
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled disassembly of azobenzene cellulose-based thin films using visible light†
K. E.
Edwards
*,
M.
Kim
,
T. H.
Borchers
and
C. J.
Barrett
 *
*
Department of Chemistry, McGill University, Montreal, Quebec, Canada. E-mail: kayrel.edwards@mail.mcgill.ca; chris.barrett@mcgill.ca; Tel: +514-398-6919
First published on 24th June 2022
Abstract
In efforts toward designing self-assembled materials that can later be disassembled easily to avoid destruction of component materials during separation and recycling to minimize waste at their end of life, we report the fabrication of robust, water resistant polymer multilayers whose disassembly could be triggered upon irradiation of wavelengths and intensities of blue visible sunlight. Self-assembled thin films for study were prepared by combining a water-soluble biodegradable polymer, sodium cellulose sulfate (NaCS), and a water-soluble azo dye photo-switch, Bismarck Brown Y (BBY) via layer-by-layer (LbL) assembly. The resultant multi-layered materials (NaCS/BBY) were held together by weak intermolecular interactions between the polymeric sulfate groups and the amino groups of the photo-switch, forming robust and water-resistant materials. The photo-disassembly of the films was assessed using ‘rainfall conditions’ and ‘sunlight’ where it was demonstrated that blue visible light could trigger the successful disassembly of the films at a rate of ka = 7 ± 1 × 10−3 absorbance units per h. Optical pump-probe experiments and confocal Raman spectroscopy supported a mechanism that the disassembly was triggered by the trans-to-cis geometric isomerization of BBY. This method of sunlight- and water-triggered disassembly can in principle allow for recovery of reusable components to be applied as next generation sustainable materials for products and recycling processes.
Introduction
Society's use of traditional disposable plastics has now clearly become a grave global environmental concern due to their overuse, non-degradability, poor reprocessibility, and toxicity. However, encouraging efforts are being made in such fields as sustainable materials science and bioengineering towards providing low- or non-impact plastic alternatives using naturally-derived polymers. Cellulose for example is one of the most abundant natural resources on Earth, and can function well as a sustainable feedstock due to its excellent recyclability, degradability, low cost and worldwide availability.1 Over the last few decades much research has focused on the engineering of cellulose into functional materials for uses in medicine and packaging, as well as the processing of nano-cellulose to bulk materials.2–6 Cellulose's ability to be used in such a wide variety of applications derives from its complex structure and versatile suite of properties. Cellulose biopolymers consist of linear chains of D-glucose (Fig. 1) repeating units which are connected through a high degree of intermolecular hydrogen bonding, rendering the polymer insoluble in water and in most organic solvents, and providing materials with excellent mechanical properties.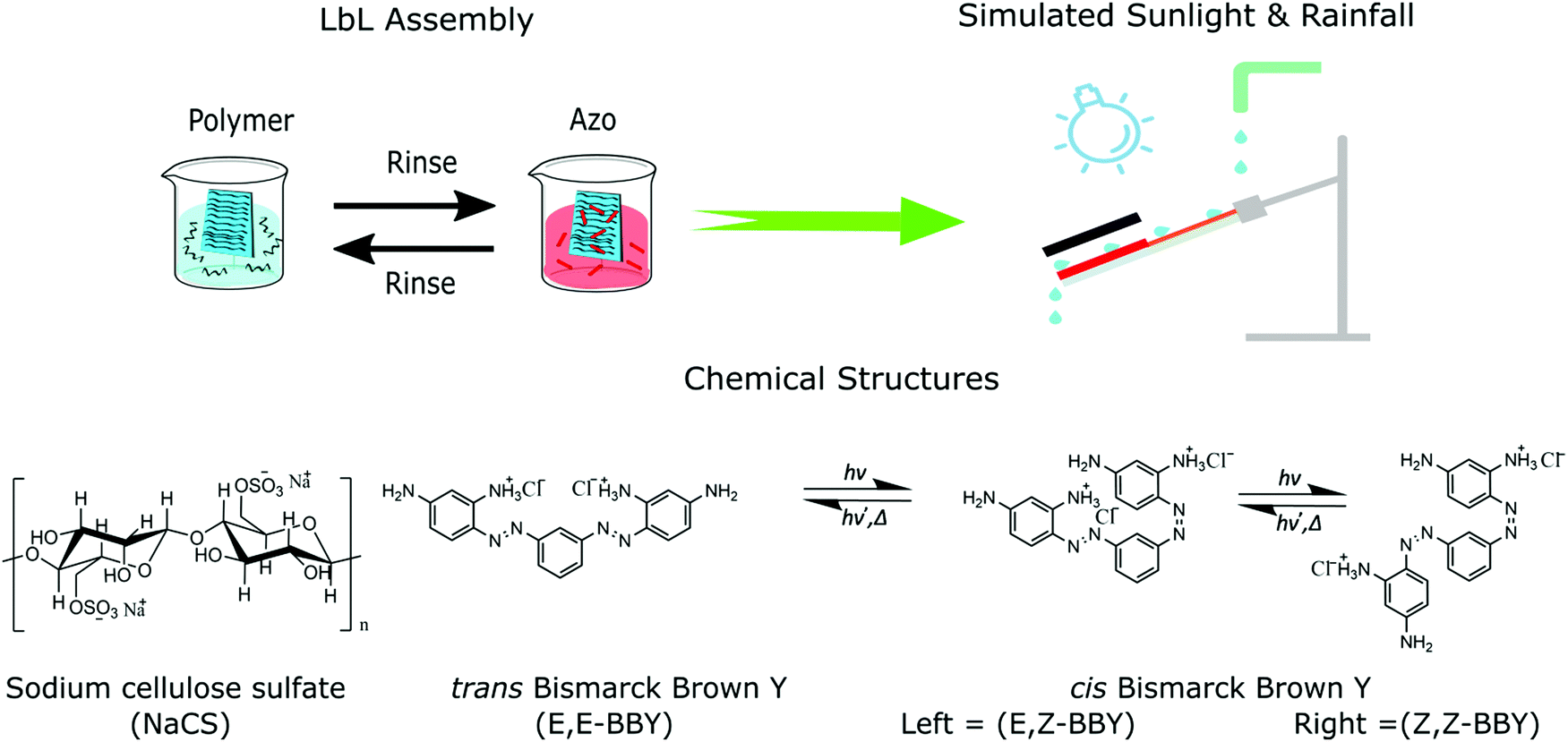 | ||
| Fig. 1 Schematic illustration of layer-by-layer (LbL) assembly to make multi-layered (NaCS/BBY) thin films, and the simulated sunlight and rainfall trigger conditions to assess the disassembly of the films and the chemical structures of the materials used. | ||
Cellulose can also be easily functionalized to create a wide array of derivatives with tailorable properties. Many of these cellulose derivatives retain the excellent properties of native cellulose such as being biodegradable and biocompatible, while adding additional benefits such as antibacterial properties, flame retardancy, and water solubility.7 Water-soluble cellulose derivatives such as commercially available methyl cellulose (MC), hydroxypropyl cellulose (HPC) and sodium cellulose sulfate (NaCS) are of particular interest, as they can be processed under more environmentally friendly conditions. Thanks to their excellent gel-forming properties, these polymers are now well-used in the fabrication of coatings, composites, films and membranes.7 NaCS in particular is interesting due to its high tensile strength in the wet state, its moldability in situ as well as its relatively simple and cost-efficient production, and biocompatibility.8,9 Recently, even novel edible films for packaging using NaCS and glycerol were developed, fabricated via solvent-casting techniques, demonstrating that NaCS could be used as potential degradable coating materials for food packaging. These dense NaCS films formed with an ordered microstructure and were transparent, flexible, and resistant to oils and fats, but not to water. Films made from water-soluble polymers can sometimes disassemble before the end of their use however, due to the hydrophilicity of their surfaces.10
One method of recent interest for developing high-value materials is through layer-by-layer (LbL) self-assembly. LbL assembly involves the sequential adsorption of alternating layers of charged polymers/molecules from aqueous solution onto a substrate via a dipping process.11 LbL assembly offers many advantages compared to other forms of film production, such as solvent casting, drop casting, and spin-coating; these include increased reproducibility and a more homogenous dispersion of the fibres resulting in more uniform films.12 This technique generally involves the combination of two water-soluble components, which when combined result in a water-resistant film. Materials are linked to each other in the films through soft, weak attractive forces such as electrostatic interactions, hydrogen bonding, or hydrophobic forces. Soft bonding interactions are of particular interest, such as electrostatic interactions and hydrogen bonding, which are both reversible and directional.13 This allows more complex systems to easily assemble and later gently and reversibly disassemble when introduced to a particular stimulus, such as pH, temperature, or salt, without breaking the covalent bonds.12 Early sets of hydrogen-bonded LbL films were commonly prepared using a carboxylic acid-containing polymer such as polyacrylic acid (PAA) and polymethacrylic acid (PMA), which allowed films to be used for the preparation of pH-sensitive materials.14 More recent hydrogen-bonded LbL films involve polymers containing hydroxyl or amino groups for use as photochromic paper, gas barriers, and flame-retardant coatings.15–19 For electrostatically-bonded LbL films, common polyanions include PAA and hyaluronic acid (HA) which can be combined with polycations poly (allylamine HCl) or poly-L-lysine, to fabricate polyelectrolyte multilayer (PEM) films that can be used as tuneable extracellular matrix materials.20
Additionally, LbL films and PEM films can be designed to be photo-responsive by covalently attaching a photo-switch to one of the polymers.21 Light is often a preferred stimulus since it can be applied precisely and remotely with minimal undesired chemical change otherwise to the host materials. One of the most effective and well-studied photo-switches is azobenzene, which can geometrically isomerize between E and Z (trans and cis) stereoisomers reversibly upon irradiation. Azobenzene derivatives exhibit a wide characteristic absorption band in their UV-Vis absorption spectra which originates from the π → π* transition of the azobenzene chromophore. Adding push-pull substituents on the aromatic rings can drastically change the optical properties of the azobenzene, giving one the ability to “tune” specific properties such as cis half-life or UV-Vis absorption profile, optimizing the photo-switching characteristics for a wide variety of specific material applications.22,23 It is also possible to load more than one isomerizing unit into an azobenzene molecule. For example, bisazobenzenes such as Bismarck Brown Y (BBY) (Fig. 1) have two azo units (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds) per molecule, which allows for higher loading of azo units in the material without aggregation and provides a larger volume change and increased geometrical disruption upon isomerization.24–26 BBY (C18H18N8·2HCl), which is the dihydrochloride of 4,4′-[1,3-phenylenebis (diazenzene-2,1-diyl)]di(benzene-1,3-diamine), is one of the few commercially available water-soluble azobenzenes. It has four intermolecular bonding donor/acceptor sites, two of which are protonated. Offering this higher number of intermolecular bonding sites also allows for increased intermolecular interactions between the photo-switch and the polymer, resulting in more stable films when self-assembled.
N bonds) per molecule, which allows for higher loading of azo units in the material without aggregation and provides a larger volume change and increased geometrical disruption upon isomerization.24–26 BBY (C18H18N8·2HCl), which is the dihydrochloride of 4,4′-[1,3-phenylenebis (diazenzene-2,1-diyl)]di(benzene-1,3-diamine), is one of the few commercially available water-soluble azobenzenes. It has four intermolecular bonding donor/acceptor sites, two of which are protonated. Offering this higher number of intermolecular bonding sites also allows for increased intermolecular interactions between the photo-switch and the polymer, resulting in more stable films when self-assembled.
The isomerization of azobenzene can result in large structural disruption, changing from a planar, nonpolar structure (E) to a polar, twisted confirmation (Z) (Fig. 1). These changes on the molecular scale can result in large changes in the entire material, and azobenzene-containing materials have been developed for a wide variety of shape-changing applications such as drug delivery via photo driven assembly/disassembly, to fabricate surface relief holographic optical gratings, employed as sunlight-driven photo-actuators, for photoalignment, and as humidity driven sensor devices.14,21,27–32 Generally, these materials are fabricated out of synthetic polymers. However, in response to the negative impact of synthetic polymers on the environment, a shift towards biodegradable and bio-based polymers has received much recent attention.
For the first time, we report here water-resistant self-assembled biobased cellulose materials that can be photo-reversibly dis-assembled and re-solubilized, prepared using a relatively simple and cost-efficient method. Thin multilayer films were prepared by the LbL assembly of two water-soluble components: NaCS, a low toxicity (edible), biobased and biodegradable polymer; and BBY, a photo-reversible azobenzene molecule. The multi-layered thin films (NaCS/BBY) were held together by electrostatic interactions between the anionic polymer and the cationic photo-switch and provided a robust and stable material that was water resistant. Upon blue visible light irradiation in running water however, we were able to trigger and control disassembly of the films, thus returning the material back to its water-soluble starting components, in principle ready for re-use. These new reversibly ‘soft-bonded’ materials could potentially be easily and inexpensively fabricated, as a new class of materials to replace some of the artificial plastics currently used for a variety of thin film or coating applications.
Results and discussion
LbL assembly of NaCS/BBY thin films
Multi-layered films (NaCS/BBY) comprising a water-soluble biodegradable polymer, sodium cellulose sulfate (NaCS), and a photo-responsive azo dye, Bismarck Brown Y (BBY) were prepared based on the LbL assembly protocols for similar compounds described previously, to produce robust and water-resistant materials.15–17 Disassembly of the thin films was then triggered by blue light under ‘simulated sunlight and rainfall’ conditions (Fig. 1). The average thickness of the obtained thin films was 390 ± 15 nm after the deposition of 50 bilayers onto a glass substrate, determined by AFM (Fig. S1a, ESI†). AFM topology measurements (Fig. S1b, ESI†), confocal Raman optical microscopy (Fig. S1c, ESI†) and SEM imaging (Fig. S1d, ESI†) confirmed a uniform film surface across various length scales. Elemental mapping of sulfur and nitrogen further confirmed an even distribution of the polymer, sulfated cellulose and the azo dye, Bismarck Brown Y, respectively, across the film's surface (Fig. S1d, ESI†). The films were also optically clear and had a deep red appearance due to the characteristic red colour of the BBY dye (Fig. 2).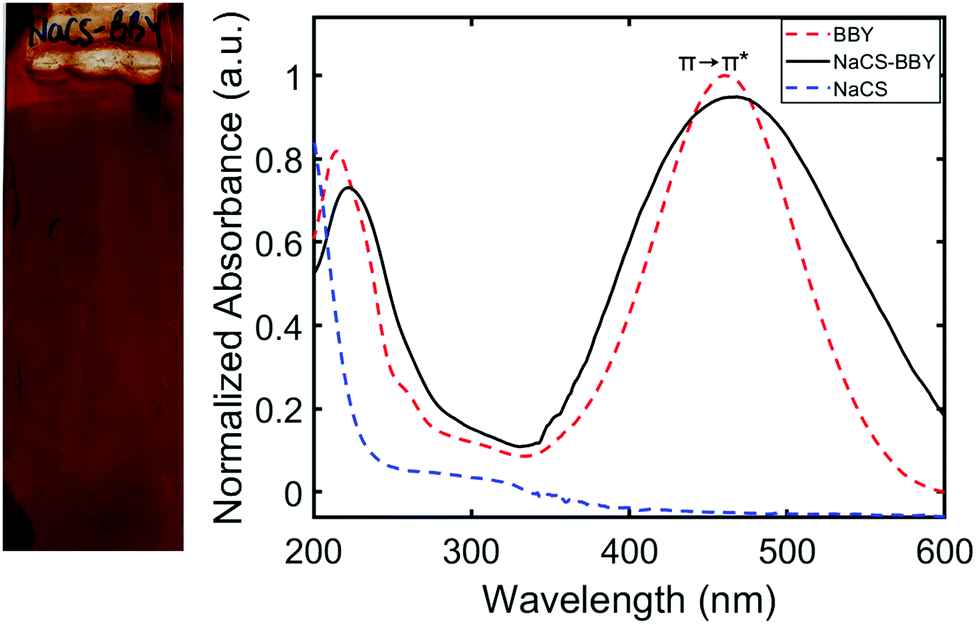 | ||
| Fig. 2 (Left) Image of multi-layered NaCS/BBY films (50 bilayers) deposited on a glass substrate with distinct red colour of BBY. (Right) UV-Vis spectra of NaCS/BBY on quartz with λmax = 450 nm, BBY in water λmax = 450 nm and NaCS in water with λmax = 200 nm. | ||
UV-Vis absorption spectroscopy confirmed that BBY displays a strong absorption band in the visible region (λmax = 450 nm, ε ≥ 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L mol−1 cm−1) where the π→ π* and n → π* transitions are overlapped (Fig. 2). In the multi-layered films the peak at λmax = 450 nm was observed to be broader than in solution. This was likely due to intermolecular interactions between the azo and the polymer which affects the electronics of the azo unit (NN bond), and helps confirm the soft-bonding interactions. UV-Vis absorption spectroscopy also showed two broad peaks for the polymer, with λmax values of approximately 200 nm and 300 nm, consistent with that of cellulose.33 These two absorption bands appear hidden below those of BBY in the multi-layered films (Fig. 2). UV-Vis spectroscopy also showed that the bilayers were deposited linearly by monitoring the increase in the peak at 450 nm which is attributed to the π →π* transition of trans BBY (E, E-BBY) as shown in the ESI† (Fig. S2). The resulting NaCS/BBY films are stable in DI water and contact angle experiments (Fig. S3, ESI†) revealed that the surface of the films was less hydrophilic than for films of the individual components: NaCS/BBY (θ = 61°, 61°), NaCS (θ = 21°, 21°) and BBY (θ = 45°, 44°). These results support the proposed mechanism that combining two water soluble components in a LbL fashion can render the material less hydrophilic and thus more water resistant, through soft-bonding of the charged hydrophilic groups employed in the electrostatic self-assembly.
000 L mol−1 cm−1) where the π→ π* and n → π* transitions are overlapped (Fig. 2). In the multi-layered films the peak at λmax = 450 nm was observed to be broader than in solution. This was likely due to intermolecular interactions between the azo and the polymer which affects the electronics of the azo unit (NN bond), and helps confirm the soft-bonding interactions. UV-Vis absorption spectroscopy also showed two broad peaks for the polymer, with λmax values of approximately 200 nm and 300 nm, consistent with that of cellulose.33 These two absorption bands appear hidden below those of BBY in the multi-layered films (Fig. 2). UV-Vis spectroscopy also showed that the bilayers were deposited linearly by monitoring the increase in the peak at 450 nm which is attributed to the π →π* transition of trans BBY (E, E-BBY) as shown in the ESI† (Fig. S2). The resulting NaCS/BBY films are stable in DI water and contact angle experiments (Fig. S3, ESI†) revealed that the surface of the films was less hydrophilic than for films of the individual components: NaCS/BBY (θ = 61°, 61°), NaCS (θ = 21°, 21°) and BBY (θ = 45°, 44°). These results support the proposed mechanism that combining two water soluble components in a LbL fashion can render the material less hydrophilic and thus more water resistant, through soft-bonding of the charged hydrophilic groups employed in the electrostatic self-assembly.
FTIR spectroscopy
Multi-layered films are generally held together by weak intermolecular interactions such as hydrogen bonding or electrostatic forces.12,20 NaCS is a water-soluble cellulose derivative consisting of hydroxyl groups and negatively charged sulfonate groups, and the degree of substitution (DS) of sulfonate groups on NaCS was determined by XPS analysis to be 1.2, which equates to 7.2 mmol g−1 (Fig. S4, ESI†). On the other hand, the azo photo-switch BBY (C18H18N8·2HCl) has four amino groups attached to the phenyl rings, two of which are protonated to form the acid salt. Titration of BBY against NaOH revealed that BBY displays a single pKb value of 8.4 (Fig. S5, ESI†). The pH of the BBY dipping solution was pH = 3, at which BBY is water soluble. At this pH two of the four amino groups are protonated, facilitating electrostatic interactions with the polymer. FTIR spectroscopy was used to assess the intermolecular interactions in the multi-layered films, with the IR spectrum of the multi-layered film deposited on an aluminium substrate shown in (Fig. 3). The spectrum displays a characteristic N–H stretching of an amine from 3310–3350 cm−1, highlighted by the black circle.15,34 The inset of the IR spectrum shows the two bands which correspond to the free N–H stretch and the intra/intermolecular N–H stretch at 3353 cm−1 and 3220 cm−1, respectively, for a solvent-cast film of BBY on an aluminium substrate (orange line). The shape and size of these peaks are drastically different in the NaCS/BBY spectrum (blue line), as both peaks are shifted to lower energies of 3340 cm−1 and 3213 cm−1, with a noticeable increase in the intensity of the latter which is assigned as the intra/intermolecular N–H stretch of the amine group. It is presumed that this is a result of increased intermolecular interactions between the polymer and the azo photo-switch due to the layer-by-layer packing in the multi-layered films. This compares well to early work published by Stockton and Rubner, where they observed similar changes in the IR spectra of polyaniline upon formation of polyaniline/PVP blends and multilayers.15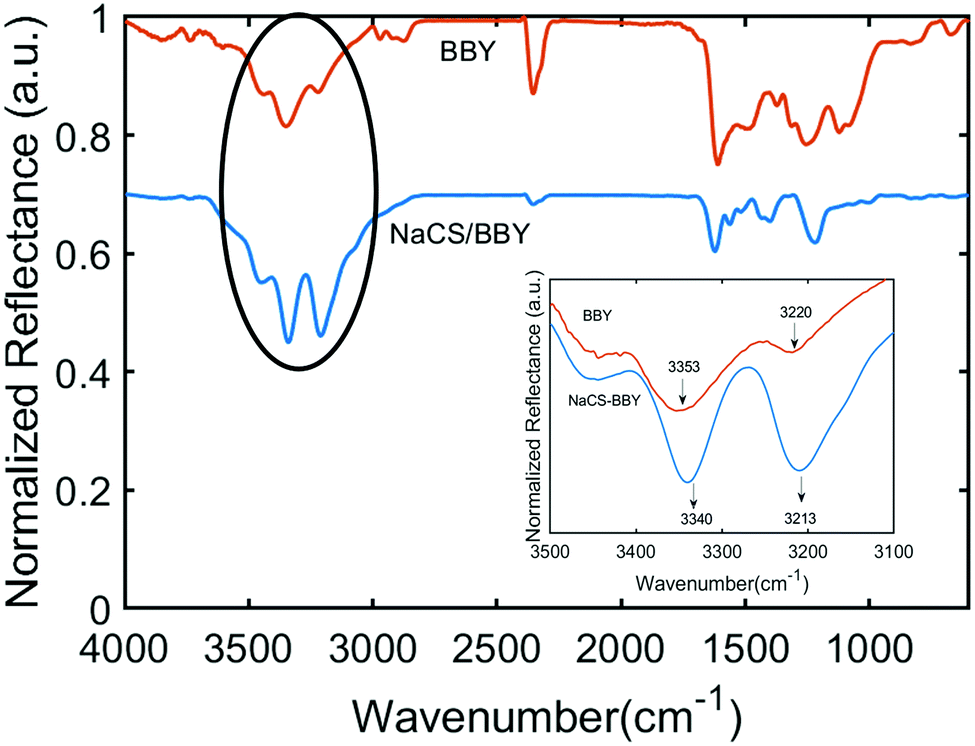 | ||
| Fig. 3 IR spectra of BBY only (orange) vs. multi-layered NaCS/BBY films (blue). Inset shows the changes in the N–H stretching mode of BBY only (orange) vs. in multi-layered NaCS/BBY films (blue). | ||
Furthermore, in Fig. 3, the peaks between 2800–3000 cm−1 and 2270–2390 cm−1 can be assigned to N–H stretching of different amine salts. Generally, amine salts display N–H stretching between 2800–3200 cm−1, 2700–3000 cm−1 and 2300–2700 cm−1 for primary (NH3+), secondary (NH2+) and tertiary (NH+) amine salts respectively.35 BBY is capable of forming all three of these amine salts, since protonation of the four NH2 groups and the two azo units (NN bond) is possible. The peaks between 2800–3200 cm−1 were more pronounced for BBY in NaCS/BBY thin films (blue line) compared to bulk BBY (orange line) likely due to electrostatic interaction between the azobenzene photo-switch and the polymer.
The IR spectrum of bulk BBY also showed peaks between 1520–1625 cm−1 which can be assigned to aromatic CC bending and/or N–H bending of primary and secondary amine salts.34,35 The peaks between 1390–1440 cm−1 were assigned to N–H bending of amines, and 1212–1274 cm−1 to C–N stretching (Fig. 3 and Fig. S7, ESI†). These peaks were also present in NaCS/BBY IR spectrum. The IR spectrum of NaCS showed strong peaks at 3318–3630 cm−1, 1217 cm−1 and 985 cm−1 which can be assigned to free O–H stretching, ether C–O stretching and alcohol C–O stretching, respectively.34 There were also two small peaks at 1396 cm−1 and 1464 cm−1 which can be assigned to sulfonate SO stretching/alkane C–H bending.34 It was difficult to identify the peaks associated with the polymer in the NaCS/BBY thin films since many of the peaks overlapped with those assigned to BBY.
Influence of pH, ionic strength, and temperature on film stability
LbL films can be sensitive to external stimuli such as pH, ionic strength of the solvent, and temperature. Before conducting the ‘sun and rain’ conditions dis-assembly experiments, we examined the effect of these stimuli on the stability of the thin films. To evaluate the effect of pH, the films were soaked in DI water adjusted from pH 2 to 12 and left out on the lab bench for 1 week under ambient conditions. Films which were immersed in pH 2–10 water did not show any signs of disassembly; the wash water remained colourless and there was no noticeable change in the UV-Vis spectrum of BBY. Conversely, the film submerged in very basic water (pH 11–12) underwent a noticeable colour change from red to orange, and the wash water clearly turned yellow (Fig. 4). There was also a significant change in the UV-Vis spectrum with the broadening of the π → π* transition absorption band from 450 nm for pH 2–10, to 385–490 nm for pH 11–12 (Fig. 4). It is proposed that at pH 2–10 most of the amino groups of BBY are protonated which favours intermolecular interactions between the polymer and azo compound, and at very basic pH values the amino groups of BBY are deprotonated which disrupts the weak intermolecular forces holding the layers together. This mechanism is supported by the titration results of BBY against NaOH where it was determined that BBY has a pKb value of 8.4 (Fig. S5, ESI†). Additionally, the deprotonation of the azo compound is responsible for the colour change from red to orange due to changes in the electrostatic character of the azo unit.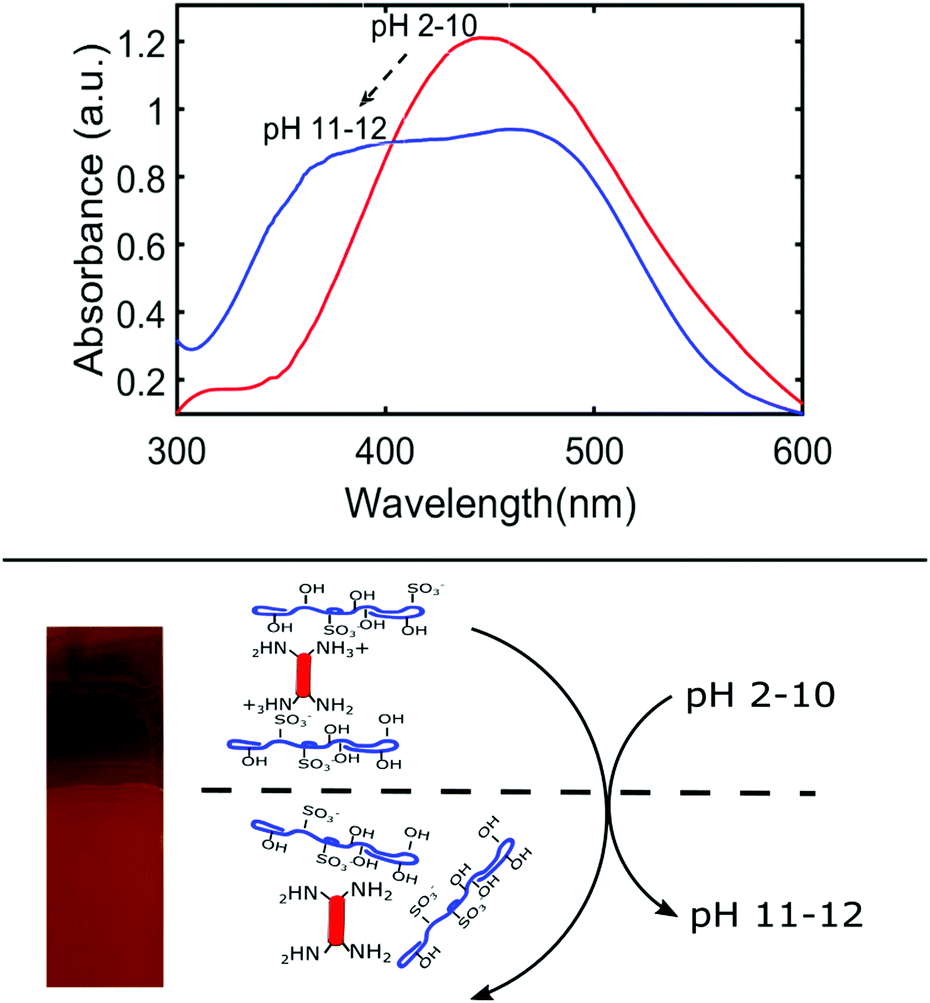 | ||
| Fig. 4 (Top) UV-Vis absorption spectra of NaCS/BBY films in response to change in pH. Between pH 2–10 the π → π* transition absorption band is at 450 nm and at pH 11–12 there is significant broadening of the absorption band to 385–490 nm. (Bottom) Image of the film showing colour change from red (pH 2–10) to orange (pH 11–12) and a schematic of the proposed molecular changes which occur in response to change in pH. | ||
The influence of salt on the stability of the thin films was evaluated by submerging the films in a beaker containing 2 M NaCl solution at RT for 1 week. There was some observed leaching of BBY into the water which turned pale yellow, as well as a slight blue shift in the π → π* transition absorption band from 450 nm to 445 nm (Fig. S7, ESI†). There was also a decrease observed in the absorbance of BBY by 32% which indicated removal of some (perhaps excess) BBY from the film (Fig. S7, ESI†), as one might expect salt to disrupt the electrostatic interactions between the positively charged protonated amino groups (NH3+) of BBY and the negatively charged sulfonate groups (SO3−) of NaCS. Lastly, to assess the impact of temperature on the stability of the thin films, the films were submerged in beakers containing DI water held at RT (21 °C), at 70 °C, and at 100 °C for 1 hour. Films were observed to remain stable at 21 °C and 70 °C but began to partially disassemble at 100 °C. There was a 35% reduction in the absorbance of BBY in the thin films exposed to this high temperature, and the wash water turned yellow (Fig. S8, ESI†). At high temperatures hydrogen bonds can suffer degradation, and it is likely that at 100 °C sufficient hydrogen bonds between the amino groups of BBY and hydroxyl groups of NaCS were broken, allowing the film to disassemble. This observation shows that the films are very stable even when exposed to relatively high temperatures, and thus any heating effect which occurs during exposure to 460 nm light in the ‘sun and rain’ conditions experiment should not be expected to play a significant contribution to the disassembly process.
Pump-probe experiments to examine isomerization of BBY
Having established the effects of pH, ionic strength, and temperature of the wash solution on the stability of the films, the trans–cis isomerization behaviour of BBY was evaluated using optical pump-probe experiments. The photo-switching of BBY could not be monitored easily with a UV-Vis spectrophotometer, since bisazobenzene photo-switches generally have fast isomerization rates, which makes it difficult to capture the thermal back relaxation of the azo compound in detail, so a separate fast laser setup was constructed. A solution of BBY in THF was pumped by irradiating with 532 nm laser light at a power of approximately 100 mW cm−2 and probed with 488 nm laser (blue light) at low power and the signal monitored over time (Fig. 5). We were able to observe switching from trans-to-cis states of BBY represented by the green box in Fig. 5A, as well as the thermal back relaxation of BBY in dark which showed a biexponential decay (Fig. 5B). In-depth analysis towards the detailed mechanism of the various BBY isomerizations is the subject of another full study forthcoming.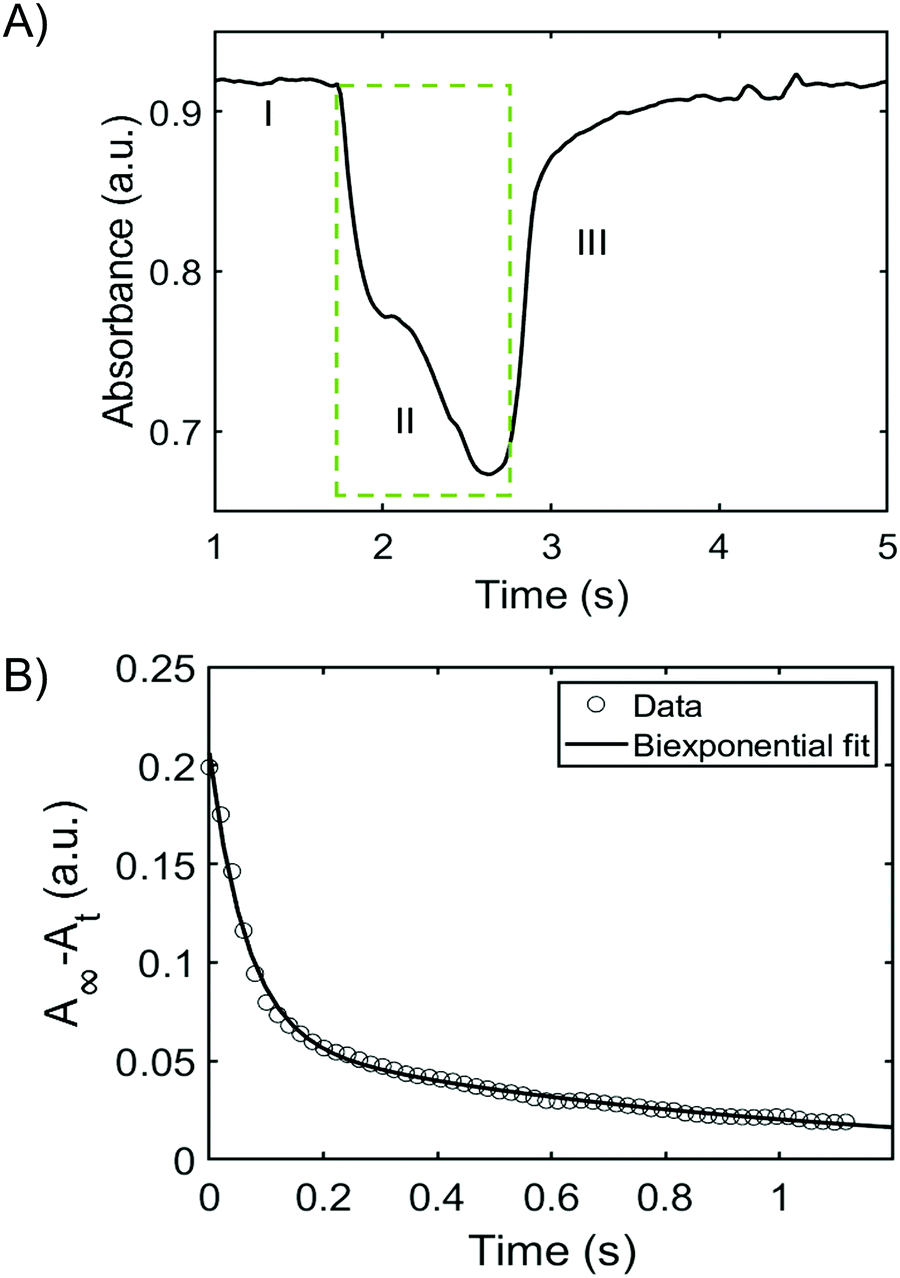 | ||
| Fig. 5 (A) Pump-probe isomerization plot showing the change in absorbance over time for BBY solution in THF: (I) Pump beam is off, (II) Green box represents when the laser is switched on for 1 s and (III) pump beam is turned off showing thermal back relaxation. (B) Decay of the cis isomer with fit with a biexponential function. | ||
Confocal Raman spectroscopy
The trans-to-cis isomerization of BBY when sandwiched in the multi-layered films was confirmed by confocal Raman spectroscopy, which was used to probe changes in the orientation of BBY upon exposure to 532 nm light. For these measurements, multi-layered films were deposited on a quartz substrate and excited with a 532 nm laser at increasing laser power, with a scan rate of 1s μm−1 and resolution of 1 spectra per μm across the width of the thin films (25 mm). Previous studies have shown that the NN Raman shift of unsubstituted trans azobenzene occurs at 1424 cm−1 and NN Raman shift of unsubstituted cis azobenzene occurs at 1510 cm−1.36 The Raman shift at 612 cm−1 corresponds to the CNNC torsion associated with cis azobenzene.36 Additionally, the Raman shift at 1283 cm−1 corresponds to C–N stretching, at 1379 cm−1 to C–C stretching, 1574 cm−1 to R–C–C stretching (where R = benzene ring), and at 1612 cm−1 to NH2 scissoring.36–38 The first scan of the multi–layered films, which was performed at a relatively low laser power of 0.1 mW, displayed Raman shifts at 482 cm−1, 997 cm−1, 1157 cm−1, 1283 cm−1, 1379 cm−1, and 1612 cm−1 as seen in Fig. 6A. As the laser power increased from 0.1 mW to 0.5 mW in 0.1 mW increments, there were notable changes in the spectra, of the appearance of Raman shifts at 612 cm−1 and 1521 cm−1 and an increase in the intensity of the peak at 1574 cm−1 with a simultaneous decrease in the intensity of the Raman shifts at 1283 cm−1 and 1612 cm−1. There was also a slight shift in the position of the peak at 1379 cm−1 to lower energy observed, and the emergence of a peak at 1296 cm−1. It is proposed that these changes in the Raman shift are due to a change in the orientation of the BBY molecules from a planar trans state to a non-planar cis state.36,37 It was also observed that if the 532 nm laser at the lowest power (0.1 mW) was focused on the same spot on a NaCS/BBY thin film for a prolonged period (t > 50 s) the Raman signal centred around the peak at 1285 cm−1, which corresponds to C–N stretching, decreased over time. The trans-to-cis photoisomerization was fit best to a biexponential function (Fig. S9, ESI†), and detailed optical characterization is the topic of a separate forthcoming paper.
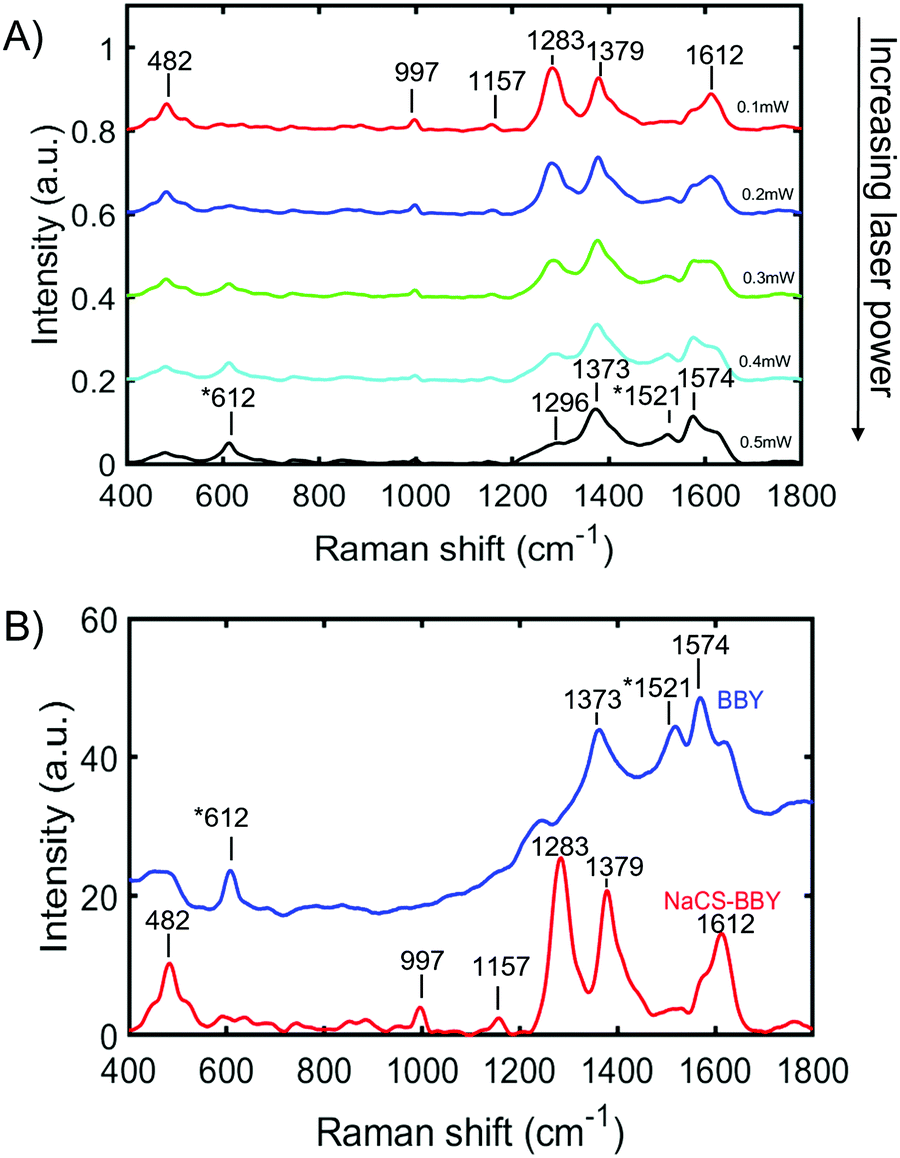 | ||
| Fig. 6 (A) Changes in the Raman spectra of BBY in multi-layered NaCS/BBY thin films with increasing 532 nm laser power (0.1–0.5 mW). (B) Raman spectra of BBY only (blue) and NaCS/BBY films (red) upon 1st scan at 0.1 mW 532 nm laser power. (*) represents the characteristic cis BBY Raman peaks. | ||
These observations confirm that BBY can undergo isomerization even when bound in multi-layered films, where the free volume is significantly reduced as compared to when in solution. Fig. 6b shows the Raman shifts of BBY molecules only, deposited onto a quartz slide (blue line), which can be considered as ‘free azo’, compared to the Raman shifts of BBY bound in the multi-layered films (red line). It was observed that even at the lowest laser power (0.1 mW) the Raman shifts associated with cis BBY at 612 cm−1 and 1521 cm−1 were present for the ‘free azo’ but were absent/less intense for BBY confined in the multi-layered films. Additionally for the films with BBY only, as the power of the laser increased, the peaks corresponding to cis-BBY increased (Fig. S10a, ESI†). It was also observed that there was considerable fluorescence interference in the Raman spectra of BBY only between 1700–4000 cm−1, not seen in the NaCS/BBY thin films (Fig. S10b, ESI†). Three peaks at 2914 cm−1, 3201 cm−1, and 3096 cm−1 were still able to be identified, can be assigned to N–H stretching.39 Figure S8c shows the Raman spectrum of NaCS which had Raman shifts at 3495 cm−1, 2959 cm−1, 1065 cm−1, and 831 cm−1 which can be assigned to O–H stretching, C–H stretching, asymmetric C–O–C, and symmetric C–O–C/SO stretching, respectively.39 The weak Raman shifts at 3203–3314 cm−1, 2910 cm−1, 993 cm−1, and 882 cm−1 confirm the presence of NaCS in the multi-layered NaCS/BBY thin films (Fig. S8c, ESI†). Due to the high fluorescence signal in the Raman spectra data could not be attained for either the NaCS/BBY thin films or BBY only films when using a red/IR probe (785 nm).
‘Sun and rain’ exposure experiment
Having established that the photo-switch, BBY, can undergo trans-to-cis isomerization when in the multi-layered films, an experiment was then constructed to mimic environmental conditions of sunlight and rainfall, to assess the disassembly of the films triggered by blue visible light. The design of the ‘sun and rain exposure’ setup is shown in Fig. 1. The films were clamped under a stream of DI water (pH = 7, T = 21–25 °C, flow rate = 1 L min−1). The upper section of the films was left exposed to light while the lower section was covered using a black mask, as a control. The films were irradiated continuously using a 460 nm LED (12 V, 299 mA) light, and the films were removed, dried, and assessed at various intervals, by photographs and UV-Vis measurements of the films taken at each time interval.
Fig. 7A shows changes in the appearance of the multi-layered films after exposure to blue light and washing with DI water for 1 week (t = 162 h). While the films did not fully disassemble there was a noticeable decrease in the intensity of the red colour of the films as well as the absorbance of BBY over time. Fig. 7C shows that the absorbance at 450 nm for the unmasked region of the films (section exposed to blue light) decreased by 65% while Fig. 7D shows that the masked region (control) was stable with a negligible decrease in BBY absorbance. Additionally, during the disassembly process the peaks related to the polymer NaCS became more pronounced (λ = 200 nm and 300 nm) as more and more of the photo-switch BBY was removed from the films. NaCS was also removed from the films evidenced by the decrease in the absorbance at (λ = 200 nm and 300 nm) from t = 66–162 h. Fig. 7B shows a rate plot of Ln absorbance over time (lnA vs. t), with slope giving the rate of disassembly which follows first order kinetics. The masked/control regions often suffered a decrease in the dark attributed to leaching of excess dye trapped in the assemblies, yet was then stable to disassembly. The average rate of disassembly for the irradiated (unmasked) section of the films was calculated to be ka = 7 ± 1 × 10−3 absorbance units per h. We also performed a thickness measurement of the unirradiated regions by AFM, further confirming the robustness of the material and water resistance when not irradiated. After 2 weeks of soaking in DI water in the dark, a height measurement of 397 nm was confirmed, from an initial height measurement of 390 nm., ±15 nm (Fig. S11, ESI†). This shows that the disassembly of the thin films could be successfully and effectively triggered only by using 460 nm (blue) light.
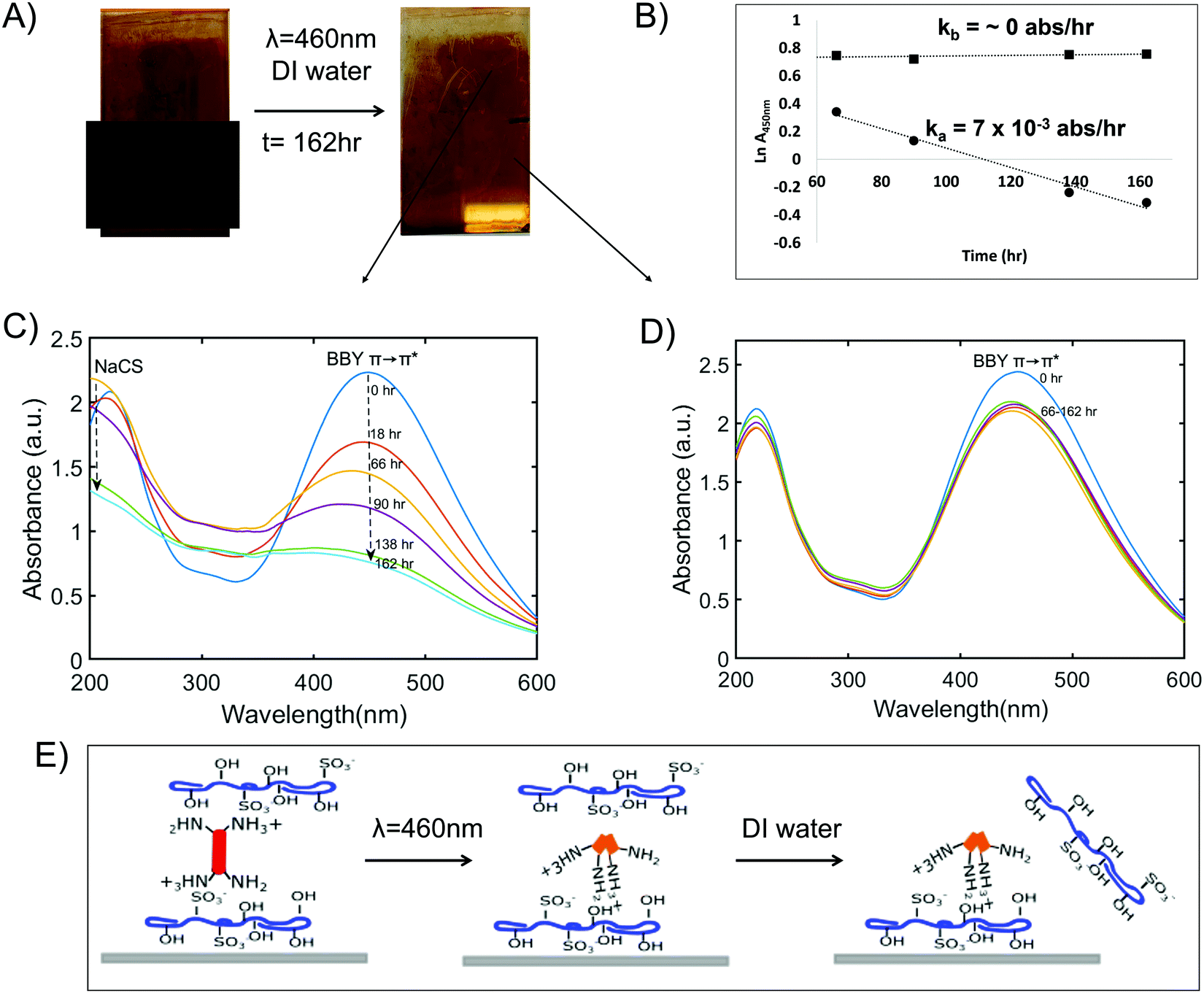 | ||
| Fig. 7 (A): Disassembly of NaCS/BBY films triggered by blue light (460 nm LED, 299 mA, 12.50 V). Upper section of films was left exposed (unmasked) while the lower section was masked. (B): Plot of ln A vs. t during disassembly process for unmasked (circles) vs. masked (squares) sections of the films. (C and D): Time resolved UV-Vis spectra of NaCS/BBY films following irradiation with blue light, washed using DI water for unmasked and masked sections, respectively (t = −162 h). (E): Schematic of disassembly of NaCS/BBY thin films under ‘simulated sunlight and rainfall’ experimental conditions. | ||
The disassembly of the thin films using (Montreal) tap water with pH = 8 (Fig. S12, ESI†) was also assessed, as this might be more relevant to a cost-effective industrial dis-assembly process. For films washed with tap water, the upper section of the films was completely disassembled while the bottom masked section remained nearly entirely intact. During the disassembly process the peaks related to the polymer NaCS became more pronounced (λ = 200 nm and 300 nm) as the photo-switch BBY was removed from the films. Complete removal of the films from the unmasked upper section was confirmed by the disappearance of absorption bands of BBY (λ = 450 nm) and NaCS (λ = 200 nm and 300 nm). Additionally, there was an observed broadening of the π → π* transition absorption band of BBY during the disassembly process, possibly due to the presence of salts, trace elements, or other ionic contaminants common in tap water, which can help screen the electrostatic charges holding the multilayer structure together. The rate of disassembly of the films when washed with tap water was also 30-fold faster than when the films were washed with DI water, during irradiation with 460 nm light. The average rates of disassembly of the films were measured to be 0.007 ± 0.001 absorbance units per h and 0.22 ± 0.02 absorbance units per h for DI water and tap water, respectively. It is speculated that the high conductivity of tap water, on the order of 1000 micro-Siemens/cm, leads to disassembly behaviour similar to high ionic strength NaCl solutions, as discussed previously.
To rationalize the observed changes in the UV-Vis spectra during the disassembly process, we propose the following mechanism: Within the multi-layered films, there are many layers of BBY molecules sandwiched between polymer layers. During irradiation with 460 nm light the upper layers consisting of trans-BBY are converted to cis-BBY which changes the polarity and geometry of BBY drastically from a planar to a non-planar, twisted molecule. Unsubstituted azobenzene can undergo a change from 9.9 Å to 5.5 Å as it undergoes trans-to-cis isomerization (a 44% reduction in length), and more importantly can lead to a significant disruption to the orientational geometry of the bonds holding the structure together.40 These geometric changes have been supported by Density Functional Theory calculations, and a detailed analysis and mechanism of these optical inter-conversions is the subject of a separate study, to be published in parallel. These changes in the geometry and alignment of BBY are proposed to disrupt the electrostatic interactions between BBY and the polymer, leading to the disassembly of the upper layers. As the upper layers are re-solubilized and are washed away, the blue light (460 nm) can penetrate more deeply to lower layers, resulting in further disassembly of the films, layer-by-layer in ‘reverse’ (Fig. 7E). It is of note that the component materials (azo dye, and cellulose polymer) should not suffer any chemical change, or other degradation during this dis-assembly process, so in principle could be completely recycled back into useable starting materials once again, providing a completely sustainable closed-loop waste-free fabrication process, if a suitable separation and recovery process could be developed, also the subject of a separate parallel study.
Conclusions
Stable multi-layered films (NaCS/BBY) formed between photo-responsive layers and layers containing a cellulose-based polymer were prepared, and their controlled disassembly when triggered by blue light was confirmed and characterized. These results demonstrate the utility of employing natural polymers in fabricating dynamic, visible light-reversible materials. The water soluble, biodegradable and bio-sourced polymer NaCS, and the water soluble azobenzene photo-switch BBY were assembled into multilayers to form robust, water-resistant, optically clear, and uniform thin films of 50 bilayers, 390 ± 15 nm thick, held together by weak intermolecular interactions between the hydroxyl and sulfonate groups of NaCS and the amino groups and amine salts of BBY. The disassembly of these LbL films however could be triggered on demand using 460 nm blue visible light, confirmed by time-dependent UV-Vis spectroscopy of the photo-disassembly in blue light vs. dark. The isomerization of BBY in solution and thin films under irradiation was confirmed by pump-probe and Confocal Raman spectroscopy experiments, respectively. The isomerization of azobenzenes results in significant microstructural changes from a planar, nonpolar structure (E) to a polar, twisted confirmation (Z), resulting in large changes in the material, leading to complete and controlled disassembly back into their water-soluble components reversibly, in principle ready for re-use. Future development work includes the preparation of free-standing films and analysis of physical and mechanical properties of the films: tensile strength, elasticity, and water vapor permeability, and will be the topics of separate forthcoming studies. It is also of great future interest to examine the mechanisms in detail to effectively separate and recover the two components from each other after the disassembly of the films, with detailed optical analysis and DFT calculations, which is being undertaken, to be published separately. Lastly, it is hoped that these new ‘reversibly soft-bonded’ materials can lead in future to a new class of materials that be easily and cheaply fabricated from water-soluble and low-toxicity ‘green’ components, recovered after dis-assembly under gentle environmental conditions, unchanged and ready for re-use, as potential alternative approaches for many of the current single-use artificial plastics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and a Molson & Hart Fellowship provided by the McGill Chemistry Department to K.E. The authors acknowledge Tobias Priemel for assistance with the confocal Raman spectroscopy measurements, Prof. T. Friscic and Group for access to the FTIR microscope, Dr Mohini Ramkaran of McGill's MIlab for the AFM measurements and analysis, Dr Lui Wong of FEMR for SEM-EDS measurements and analysis, and Dr Lihong Shang of MIAM McGill for XPS analysis. The authors also thank Setalé Eiyegbenin of McGill Chemistry for assistance with materials characterization.Notes and references
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- P. Sonkaew, A. Sane and P. Suppakul, J. Agric. Food Chem., 2012, 60, 5388–5399 CrossRef CAS PubMed.
- Q. Tarrés, P. Mutjé and M. Delgado-Aguilar, Cellulose, 2019, 26, 6917–6932 CrossRef.
- J. Wang, D. J. Gardner, N. M. Stark, D. W. Bousfield, M. Tajvidi and Z. Cai, ACS Sustainable Chem. Eng., 2017, 6, 49–70 CrossRef.
- F. Li, P. Biagioni, M. Bollani, A. Maccagnan and L. Piergiovanni, Cellulose, 2013, 20, 2491–2504 CrossRef CAS.
- K. Torvinen, J. Sievänen, T. Hjelt and E. Hellén, Cellulose, 2012, 19, 821–829 CrossRef CAS.
- Y. Wang, X. Wang, Y. Xie and K. Zhang, Cellulose, 2018, 25, 3703–3731 CrossRef CAS.
- G. Chen, B. Zhang, J. Zhao and H. Chen, Carbohydr. Polym., 2013, 95, 332–337 CrossRef CAS PubMed.
- Q.-X. Wu, Y.-X. Guan and S.-J. Yao, Front. Chem. Sci. Eng., 2018, 13, 46–58 CrossRef.
- G. Chen, B. Zhang, J. Zhao and H. Chen, Food Hydrocolloids, 2014, 35, 476–483 CrossRef CAS.
- G. Decher and J.-D. Hong, Makromol. Chem., Macromol. Symp., 1991, 46, 321–327 CrossRef CAS.
- E. Kharlampieva, V. Kozlovskaya and S. Sukhishvili, Adv. Mater., 2009, 21, 3053–3065 CrossRef CAS.
- C. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- Q. Yi and G. B. Sukhorukov, Soft Matter, 2014, 10, 1384–1391 RSC.
- W. B. Stockton and M. F. Rubner, Macromolecules, 1997, 30, 2717–2725 CrossRef CAS.
- Q. Hou, X. Wang and A. J. Ragauskas, Cellulose, 2019, 26, 4787–4798 CrossRef CAS.
- X. Tian, B. Wang, J. Li, J. Zeng and K. Chen, Carbohydr. Polym., 2017, 157, 704–710 CrossRef CAS PubMed.
- F. Li, P. Biagioni, M. Finazzi, S. Tavazzi and L. Piergiovanni, Carbohydr. Polym., 2013, 92, 2128–2134 CrossRef CAS PubMed.
- F. Carosio, M. Ghanadpour, J. Alongi and L. Wagberg, Carbohydr. Polym., 2018, 202, 479–487 CrossRef CAS PubMed.
- M. Landry, K. Gu, S. Harris, L. Al-Alwan, L. Gutsin, D. De Biasio, B. Jiang, D. Nakamura, T. C. Corkery, T. T. Kennedy and C. J. Barrett, Macromol. Biosci., 2019, 19, 1900036 CrossRef PubMed.
- Q. Bian, M. Jin, S. Chen, L. Xu, S. Wang and G. Wang, Polym. Chem., 2017, 8, 5525–5532 RSC.
- H. Rau and S. Yu-Quan, J. Photochem. Photobiol., A, 1988, 42, 321–327 CrossRef CAS.
- C. J. Barrett, J. I. Mamiya, K. G. Yager and T. Ikeda, Soft Matter, 2007, 3, 1249–1261 RSC.
- C. Slavov, C. Yang, L. Schweighauser, C. Boumrifak, A. Dreuw, H. A. Wegner and J. Wachtveitl, Phys. Chem. Chem. Phys., 2016, 18, 14795–14804 RSC.
- M. F. S. Teixeira, M. M. Barsan and C. M. A. Brett, RSC Adv., 2016, 6, 101318–101322 RSC.
- S. Sun, S. Liang, W.-C. Xu, G. Xu and S. Wu, Polym. Chem., 2019, 10, 4389–4401 RSC.
- K. G. Yager and C. J. Barrett, Macromolecules, 2006, 39, 9320–9326 CrossRef CAS.
- M. Matsumori, A. Takahashi, Y. Tomioka, T. Hikima, M. Takata, T. Kajitani and T. Fukushima, ACS Appl. Mater. Interfaces, 2015, 7, 11074–11078 CrossRef CAS PubMed.
- S. Schoelch, J. Vapaavuori, F. G. Rollet and C. J. Barrett, Macromol. Rapid Commun., 2017, 38, 1600582 CrossRef PubMed.
- Q. Si, Y. Feng, W. Yang, L. Fu, Q. Yan, L. Dong, P. Long and W. Feng, ACS Appl. Mater. Interfaces, 2018, 10, 29909–29917 CrossRef CAS PubMed.
- C. Qin, Y. Feng, H. An, J. Han, C. Cao and W. Feng, ACS Appl. Mater. Interfaces, 2017, 9, 4066–4073 CrossRef CAS PubMed.
- Y. Xiong, L. Zhang, P. Weis, P. Naumov and S. Wu, J. Mater. Chem. A, 2018, 6, 3361–3366 RSC.
- H. Orelma, A. Hokkanen, I. Leppänen, K. Kammiovirta, M. Kapulainen and A. Harlin, Cellulose, 2020, 27, 1543–1553 CrossRef CAS.
- MilliporeSigma IR Spectrum Table & Chart, https://www.sigmaaldrich.com/CA/en/technical-documents/technical-article/analytical-chemistry/photometry-and-reflectometry/ir-spectrum-table, accessed January 4th, 2022.
- B. Smith, Spectroscopy, 2019, 34, 30–37 Search PubMed.
- C. M. Stuart, R. R. Frontiera and R. A. Mathies, J. Phys. Chem. A, 2007, 111, 12072–12080 CrossRef CAS PubMed.
- N. S. Chong, K. Donthula, R. A. Davies, W. H. Ilsley and B. G. Ooi, Vib. Spectrosc., 2015, 81, 22–31 CrossRef CAS.
- T. I. Burganov, S. A. Katsyuba, T. A. Vakhonina, A. V. Sharipova, O. D. Fominykh and M. Y. Balakina, J. Phys. Chem. C, 2018, 122, 1779–1785 CrossRef CAS.
- Horiba Raman data analysis- Raman bands, https://static.horiba.com/fileadmin/Horiba/Technology/Measurement_Techniques/Molecular_Spectroscopy/Raman_Spectroscopy/Raman_Academy/Raman_Tutorial/Raman_bands.pdf, accessed January 4th, 2022.
- F. P. Nicoletta, D. Cupelli, P. Formoso, G. De Filpo, V. Colella and A. Gugliuzza, Membranes, 2012, 2, 134–197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, AFM image, UV-Vis spectra, contact angle, XPS spectrum, titration curve, FTIR spectra, Raman spectra, images of disassembly of thin films using tap water and 460 nm light. See DOI: https://doi.org/10.1039/d2ma00387b |
| This journal is © The Royal Society of Chemistry 2022 |