
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantifying the removal of stabilizing thiolates from gold nanoparticles on different carbon supports and the effect on their electrochemical properties†
Emil
Dieterich
,
Simon-Johannes
Kinkelin
,
Matthias
Steimecke
and
Michael
Bron
 *
*
Institut für Chemie, Technische Chemie I, Martin-Luther-Universität Halle-Wittenberg, Von-Danckelmann-Platz 4, 06120 Halle, Germany. E-mail: michael.bron@chemie.uni-halle.de
First published on 3rd November 2022
Abstract
Gold nanoparticles <10 nm in size are typically prepared using stabilizing agents, e.g. thiolates. Often standard recipes from literature are used to presumably remove these stabilisers to liberate the surface, e.g. for catalytic or electrocatalytic applications, however the success of these procedures is often not verified. In this work, thiolate-stabilised AuNPs of ca. 2 nm in size were synthesized and supported onto three different carbon supports, resulting in loadings from 15 to 25 wt% Au. These materials were post treated using three different methods in varying gas atmospheres to remove the stabilizing agent and to liberate the surface for electrochemical applications. Using thermogravimetry – mass spectroscopy (TG-MS), the amount of removed stabilizer was determined to be up to 95%. Identical location scanning transmission electron microscopy (il-(S)TEM) measurments revealed moderate particle growth but a stable support during the treatments, the latter was also confirmed by Raman spectroscopy. All treatments significantly improved the electrochemically accessible gold surface. In general, the results presented here point out the importance of quantitatively verifying the success of any catalyst post treatment with the aim of stabilizer removal.
Introduction
Supported gold nanoparticles (AuNPs) are of particular interest for a wide range of applications, including catalysis. Factors determining their catalytic properties are, among others, size, shape and dispersion on a support, metal–support-interactions as well as surfactants. Therefore, there is a major interest in tunable and controllable synthesizes of supported nanoparticles.1–4 For AuNPs below 10 nm the share of surface atoms overtakes the amount of bulk atoms, which results in new optical and electronic properties. A large number of studies addresses the size and shape of AuNPs depending on the chosen synthesis.2,4,5 The characteristics of the materials directly depend on the chosen stabilizing agent, the synthesis conditions and the support material.For the application as (electro)catalysts, the AuNPs are usually immobilized onto support materials like carbon nanotubes,6 multi-walled carbon nanotubes,6 carbon blacks7 and metal oxides like ZnO,8 TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 and SiO2.8 Supporting typically results in higher stability, ease of use and synergetic effects with the support material. AuNP which are synthesized as colloids before their immobilization are usually covered by surfactants which are also referred to as stabilizers. The stabilizer has the crucial role of protecting the colloids against aggregation and growth, because of their higher surface energy. Typical stabilizing agents used in the AuNP synthesis are polyvinyl alcohol (PVA), polyvinylpyrrolidone (PVP), thiolates etc.10 However, it is often unclear to which extent the stabilizing agent is influencing the activity, selectivity and stability of the AuNPs in catalytic reactions. For example, PVA exhibits a shielding effect in the liquid phase glycerol oxidation on Au/TiO2, where the remaining PVA entails a negative impact on the activity.11 Alkanethiolates are widely used stabilizing agents for various kinds of metals since they form self-assembling monolayers (SAMs) on the metal surfaces, e.g. Au,12 Ag,13 Cu,14 Pt15 and Pd.16 A different surface behavior is observed for Au compared to Pt group metals in the surface binding of thiolate, where the bonding structure on Au is more ordered compared to Pt.16,17 For palladium nanoparticles stabilized by alkanethiolates, a Pd0 core with a PdSx shell has been found, which is poisoning the nanoparticles with respect to hydrogenation and dehydrogenation.16 It is also shown that the d-electron distribution on AuNPs is altered by thiol capping agents and the stabilized Au atoms are depleted in 5d-electrons because of the strong S–Au interaction.18 Stabilizing agents are thus modifying the electronic structure and are therefore having a great impact on activity and selectivity. Furthermore, the stabilizer might react under catalytic conditions, which can damage and change the structure of the catalyst during the reaction, which might result in a change of the catalytic properties.19 Therefore, the removal of stabilizing agents is of particular interest. However, the removal of any stabilizer does not necessarily imply a fully liberated metal surface.
9 and SiO2.8 Supporting typically results in higher stability, ease of use and synergetic effects with the support material. AuNP which are synthesized as colloids before their immobilization are usually covered by surfactants which are also referred to as stabilizers. The stabilizer has the crucial role of protecting the colloids against aggregation and growth, because of their higher surface energy. Typical stabilizing agents used in the AuNP synthesis are polyvinyl alcohol (PVA), polyvinylpyrrolidone (PVP), thiolates etc.10 However, it is often unclear to which extent the stabilizing agent is influencing the activity, selectivity and stability of the AuNPs in catalytic reactions. For example, PVA exhibits a shielding effect in the liquid phase glycerol oxidation on Au/TiO2, where the remaining PVA entails a negative impact on the activity.11 Alkanethiolates are widely used stabilizing agents for various kinds of metals since they form self-assembling monolayers (SAMs) on the metal surfaces, e.g. Au,12 Ag,13 Cu,14 Pt15 and Pd.16 A different surface behavior is observed for Au compared to Pt group metals in the surface binding of thiolate, where the bonding structure on Au is more ordered compared to Pt.16,17 For palladium nanoparticles stabilized by alkanethiolates, a Pd0 core with a PdSx shell has been found, which is poisoning the nanoparticles with respect to hydrogenation and dehydrogenation.16 It is also shown that the d-electron distribution on AuNPs is altered by thiol capping agents and the stabilized Au atoms are depleted in 5d-electrons because of the strong S–Au interaction.18 Stabilizing agents are thus modifying the electronic structure and are therefore having a great impact on activity and selectivity. Furthermore, the stabilizer might react under catalytic conditions, which can damage and change the structure of the catalyst during the reaction, which might result in a change of the catalytic properties.19 Therefore, the removal of stabilizing agents is of particular interest. However, the removal of any stabilizer does not necessarily imply a fully liberated metal surface.
After surfactant removal, the NPs are typically stabilized by interaction with the support material and by small molecules from the surrounding. These small molecules often are more easy to displace and therefore less critical for the catalytic reaction.10 During the removal, there is usually a change in shape as well as a growth of the nanoparticles. This growth is affected by the conditions used during stabilizer removal as well as the support material.20,21 The removal is a very delicate process and depending on the stabilizer there are different common methods to remove the stabilizer, such as washing with different solvents and acids, UV-ozone irradiation treatment, calcination or heat treatment in different gas atmospheres.7,22–25
Concerning the supporting process of AuNPs stabilized by thiolates, different methods are suggested, which are to choose with caution depending on the chosen metal, since, as already mentioned above, the alkanethiolates-metal surface behavior varies. Commonly, the AuNPs are stirred with the support, followed by a drying step (50–120 °C, air or argon atmosphere). To actively remove the stabilizing thiolates, various treatment processes were reported: (I) annealing for 10 h in air at temperatures between 120 and 185 °C,26 (II) heating in vacuum for 1 h8 to overnight27 at 150–180 °C, (III) treating up to 300 °C in N228 and (IV) treatment at 300 °C with 20% O2/N2 followed by 400 °C with 15% H2/N2.29 Depending on the treatment and support material, particle growth is to be expected.4
As already mentioned, in addition to the activity, the stability towards a given application is also influenced by the presence or absence of stabilizers or their residues. Typical degradation mechanisms during electrochemical stress are particle growth by Ostwald ripening or agglomeration, dissolution, detachment of particles and loss of electric contact.30–32 The stability also depends on the particle size, with smaller particles being less stable. As shown in33,34 particles below 5 nm show a significantly lower stability against repeated oxidation and reduction in acid media because of dissolution.
In this work, an in-depth investigation of the effect of different heat treatment processes (300 °C 20% O2/Ar, 400 °C 15% H2/Ar, both individually or combined) onto thiolate-protected AuNPs in comparison to untreated AuNPs on different carbon supports is made. In particular, we want to analyze the amount of removed stabilizer determined by TG-MS as well as the influence of treatments onto the morphological characteristics examined by identical location-(S)TEM, Raman spectroscopy and cyclic voltammetry. Furthermore, the stability towards electrochemical stress in alkaline and acidic media is investigated by cyclic voltammetry. This work will demonstrate that care must be taken when removing thiolate capping agents from Au catalysts and that both temperature and gas atmosphere during removal are important.
Methods
Chemicals
The following chemicals and materials were used as received: ethanol (abs. HPLC, Th Geyer), DI H2O (SG water ultra clear UV ultrapure water, 0.055 μS cm−1), HAuCl4·3 H2O (ACS 99.99% metal basis, Au 49.0% min, Alfa Aesar), NaBH4 (99%, Acros Organics), 1-dodecanethiol (>98%, Sigma Aldrich), toluene (>99.5% for synthesis, ROTH), n-hexane (>99% p.a. ACS, ROTH), tetra-n-octylammonium bromide (98+%, Alfa Aesar), Vulcan XC72 (carbon black, Cabot), Nafion 117 (sol. 5%, Sigma Aldrich), Baytubes C 150 P (carbon nanotubes, C-purity >95%, number of walls 3–15, Bayer Material Science), Black Pearls 2000 (carbon black, Cabot), argon (99.999%, Air Liquide), oxygen (99.998%, Air Liquide), hydrogen (99.999%, Air Liquide), potassium hydroxide (99.98%, ROTIMETIC, 3N8), perchloric acid (Supra 70%, ROTH), CO2 (99.995%, Air Liquide).Instruments and measurements
Instruments used during the synthesis of nanoparticles include a rotary evaporator (Heidolph G3) and a centrifuge (Eppendorf 5804). For the carbon pre-treatment a furnace (Nabatherm RHTH 80-300/18) and for the AuNPs on carbon treatment a tube furnace (Carbolite MTF 12/38/250) were used.Transmission electron microscopy (TEM) was performed using a Zeiss Leo 912 Omega (120 keV, specific point-resolution 0.37 nm) and a scanning transmission electron microscope (STEM) (Zeiss GeminiSEM 500 with EDX (Oxford Ultim Max & Oxford Extreme and EBSD, Oxford C-Nano), resolution: 0.5 nm at 15 kV; 0.9 nm at 1 kV, 1.0 nm at 500 V, acceleration voltage: 0.02–30.0 kV, magnification: 50 times to 2000000 times, high efficiency Inlens secondary detector for ultra-high resolution surface information). For the measurements the sample was suspended in ethanol and drop-casted onto carbon-coated copper grids (PLANO GmbH, carbon holey film, 2.05 mm Cu-net 300 mesh), followed by drying at room temperature. For the identical location measurements, the same preparation was used, but instead of the carbon-coated copper grids, carbon-coated gold finder grids (PLANO GmbH, carbon holey film on Au-finder grids, net type H7) were used.
Raman spectroscopy was performed with an InVia Raman spectrometer setup (Renishaw), which is equipped with a microscope with a 100× objective (both Leica), a Cobolt CW DPSS laser (532 nm excitation wavelength), a 1800 l mm−1 grating and a Peltier cooled charge coupled device (CCD) camera detecting the Raman scattering. The Raman spectrometer was calibrated to a silicon reference peak and the signal was adjusted to 520.4 cm−1 before the measurements. For every sample at least three Raman mapping experiments with 42 to 110 measurement points each and a distance of ∼1 μm in x- and y-direction between the measurement points were performed. The spectra were recorded between 800 and 1900 cm−1 with 1.0% laser intensity and 15 s integration time for all CNT samples and the untreated as well as the H2 and O2 treated Vulcan samples. All BP2000 and the Vulcan sample with combined treatment were measured with 0.5% laser intensity and 20 s integration time since these samples were damaged by irradiating with higher intensities.
Thermogravimetric measurements combined with mass spectroscopy analysis (TG-MS) were used to evaluate the gold loading on the supported catalysts and the amount of removed stabilizer. The measurements were performed with a Netzsch STA 449 F1 thermobalance. ∼5 mg of sample where placed in a crucible (Al2O3) followed by heating (10 K min−1) in 20% O2/Ar up to 1000 °C. 500 μL of CO2 were pulsed using a sample loop, at 5 min and 83 min, as an internal reference, and data were normalized to the average area of these peaks to compare the individual measurements. The mass spectrometer (Netzsch QMS 403C Aëolos) was probing the exhaust stream for specific masses (44 g mol−1 (CO2), 28 g mol−1 (CO), 64 g mol−1 (SO2), 29 g mol−1 (COH+), 45 g mol−1 (COOH−), 91 g mol−1 (unknown)35).
The specific surface area (SSA) was determined with a Sorptomatic 1990 (Thermo Finnigan). Before each measurement, the sample was heated under vacuum (10−5 mbar) at 120 °C for 24 h. The adsorption and desorption isotherms were measured at 77 K using nitrogen as adsorbate at relative pressures p/p0 of 0.05 to 0.3. The evaluation of the adsorption data was done using the Brunauer–Emmett–Teller (BET) theory.
Cyclic voltammetry was carried out with a Metrohm Autolab PGSTAT 128N potentiostat. A four-neck one-compartment glass cell with three electrodes immersed into 0.1 M KOH or 0.1 M HClO4 electrolyte solution, flushed with Ar for 20 min, was used. An Ag|AgCl|KClsat. electrode (Meinsberger Elektroden) served as reference electrode, an Au-mesh (Au-net 99.9%, Goodfellow) as counter electrode and a freshly polished (1 μm, 0.3 μm Al2O3 powder and DI H2O on fleece) catalyst coated glassy carbon electrode (GCE, A = 0.126 cm2) as working electrode. The working electrode was prepared by dispersing the sample in EtOH/Nafion 117 solution (ratio v/v 977/23) with a loading of 3 mg ml−1 under sonication (Bandelin Sonocool, 75% intensity) for 5 min. Then 3.14 μL of this suspension were drop-casted onto the GCE four times and dried at room temperature in between (resulting loading was 300 μg cm−2). Cyclic voltammetry was performed between 0.3 V and 1.65 V vs. RHE in HClO4 and 0.05 V to 1.65 V vs. RHE in KOH electrolyte solution. The following procedure was applied: the internal resistance was determined using the current interrupt method, followed by 1000 CV cycles at a scan rate of 50 mV s−1. For the electrochemical degradation measurements on identical location Au-finder grids the identical setup was used but instead of 1000 CVs, 10 CVs where performed. The sample holder for the Au-finder grids is displayed in the ESI, paragraph 1.4.†
Synthesis
All glassware was pre-cleaned with aqua regia, as detailed in the ESI, paragraph 1.1† and for solution preparation DI water was used. All experiments were carried out at least three times to ensure reproducibility.For AuNP synthesis a modified36 procedure according to Brust et al.12 was used. Briefly, a solution of 184.5 mg tetraoctylammonium bromid (TOAB) in 6.75 ml toluene was prepared followed by dropwise addition of 3.75 ml freshly prepared 0.03 M HAuCl4·3 H2O aqueous solution. The dispersion was stirred for 10 min, followed by the addition of 81 μL 1-dodecanethiol (1-DDT). 3125 μL of freshly prepared 0.4 M NaBH4 aqueous solution was added. The dispersion was stirred until the aqueous phase became clear, and the aqueous phase was decanted to remove excess educts. The organic phase was reduced to a tenth of its volume with a rotary evaporator. Then 40 ml of ethanol was added and the suspension was stored at 4 °C for 16 h. The precipitate was separated by centrifugation (5000 rpm, 30 min) and washed with ethanol twice for removal of excess stabilizing agent. Then the product was dispersed in 3 ml toluene and 7 ml ethanol were added. The suspension was stored at 4 °C.
AuNP were supported onto carbon (Vulcan XC72 as well as Black Pearls 2000 and Baytubes (both pre-treated, see below)), according to a modified method presented in,29 if not mentioned otherwise. The required amount of carbon black was suspended in n-hexane (20 ml/80–100 mg n-hexane/carbon black) for 5:30 h with the aid of an ultrasonic bath. The AuNP suspension was agitated for 30 min (ultrasonic bath). The latter suspension was then added to the carbon black suspension, sonicated again for 30 min and stirred overnight. The solid was separated by centrifugation (5000 rpm, 30 min), washed with ethanol to remove any remaining impurities from the colloid synthesis and dried at 60 °C and ∼500 mbar overnight. The product was a black powder labelled Au/C and the calculated Au-loading was 20 wt% Au.
For the pre-treatment of Baytubes and Black Pearl 2000 the material was placed in a quartz glass vessel in a furnace, which was flushed before (∼30 min) and during the treatment with Ar (10 L h−1). Then the support material was heated to 800 °C (5 K min−1) for 1 h to achieve a homogenous starting material.
Catalyst treatments
Different treatment methods are suggested in literatur.8,12,26,27 Accordingly, in this work the various samples were treated in a tube-furnace under different conditions. Treatment processes at 300 °C in 20% O2/Ar for 1 h, 400 °C in 15% H2/Ar for 1 h, and both treatments combined (the oxygen followed by the hydrogen treatment) were compared with the samples without treatment. The total gas flow was 6 L h−1 with a heating ramp of 10 K min−1. For the treatment a tube-furnace was used with a quartz glass tube (ø 35 mm) and a quartz glass vessel. Before starting the heating ramp, the quartz glass tube containing the sample was flushed with Ar for at least 30 min. For the combined treatment, in between the oxygen and the hydrogen treatment the system was flushed with Ar for 30 min. These treatments were carried out both on the powdery samples as well as on those deposited onto Au finder grids.Results and discussion
To unravel the influence of the various treatment procedures on the size and distribution of the AuNP and the structure of the support material, transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) images were recorded before and after the treatments. These investigations were carried out on the powder samples but also for Au/Vulcan of samples deposited onto Au-finder grids for identical location-(S)TEM (il-(S)TEM) investigations.All supported Au samples in this work were prepared with colloidal AuNPs, the synthesis and properties of which are discussed in detail in our previous work (for a representative TEM image and size distribution see ESI Fig. 2†).36 An average size of 1.9 nm (±0.4 nm) with a size distribution in the range of 0.6–7.5 nm was found. In the present work, besides Vulcan XC72 (“Vulcan”), Baytubes (“CNTs”) and Black Pearls 2000 (“BP2000”) were used as support. A negligible particle growth, if any, is observed during the supporting procedure as can be evaluated from the TEM images in Fig. 1(a), (c), (e) and ESI Fig. 3(a), (e), (i),† as well as the corresponding average particle sizes and standard deviations in Fig. 2(a) (blue bars).
 | ||
| Fig. 1 Identical location (S)TEM images of Au/Vulcan, (a), (c), (e) untreated and (b) O2-, (d) H2- and (f) O2 + H2-treated. | ||
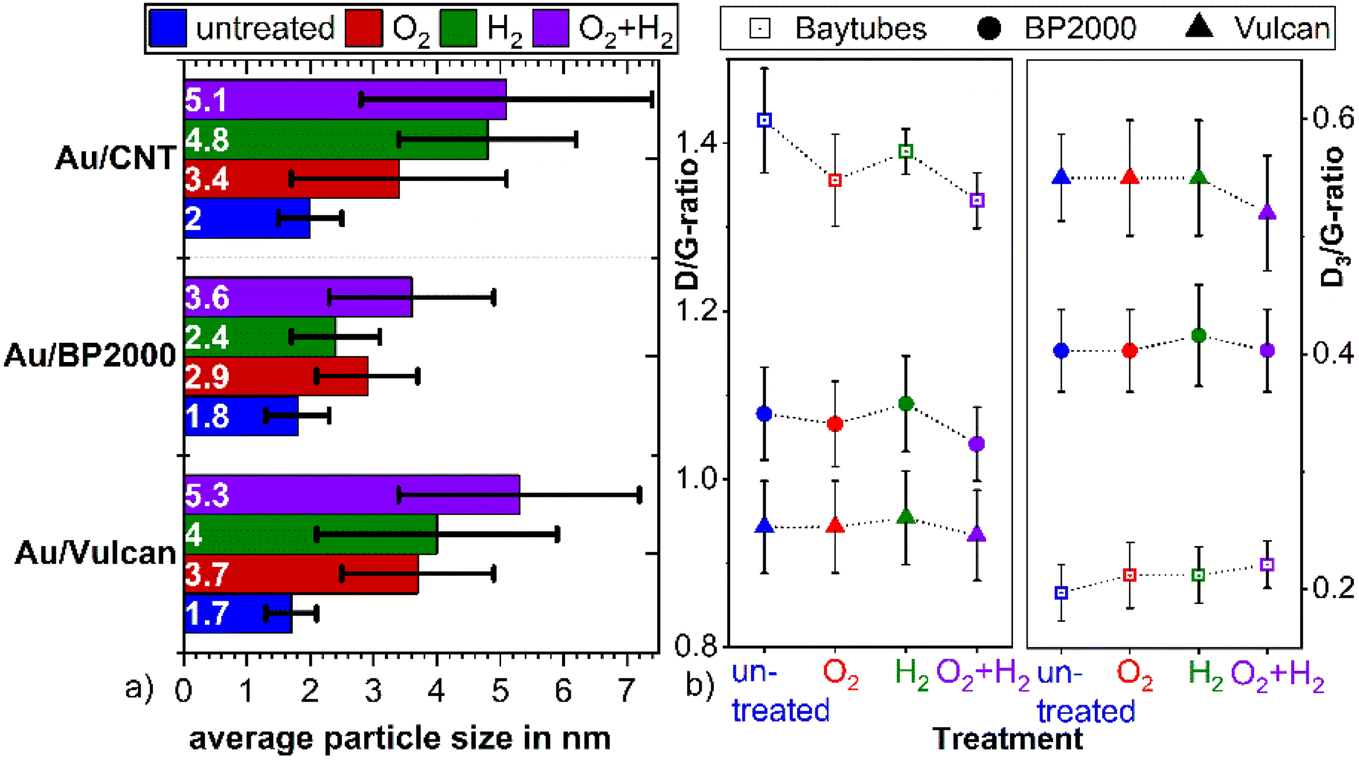 | ||
| Fig. 2 (a) Particle size (as noted at the base of the bars) and standard deviation depending on the specific treatment of the powder samples, (b) D/G-band intensity ratio and the D3/G-band intensity ratio of the differently treated samples, as obtained from Raman measurements. | ||
For all samples the average particle sizes and standard deviations after the various treatment processes are shown in Fig. 2(a). Corresponding selected (S)TEM images of the powdery samples after the various treatment procedures are shown in ESI Fig. 3,† while the evaluation of the (S)TEM images and the Au-loading measured by TG-MS are summarized in ESI Table 1.† For a better visualization, the particle size distribution is additionally shown in ESI Fig. 4.† The results clearly indicate particle growth specific for each treatment and support. AuNPs on Vulcan and CNTs show a similar behavior with a significant particle growth that increases in the O2- < H2- < O2 + H2-treated sample. In contrast, on BP2000 the particle growth is less pronounced, and also the order of the particle growth changes to H2- < O2- < O2 + H2-treatment. This different behavior supposedly is caused by different surface areas and defect structures of the supports.20,21,37 While Vulcan and CNT have a similar specific surface area (SSABET), BP2000 has a significantly higher SSABET (Vulcan – 217 m2 g−1, CNT – 200 m2 g−1 and BP2000 – 1357 m2 g−1). Despite Vulcan and CNTs are similar in terms of SSA, clearly there is a difference in their structure, as Vulcan has a greater share of disordered carbon whereas CNTs should possess and increased amount of graphitic surface carbon, albeit certainly with defects. Vulcan and BP2000 show a similar structure in terms of their share of disordered and graphitic structured carbon. Thus, the differences in particle growth likely can be attributed to the different SSA.
Raman measurements indicate only minor changes of the carbon support by the treatment processes as shown in Fig. 2(b) and ESI Fig. 5.† The ratio between the so-called D- and G-band (D/G ratio), which is indicative of the ratio between disordered and graphitic carbon, slightly changes. It seems to be slightly reduced for the O2- and the O2 + H2-treated samples, which might indicate removal of amorphous/disordered carbon by the oxygen treatment, however these changes are within the error bars of the measurements and should not be over-interpreted.
Besides the general assessment of particle growth of the powdery samples, as discussed above, il-(S)TEM allows for much deeper insight into the local changes. Fig. 1 and 3 display il-(S)TEM images of Au/Vulcan before and after the treatment procedures with a focus on the Au particles (Fig. 1) and on the carbon support (Fig. 3). It can be seen that the particle growth is not homogeneous and that in areas of higher particle density there is obviously an increased growth. Furthermore, larger particles in particular remain intact and only change their position slightly, while many smaller particles disappear. Therefore, we suggest a particle surface mobility during the treatments that leads to agglomeration. This particle mobility increases with decreasing particle size. That would also explain the lower growth of the AuNP on BP2000 support compared to Vulcan and CNTs resulting from a higher particle-to-particle distances because of the higher SSA (see above), although there is a greater particle mobility due to the smaller particles size. No influence of the growth behavior was found whether treating the samples as powder (∼100 mg) or onto the Au-finder grids.
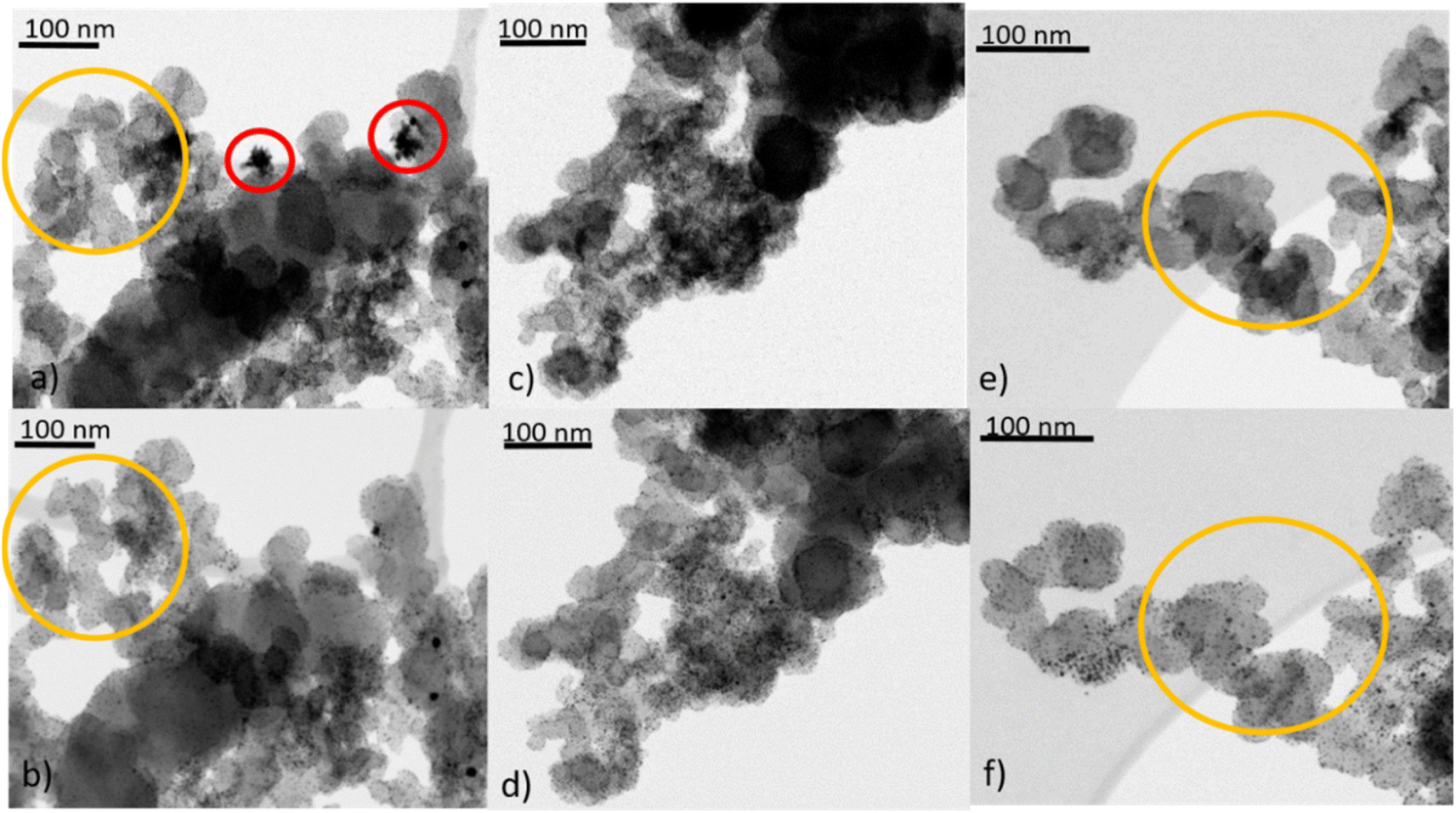 | ||
| Fig. 3 il-(S)TEM images of Au/Vulcan, top row (a), (c), (e) untreated and (b) O2-, (d) H2- and (f) O2 + H2-treated. Yellow circles to highlight areas of changes of Vulcan during treatments. Red circles for removal of impurities. | ||
il-(S)TEM has further been used to unravel possible changes which might occur at the support material during heat treatment. Fig. 3 indicates, that there are only minor changes in the support material (Fig. 3(a), (b), (e) and (f), yellow circles) for the O2- and the O2 + H2-treated samples, in accordance with the Raman results. For the H2-treated samples no changes in the support material are observed at all by il-(S)TEM. However, there are some crystal-like structures of higher density observable (Fig. 3(a) red circles), which are removed by the treatments. Since it is unlikely that these structures are gold particles, because they are removed by the treatments, we assume that they consist of impurities from the carbon support. These structures can be observed in all untreated samples and are always removed during the treatments. As shown in ESI Fig. 6† we can exclude excess stabilizer as this impurities because of the different appearance in TEM images.
The total amount of stabilizer removed by the different treatments was determined by TG-MS measurements. To ensure the effect of excess stabilizer in the catalysts is as small as possible the products where repeatedly washed with ethanol after the synthesis. ESI Fig. 6† shows the appearance of 1-DDT which is not present in the (S)TEM images of our samples. As already discussed, the stabilizing agents respectively surfactants have a crucial role in determining the catalytic properties of metallic nanoparticles and for most applications there is a need of stabilizer free particles. Fig. 4(a) and (b) present TG curves of Vulcan and Au/Vulcan as well as the corresponding MS signals of CO2 (Fig. 4(a)) and of SO2 (Fig. 4(b)) of the different treated and untreated samples as well as the bare support material. To compare the un-/treated samples in terms of amount of removed stabilizer, the SO2 MS-signal was normalized by the area of the CO2 injection peaks (see Experimental part) and then corrected by the normalized SO2 MS-signal of the related bare un-/treated support (see also ESI paragraph 1.3†). While the SO2 quotient of the untreated sample is related to an Au surface fully covered with stabilizing agent plus the sulphur impurities in the support, the SO2 quotient from the support is directly proportional to the amount of sulphur impurities in the carbon support. The resulting unit-less quotient after correction with by support is therefore proportional to the amount of SO2 on the Au surface. The normalization towards a standard was necessary due to the fluctuations of the MS signal, which are obvious for the different samples as shown for the CO2 signal in Fig. 4(a) (green circles).
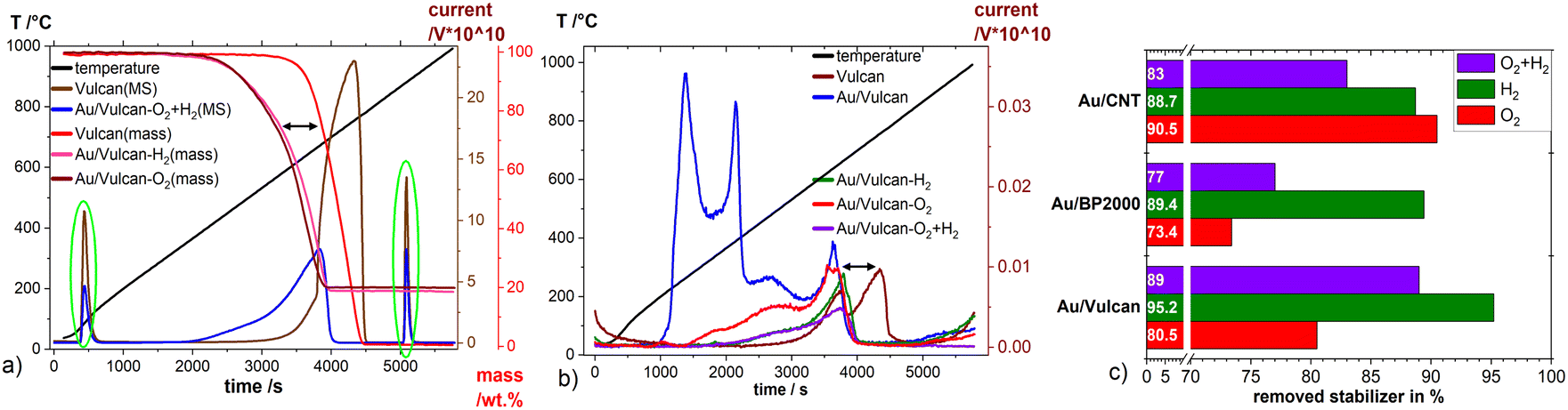 | ||
| Fig. 4 (a) TG-MS measurements with currents normalized to the CO2 (m/z = 44u) peak area, baseline corrected, (b) SO2 (m/z = 64u) currents multiplied ×1000, green circles in (a) CO2 injections for normalization, (c) amount (as noted at base of the bars) of removed stabilizer from AuNP by the different treatments (see text). | ||
The SO2 formation (comp. Fig. 4(b)) of the untreated sample (Au/Vulcan) starts around 200 °C, while it shifts to higher temperatures for the treated samples. It is evident that the main part of the sulfur is removed at higher temperatures (200–400 °C), indicating that treatment processes below 200 °C in air or Argon are not sufficient for stabilizer removal, while temperatures and atmospheres used in this study are (see below). There is also displacement of the decomposition curve from TG (∼90 °C, (Fig. 4(a) black arrow) as well as for the SO2 MS signal (Fig. 4(b)) black arrow) between the support and the samples, which suggest that the AuNP are catalyzing the thermal carbon oxidation of the support.
Fig. 4(c) summarizes the amount of stabilizer removed from the different Au/C samples by the various treatments. For Au/BP2000 and Au/Vulcan the H2-treated samples retain the lowest amount, while the O2- and the O2 + H2-treated samples show much higher amounts of remaining stabilizer (or its decomposition/oxidation products). For Au/CNTs a different behavior is observed. Here, the O2- and the H2-treated samples retain a comparable low amount, while the combined treatment surprisingly leaves a higher amount of stabilizer in the sample. In contrast to the Au/CNT sample, where these observations cannot be finally clarified, the behavior of Au/Vulcan and Au/BP2000 can be plausibly explained. For Au/BP2000 and Au/Vulcan the combined treatment is showing a higher amount of remaining stabilizer then the H2-treatment. We assume that during the O2-treatment the stabilizer is oxidized to a species that cannot be removed by H2-treatment; in contrast, 1-DDT is substantially removed by the H2-treatment.
To further characterize the different carbon-supported Au samples and to analyze the influence of the different treatment procedures towards the surface properties, electrochemical methods, here cyclic voltammetry, in two different electrolyte solutions (0.1 M KOH, 0.1 M HClO4) were applied. ESI Fig. 7(a)–(e)† show cyclic voltammograms (CVs, Fig. 5(a)) of the samples as well as the Au-mass specific reduction charge (Fig. 5(b)) and the double layer capacity (ESI Fig. 7(f)†). The CVs show Au-oxidation (forward scan ∼0.8 V in HClO4, ∼0.1 V in KOH) which is overlapping with corrosion of the supporting carbon material. The Au-reduction peak (backward scan ∼0.8–0.9 V in HClO4, ∼−0.02 to 0.1 V in KOH) is proportional to the Au surface and therefore used to determine the impact of the various treatments on the accessible Au surface.36 All samples have in common that the untreated samples display significantly less intense Au redox features in comparison to the treated ones, which is a result of surface blocking by the stabilizing agent, underpinning again the necessity of stabilizer removal. On closer inspection of the CVs, the Au reduction peak of the untreated samples is shifted (∼60 mV in HClO4; ∼100 mV in KOH) to lower potentials in comparison to the treated samples in all cases. The same holds true for the Au-oxidation, which is shifted to higher potentials and inseparably accompanied by carbon oxidation.36 Thus, while blocking the surface, the stabilizer also protects the gold from being oxidized. For a quantitative evaluation of the redox features, the Au-reduction peak was integrated and its charge was normalized to the Au mass loading for all samples (cf.Fig. 5(b)). As mentioned above, a higher reduction charge equivalent to a higher free surface area can be deduced for all samples. However, a noticeable higher mass-specific charge of untreated Au/CNT in comparison to the untreated samples of Au/Vulcan and Au/BP2000 is also apparent. Furthermore, the increase of the mass specific charge of Au/CNT after all treatments is significantly lower than for all other samples, although the change in particle size is comparable to Au/Vulcan (cf.Fig. 3(a)). To understand this issue, it should be recalled that the particles of the untreated samples are smaller than those of the corresponding treated samples, which in principle should result in larger free surface area (i.e. redox features) than the treated ones. The observation that the larger particles show more intense redox features indicates that also for the CNT-supported samples there is a significant effect of the treatment, and for the other samples the removal effect is even more pronounced than suggested by Fig. 5(b). However, a second effect should be considered here, which is a possibly poor electrical contact through the protective stabilizing agent to the support material, which might be different in case of Au/CNT with the more ordered, graphitic support, which could lead to a better contact and thus higher mass specific charge of the untreated sample. However, as already mentioned, the Au-oxidation and -reduction is shifted, which means that a higher potential is needed for the reaction and the Au-reduction charge is smaller, thus probably both effects are responsible.
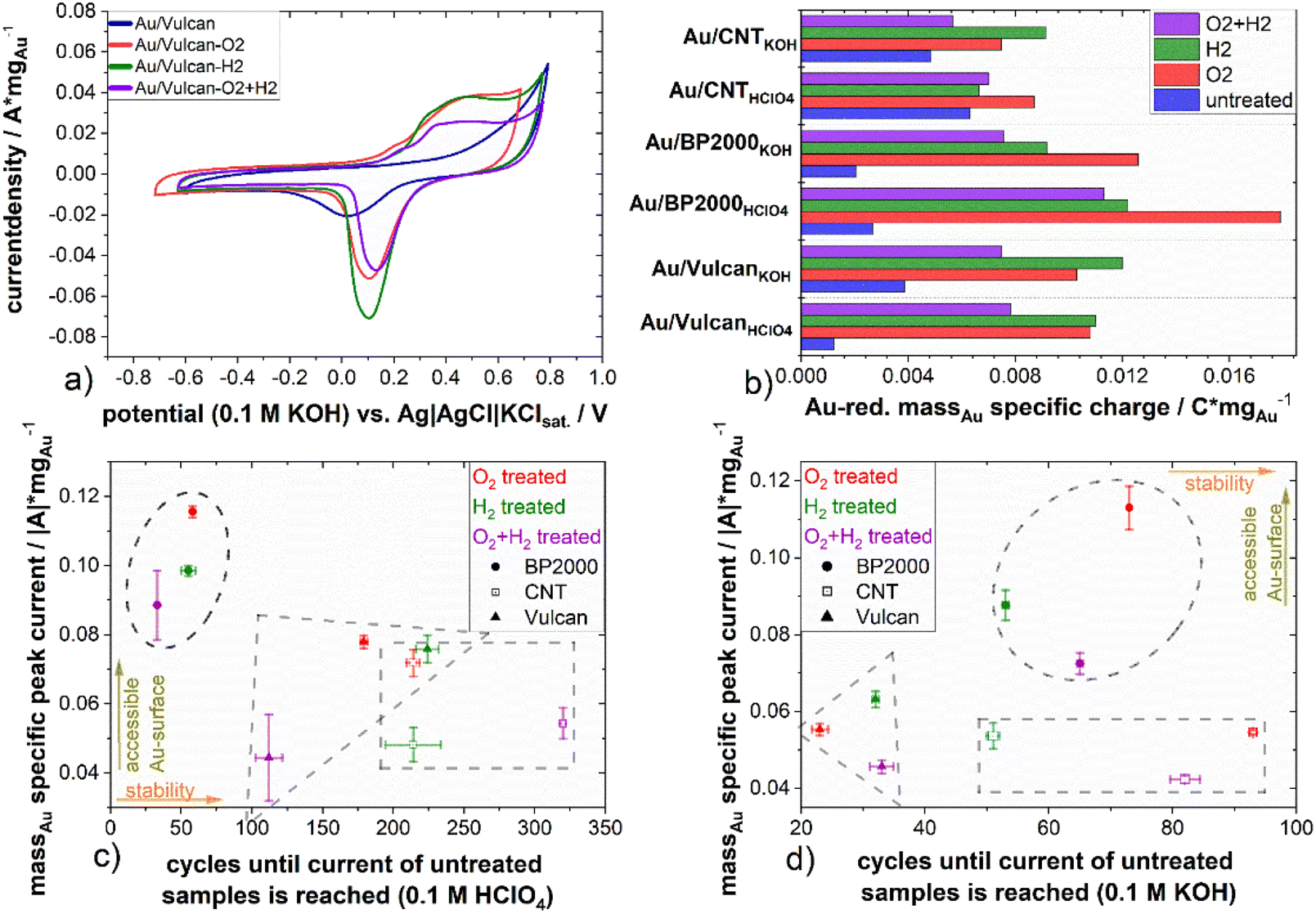 | ||
| Fig. 5 (a) Cyclic voltammograms (3rd cycle) of the untreated and differently treated AuNP on Vulcan in 0.1 M HClO4, (b) the Au-reduction peak area normalized by the Au-loading (Au-mass specific charge), (c) and (d) absolute minimum currents of the Au-reduction peaks over the number of cycles until the absolute minimum current of the untreated sample is reached for degradation measurements in (c) 0.1 M HClO4 and (d) 0.1 M KOH, all evaluated currents where corrected by the capacitive current through baseline correction. | ||
Comparing the different treatment processes, the samples treated in a single step (only H2 or O2) display higher mass specific reduction charge than the samples that underwent the combined treatment, indicating a higher electrochemically available gold surface area.
The double layer capacity (ESI Fig. 7(f)†), which is indicative of the whole catalysts surface area and not just that of the gold, is increasing with all treatments. We assume that the increase by the H2 treatment is solely due to the removal of stabilizer, which blocks most of the Au and some support surface. For the O2 treatments, we expect a higher surface functionalization of the support in addition to the effects we assumed for the H2 treatments. For the combined treatment, a mixture of the effects can be assumed. The high increase of the double layer capacity of the BP2000 samples after the treatment indicates a particularly high surface functionalization and defect formation in these samples during the treatment as well as the smaller AuNPs with higher Au surface.
With respect to a possible application of these materials in various electrochemical conversion reactions, the stability during potential cycling is of interest. In particular, the influence of the presence of stabilizing material should be addressed. Thus, all samples were exposed to repeated (up to 1000 cycles) electrochemical stress by cyclic voltammetry. The mass specific current of the Au-reduction peak in KOH and HClO4 was taken for quantitative evaluation of the experiment and is shown for the treated samples at the y-axis in Fig. 5(c) and (d). Selected CVs of the stress cycling can be found in ESI Fig. 8 and 9.† As already mentioned, the untreated samples show the lowest Au-reduction current. Therefore, the treated samples were analyzed in relation to the untreated sample to emphasize the effect of the treatment and the number of cycles that are needed to reach the current of the untreated sample, which was taken as a measure for evaluation. Fig. 5(c) and (d) present at the x-axis the number of cycles which are needed until the Au-reduction current of the treated sample reaches the Au-reduction current of the untreated sample. At the y-axis the mass specific current of all treated samples in HClO4 and KOH electrolyte, respectively is presented. The measurements in acidic electrolyte indicate a higher cycling stability (up to 320 cycles) compared to the measurements in alkaline electrolyte. This is also supported by ESI Fig. 10,† which shows the mass specific reduction current minimum over the number of cycles. The Au/BP2000 samples show the highest mass specific reduction current while the Au/Vulcan and Au/CNT samples are performing similarly, still in average the Au/CNT samples have the lowest mass specific current, which agrees with the particle size of the treated sample and the initial CVs. Au/CNTs is showing the highest average cyclic stability. The higher stability of Au/CNTs is assumed to be a results of the structural difference of the CNTs compared with BP2000 and Vulcan and also stronger particle–support interaction. Furthermore, it is apparent that for the untreated samples, there is a strong increase in reduction current during the initial cycles, which might indicate stabilizer removal and surface liberation. However, it must also be stated that there seems to be no straightforward correlation between treatment procedure and stability.
Additionally, il-STEM of the untreated and treated Au/Vulcan sample was performed before and after electrochemical stress to further investigate the electrochemical stability and the degradation mechanism. Fig. 6 shows the il-(S)TEM images of Au/Vulcan untreated and treated before and after electrochemical stress. There is an obvious loss of particles in the il-(S)TEM images and furthermore a growth of the remaining particles. While the growth appears to occur predominantly in areas with larger particles and higher particle density, a greater loss of particles appears in the area of smaller particles (Fig. 6 and ESI Fig. 11–13†). It is also noticeable that there is no apparent carbon loss or drastically changes in the support structure in the il-(S)TEM images (ESI Fig. 12†) nor a change of the double layer capacity during the electrochemical stress (ESI Fig. 8 and 9†).
 | ||
| Fig. 6 il-(S)TEM images of Au/Vulcan before being subjected to electrochemical stress in 0.1 M KOH, untreated (a), O2- (c), H2- (e) and O2 + H2-treated (g) and after subjection to electrochemical stress un- (b), O2- (d), H2- (f) and O2 + H2-treated (h). | ||
Conclusions
This study examines typical treatment procedures of thiolate stabilized AuNP on three widely used carbon supports (Vulcan XC72, Baytubes and Black Pearls 2000) towards the amount of removed stabilizer, the particle growth and change of the carbon support during the treatment as well as the electrochemically accessible Au surface and the stability against electrochemical stress. A combination of a treatment with 20% O2/Ar at 300 °C followed by 15% H2/Ar at 400 °C is such an often-used treatment in literature. This work, however, sheeds light on the processes occurring during such treatment, which are often not considered in literature.With this treatment at least 77% (Au/CNT) of the stabilizer were removed (77–89%). The single H2-treatment is removing a higher amount stabilizer (89–95%) while the O2-treatment leads to a broad variation (73–91% of removed stabilizer) depending on the support material. il-(S)TEM investigations show the influence of treatments and support specific particle growth. The particle growth was more intense for the combined treatment in comparison to the single treatments. The results lead us to the assumption that the support specific growth results mainly from the different SSA of the support material, therefore a higher SSA leads to lower particle growth. The electrochemical investigations show that the gold reduction charge is not direct proportional to the particle size and also there is a shift in the Au oxidation and reduction potential which let us assume that there is poor or no electrical contact between AuNP and support material for the untreated samples. Also there is an increase of the double layer capacity which likely stems from carbon surface functionalization and stabilizer removal of the Au surface. There are only slight changes of the support materials by the various treatments. With il-(S)TEM small changes were observed for the O2- and the O2 + H2-treated sample. This is consistent with the Raman measurements, which show slightly higher changes after these treatments compared with the H2 treatment. Also there are negligible specific structural changes observed.
Furthermore, the stability against electrochemical stress was examined, and compared to the untreated samples. The electrochemical stability depends mainly on the electrolyte and the support material. The il-(S)TEM examination of electrochemically stressed Au/Vulcan displays a growth of the particles predominantly in the area of high particle density. Also the loss of NP is mainly restricted to smaller particles. Finally, there is no observable loss of support or change in the double layer capacity.
In conclusion, we were demonstrating that the single treatments used by us are better in terms of less intense particle growth. Furthermore, the single H2-treatment removed most stabilizer with up to 95% in comparison to the combined treated samples and might be the best choice in removing thiolates from a gold surface. However, all treated samples showed higher electrochemical available surface area. For the particle growth, a dependence of the SSA of the support can be found as well as a particle surface mobility, which is higher for smaller particles. This leads to the specific loss of smaller particles mainly by agglomeration.
Author contributions
E. D. synthesized the catalysts and performed the treatments, conducted the electrochemical measurements, analyzed the TEM, STEM, TG-MS and electrochemical measurements and wrote the draft. S.-J. K. performed parts of the Raman measurements and evaluated them. M. S. contributed with in-depth scientific discussions. The work was supervised by M. B. The draft was proofread by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Annett Quetschke (Institut für Chemie, Technische Chemie I, Martin-Luther-Universität Halle Wittenberg) for performing TEM and TG-MS measurements, Eik Koslowski (Institut für Chemie, Technische Chemie I, Martin-Luther-Universität Halle Wittenberg) for performing Raman measurements and Frank Syrowatka (Interdisziplinäres Zentrum für Materialwissenschaften, Martin-Luther-Universität Halle-Wittenberg) for the STEM measurements. Martin-Luther-University Halle-Wittenberg helped in meeting the publication cost of this article.References
- Nanoparticles and catalysis, ed. D. Astruc, Wiley-VCH, Weinheim, 2008 Search PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320–321, 238–250 CrossRef CAS.
- L. Prati and A. Villa, Acc. Chem. Res., 2014, 47, 855–863 CrossRef CAS PubMed.
- L. Prati and A. Villa, Gold catalysis. Preparation, characterization, and applications, Pan Stanford Publishing, Singapore, 2016 Search PubMed.
- R. Zhang, M. Hummelgård and H. Olin, Mater. Sci. Eng., 2009, 158, 48–52 CrossRef CAS.
- J. Luo, M. M. Maye, N. N. Kariuki, L. Wang, P. Njoki, Y. Lin, M. Schadt, H. R. Naslund and C.-J. Zhong, Catal. Today, 2005, 99, 291–297 CrossRef CAS.
- G. Li, D. Jiang, C. Liu, C. Yu and R. Jin, J. Catal., 2013, 306, 177–183 CrossRef CAS.
- A. Primo, A. Corma and H. García, Phys. Chem. Chem. Phys., 2011, 13, 886–910 RSC.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- A. Villa, Di Wang, G. M. Veith, F. Vindigni and L. Prati, Catal. Sci. Technol., 2013, 3, 3036 RSC.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- M. M. Oliveira, D. Ugarte, D. Zanchet and A. J. G. Zarbin, J. Colloid Interface Sci., 2005, 292, 429–435 CrossRef CAS PubMed.
- T. P. Ang, T. S. A. Wee and W. S. Chin, J. Phys. Chem. B, 2004, 108, 11001–11010 CrossRef CAS.
- T. Laiho, J. Leiro and J. Lukkari, Appl. Surf. Sci., 2003, 212–213, 525–529 CrossRef CAS.
- G. Corthey, A. A. Rubert, A. L. Picone, G. Casillas, L. J. Giovanetti, J. M. Ramallo-López, E. Zelaya, G. A. Benitez, F. G. Requejo, M. José-Yacamán, R. C. Salvarezza and M. H. Fonticelli, J. Phys. Chem. C, 2012, 116, 9830–9837 CrossRef CAS.
- P. Carro, G. Corthey, A. A. Rubert, G. A. Benitez, M. H. Fonticelli and R. C. Salvarezza, Langmuir, 2010, 26, 14655–14662 CrossRef CAS PubMed.
- P. Zhang and T. K. Sham, Appl. Phys. Lett., 2002, 81, 736–738 CrossRef CAS.
- J. Xian, Q. Hua, Z. Jiang, Y. Ma and W. Huang, Langmuir, 2012, 28, 6736–6741 CrossRef CAS PubMed.
- L. Prati, A. Villa, C. E. Chan-Thaw, R. Arrigo, D. Wang and D. S. Su, Faraday Discuss., 2011, 152, 353–365 RSC , discussion 393–413.
- L. Prati, A. Villa, A. R. Lupini and G. M. Veith, Phys. Chem. Chem. Phys., 2012, 14, 2969–2978 RSC.
- S. M. Ansar, F. S. Ameer, W. Hu, S. Zou, C. U. Pittman and D. Zhang, Nano Lett., 2013, 13, 1226–1229 CrossRef CAS PubMed.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- N. Naresh, F. G. S. Wasim, B. P. Ladewig and M. Neergat, J. Mater. Chem. A, 2013, 1, 8553 RSC.
- J. Wu, J. Zhang, Z. Peng, S. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984–4985 CrossRef CAS PubMed.
- C. Rogers, W. S. Perkins, G. Veber, T. E. Williams, R. R. Cloke and F. R. Fischer, J. Am. Chem. Soc., 2017, 139, 4052–4061 CrossRef CAS PubMed.
- K. Sun, T. Cheng, L. Wu, Y. Hu, J. Zhou, A. Maclennan, Z. Jiang, Y. Gao, W. A. Goddard and Z. Wang, J. Am. Chem. Soc., 2017, 139, 15608–15611 CrossRef CAS PubMed.
- Z. Zhuang and W. Chen, J. Colloid Interface Sci., 2019, 538, 699–708 CrossRef CAS PubMed.
- J. Luo, M. M. Maye, N. N. Kariuki, L. Wang, P. Njoki, Y. Lin, M. Schadt, H. R. Naslund and C.-J. Zhong, Catal. Today, 2005, 99, 291–297 CrossRef CAS.
- K. J. Mayrhofer, J. C. Meier, S. J. Ashton, G. K. Wiberg, F. Kraus, M. Hanzlik and M. Arenz, Electrochem. Commun., 2008, 10, 1144–1147 CrossRef CAS.
- F. J. Perez-Alonso, C. F. Elkjær, S. S. Shim, B. L. Abrams, I. E. Stephens and I. Chorkendorff, J. Power Sources, 2011, 196, 6085–6091 CrossRef CAS.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46, 285–305 CrossRef CAS.
- J. T. Steven, V. B. Golovko, B. Johannessen and A. T. Marshall, Electrochim. Acta, 2016, 187, 593–604 CrossRef CAS.
- M. Smiljanić, U. Petek, M. Bele, F. Ruiz-Zepeda, M. Šala, P. Jovanovič, M. Gaberšček and N. Hodnik, J. Phys. Chem. C, 2021, 125, 635–647 CrossRef PubMed.
- M. Iqbal, J. McLachlan, W. Jia, N. Braidy, G. Botton and S. H. Eichhorn, J. Therm. Anal. Calorim., 2009, 96, 15–20 CrossRef CAS.
- E. Dieterich, S.-J. Kinkelin and M. Bron, ChemNanoMat, 2022, 8, e202200098 CrossRef CAS.
- S. Campisi, C. E. Chan-Thaw, Di Wang, A. Villa and L. Prati, Catal. Today, 2016, 278, 91–96 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, TEM images as well as Raman end electrochemical data. See DOI: https://doi.org/10.1039/d2na00561a |
| This journal is © The Royal Society of Chemistry 2022 |