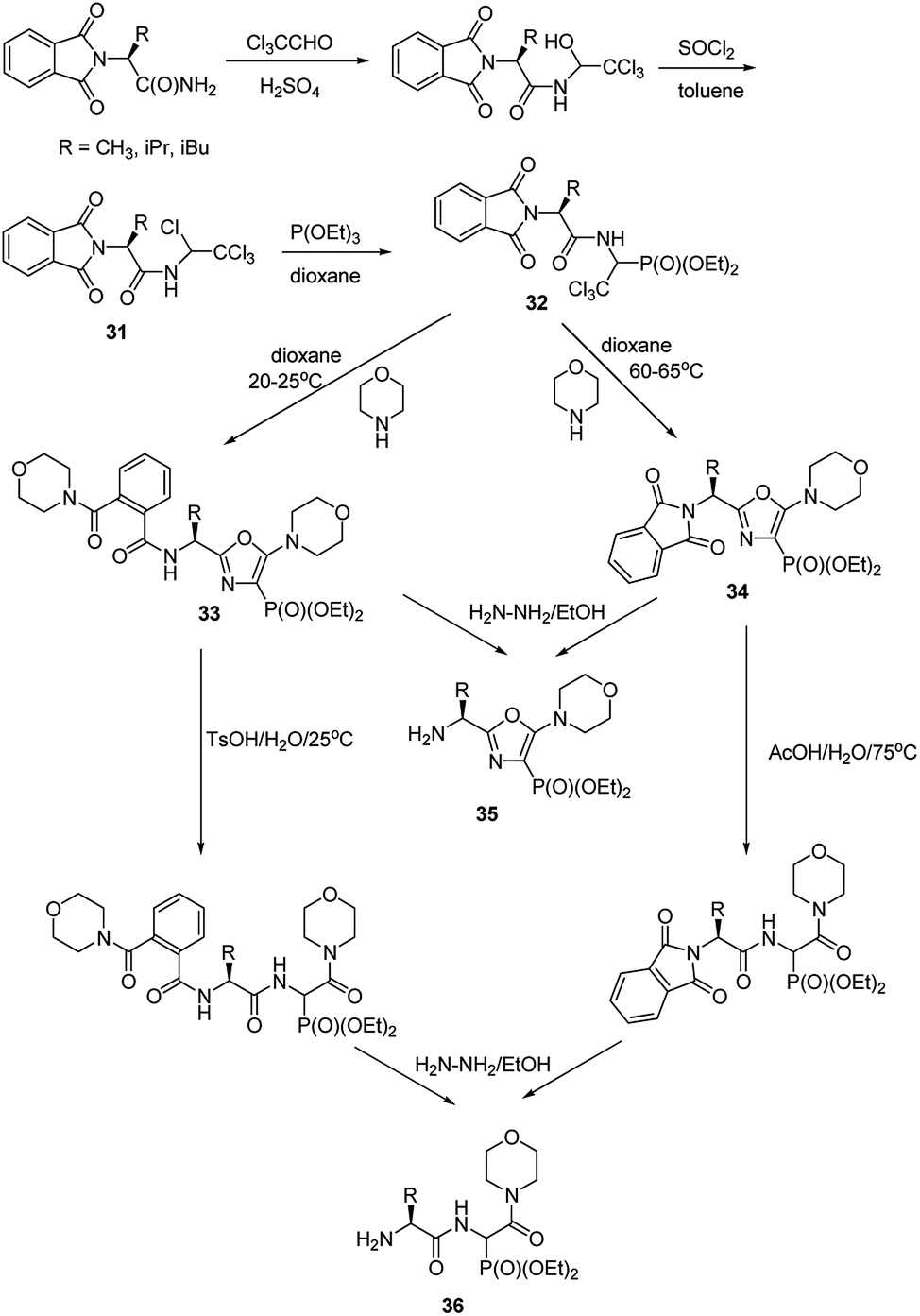 | ||
| Fig. 11 Synthesis of phosphonopeptides via 5-amino-1,3-oxazol-4-ylphosphonates. | ||
A preliminary study on the addition of thiols and amines to double bonds of quite readily affordable phosphono dehydropeptide 37 (Fig. 12) indicated that this procedure could be a new mean to produce phosphono peptides containing non-typical side chains (compounds 38 and 39).54,55
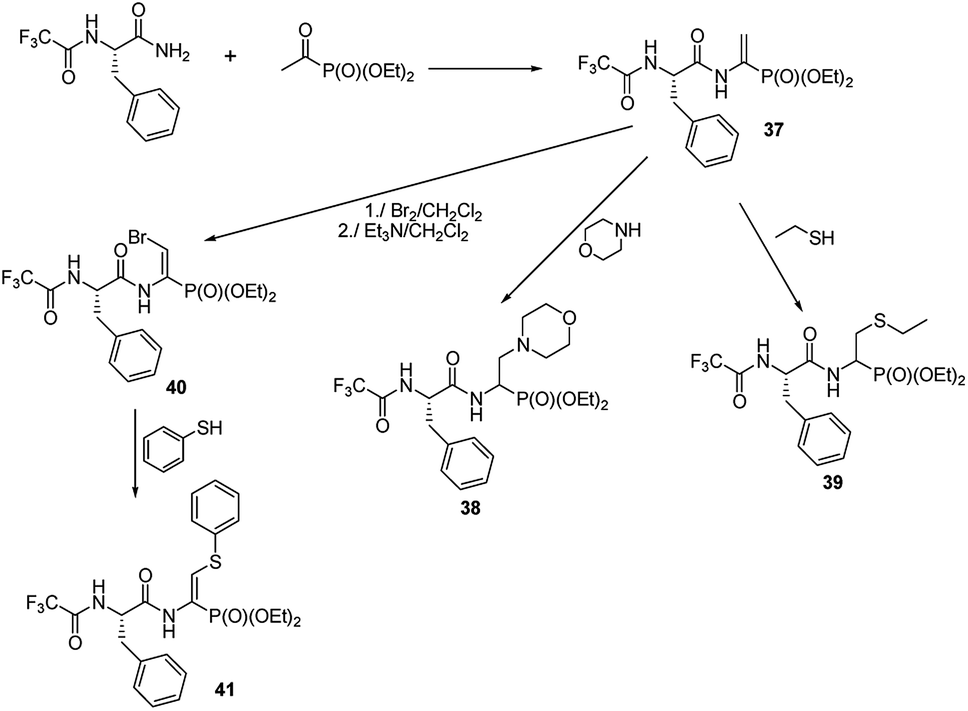 | ||
| Fig. 12 Functionalization of phosphonodehydropeptide. | ||
The peptide 37 is also a good entry for compounds 40 by means of addition–elimination reaction upon action of bromine.55 Upon reaction of 40 with thiophenol functionalized dehydropeptide 41 was obtained. However, the further studies are required to validate general utility of these reactions.
C Peptides with non-hydrolysable analogues of phosphoamino acids
Post-translational modification of proteins by phosphorylation is an integral component of cellular physiology. Hundreds of thousands of sites of protein phosphorylation have been discovered but the understanding of the functions of the vast majority of these post-translational modifications is lacking. In that respect transfer of a phosphoryl group to an amino acid side chain and formation of phosphoamino acid residues (phosphoserine, phosphothreonine, phosphotyrosine, phosphohistidine, phospholysine and/or phosphoarginine) cause that they quite often serve as molecular switches to control protein–protein interactions.56 Physiological functions of these cell cycle proteins and their relevance for cancer stimulate the studies on their physiologic role and makes them the possible targets in cancer control. Therefore, it is not surprising that syntheses and biological evaluation of non-hydrolysable mimics of these amino acids and their peptides is an active field of research.57–59It is worth to notice that hydrolysis of natural antibiotics rhizocticins and plumbemycins in bacterial cells provides an analogue of phosphothreonine (compound 42, Fig. 13),20 a potent inhibitor of threonine synthase, which exerts antibacterial action inside the cell. Unfortunately, this analogue, as well as its peptides, has never been tested as possible cell cycle proteins regulator.
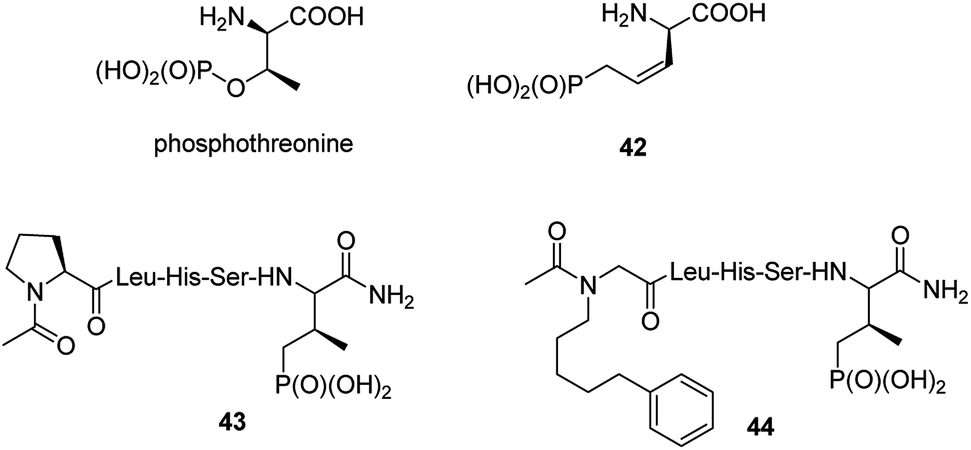 | ||
| Fig. 13 Mimetics of phosphothreonine and its peptides. | ||
Phosphoproteins and phosphopeptides are considered as useful tools for elucidating biological functions of phosphorylations. However, the hydrolytic liability of phosphates in a cellular context limits their use in such studies. Thus, phosphatases inert mimetics, in which phosphate methylene or difluoromethylene groups replace oxygen, have been developed and used in many biological studies and therapeutic designs.59,60 It is worth to note that difluoromethylene group is a better mimetic of the oxygen atom in phosphopeptides than methylene moiety in terms of basicity and charge distribution.61
The cell cycle regulatory serine/threonine-specific polo-like kinase is recognized as an oncogenic target. Peptides containing analogues, in which phosphate moiety of phosphothreonine is replaced by its non-hydrolysable analogue (compounds 43 and 44, Fig. 13), appeared to be potent effectors of this cell cycle regulatory protein and may serve as a tool to study its role in cancerogenesis.62–65 Masking the phosphonate moiety of peptide 43 by esterase labile pivaloyloxymethyl groups provided interesting prodrug.63
The major challenge in the studies on these mimetics is their synthesis. In most cases threonine and serine serve as substrates of choice in the elaborated procedures.66–69 One of the most effective is shown in Fig. 14. Thus, methyl threoninate upon action of acetic acid anhydride was fully acylated and then converted to Z form of dehydroaminobutyrate (compound 45). Reaction with N-bromosuccinimide (NBS) followed by treatment with strong base resulted in bromo derivative (compound 46), which was coupled with diethyl difluoromethylenephosphonate providing E-prochiral substrate 47. This compound was asymmetrically reduced using chiral catalyst and finally converted into compound 48 suitable for introduction into peptide chain.64 Preparation of the desired peptides was achieved easily by using classical procedures of both liquid- and solid-phase peptide syntheses.
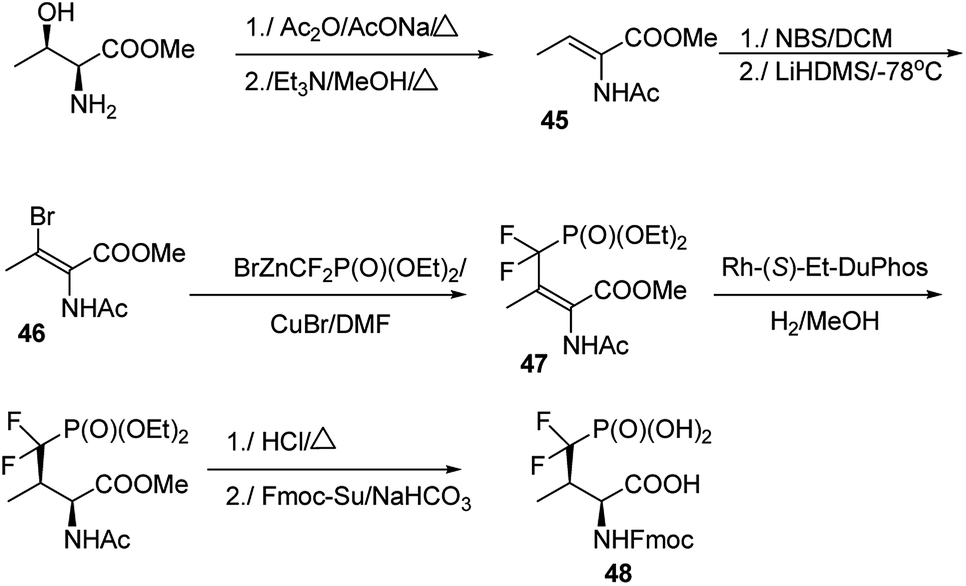 | ||
| Fig. 14 Representative synthetic route to mimetic of phosphothreonine. | ||
The 14-3-3 proteins were the first phosphoserine/phosphothreonine binding proteins to be discovered. They are a family of highly conserved dimeric acidic proteins that are found in all eukaryotic organisms that regulate essentially every major cellular function. They integrate and control multiple signalling pathways and these interactions place them at the center of the signalling hub that governs critical processes in cancer, including apoptosis, cell cycle progression, autophagy, glucose metabolism, and cell motility. In human cells, this family of proteins consists of seven distinct but highly homologous 14-3-3 isoforms: β, ε, η, γ, σ, τ, ζ. Therefore, peptides able to bind selectively these proteins have been selected and used for elucidation of physiologic roles of certain 14-3-3 proteins. Such molecules containing phosphatase-resistant phosphopeptide mimetics were synthesized and their affinities to isoforms: ζ (peptides 49 and 50, Fig. 15), and β (peptide 51) had been evaluated.61,66–70 They might be used as valuable in vivo tools for probing the role of phosphorylations.
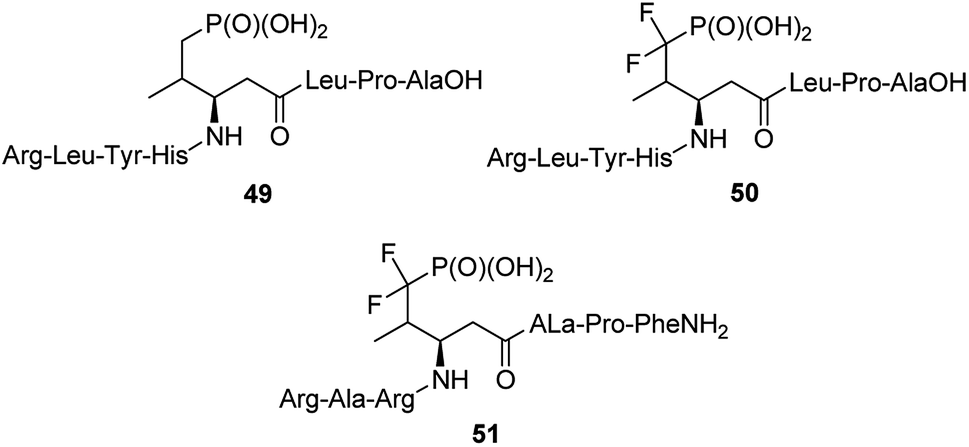 | ||
| Fig. 15 Peptides containing non-hydrolysable mimetics of phosphotyrosine designed to bind 14-3-3-proteins. | ||
Another possibility is genetically directed incorporation of phosphonic mimetics into proteins involved in cell regulation processes. Preliminary studies indicate that it is still challenging task.71,72
Albeit being effective in vitro low-molecular weight inhibitors of phosphoserine/phosphothreonine binding proteins could not be used in cellular systems. It is mainly because the intracellular delivery of drugs that contain the necessary doubly charged phosphonate group is difficult because of the character of cell membrane. In order to get around this problem a very elegant system of transportation of affinity probe 52 was designed (Fig. 16). Ester moiety of the prodrug 53 is readily cleaved in the cell by enzymatic reduction followed by the hydrolysis of protonated phosphonamide and release of suitable inhibitor.62,69
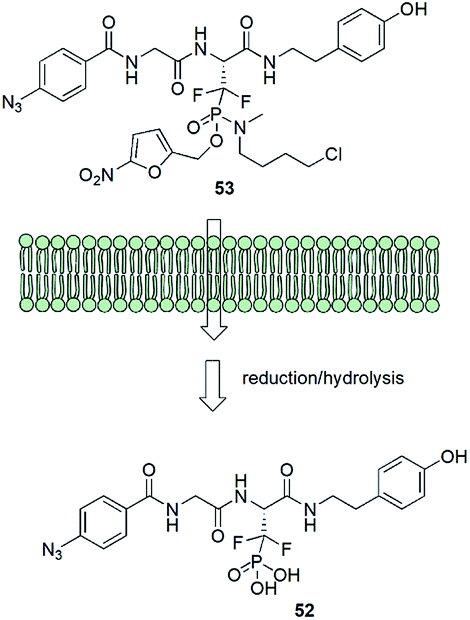 | ||
| Fig. 16 Prodrug design for transportation of affinity probe through cell membranes. | ||
Penta- to decapeptides containing non-hydrolysing analogue of pyrophosphoserine (see the representative structure 54 in Fig. 17) were obtained by solid-phase synthesis from bisphosphonate analogue 55. They exerted enhanced stability in cell lysates and plasma, and therefore may serve as tools for studying, the marginally characterized to date, role of pyrophosphorylation.73 In order to generate antibodies they were ligated with bovine serum albumin. The ligations were carried out: (i) by reaction of mimetic-hydrazide with N-terminal cysteine of chosen oligopeptide followed by radical desulfurization (formation of alanine); (ii) by reaction of N-terminal cysteine of the mimetic peptide with maleimide-activated bovine albumin.73
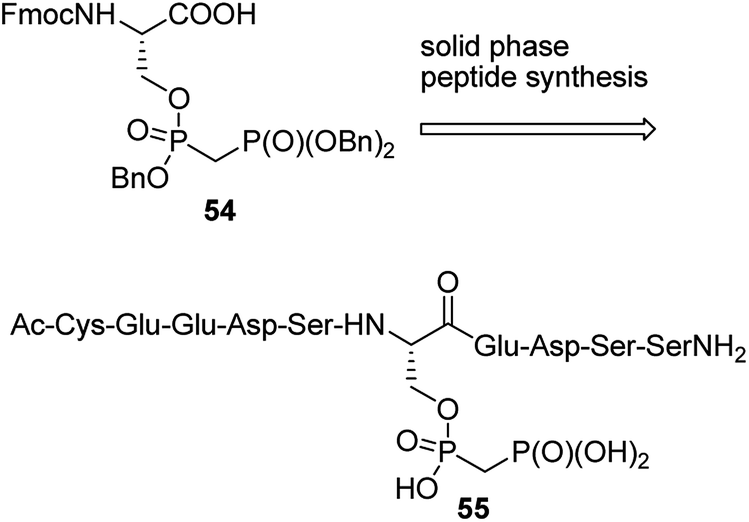 | ||
| Fig. 17 Peptides containing mimetic of pyrophosphate-substituted serine. | ||
N-Carboxyanhydrides of diisopropyl phosphonoalanine and homoalanine (compounds 56 and 57) underwent metallo-catalysed living polymerization yielding peptides of narrow chain length distribution and chain lengths to over 150 residues (Fig. 18). Copolymerization of compound 56 with appropriate N-carboxyanhydride of protected lysine provided block copolymers in high yields and low polydispersity indices. The obtained peptide exhibited pH-responsive conformational changes (Fig. 18).74
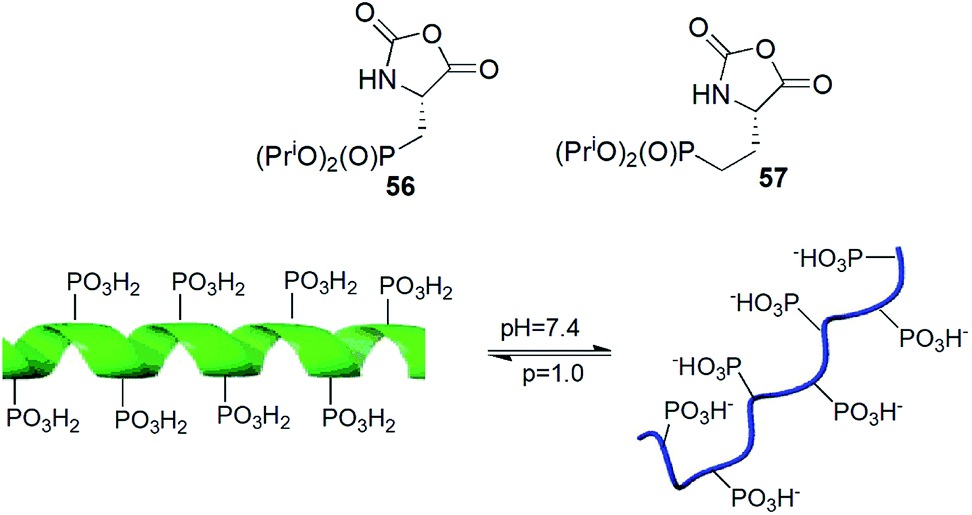 | ||
| Fig. 18 pH-responsive polymers of phosphoserine mimetics. | ||
Non-hydrolysable phosphonic mimetics of phosphotyrosine were designed as molecular probes to study Src SH2 homology domains and tyrosine phosphatases. p-(Phosphonomethyl)-L-phenylalanine (compound 58, Fig. 19) and its difluoro-derivative 59 are the most popular here.75,76 The latter one better mimics electronic structure and acidity of mother counterpart. In last decade, however, the number of papers devoted to phosphonic analogues of phosphotyrosine decreased significantly and they relate rather to design of new effectors of specific proteins or their use in construction of diagnostic tools.
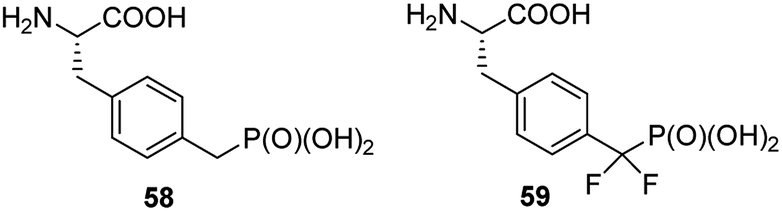 | ||
| Fig. 19 Analogues of phosphotyrosine designed to study Src SH2 homology domains and tyrosine phosphatases. | ||
Benzoylphosphonate analogue 60 (Fig. 20) appeared to act as photoactive, stable isoster of phosphotyrosine. It was introduced into two inhibitors (compounds 61 and 62, Fig. 20), which are derived from phosphopeptide substrates of protein tyrosine phosphatase PTP1B. Photo-activation of these inhibitors results in the formation of triplet biradical intermediates of the benzoylphosphonate moiety, which deactivate PTP1B by an oxidative radical oxidation of active site Cys125.77 These peptides if used in cell lysates or even living cells might possibly be useful tools in the time- and spatially resolved switching of PTP activities.
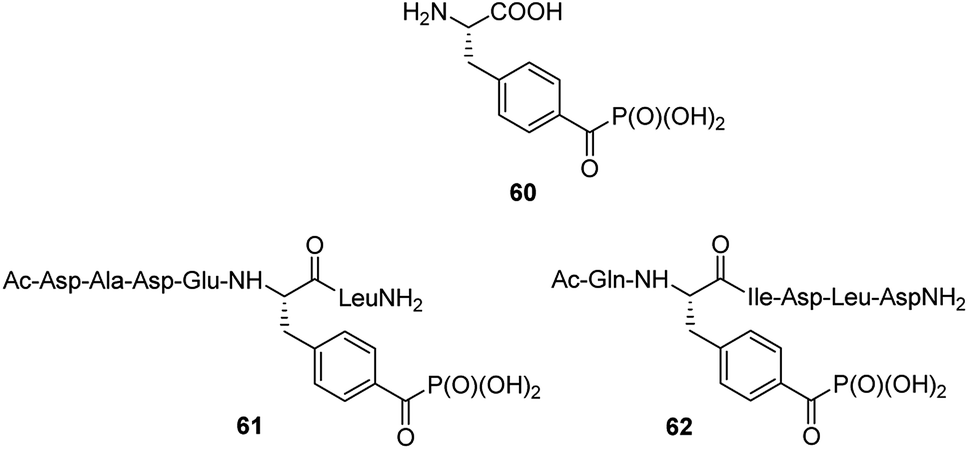 | ||
| Fig. 20 Photoactive analogue of phosphotyrosine and its peptides. | ||
In response to cytokines and growth factors, STAT protein family members are phosphorylated by the receptor-associated kinases, and then form homo- or heterodimers that translocate to the cell nucleus where they act as transcription activators.
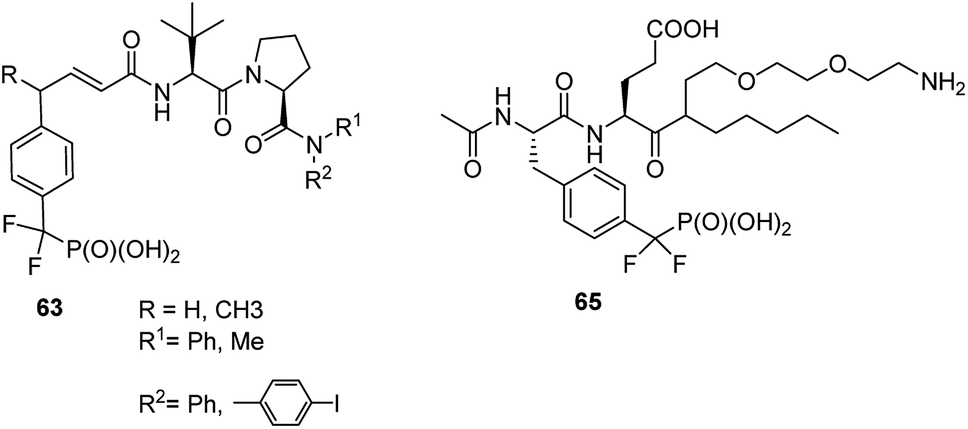
STAT6 plays a central role in exerting interleukins 4 and 13 (IL-4 and IL-13) mediated biological responses and thus modulate immunological response. Specific inhibitor of STAT6 containing compounds 63 (Fig. 21) were designed and synthesized.78 They block recruitment to phosphotyrosine residues on IL-4 or IL-13 receptors and subsequent Tyr641 phosphorylation, which in turn results in inhibition of the expression of genes contributing to asthma.
 | ||
| Fig. 21 Peptides containing non-hydrolysable analogue of phosphotyrosine. | ||
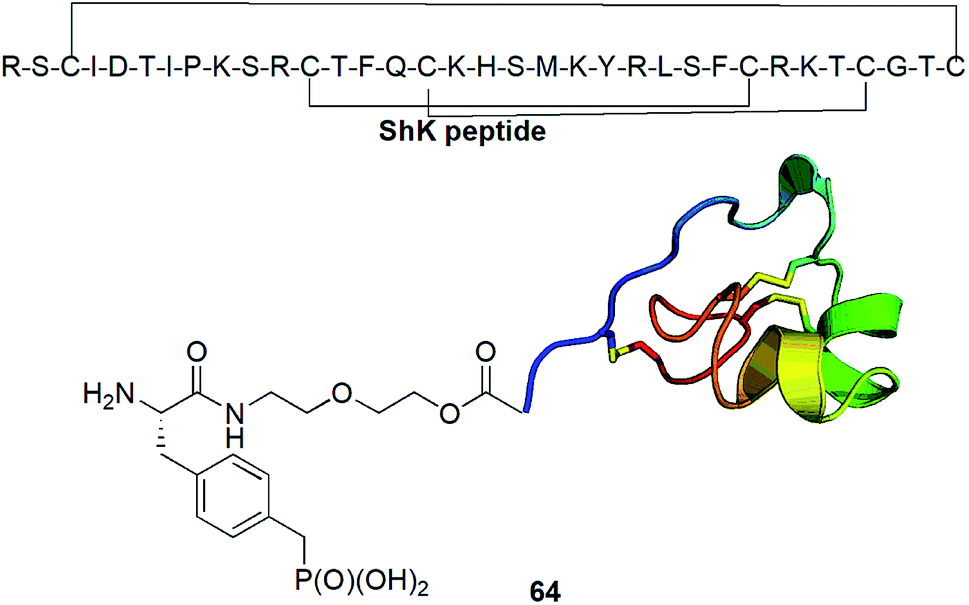
Kv1.3 is one of 76 human potassium homotetrameric channels, which regulate membrane potential and calcium signalling in human T cells. It is considered as target for therapeutics for multiple sclerosis. Recently a 35-residue disulfide-rich ShK peptide that blocks the voltage-gated potassium channels Kv1.3 and Kv1.1 has been isolated from sea anemone Stichodactyla helianthus. Modification of N-terminus of this peptide with p-(phosphonomethyl)phenylalanine (conjugate 64, Fig. 22) resulted in highly selective picomolar blocker of this channel.79
 | ||
| Fig. 22 Modified Stichodactyla helianthus peptide as potential agent against multiple sclerosis. | ||
Identification of particular SH2 domains is of importance since it may be used to shape the diagnosis. Consequently, the first step here is the enrichment of all (or at least majority) SH2 proteins present in biological material. Constructing an affinity resin containing general affinity probe could be a resolution of this problem. The preliminary studies with compound 65 (Fig. 21) have shown that this is possible since this compound was used for successful enrichement of 22 SH2 proteins from mixed cell lysates containing 50 of these proteins.80
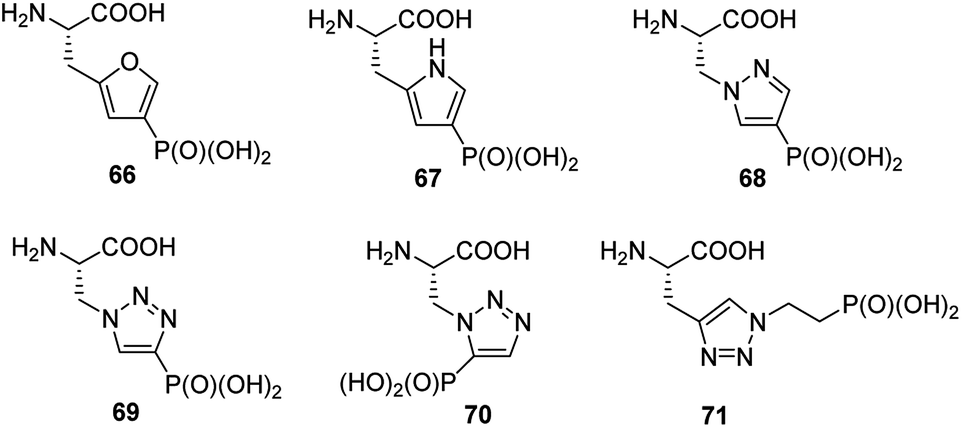
Phosphorylation of histidine is recognized as being critical to signaling processes in bacteria, fungi and plants. Their role in eukaryotes is far less understood. In order to overcome the problems of low stability and isomerism observed for phosphohistidine (Fig. 24) their stable analogues have been synthesized and used as tools (compounds 66–71, Fig. 23). Their use and utility has been recently reviewed.81–83
 | ||
| Fig. 23 Non-hydrolysable analogues of phosphohistidine. | ||
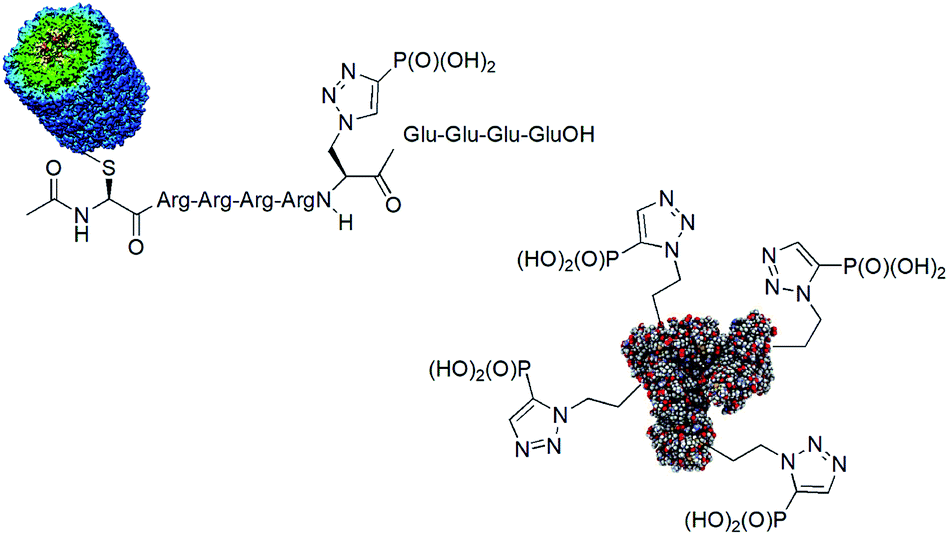
One of the drawbacks in studies on the role of histidine phosphorylation in mammals is a lack of specific antibodies to both isomers of phosphohistidine. Some studies on the use of phosphonate mimics of isomers of phosphohistidine have already been published. Such antibodies were raised by using oligopeptides containing these mimetics conjugated with bovine serum albumin (BSA)84,85 and keyhole limpet hemocyanin (KLH).86,87 The structures of the representative antigens obtained are shown in Fig. 24. The preliminary results of the cell studies are promising.
 | ||
| Fig. 24 Representative antigens containing mimetics of phosphohistidine. | ||
Phosphohistidine has two isomeric forms (1-phosphohidstidine and 3-phosphohidtidine, Fig. 25) and both of them exist in nature. Phosphoryltriazoylalanine analogues (compounds 69 and 70, Fig. 23) represent nonhydrolysable and nonisomerisable analogues of these isomers and both were used for construction of the above-mentioned antibodies (Fig. 24). They are readily available thanks to application of Huisgen dipolar cycloaddition (click reaction) of the commercially available ethynylphosphonate (compound 72) and N-protected serine-derived azides (for example, Fmoc protected compound 73). Regioselectivity of the reaction is strongly affected by applied catalyst and, to a lesser extent, by carboxy-protected group (Fig. 25). Importantly, click reaction could be also applied using peptides containing diaminopropionic acid inside the peptide chain.81,88,89
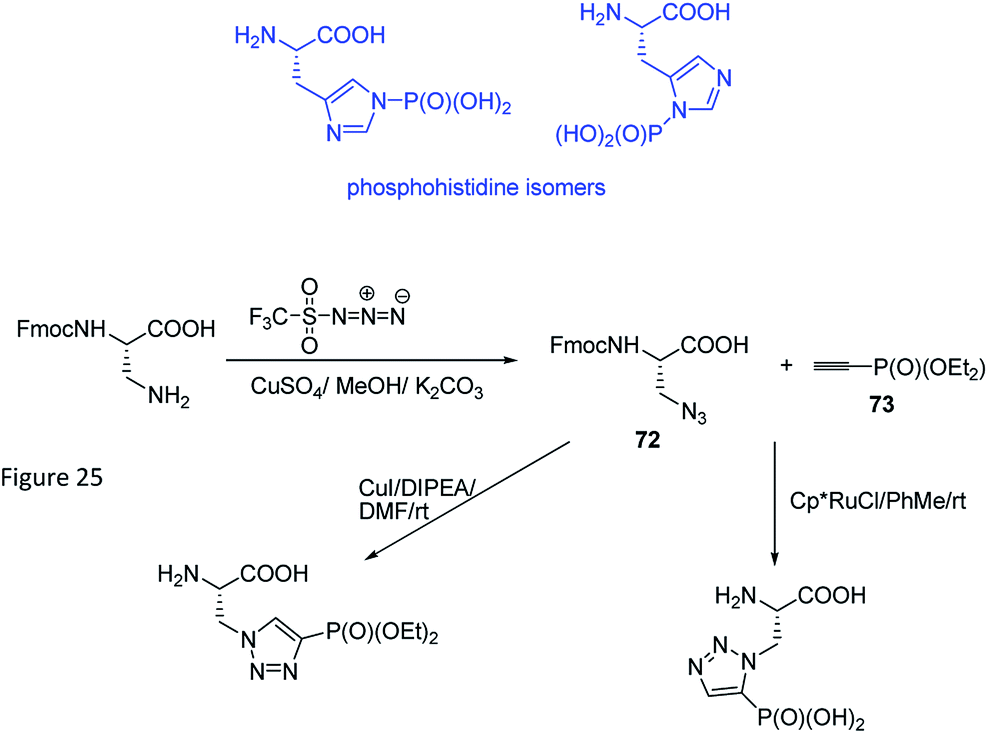 | ||
| Fig. 25 Synthesis of analogues of phosphohistidine via click reaction. | ||
Synthesis of phophonopyrazol-4-yl alanine (compound 68, Fig. 23) represents second type of general syntheses of phosphohistidine mimetics (Fig. 26). It starts from the ring opening of the Vederas β-lactone 74 with appropriate histamine analogue (in this case 4-iodopyrazole) yielding the acid 75, which after protection is phosphonylated by means of Hirao cross-coupling reaction. The two-step deprotection of the resulting product (compound 76) yields desired compound 68.87
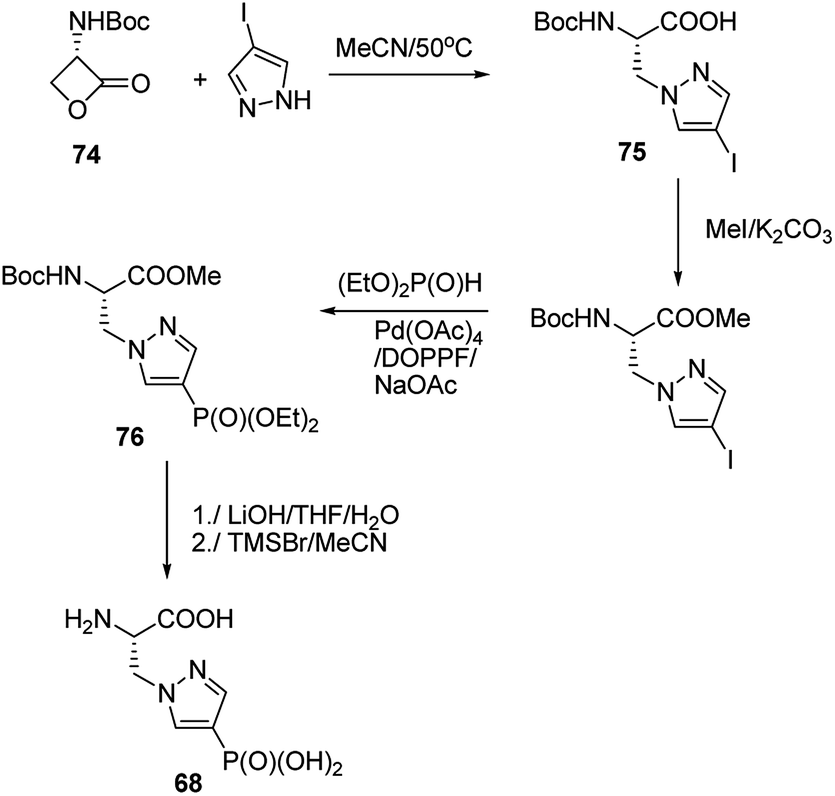 | ||
| Fig. 26 Synthesis of phosphohistidine analogue 68. | ||
Homologue of phosphonic mimetic of phosphohistidine (compound 77, Fig. 27) was found to be inactive. However, comparison of its structure with phosphotyrosine revealed that it might be an interesting mimetic of the latter. Indeed after inclusion into peptide chain (compound 78, Fig. 27) it was found to bind SH2 domain of eukaryotic cell signalling protein Grb2. Since its synthesis is relatively simple it is considered as more accessible mimetic of phosphotyrosine than current alternatives.89
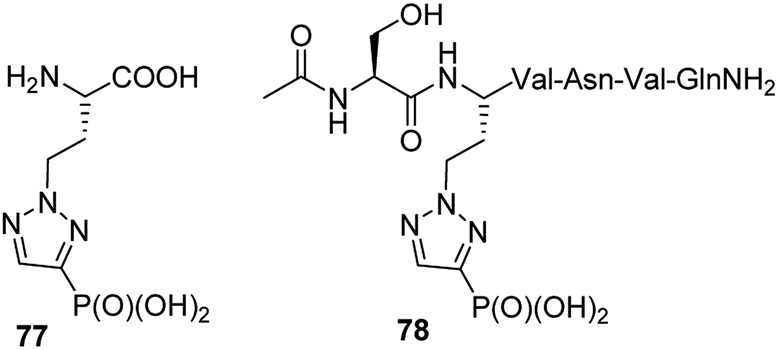 | ||
| Fig. 27 Peptide containing analogue of phosphohistidine 76, which acts as mimic of phosphotyrosine. | ||
D Peptides and proteins functionalized with phosphonic acid group
In this paragraph some dispersed data on synthesis of peptides and proteins containing phosphonic acid groups bound via different linkers is described. These entities were usually designed on purpose and were devoted to study selected physiologic processes.CheY protein is involved in the transmission of sensory signals from the chemoreceptors to the bacterial flagellar motors because its binding to a switch component induces a change from counterclockwise to clockwise flagellar rotation and thus influence of bacterial chemotaxis. Better understanding of this process should lead to additional insights into bacterial pathogenicity. Phosphorylation of CheY promotes this association. However, biochemical studies of activated CheY-phosphate have been challenging due to the rapid hydrolysis of the aspartyl-phosphate in vitro. Therefore, mutated CheY, containing cysteine in place of active aspartyl acid, was alkylated with phosphonomethyltriflate providing non-hydrolysable analogue of phosphorylated CheY (conjugate 79, Fig. 28).90 It was found to bind effectively to the switch component in similar manner as CheY-phosphate.
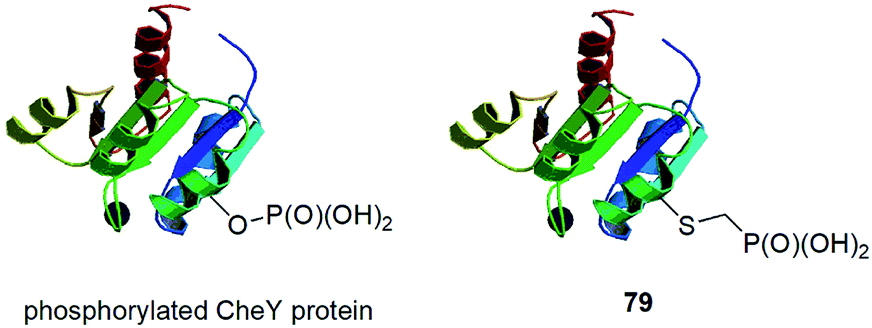 | ||
| Fig. 28 CheY protein functionalized with phosphorus fragments. | ||
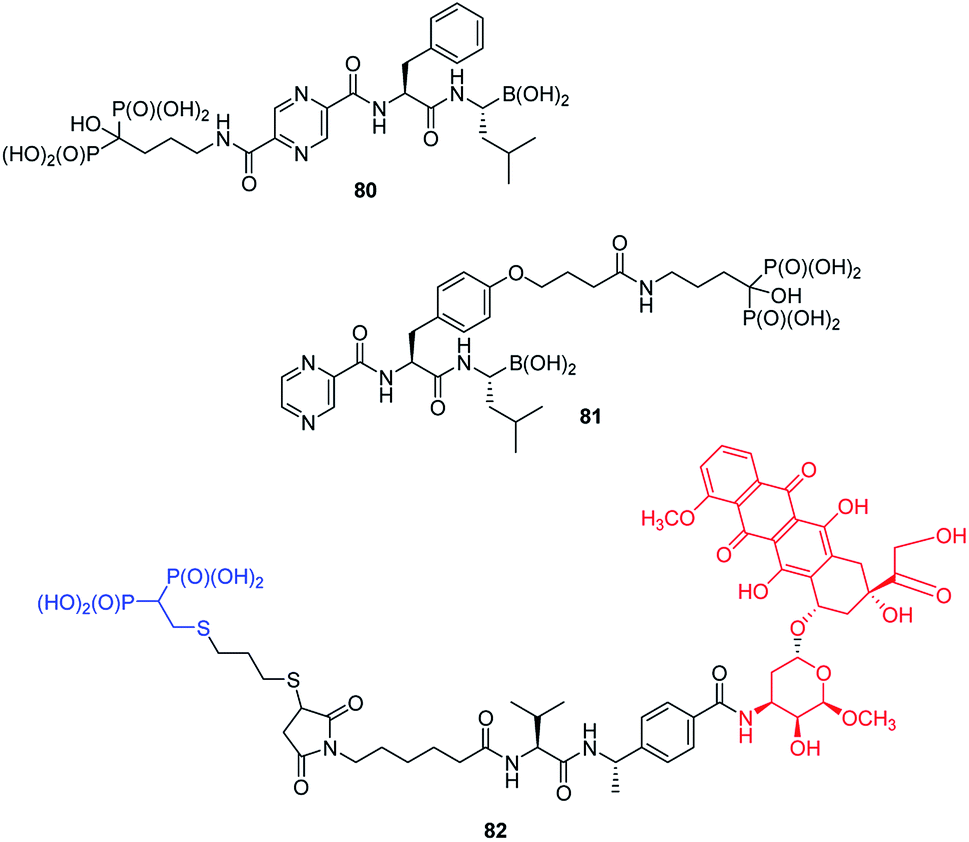
Functionalization of peptides with bisphosphonic acid rises as a standard procedure for delivery of drugs to bone tissue. Thus, conjunction of potent inhibitor of proteasome with potent antiosteoporotic alendronate yielded two types of compounds: 80 and 81 (Fig. 29) designed as drugs against multiple myelanoma,91 whereas conjunction of doxorubicin with bisphosphonate using peptidyl linker provided compound 82, which is a potential drug against bone metastases.92
 | ||
| Fig. 29 Bisphosphonates as drug carriers. | ||
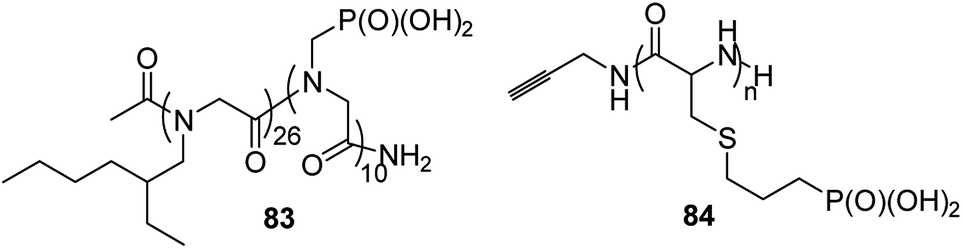
Polymers that conduct protons are of crucial importance in clean energy research, since they are an ideal material for constructing hydrogel gels or mimetics of photosynthesis. A series of diblock copolymers 83 (Fig. 30) of poly-N-(2-ethyl)hexylglycine and poly-N-phosphonmethylglycine upon hydration (8 water molecules per phosphonate moiety) effectively conduct protons, which makes them particularly attractive for electrochemical applications.93
 | ||
| Fig. 30 Polymeric peptides of phosphonate holding amino acids. | ||
Phosphopolypeptides and their non-hydrolysable analogues are synthesized as mimetics of phosphorylated proteins that are involved in biomineralization. Such peptides (compounds 84, Fig. 30)94 were synthesized starting from N-propylphospho-cysteine N-carboxyanhydride by simple polymerization upon action of primary amines.
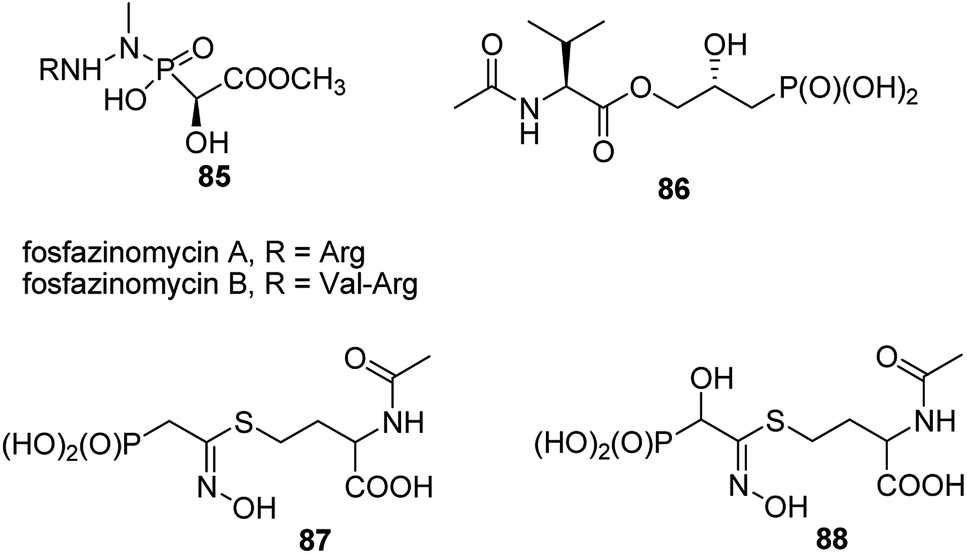
A very specific cases worth to mention are: antifungal and antibacterial fosfazinomycins (compounds 85, Fig. 31)95–97 and valinophos (compound 86).23 Fosfazomycins are azapeptidyl analogues of phosphinopeptides and are rather representative of compounds with phosphonic group replacing peptide bond, whereas valinophos may be classified as phosphonodepsipeptide. Also recently isolated phosphonocystoximate and its hydroxyl-derivative (compounds 87 and 88, Fig. 29)23 could be considered as specific peptidomimetics.
 | ||
| Fig. 31 Naturally occurring phosphonate peptidomimetics. | ||
E Conclusions
Both groups of reviewed phosphonopeptides – those containing phosphonic acid moiety at C-terminus and those being nonhydrolysable mimetics of phosphonoamino acids, were discovered in 70s last century. Progress of the studies on these compounds has been quite slow, however, in last decade significant acceleration was noticed. In the case of antibacterial peptides it resulted from both, discovery of novel antibiotics by innovative and elegant genomic approach and from repurposing of old or abandon antibacterial agents, which is stimulated by fast growth of number of resistant bacterial strains. Effective methods for the synthesis of these compounds had been elaborated and optimized in last century. However, they require accessibility to esters of aminophosphonic acids, which usually are not readily available and have to be synthesized by independent procedures. Thus, modifications of simple phosphonopeptide side chains might be considered as useful alternative.In the case of analogues of phosphoamino acids their possible functioning as molecular probes and generation of specific antibodies used as tolls for more detailed study on the role and physiological consequences of phosphorylations at cellular level is a driving force of their synthesis. Thus, research described so far was concentrated on the elaboration of simple and effective procedures for the synthesis of structurally variable mimetics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Science Centre of Poland (NCN) grant number 2016/21/B/ST5/00115, supported this work.Notes and references
- Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity, ed. V. Kukhar and H. Hudson, John Wiley & Sons, New York, 2000 Search PubMed.
- J. G. Allen, F. R. Atherton, M. J. Hall, C. H. Hassal, S. W. Holmes, R. W. Lambert, L. J. Nisbet and P. S. Ringrose, Nature, 1978, 272, 56–58 CrossRef CAS PubMed.
- E. Bayer, K. H. Gugel, K. Hägele, H. Hagenmaier, S. Jessipow, W. A. König and H. Zähner, Helv. Chim. Acta, 1972, 55, 224–239 CrossRef CAS PubMed.
- Y. Ogawa, T. Tsuruoka, S. Inouye and T. Niida, Sci. Rep. Meiji Seika Kaisha, 1973, 13, 42–48 Search PubMed.
- M. Maślanka and A. Mucha, Pharmaceuticals, 2019, 12, 86 CrossRef PubMed.
- E. Burchacka and M. Sieńczyk, Curr. Pharm. Des., 2018, 37, 4445–4465 Search PubMed.
- R. Grzywa and M. Sieńczyk, Curr. Pharm. Des., 2013, 19, 1154–1178 CrossRef CAS PubMed.
- A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed.
- A. Łupicka-Słowik, M. Psurski, R. Grzywa, M. Cuprych, J. Ciekot, W. Goldeman, E. Wojaczyńska, J. Wojaczyński, J. Oleksyszyn and M. Sieńczyk, Invest. New Drugs, 2020, 38 DOI:10.1007/s10637-020-00923-4.
- S. McGowan, C. J. Porter, J. Lowther, C. M. Stack, S. J. Golding, T. S. Skinner-Adams, K. R. Trenholme, F. Teuscher, S. M. Donnelly, J. Grembecka, A. Mucha, P. Kafarski, R. Degori, A. M. Buckle, D. L. Gardiner, J. C. Whisstock and J. P. Dalton, Proc. Natl. Acad. Sci., 2009, 106, 2537–2542 CrossRef CAS PubMed.
- T. R. Burke, M. S. Smyth, M. Nomizu, A. Otaka and P. R. Roller, J. Org. Chem., 1993, 58, 1336–1340 CrossRef CAS.
- M. Talma and A. Mucha, Biomolecules, 2020, 10, 659 CrossRef CAS PubMed.
- M. Talma, M. Maślanka and A. Mucha, Bioorg. Med. Chem. Lett., 2019, 29, 1031–1042 CrossRef CAS PubMed.
- M. M. Abdou, P. M. O'Neill, E. Amigues and M. Matziari, Drug Discovery Today, 2019, 24, 916–929 CrossRef CAS PubMed.
- D. Georgiadis and V. Dive, Top. Curr. Chem., 2015, 360, 1–38 CAS.
- A. Mucha, Molecules, 2012, 17, 13530–13568 CrossRef CAS PubMed.
- F. R. Atherton, M. J. Hall, C. H. Hassal, R. W. Lambert, W. J. Lloyd and P. S. Ringrose, Antimicrob. Agents Chemother., 1979, 15, 696–705 CrossRef CAS PubMed.
- M. Leason, D. Cunliffe, D. Parkin, P. J. Lea and B. J. Miflin, Phytochemistry, 1982, 21, 855–857 CrossRef CAS.
- S. Omura, M. Murata, H. Hanaki, K. Hinotozawa, R. Oiwa and H. Tanaka, J. Antibiot., 1984, 37, 829–835 CrossRef CAS PubMed.
- M. Gahungu, A. Arguelles-Arias, P. Fickers, A. Zervosen, B. Joris, C. Damblon and A. Luxen, Bioorg. Med. Chem., 2013, 4958–4967 CrossRef CAS PubMed.
- R. D. Johnson, R. M. Kastner, S. H. Larsen and E. E. Ose, US Pat. 4,482,488, 1984.
- B. T. Circello, C. G. Miller, J.-H. Lee, W. A. van der Donk and W. W. Metcalf, Antimicrob. Agents Chemother., 2011, 55, 3357–3362 CrossRef CAS PubMed.
- K.-S. Ju, J. Gao, J. R. Doroighazi, K.-K. A. Wang, C. J. Thibodeaux, S. Li, E. Metzger, J. Fudala, J. Su, J. K. Zhang, J. Lee, J. P. Cioni, B. S. Evans, R. Hirota, D. P. Labeda, W. A. van der Donk and W. W. Metcalf, Proc. Natl. Acad. Sci., 2015, 112, 12175–12180 CrossRef CAS PubMed.
- M. Yamato, T. Koguchi, R. Okachi, K. Yamada, K. Nakayama, H. Kase, A. Karasawa and K. Shuto, J. Antibiot., 1986, 39, 44–52 CrossRef CAS PubMed.
- G. J. Kramer, A. Mohd, L. S. U. Schwager, G. Masuyer, K. R. Acharya, E. D. Sturrock and B. O. Bachmann, ACS Med. Chem. Lett., 2014, 5, 346–351 CrossRef CAS PubMed.
- A. Mucha and P. Kafarski, Methods Mol. Biol., 2020, 2103, 287–301 CrossRef CAS PubMed.
- N. Rabasso and N. Fadel, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 1811–1819 CrossRef CAS.
- J. M. Cervera-Villanueva Jr, J. L. Viveros-Ceballos, I. Lizanga-Elizalde and M. Ordóñez, J. Pept. Sci., 2015, 22, 70–75 CrossRef PubMed.
- K. V. Turcheniuk, K. O. Poliashko, V. P. Kukhar, A. B. Rozhenko, V. A. Soloshonok and A. F. Sorochinsky, Chem. Commun., 2012, 48, 11519–11521 RSC.
- M. Cortes-Clerget, O. Gager, M. Monteil, J.-L. Pirat, E. Migianu-Griffoni, J. Deschamp and M. Lecouvey, Adv. Synth. Catal., 2016, 358, 34–40 CrossRef CAS.
- M. Cortes-Clerget, J. Jover, J. Dussart, E. Kolodziej, M. Monteil, E. Migianu-Griffoni, O. Gager, J. Deschamp and M. Lecouvey, Chem.–Eur. J., 2017, 23, 6654–6662 CrossRef CAS PubMed.
- M. O. Clarke, X. Chen, A. Cho, W. E. Delaney IV, E. Doerffler, M. Fardis, M. Je, M. Mertzman, R. Pakdaman, H.-J. Pyun, T. Rowe, C. Y. Yang, X. C. Sheng and C. U. Kim, Bioorg. Med. Chem. Lett., 2011, 21, 3568–3572 CrossRef CAS PubMed.
- E. C. L. Marss, L. Varadi, A. F. Bedernjak, K. M. Day, M. Gray, A. L. Jones, S. P. Cummings, R. J. Anderson and J. D. Perry, Molecules, 2020, 25, 1445 CrossRef PubMed.
- K. T. Ng, J. D. Perry, E. C. L. Marss, S. Orenga, R. J. Anderson and M. Grey, Molecules, 2020, 25, 1557 CrossRef CAS PubMed.
- D. J. Bougioukou, C. P. Ting, S. C. Peck, S. Mukherjee and W. A. van der Donk, Org. Biomol. Chem., 2019, 17, 822–829 RSC.
- M. M. Jiménez-Andreu, F. J. Sayago and C. Cativiela, Eur. J. Org. Chem., 2018, 3965–3973 CrossRef.
- V. P. Kukhar and V. D. Romanenko, in Amino Acids, Peptides and Proteins in Organic Chemistry: Modified Amino Acids, Organocatalysis and Enzymes, ed. A. B. Hughes, Wiley-VCH Verlag GmbH & Co, Weinheim, 2010, Ch. 5, vol. 2 Search PubMed.
- R. A. Shastri, Chem. Sci. Trans., 2019, 8, 359–367 CAS.
- B. Sun and J. Xu, Synthesis, 2015, 47, 1479–1487 CrossRef CAS.
- B. Sun and J. Xu, J. Pept. Sci., 2015, 21, 615–619 CrossRef CAS PubMed.
- J. Xu, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 487–492 CrossRef CAS.
- F. Meng, F. He, X. Song, L. Zhang, W. Hu and G. Liu, Amino Acids, 2012, 43, 423–429 CrossRef CAS PubMed.
- B. Sun, M. K. Sundaraneedi, H. M. Southam, R. K. Poole, I. F. Musgrave, F. R. Keene and J. G. Collins, Dalton Trans., 2019, 48, 14505–14515 RSC.
- J. Liu, P. Liao, J. Hu, H. Zhu, Y. Wang, Y. Li, Y. Li and B. He, Molecules, 2017, 16, 238 CrossRef PubMed.
- P. Liao, S.-Q. Hu, H. Zhang, L.-B. Xu, J.-Z. Liu, B. He, S.-G. Liao and Y.-J. Li, Bull. Korean Chem. Soc., 2018, 39, 300–304 CrossRef CAS.
- M. Drąg, E. Wieczerzak, M. Pawełczak, Ł. Berlicki, Z. Grzonka and P. Kafarski, Biochimie, 2013, 95, 1640–1649 CrossRef PubMed.
- A. F. G. Gargano, S. Buchinger, M. Kohout, W. Lindner and M. Lämmerhoffer, J. Org. Chem., 2013, 78, 10077–10087 CrossRef CAS PubMed.
- A. F. G. Gargano, T. Leek, W. Lindner and M. Lämmerhoffer, J. Chromatogr. A, 2013, 1317, 12–21 CrossRef CAS PubMed.
- K. M. Kondratyuk, E. I. Lukashuk, A. V. Golovchenko, E. B. Rusanov and V. S. Brovarets, Russ. J. Gen. Chem., 2012, 28, 643–651 CrossRef.
- K. M. Kondratyuk, E. I. Lukashuk, A. V. Golovchenko, I. V. Komarov, V. S. Brovarets and V. P. Kukhar, Tetrahedron, 2013, 69, 6251–6261 CrossRef CAS.
- O. I. Lukashuk, E. R. Abdurakhamova, K. M. Kondratyuk, O. V. Golovchenko, K. V. Khokhlov, V. S. Brovarets and V. P. Kukhar, RSC Adv., 2015, 5, 11198–11206 RSC.
- E. I. Lukashuk, E. R. Abdurakhmanova, K. M. Kondratyuk, A. V. Golovchenko and V. S. Brovarets, Russ. J. Gen. Chem., 2015, 85, 71–74 CrossRef CAS.
- E. R. Abdurakhmanova, O. I. Lukashuk, A. V. Golovchenko and V. S. Brovarets, Russ. J. Gen. Chem., 2016, 86, 1206 CrossRef CAS.
- B. Vanderhoydoncky and C. V. Stevens, Tetrahedron, 2007, 63, 7679–7689 CrossRef.
- P. Lenartowicz, B. Dziuk, B. Zarychta, M. Makowski and P. Kafarski, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 706–712 CrossRef CAS.
- F. Ardito, M. Giuliani, D. Perrone, G. Troiano and L. Lo Muzio, Int. J. Mol. Med., 2017, 40, 271–280 CrossRef CAS PubMed.
- Z. Chen and P. A Cole, Curr. Opin. Chem. Biol., 2015, 28, 115–122 CrossRef CAS PubMed.
- K. N. Chuh, A. R. Batt and M. R. Pratt, Cell Chem. Biol., 2016, 23, 86–107 CrossRef CAS PubMed.
- M. Arribat, F. Cavalier and E. Rémond, RSC Adv., 2020, 10, 6678–6724 RSC.
- A. M. Marmelstein, J. Moreno and D. Fiedler, Top. Curr. Chem., 2017, 375, 20 CrossRef PubMed.
- K. Panigrahi, D. L. Nelson and D. P. Berkovitz, Chem. Biol., 2012, 19, 666–667 CrossRef CAS PubMed.
- F. Liu, J.-E. Park, W.-J. Qian, D. Lim, A. Scharow, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke, ACS Chem. Biol., 2012, 7, 805–810 CrossRef CAS PubMed.
- F. Liu, J.-E. Park, W.-J. Qian, D. Lim, A. Scharow, T. Berg, M. B. Yaffe, K. S. Lee and T. R. Burke, ChemBioChem, 2012, 13, 1291–1296 CrossRef CAS PubMed.
- W.-J. Qian and T. R. Burke, Amino Acids, 2013, 45, 1143–1148 CrossRef CAS PubMed.
- D. Hymel and T. R. Burke Jr, ChemMedChem, 2017, 12, 202–206 CrossRef CAS PubMed.
- H.-Z. Duan, H.-X. Chen, Q. Yu, J. Hu, J.-M. Li and Y.-X. Chen, Org. Biomol. Chem., 2019, 17, 2099–2102 RSC.
- H.-X. Chen, J. Kang, R. Chang, Y.-L. Zhang, H.-Z. Duan, Y.-M. Li and Y.-X. Chen, Org. Lett., 2018, 20, 3278–3281 CrossRef CAS PubMed.
- J. Kang, H.-X. Chen, S.-Q. Huang, Y.-L. Zhang, R. Chang, F.-Y. Li, Y.-M. Li and Y.-X. Chen, Tetrahedron Lett., 2017, 58, 2551–2553 CrossRef CAS.
- A. A. Arrendale, K. Kim, J. Y. Choi, W. Li, R. L. Geahlen and R. F. Borch, ACS Chem. Biol., 2012, 19, 764–771 CrossRef CAS PubMed.
- K. Ebisuno, M. Denda, K. Oguro, T. Inokuma, A. Shigenaga and A. Otaka, Bioorg. Med. Chem., 2014, 22, 2984–2991 CrossRef CAS PubMed.
- V. Beránek, C. D. Reinkemeier, M. S. Zhang, A. D. Liang, G. Kym and J. W. Chin, Cell Chem. Biol., 2018, 25, 1067–1074 CrossRef PubMed.
- R. Klingberg, J. O. Jost, M. Schümann, K. A. Gelato, W. Fischle, E. Krause and D. Schwarzer, ACS Chem. Biol., 2015, 10, 138–145 CrossRef CAS PubMed.
- L. M. Yates and D. Fiedler, ACS Chem. Biol., 2016, 11, 1066–1073 CrossRef CAS PubMed.
- I. Yakovlew and T. J. Deming, ACS Macro Lett., 2014, 3, 378–381 CrossRef.
- R. A. Cerulli and J. A. Kritzer, Org. Biomol. Chem., 2020, 18, 583–605 RSC.
- P. Morlacchi, F. M. Robertson, J. Klostergaard and J. S. Mac Murray, Future Med. Chem., 2014, 6, 1909–1926 CrossRef CAS PubMed.
- S. Wagner, A. Schütz and J. Rademann, Bioorg. Med. Chem., 2015, 23, 2839–2847 CrossRef CAS PubMed.
- P. K. Mandal, P. Moriacchi, J. M. Knight, T. M. Link, G. R. LeeIV, R. Nurieva, D. Singh, A. Dhanik, L. Kavaraki, D. B. Corry, J. E. Ladbury and J. S. McMurray, J. Med. Chem., 2015, 58, 8970–8984 CrossRef CAS PubMed.
- M. W. Pennington, S. C. Chang, S. Chauhan, R. Huq, R. B. Tajhya, S. Chhabra, R. S. Norton and C. Beeton, Mar. Drugs, 2015, 13, 529–542 CrossRef PubMed.
- M. Höfener, S. Heinzmleir, B. Kuster and N. Sewald, Proteome Sci., 2014, 12, 41 CrossRef PubMed.
- T. E. McAllister, M. G. Nix and M. E. Webb, Chem. Commun., 2011, 47, 1297–1299 RSC.
- M. V. Makwana, R. Muimo and R. F. W. Jackson, Lab. Invest., 2018, 98, 291–303 CrossRef CAS PubMed.
- T. E. McAllister, J. J. Hollins and M. E. Webb, Biochem. Soc. Trans., 2013, 41, 1072–1077 CrossRef CAS PubMed.
- J.-M. Kee, R. C. Oslund, D. H. Perlman and T. W. Muir, Nat. Chem. Biol., 2013, 9, 416–421 CrossRef CAS PubMed.
- S. R. Fuhs, J. Meisenhelder, A. Aslanian, L. Ma, A. Zagorska, M. Stankova, A. Binnie, F. Al-Obeidi, J. Mauger, G. Lemke, J. R. Yates III and T. Hunter, Cell, 2015, 162, 198–210 CrossRef CAS PubMed.
- J.-M. Kee, R. C. Oslund, A. D. Couvillon and T. W. Muir, Org. Lett., 2015, 17, 187–189 CrossRef CAS PubMed.
- M. Lilley, B. Mambwe, M. J. Thompson, R. F. W. Jackson and R. Muimo, Chem. Commun., 2015, 51, 7305–7308 RSC.
- J.-M. Kee, B. Villani, L. R. Carpenter and T. W. Muir, J. Am. Chem. Soc., 2010, 132, 14327–14329 CrossRef CAS PubMed.
- T. E. McAllister, K. A. Horner and M. E. Webb, ChemBioChem, 2014, 15, 1088–1091 CrossRef CAS PubMed.
- M. S. Beyersdorf, R. Sircar, D. B. Lookadoo, C. J. Bottone, M. J. Lynch, B. R. Crane and C. J. Halkides, Protein Sci., 2017, 26, 1547–1554 CrossRef CAS PubMed.
- J. K. Agyin, B. Santhamma and S. S. Roy, Bioorg. Med. Chem. Lett., 2013, 23, 6355–6458 CrossRef PubMed.
- K. Hochdörffer, K. Abu Ajaj, C. Schäfer-Obodozie and F. Kratz, J. Med. Chem., 2012, 55, 7501–7512 CrossRef PubMed.
- J. Sun, X. Jing, A. Siegmund, M. D. Conolly, K. H. Downing, N. P. Balsara and R. N. Zuckermann, Macromolecules, 2016, 49, 3083–3090 CrossRef CAS PubMed.
- S. Das, M. Kar and S. S. Gupta, Polym. Chem., 2013, 4, 4087–4091 RSC.
- S. Gunji, K. Arima and T. Beppu, Agric. Biol. Chem., 1983, 41, 2016–2069 Search PubMed.
- Z. Huang, K.-K. A. Wang and W. A. van der Donk, Chem. Sci., 2016, 7, 5219–5223 RSC.
- J. Gao, K.-S. Ju, X. Yu, J. E. Velásques, S. Mukherjee, J. Lee, C. Zhao, B. S. Evans, J. R. Doroghazi, W. W. Metcalf and W. A. van der Donk, Angew. Chem., Int. Ed., 2014, 53, 1334–1337 CrossRef CAS PubMed.
Footnote |
| † This article is dedicated in honour of Professor Zbigniew Czarnocki on his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2020 |