
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVinylazaarenes as dienophiles in Lewis acid-promoted Diels–Alder reactions†
Anna E.
Davis
,
Jared M.
Lowe
and
Michael K.
Hilinski
 *
*
Department of Chemistry, University of Virginia, Charlottesville, Virginia 22904-4319, USA. E-mail: hilinski@virginia.edu
First published on 24th November 2021
Abstract
Described are the first examples of Lewis acid-promoted Diels–Alder reactions of vinylpyridines and other vinylazaarenes with unactivated dienes. Cyclohexyl-appended azaarenes constitute a class of substructures of rising prominence in drug discovery. Despite this, thermal variants of the vinylazaarene Diels–Alder reaction are rare and have not been adopted for synthesis, and Lewis acid-promoted variants are virtually unexplored. The presented work addresses this gap and in the process furnishes increased scope, dramatically higher yields, improved regioselectivity, and high levels of diastereoselectivity compared to prior thermal examples. These reactions provide scalable access to druglike scaffolds not readily available through other methods. More broadly, these studies establish a useful new class of dienophiles that, based on preliminary mechanistic studies, should be amenable to conventional strategies for enantioselective catalysis.
Introduction
The Diels–Alder reaction is widely regarded as one of the most useful and essential transformations in complex molecule synthesis,1,2 enabling the predictable construction of complex regio- and stereoselectively substituted rings with perfect atom economy. Nearly a century after the initial report, numerous classes of compounds have emerged as useful dienes and dienophiles for these reactions.3 Synthetic applications have also been aided by the development of dienophile activation strategies, such as the use of Lewis acids to promote sluggish reactions and influence regiochemical and stereochemical outcomes.4 However, because of the explosion of new Diels–Alder variants beginning in the latter half of the last century, the present-day identification of unexploited dienes and dienophiles that are structurally simple and readily available is rare. In this context, unactivated vinylazaarenes have largely eluded development as useful dienophiles despite early promise. In seminal work on this topic, Meek and Cristol and, subsequently, Doering and Rhoads, demonstrated that vinylpyridines could undergo thermal [4 + 2] cycloaddition with unactivated dienes such as butadiene and isoprene (Fig. 1A), albeit in low yield.5,6 In the intervening years, the adoption of these transformations for synthetic application has been notably absent from the literature. This is not due to lack of potential utility; cyclohexyl-appended azaarenes have proven to be useful structural motifs in drug discovery, with at least one member of this class (LGH-447) able to target the SARS-CoV-2 RNA dependent RNA polymerase (Fig. 1B).7,8 Current synthetic approaches to these compounds involve aryl-Csp3 C–C bond formation via cross coupling or nucleophilic addition, which generally depend on access to an appropriately substituted cyclohexanone or other prefunctionalized cyclohexane.9 Therefore, a synthetically useful Diels–Alder protocol would have the potential to transform the way these valuable scaffolds are synthesized, increasing the accessible structural diversity of the appended cyclohexane in a way that could be readily applied to the development of new medicines. Herein, we report a method for Diels–Alder reactions of several different classes of vinylazaarenes via Lewis acid activation (Fig. 1A). These processes represent the first examples of reagent control of reactivity, regioselectivity, and stereoselectivity in cycloadditions of this class of dienophiles with unactivated dienes.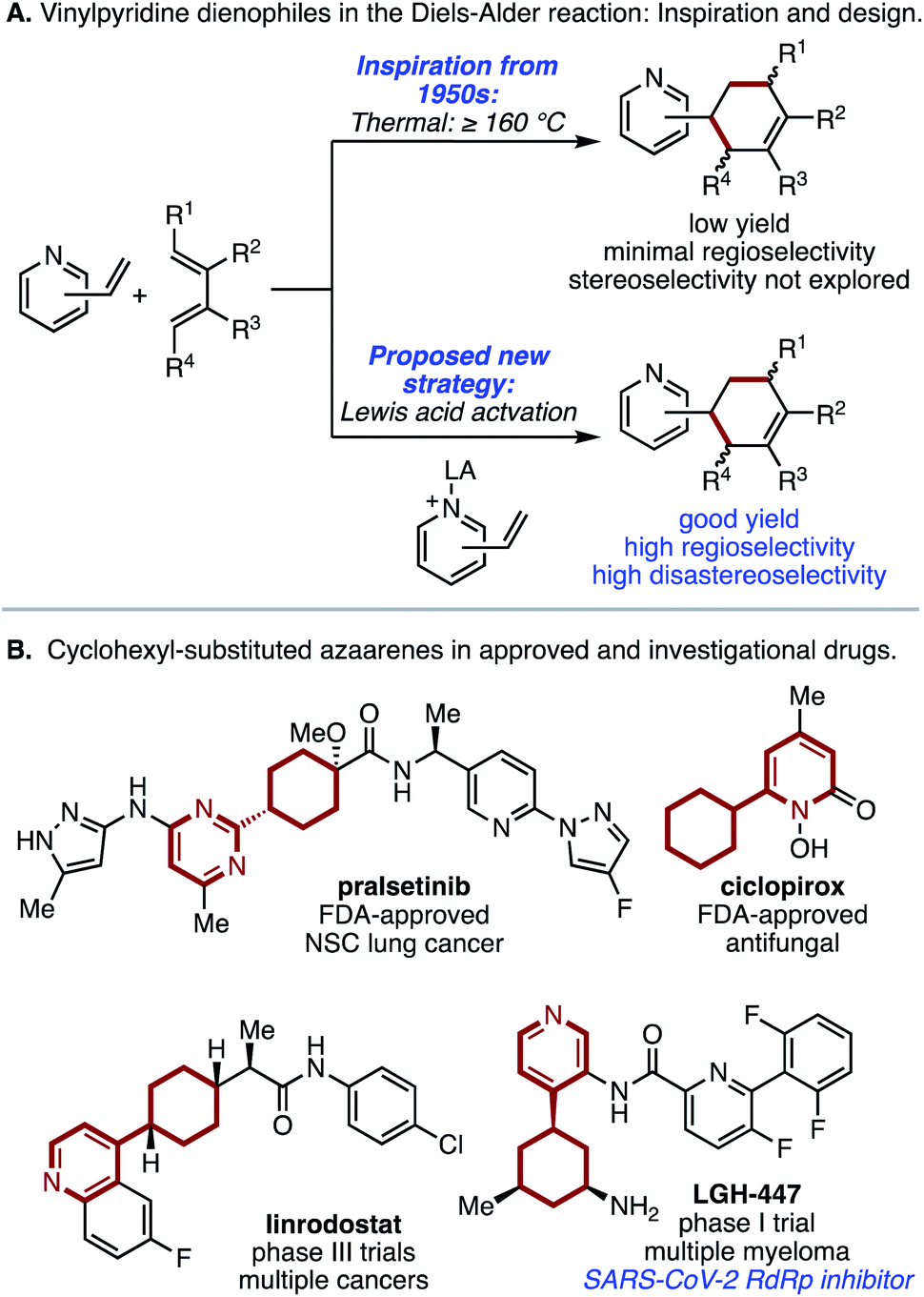 | ||
| Fig. 1 (A) Overview of vinylpyridine Diels–Alder variants. (B) Relevance of cyclohexylazaarenes to medicine. | ||
Results and discussion
We hypothesized that the strategy of Lewis acid activation of Diels–Alder dienophiles could be extended to vinylazaarenes, which might address deficiencies observed for thermal cycloadditions. Given the historical context, and the fact that pyridines are the second most common ring system of any type in FDA-approved drugs,10 we began by investigating the Diels–Alder reaction with vinylpyridine. A significant drawback to the thermal reaction is the reliance on the intrinsic reactivity of the diene and dienophile, leading to elevated reaction temperatures, low yields, and minimal regioselectivity.5,6,11 In our hands, the thermal Diels–Alder reaction between 4-vinylpyridine and isoprene provided the cycloadducts 3a and 3b in 9% combined yield and with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioselectivity favoring the 1,4-isomer, with minimal improvement in yield observed upon longer reaction times (Table 1, entries 1 and 2).12 In contrast, reports of catalyst- or reagent-controlled variants are almost entirely absent from the literature, including low-valent transition metal-catalyzed [4 + 2] cycloadditions. We have identified only one such example involving vinylpyridines – a Ni(0) catalyzed cycloaddition that, for the cycloaddition between 1 and 2, leads to a nonselective, statistical mixture of regioisomers in 30% yield.13 Both this process and a non-selective Cp2ZrCl2-promoted reaction reported alongside the Ni(0) work require nearly the same temperature as the thermal reaction (140–150 °C).
:1 regioselectivity favoring the 1,4-isomer, with minimal improvement in yield observed upon longer reaction times (Table 1, entries 1 and 2).12 In contrast, reports of catalyst- or reagent-controlled variants are almost entirely absent from the literature, including low-valent transition metal-catalyzed [4 + 2] cycloadditions. We have identified only one such example involving vinylpyridines – a Ni(0) catalyzed cycloaddition that, for the cycloaddition between 1 and 2, leads to a nonselective, statistical mixture of regioisomers in 30% yield.13 Both this process and a non-selective Cp2ZrCl2-promoted reaction reported alongside the Ni(0) work require nearly the same temperature as the thermal reaction (140–150 °C).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Temp. (°C) | Time (h) | Equiv. of 2 | Lewis acid | Yield 3a + 3b (%) |
3a:3b |
| a All reactions were conducted on 2 mmol scale using acetonitrile (4 mL) as the solvent. Yields and regioselectivities determined by gas chromatography. See ESI for details and additional data. b Isolated yield. | ||||||
| 1 | 170 | 24 | 1 | — | 9 | 2:1 |
| 2 | 170 | 72 | 1 | — | 13 | 2:1 |
| 3 | 40 | 24 | 1 | Zn(NO3)2 (0.025 equiv.) | <1 | — |
| 4 | 82 | 24 | 1 | Zn(NO3)2 (1 equiv.) | <1 | — |
| 5 | 70 | 24 | 1 | BF3·OEt2 (0.5 equiv.) | 18 | 5:1 |
| 6 | 70 | 24 | 1 | — | <1 | — |
| 7 | 70 | 72 | 2 | BF 3 ·OEt 2 (0.5 equiv.) | 54 |
5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 8 | 70 | 24 | 2 | BF3·OEt2 (0.5 equiv.) | 35 | 4:1 |
| 9 | 70 | 24 | 2 | BF3·OEt2 (1 equiv.) | 37b | 5:1 |
| 10 | 70 | 24 | 2 | BF3·OEt2 (0.4 equiv.) | 12 | 4:1 |
| 11 | 70 | 24 | 2 | AlCl3 (0.5 equiv.) | 12 | 4:1 |
We envisioned that Lewis acid complexation of the pyridine nitrogen would lower the activation energy for cycloaddition and, characteristically, promote enhanced regio- and stereoselectivity compared to thermal examples. To our knowledge, there are no reported studies of this type, with vinylpyridines more frequently reported for their reactions as dienes rather than dienophiles.14 Competing reactions of potential vinylpyridine dienophiles with electron-rich dienes, such as conjugate addition and ene reactions, as well as polymerization, substantially interfere with successful production of Diels–Alder products.14 We have found only one reported instance of Lewis acid promotion of dienophile reactivity in the literature – an observation by Williams, in the course of an unrelated study, of a Zn(NO3)2·6H2O-catalyzed cycloaddition between either 2-vinylpyridine or 4-vinylpyridine and the highly reactive diene pentamethylcyclopentadiene.15 In light of this, we continued our efforts by assessing these conditions using other hydrocarbon dienes of varying reactivity, but found that those less reactive than cyclopentadiene (e.g. isoprene) provided no observable cycloadducts, even when the temperature was increased and an equimolar amount of Zn(NO3)2·6H2O was used (Table 1, entries 3 and 4). In contrast, upon investigation of the use of other Lewis acids, we found that aluminum and boron-based Lewis acids were well-suited to the desired reagent-controlled reaction, with the use of BF3·OEt2 providing the highest yields. Using the optimal amount (0.5 equiv.), at a temperature (70 °C) significantly below the lower limit for a productive thermal reaction, the cycloadduct is produced in 18% yield after 24 h (entries 5 and 6). By optimizing stoichiometry and extending the reaction time to 72 hours, a synthetically useful yield of a 5:1 mixture of 3a:3b was obtained (54%, entry 7).
As we hypothesized, the use of a Lewis acid also led to improved regioselectivity (5:1 favoring 3a), which we attribute to increased polarization of the vinyl group upon acid–base complexation.16 Overall, this initial success was highly encouraging since a particularly challenging reaction was selected for evaluation – isoprene is 600-fold less reactive than cyclopentadiene towards representative dienophile maleic anhydride17 and, as demonstrated, does not exhibit a particularly strong inherent regioselectivity preference. In addition, the extended conjugation and distance between the 4-vinylpyridine nitrogen and olefin relative to another choice of dienophile, such as 2-vinylpyridine, would be expected to limit both inductive and resonance effects of Lewis acid activation on reaction rate and regioselectivity.
The scope of this process with respect to both diene and dienophile was examined. In contrast to the reaction of 4-vinylpyridine with isoprene, most reactions provided good to excellent yield of Diels–Alder cycloadducts after 24 h (Fig. 2). Illustrating the strategic value of this method for synthetic planning, the reaction of 4- and 2-vinylpyridines with a variety of benchmark dienes follows predictable trends of reactivity and regioselectivity commonly observed with conventional dienophiles (Fig. 2a and b).17 Regarding the former, this is illustrated by the difference in the rate of formation of cyclopentadiene (7, 13) vs. 1,3-cyclohexadiene cycloadducts (8, 14), which required extended reaction times to achieve comparatively modest conversion. As expected, we found 2-vinylpyridine to be superior to 4-vinylpyridine when directly compared, with the former providing higher yields of similar cycloadducts while requiring shorter reaction times (e.g.8vs.14), and proceeding with considerably higher regioselectivity (e.g.5vs.11).
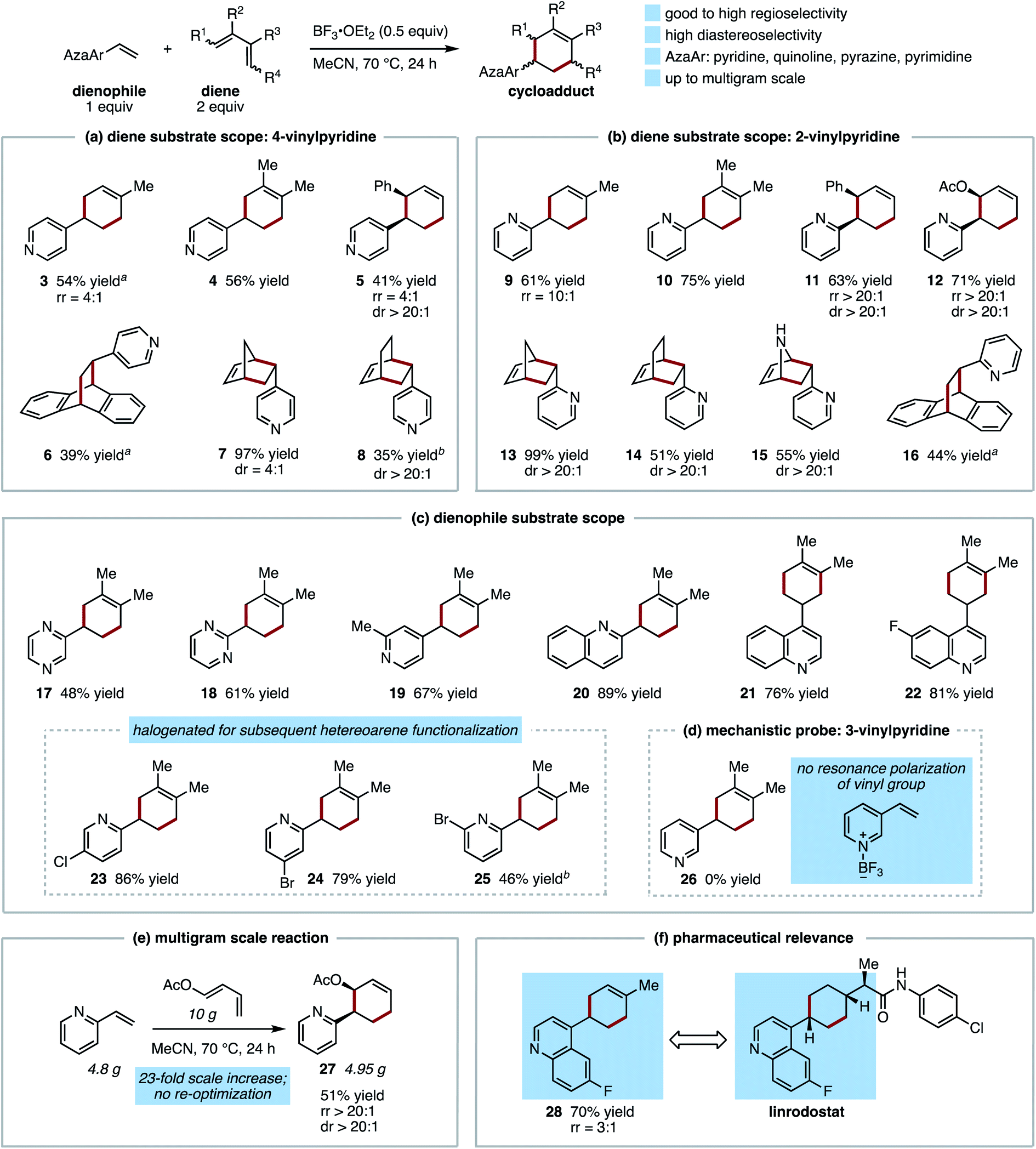 | ||
| Fig. 2 Scope of the vinylazaarene Diels–Alder reaction. All reactions conducted on 2 mmol scale and for 24 h unless otherwise noted; yields are of isolated products; reported regioselectivities and diastereoselectivities were determined via1H NMR integration of the generally inseparable pure product mixtures; major product isomers are shown. a Reaction time = 72 h. c Reaction time = 48 h. | ||
Generally high degrees of diastereoselectivity were also obtained, with endo diastereomers predominating. The selective construction of vicinal tertiary stereocenters in products 5 and 11, and an additional stereocenter in bridged bicyclic products 6–8 and 13–16, provides diastereoenriched motifs that are rare or previously unknown. These scaffolds are primed for additional stereoselective elaboration to cyclic or linear products. In addition, norbornenyl and bicyclo[2.2.2] octenyl pyridines 7, 8, 13, and 14 may find use as bioisosteres of phenylpyridines.18 Notably, even for dienes that are considerably more reactive than isoprene (e.g. cyclopentadiene), yields and diastereoselectivities are improved for the Lewis acid-promoted reaction (97% yield, 4:1 dr for 7) versus the thermal cycloaddition (24% yield, 3:1 dr for 7 after 24 h at 170 °C).
Evaluation of other vinylazaarenes with a focus on the nitrogen heterocycle employed suggests the potential for broad applicability of this protocol with respect to the dienophile (Fig. 2c). In addition to 2- and 4-vinylpyridines, 2-vinylpyrazine (17), 2-vinylpyrimidine (18), and 2- and 4-vinylquinolines (20–22) formed cycloadducts with 2,3-dimethylbutadiene. Of these, to our knowledge only vinylquinolines have been previously reported to undergo formal [4 + 2] cycloadditions, requiring the use of highly activated dienamines, in contrast to the unactivated hydrocarbon diene demonstrated herein.19 The 4-cyclohexenyl-quinoline (21) and 2-cyclohexenylpyrimidine (18) moieties relate closely to substructures found in representative drug molecules (Fig. 1b). Halogenation of the dienophile is also well tolerated; fluorine substitution (product 22) is particularly valued for drug discovery,20 and in addition, the tolerance of chlorine and bromine substitution at multiple positions on the 2-vinylpyridine scaffold (23–25) is expected to be useful for subsequent functionalization via cross coupling or other approaches.21,22
Limitations of dienophile scope (Fig. 2d) are in line with expected trends and consistent with a mechanism of Lewis acid activation of the azaarene. For example, under the optimized conditions, no reaction was observed between 2,3-dimethylbutadiene and 3-vinylpyridine, in which the effect of BF3 cannot be transmitted to the vinyl group through the π-system (product 26). This contrasts with observations made for thermal reactions, in which 3-vinylpyridine and 2-vinylpyridine exhibit nearly the same reactivity.6 In opposition to expected trends, the 1,1-disubstituted 2-isopropenylpyridine was unreactive, but this is likely due to an increased steric penalty associated with alkene-arene coplanarity. Under the optimized conditions 2-(prop-1-en-1-yl)pyridine (as a 3:1 mixture of E:Z isomers) also failed to react, but this is in line with expectations.17,23 These preliminary results do not preclude the possibility of successful Diels–Alder reactions of these dienophiles at higher temperatures or with more reactive dienes.
Scalability of the protocol and relevance to known pharmaceutical scaffolds are pertinent to potential future applications of this approach. Without reoptimizing conditions for multigram scale, cycloaddition between 4.68 grams of 2-vinylpyridine and 10 grams of trans-1-acetoxy-1,3-butadiene produces product 12 with only a modest reduction in yield (51% vs. 71%) and no change in regio- and diastereoselectivity (Fig. 2e). Linrodostat, an indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor that has advanced as far as phase III clinical trials in the immuno-oncology space,7c contains a 4-alkylcyclohexyl-substituted fluoroquinoline (Fig. 2f). A regioselective Diels–Alder reaction between 6-fluoro-4-vinylquinoline and isoprene provides selective and atom-economical access to this scaffold (product 28), the commercial synthesis of which requires nucleophilic addition to a cyclohexanone under cryogenic conditions.9b
To develop a more detailed mechanistic understanding of these reactions, we performed DFT calculations for thermal and BF3-promoted cycloadditions between 2-vinylpyridine and trans-1-phenyl-1,3-butadiene (Scheme 1a), a reaction that proceeds both regioselectively and stereoselectively. For each set of conditions, concerted asynchronous transition states (TS‡11a) were located for endo cycloadditions leading to major product 11a (Scheme 1b). In support of our initial hypothesis, we found that the use of BF3 lowers the overall reaction barrier by 14.3 kcal mol−1 and the activation energy for the rate-determining step (29 → TS‡11a·Δ vs.29·BF3 → TS‡11a·BF3) by 4.5 kcal mol−1. This is consistent with additional data showing a smaller HOMO–LUMO gap between the diene and the BF3-dienophile complex.24 In addition, the endo transition state for the BF3-promoted reaction was found to be lower in energy than exo, consistent with our experimental observations.24 Unexpectedly, for the thermal reaction the calculations predict that the exo transition state should be favored by 1.2 kcal mol−1. To evaluate this experimentally, we performed a thermal cycloaddition between 29 and trans-1-phenyl-1,3-butadiene at 70 °C. The low yield of this reaction (≤3% for all products as determined by GC) is clearly in line with the higher calculated barrier. Moreover, the regioselectivity of the reaction, favoring exo-11a, verified the computational results and revealed an unanticipated effect of the BF3-promoted conditions in enabling a reversal of stereochemical preference (Scheme 1c).
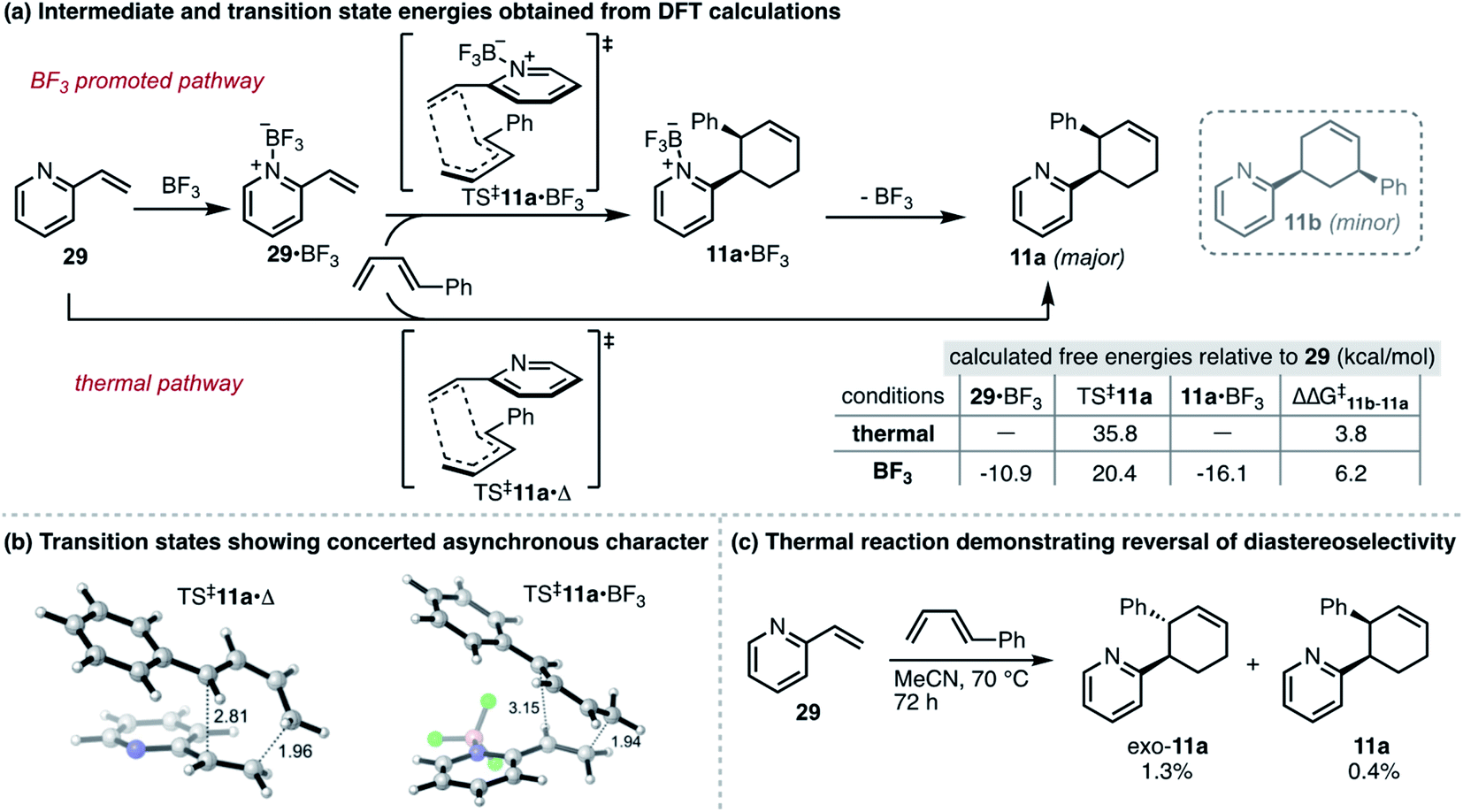 | ||
| Scheme 1 (a) Thermal vs. BF3-promoted pathways: summary of DFT calculations for formation of 11a. Transition state energies provided reflect concerted, endo cycloadditions consistent with observed products. Calculations carried out using Gaussian 16, using the B3LYP functional with the 6-311+G(d) basis set with a solvent model using the dielectric constant for acetonitrile. See ESI† for details. (b) Transition states for endo cycloadditions of 2-vinylpyridine and 1-phenylbutadiene, showing half-formed σ-bonds as dashed lines, with bond lengths in Å. Hydrogens in white, carbons in grey, nitrogen in blue, boron in pink and fluorine in green. (c) Results for thermal cycloaddition between 29 and 1-phenylbutadiene at 70 °C. | ||
To further understand the factors governing selectivity, natural bond orbital analysis (NBO)25 was used to obtain partial atomic charges.24 They indicate a greater degree of polarization of the pyridine vinyl group toward the heteroarene during the transition state of the BF3-promoted reaction, and an attendant increase in the polarization of the diene. This is reflected in an increase of 2.4 kcal mol−1 for ΔΔG‡ between 11b and 11a for the BF3-promoted reaction. These results, along with the greater asynchronicity observed in TS‡11a·BF3 compared to TS‡11a·Δ (Fig. 1b), are consistent with the observed regioselectivity and the increased selectivity when the Lewis acid is employed in this and other reactions.
Of note, the calculations also show that the product-BF3 complex is favored by 5.2 kcal mol−1 over the 2-vinylpyridine-BF3 complex. Based on this prediction, we surmised that product inhibition might contribute to lower yields observed when less than 0.5 equivalents of the Lewis acid are used (Table 1, entry 10). Therefore, we performed an experiment to evaluate the outcome of the reaction between 2-vinylpyridine and 2,3-dimethylbutadiene in the presence of 0.2 equivalents of product 10. After 24 h, this reaction provided 53% yield of new cycloadduct compared to 75% in the absence of added 10, providing support for the product inhibition hypothesis.24 Overall, these preliminary calculations establish a theoretical foundation for this work and suggest that catalytic strategies that are well established for more conventional dienophiles, including enantioselective variants, should also apply to vinyl-azaarenes.4,26 Additional studies will be required to fully realize these possibilities.
Conclusions
In summary, the first definitive examples of Lewis acid-promoted Diels–Alder reactions of vinylazaarene dienophiles have been demonstrated. Our discovery that BF3·OEt2 can promote Diels–Alder cycloadditions of vinyl pyridines, pyrazines, pyrimidines, and quinolines with unactivated dienes, with predictably high levels of regioselectivity and diastereoselectivity, makes these transformations, previously only curiosities, synthetically useful for the first time. Furthermore, the observed acceleration of reaction rates in the presence of BF3, supported by DFT calculations, provides a firm foundation for the future development of catalytic and enantioselective variants. Finally, this new method enables rapid access to cyclohexenyl- and cyclohexyl-appended azaarenes, and thus should facilitate the discovery of new medicines incorporating these noteworthy structures.Data availability
Detailed synthetic procedures, complete characterization data for all new compounds, computational data, and supplementary figures and tables can be found in the ESI.†Author contributions
M. K. H. and A. E. D. conceived the project. A. E. D. and J. M. L. conducted the experiments. A. E. D., J. M. L., and M. K. H. analyzed the experimental results. J. M. L. conducted the computational experiments. J. M. L. and M. K. H. analyzed the computational results. A. E. D., J. M. L., and M. K. H. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Virginia and the National Science Foundation (NSF CAREER 1845219) for financial support.Notes and references
- A. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98 CrossRef.
- (a) K. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef CAS; (b) L. Min, Y.-J. Hu, J.-H. Fan, W. Zhang and C.-C. Li, Chem. Soc. Rev., 2020, 49, 7015 RSC; (c) K. C. Nicolaou and J. S. Chen, Classics in Total Synthesis III, Wiley-VCH, 2011 Search PubMed; (d) K. C. Nicolaou and S. A. Snyder, Classics in Total Synthesis II, Wiley-VCH, 2003 Search PubMed; (e) K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis, Wiley-VCH, 1996 Search PubMed.
- For selected recent examples see: (a) I. Dissanayake, J. D. Hart, E. C. Becroft, C. J. Sumby and C. G. Newton, J. Am. Chem. Soc., 2020, 142, 13328 CrossRef CAS PubMed; (b) J. S. Barber, M. M. Yamano, M. Ramirez, E. R. Darzi, R. R. Knapp, F. Liu, K. N. Houk and N. K. Garg, Nat. Chem., 2018, 10, 953 CrossRef CAS PubMed; (c) M. E. Abbasaov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492 CrossRef PubMed; (d) X. Li and S. J. Danishefsky, J. Am. Chem. Soc., 2010, 132, 11004 CrossRef CAS PubMed.
- For selected useful reviews on this topic see: (a) X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515 CrossRef CAS PubMed; (b) S. Raymond and J. Cossy, Chem. Rev., 2008, 108, 5359 CrossRef PubMed; (c) D. A. Evans and J. S. Johnson, Comprehensive Asymmetric Catalysis, Springer, 1999, vol. 3, p. 1177 Search PubMed; (d) H. B. Kagan and O. Riant, Chem. Rev., 1992, 92, 1007 CrossRef CAS.
- J. S. Meek, R. T. Merrow and S. J. Cristol, J. Am. Chem. Soc., 1952, 74, 2667 CrossRef CAS.
- W. V. E. Doering and S. J. Rhoads, J. Am. Chem. Soc., 1953, 75, 4738 CrossRef CAS.
- (a) A. Markham, Drugs, 2020, 80, 1865 CrossRef CAS PubMed; (b) A. Subissi, D. Monti, G. Togni and F. Mailland, Drugs, 2010, 70, 2133 CrossRef PubMed; (c) G. Sonpavde, A. Necchi, S. Gupta, G. D. Steinberg, J. E. Gschwend, M. S. Van Der Heijden, N. Garzon, M. Ibrahim, B. Raybold, D. Liaw, M. Rutstein and M. D. Galsky, Future Oncol., 2020, 16, 4359 CrossRef CAS PubMed; (d) P. Moreau and R. V. Rajkuma, Lancet, 2016, 388, 111 CrossRef PubMed.
- W. D. Jang, S. Jeon, S. Kim and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024302118, DOI:10.1073/pnas.2024302118.
- (a) X. Liang, Q. Yang, P. Wu, C. He, L. Yin, F. Xu, Z. Yin, G. Yue, Y. Zou, L. Li, X. Song, C. Lu, W. Zhang and B. Jing, Bioorg. Chem., 2021, 113, 105011 CrossRef CAS PubMed; (b) K. J. Fraunhoffer, A. J. Delmonte, G. L. Beutner, M. S. Bultman, K. Camacho, B. Cohen, D. D. Dixon, Y. Fan, D. Fanfair, A. J. Freitag, A. W. Glace, F. Gonzalez-Bobes, M. Gujjar, M. W. Haley, M. R. Hickey, J. Ho, V. Iyer, P. Maity, S. Patel, V. W. Rosso, M. A. Schmidt, J. M. Stevens, Y. Tan, C. Wilbert, I. S. Young and M. Yu, Org. Process Res. Dev., 2019, 23, 2482 CrossRef CAS; (c) M. T. Burger, G. Nishiguchi, W. Han, J. Lan, R. Simmons, G. Atallah, Y. Ding, V. Tamez, Y. Zhang, M. Mathur, K. Muller, C. Bellamacina, M. K. Lindvall, R. Zang, K. Huh, P. Feucht, T. Zavorotinskaya, Y. Dai, S. Basham, J. Chan, E. Ginn, A. Aycinena, J. Holash, J. Castillo, J. L. Langowski, Y. Wang, M. Y. Chen, A. Lambert, C. Fritsch, A. Kauffmann, E. Pfister, L. G. Vanasse and P. D. Garcia, J. Med. Chem., 2015, 58, 8373 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5485 CrossRef PubMed.
- J. A. Bristol and R. Brambilla, J. Org. Chem., 1979, 44, 1889 CrossRef CAS.
- The intrinsic reactivity of the vinylpyridine subunit can be improved by structural changes, such as in 3-pyridinylacrylates, but these are better thought of as pyridine-substituted variants of an acrylate dienophile, and they react accordingly, providing the opposite regioselectivity to vinylpyridines. The related 2-pyridinylacrylate cycloadditions have not been reported, to our knowledge. See: (a) J. Ward, A. B. Johnson, G. R. Clark and V. Caprio, Synthesis, 2009, 20, 3411 Search PubMed; (b) E. J. Corey, T. P. Loh, S. Achyutha Rao, D. C. Daley and S. Sarshar, J. Org. Chem., 1993, 58, 5600 CrossRef CAS; (c) J. Bourguignon, G. Le Nard and G. Queguiner, Can. J. Chem., 1985, 63, 2354 CrossRef CAS.
- F. A. Selimov, O. A. Ptashko, A. A. Fatykhov, N. R. Khalikova and U. M. Dzhemilev, Russ. Chem. Bull., 1993, 42, 872 CrossRef.
- J. Sepúlveda-Arques, B. Abarca-González and M. Medio-Simón, Adv. Heterocycl. Chem., 1995, 53, 339 CrossRef.
- S. Abou-Shehada, M. C. Teasdale, S. D. Bull, C. E. Wade and J. M. J. Williams, ChemSusChem, 2015, 8, 1083 CrossRef CAS PubMed.
- K. N. Houk and R. W. Strozier, J. Am. Chem. Soc., 1973, 95, 4094 CrossRef CAS.
- R. Huisgen, R. Grashey and J. Sauer, The Chemistry of Alkenes, John Wiley & Sons, 1964 Search PubMed.
- E. G. Tse, S. D. Houston, C. M. Williams, G. P. Savage, L. M. Rendina, I. Hallyburton, M. Anderson, R. Sharma, G. S. Walker, R. S. Obach and M. H. Todd, J. Med. Chem., 2020, 63, 11585 CrossRef CAS PubMed.
- J. Chen, Y. Fu, Y. Yu, J.-R. Wang, Y.-W. Guo, H. Li and W. Wang, Org. Lett., 2020, 22, 6061 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- K. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3358 CrossRef CAS PubMed.
- R. Chinchilla, C. Nájera and M. Yus, Chem. Rev., 2004, 104, 2667 CrossRef CAS PubMed.
- Similarly, (E)-2-styrylpyridine, as a probe of reactivity of 1,2-disubstituted dienophiles, failed to form a cycloadduct with 2,3-dimethylbutadiene at 70 °C.
- See ESI† for details.
- E. D. Glendening, A. E. Reed, J. E. Carpenter, and F. Weinhold, NBO Version 3.1, 2003 Search PubMed.
- For other examples of BF3·OEt2 activation of pyridines in a different context, see: (a) S. V. Kessar and P. Singh, Chem. Rev., 1997, 97, 721 CrossRef CAS PubMed; (b) S. V. Kessar, P. Singh, K. N. Singh and M. Dutt, J. Chem. Soc., Chem. Commun., 1991, 570 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization data, and spectra for all new compounds. See DOI: 10.1039/d1sc05095h |
| This journal is © The Royal Society of Chemistry 2021 |