DOI:
10.1039/D2RA02891C
(Paper)
RSC Adv., 2022,
12, 16576-16580
Total syntheses of ent-hypocoprin A and ent-hypocoprin B†
Received
7th May 2022
, Accepted 26th May 2022
First published on 6th June 2022
Abstract
This study reports the stereoselective total syntheses of the antipodes of the unique 3/10 bicyclic skeletal sesquiterpenoids, namely, hypocoprin A and hypocoprin B. The synthesis involved conjugate addition accelerated by trimethylsilyl chloride, construction of the ten-membered ring via the intramolecular SN2 reaction promoted by 1,8-diazabicyclo[5.4.0]undec-7-ene, and osmium-mediated π-facial selective dihydroxylation to functionalize the 1,1-disubstituted alkene.
Introduction
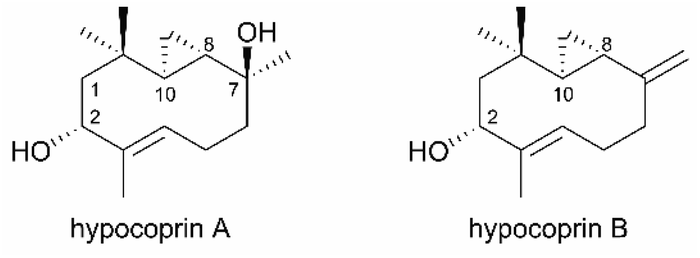
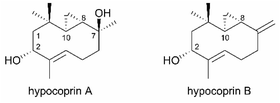
Hypocoprin A and hypocoprin B, isolated from the coprophilous fungus Hypocopra rostrata on horse dung, are sesquiterpenoids with an unprecedented 3/10 bicyclic ring system (Fig. 1).1 The 3/10 bicyclic scaffold of these sesquiterpenoids is thought to arise biosynthetically by 8,11-cyclization of the trans-humulyl cation derived from farnesyl diphosphate. The only other examples with the same scaffold are the marine diterpenoid palmatol2 (from the Mediterranean octocoral Alcyonium palmatum) and pacificins3,4 (from the Formosan soft coral Nephthea sp.). Consequently, the biosynthetic origins of the bicyclic system in hypocoprins are largely unknown. Hypocoprin A moderately inhibits the growth of the Gram-positive bacteria Staphylococcus aureus and Bacillus subtilis but is ineffective against Candida albicans or the Gram-negative bacterium Escherichia coli.1 To explore the unreported biological properties of hypocoprins A and B with novel bicyclic scaffolds, we describe novel total syntheses of ent-hypocoprin A (1) and ent-hypocoprin B (2) starting from the optically active acetonide 3.
 |
| | Fig. 1 Structures of natural hypocoprin A and hypocoprin B. | |
Results and discussion
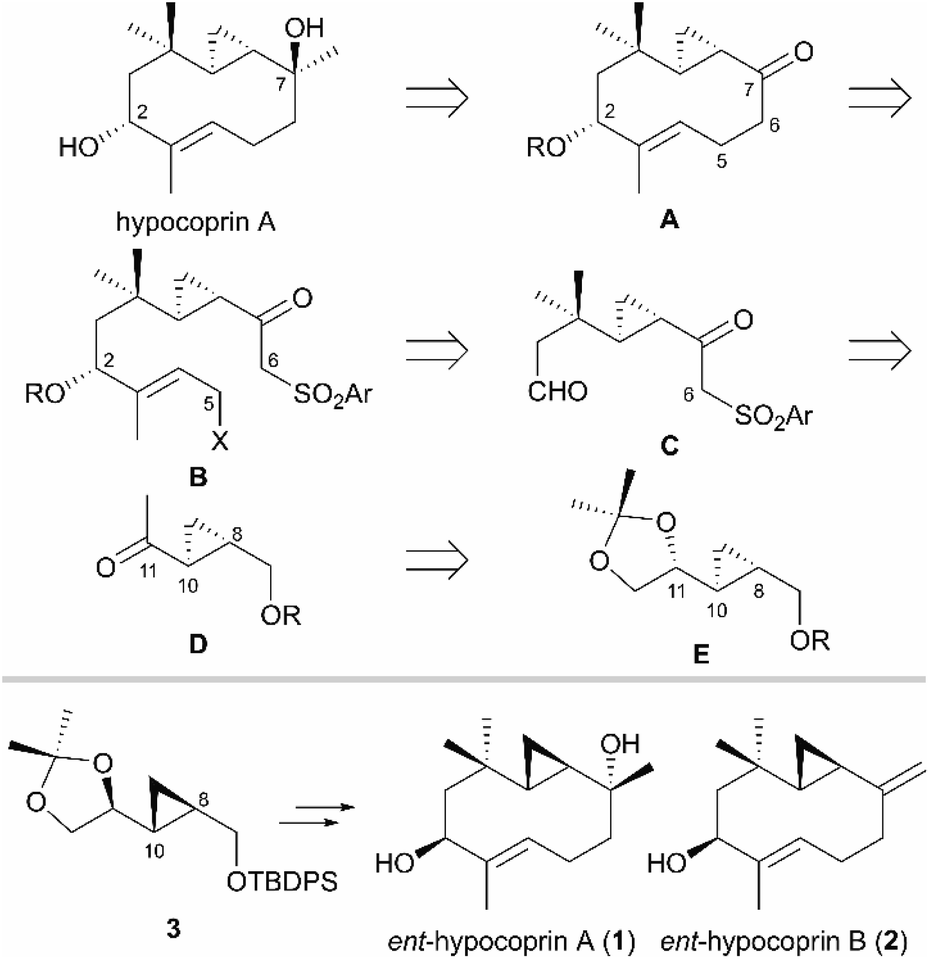
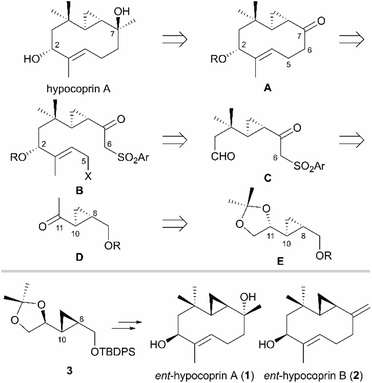
Our retrosynthetic analysis of hypocoprin A is depicted in Scheme 1. In the initial planning stage, we recognized that the C-7 tertiary alcohol moiety can be constructed in hypocoprin A via diastereoselective addition of the C1-unit to bicyclic ketone A. We thus planned the synthesis of bicyclic ketone A with a core ten-membered ring from β-ketosulfone B via an intramolecular SN2 reaction followed by reductive desulfonylation. We assumed that β-ketosulfone B would arise from the chemoselective addition of a vinyllithium species to ketoaldehyde C and the introduction of a halogen (X) atom. The β,β-disubstituted aldehyde moiety in β-ketosulfone C can be constructed by the Horner–Wadsworth–Emmons homologation of cyclopropyl ketone D followed by conjugate addition with an organocopper reagent. Cyclopropyl ketone D can be obtained from acetonide E, which possesses C-8 and C-10 stereochemistry established by substrate-controlled stereoselective cyclopropanation. We established a synthetic route for the antipodes of hypocoprins A and B from acetonide 3, which corresponds to the enantiomer of acetonide E synthesized from D-mannitol, an inexpensive and ideal chiral pool for total synthesis. The total syntheses of ent-hypocoprin A (1) and ent-hypocoprin B (2) were initiated from acetonide 3 via a retrosynthetic analysis.
 |
| | Scheme 1 Retrosynthetic analysis of ent-hypocoprin A (1) and ent-hypocoprin B (2). | |
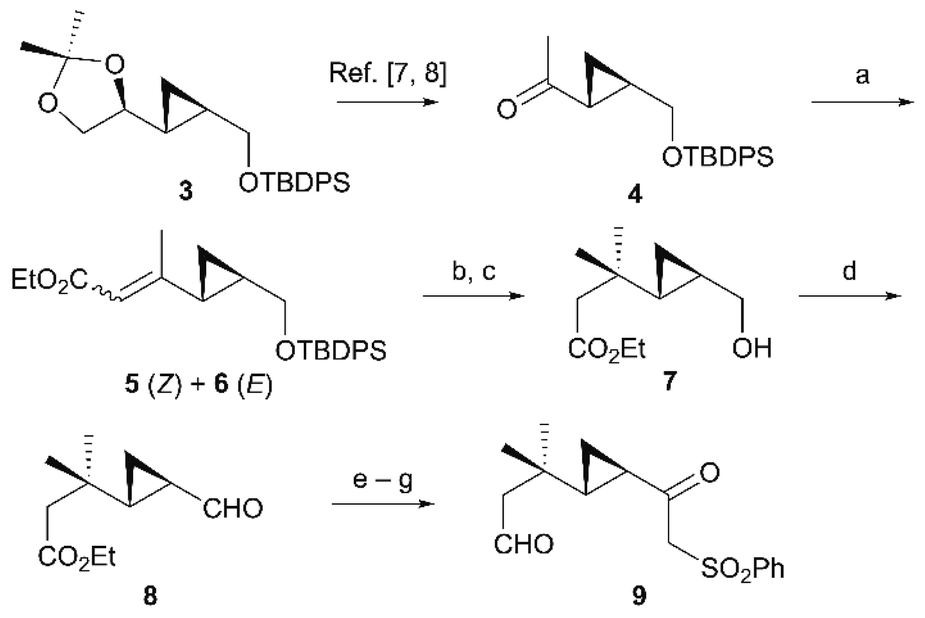
Our synthesis commenced from acetonide 3 (>97% ee),5 a known enantiopure compound prepared from D-mannitol (Scheme 2).6 After sequential acidic deprotection of the isopropylidene group of 3 and oxidative cleavage with periodic acid,7 the aldehyde was methylated with MeMgBr and the resulting secondary alcohol was oxidized with 2-iodoxybenzoic acid (IBX) to afford cyclopropyl ketone 4.8 C2-Homologation of cyclopropyl ketone 4 by the Horner–Wadsworth–Emmons reaction afforded α,β-unsaturated esters 5 and 6 in 95% yield (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 9:1). We next examined various conditions for the conjugate addition of 5 and 6, but unexpectedly, the starting material was recovered with only trace amounts of the desired adducts, presumably because the conjugate addition of organocopper reagents was inhibited by steric hindrance of the adjacent cyclopropyl group. To improve the reactivity, we extensively screened the solvents, temperatures, and addition of Lewis acid. The conjugate addition proceeded efficiently under a combined lithium dimethylcuprate–chlorotrimethylsilane reagent9 in CH2Cl2/Et2O followed by desilylation, delivering primary alcohol 7 in high yields (85% overall yield over two steps). IBX oxidation converted the primary alcohol 7 into aldehyde 8 (88% yield). Aldehyde 8 was subsequently treated with the α-sulfonyl carbanion derived from methyl phenyl sulfone and butyllithium (BuLi), providing secondary alcohols as a diastereomeric mixture. The resulting alcohols were reduced with diisobutylaluminium hydride (DIBAH) followed by oxidation with IBX to give β-ketosulfone 9 (77% overall yield over three steps).
:Z = 9:1). We next examined various conditions for the conjugate addition of 5 and 6, but unexpectedly, the starting material was recovered with only trace amounts of the desired adducts, presumably because the conjugate addition of organocopper reagents was inhibited by steric hindrance of the adjacent cyclopropyl group. To improve the reactivity, we extensively screened the solvents, temperatures, and addition of Lewis acid. The conjugate addition proceeded efficiently under a combined lithium dimethylcuprate–chlorotrimethylsilane reagent9 in CH2Cl2/Et2O followed by desilylation, delivering primary alcohol 7 in high yields (85% overall yield over two steps). IBX oxidation converted the primary alcohol 7 into aldehyde 8 (88% yield). Aldehyde 8 was subsequently treated with the α-sulfonyl carbanion derived from methyl phenyl sulfone and butyllithium (BuLi), providing secondary alcohols as a diastereomeric mixture. The resulting alcohols were reduced with diisobutylaluminium hydride (DIBAH) followed by oxidation with IBX to give β-ketosulfone 9 (77% overall yield over three steps).
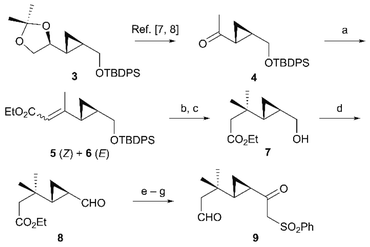 |
| | Scheme 2 Synthesis of aldehyde 9. Reagents and conditions (a) (EtO)2POCH2CO2Et, NaH, toluene, reflux, 95% (E:Z = 9:1); (b) MeLi, CuI, TMSCl, CH2Cl2, Et2O, 0 °C; (c) TBAF, THF, r.t., 85% (2 steps); (d) IBX, DMSO/THF, r.t., 88%; (e) MeSO2Ph, BuLi, THF, −78 °C; (f) DIBAH, CH2Cl2, –78 °C; (g) IBX, DMSO/THF, r.t., 77% (3 steps). | |
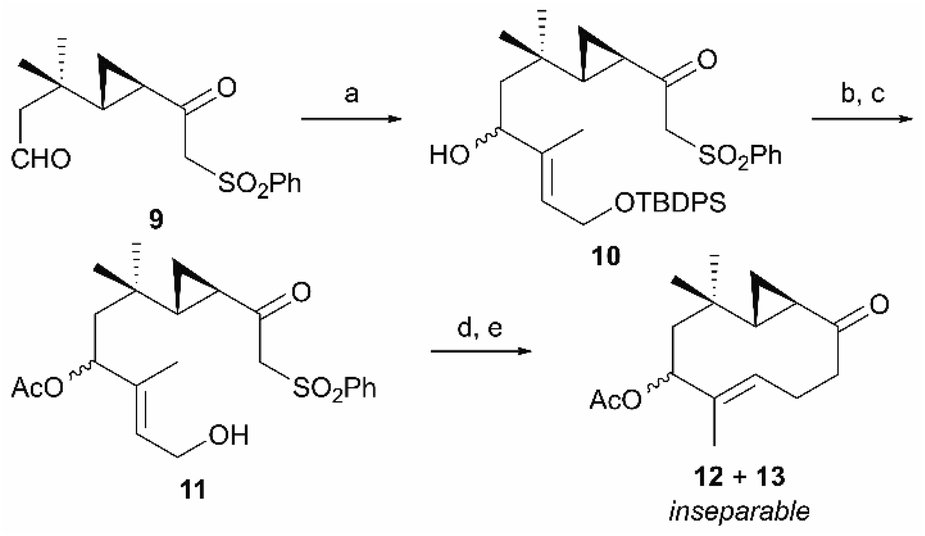
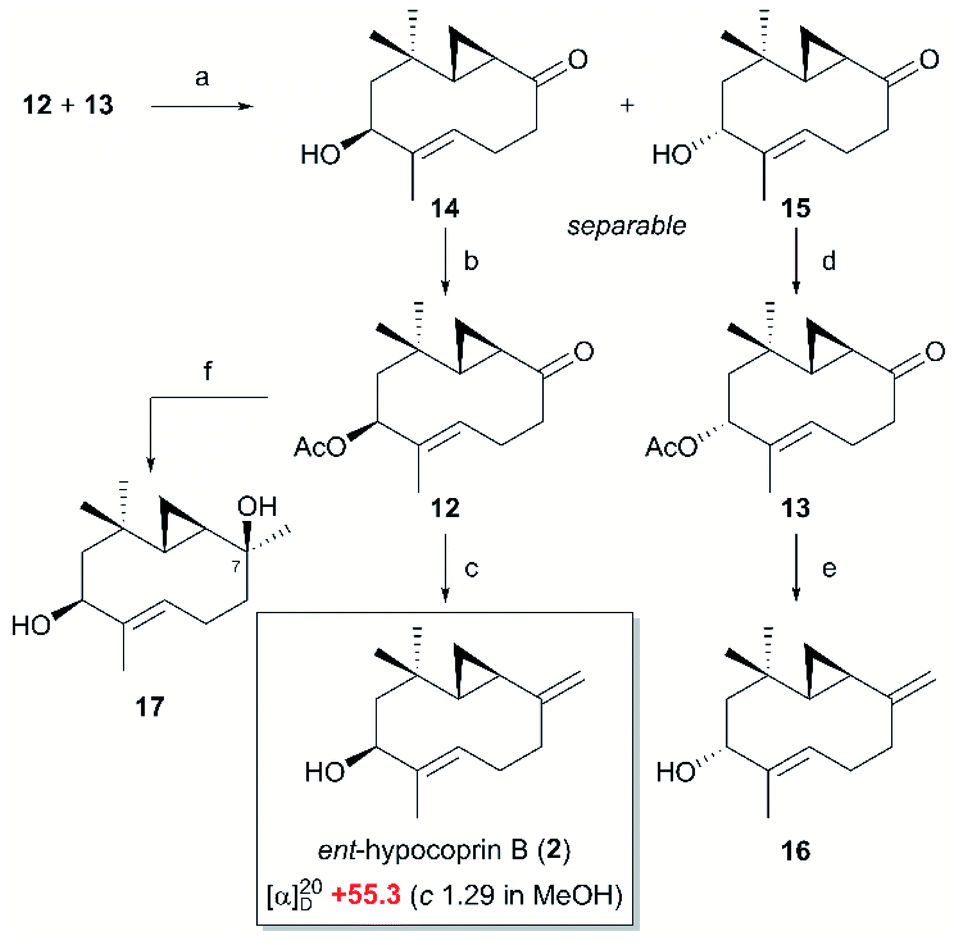
Having obtained β-ketosulfone 9, which corresponds to the retrosynthetic intermediate C shown in Scheme 1, we introduced a trisubstituted alkene moiety followed by cyclization of the ten-membered ring (Scheme 3). The trisubstituted alkene moiety was introduced to β-ketosulfone 9 by aldehyde-selective addition of the vinyllithium species derived from (E)-tert-butyl((3-iodobut-2-en-1-yl)oxy)diphenylsilane10 and tBuLi, forming the secondary alcohol 10 as a pair of inseparable diastereomers (79% yield, dr = 1:1). After acetylating the secondary alcohol 10, primary alcohol was selectively unmasked to allylic alcohol 11 (89% overall yield over two steps), which was converted to allylic bromide with CBr4 and Ph3P. The resulting allylic bromide was an important precursor for cyclizing the ten-membered ring, but was unstable. Therefore, after confirming the disappearance of the starting material by thin-layer chromatography, we immediately added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). To our delight, the intramolecular SN2 alkylation proceeded smoothly, followed by SmI2-mediated reductive desulfonylation11 to give a pair of ten-membered cyclic ketones (12 and 13) as an inseparable diastereomeric mixture (76% overall yield over two steps).
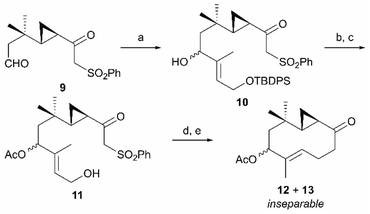 |
| | Scheme 3 Closure of the ten-membered ring via the intramolecular SN2 reaction. Reagents and conditions (a) (E)-tert-butyl((3-iodobut-2-en-1-yl)oxy)diphenylsilane, tBuLi, THF/Et2O, −78 °C, 79% (dr = 1:1); (b) Ac2O, pyridine, r.t.; (c) TBAF, THF, r.t., 89% (2 steps); (d) CBr4, Ph3P, CH2Cl2, 0 °C then DBU, benzene, 0 °C to r.t.; (e) SmI2, THF, 0 °C, 76% (2 steps). | |
The ten-membered cyclic ketones 12 and 13 were deacetylated and separated as secondary alcohols 14 and 15, respectively, and then re-acetylated to give ketones 12 and 13 as single stereoisomers (Scheme 4). The ten-membered cyclic ketones 12 and 13 were exposed to Wittig methylenation, providing the exo-olefins 2 and 16 in 92% and 90% yield, respectively. Comparing the 1H and 13C NMR spectral data of the exo-olefins 2 and 16 with the reported data, the NMR spectrum of 2 was found to be consistent with that of natural hypocoprin B.1 However, whereas the specific rotation should be opposite to that of natural hypocoprin B ([α]20D +13 (c 0.58 in MeOH)),1 it was instead consistent with that of synthesized 2 ([α]20D +55.3 (c 1.29 in MeOH)). In the previous isolation paper,1 the absolute configuration of natural hypocoprin B was determined by conversion from hypocoprin A, whose absolute configuration was determined by the Mosher method. Therefore, withholding our doubts on stereochemistry at this stage, we continued with the synthesis of ent-hypocoprin A (1). Toward the completion of the total synthesis of ent-hypocoprin A (1), we first attempted a direct nucleophilic addition of MeLi to the intermediate ten-membered cyclic ketone 12, obtaining the tertiary alcohol 17 as a single diastereomer in 98% yield. Unfortunately, the 1H and 13C NMR spectra of 17 were inconsistent with those of natural hypocoprin A.1 Although the relative configuration of 17 could not be determined by nuclear Overhauser effect (NOE) spectroscopy, most of the substrate was converted to ent-hypocoprin B (2) by dehydration during three-weeks' storage of 17 in CDCl3. Consequently, compound 17 was inferred as the C-7 epimer of ent-hypocoprin A (1).
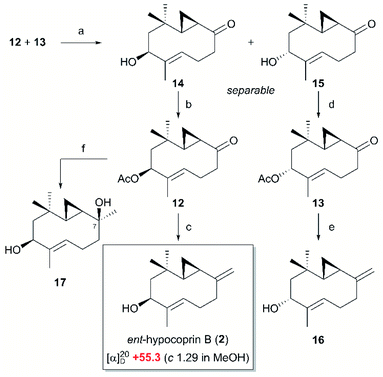 |
| | Scheme 4 Synthesis of ent-hypocoprin B (2) and 7-epi-ent-hypocoprin A (17). Reagents and conditions (a) K2CO3, MeOH, r.t., 14 48%, 15 46%; (b) Ac2O, pyridine, r.t., 96%; (c) Ph3PCH3I, BuLi, THF, r.t., 99%; (d) Ac2O, pyridine, r.t., 95%; (e) Ph3PCH3I, BuLi, THF, r.t., 90%; (f) MeLi, Et2O/THF, 0 °C, 95%. | |
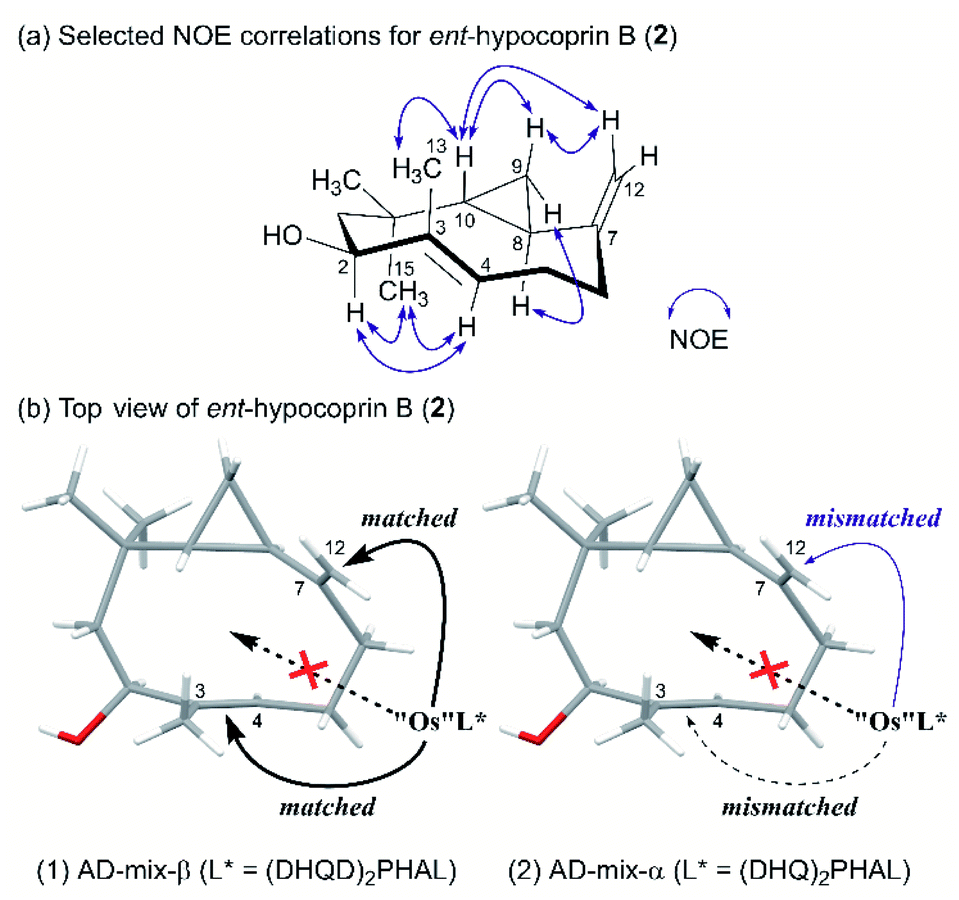
The exclusive diastereoselectivity of alkylation was attributable to steric congestion inside the ten-membered ring. We then estimated the conformation of ent-hypocoprin B (2) based on the observed NOE correlations (Fig. 2a). The results suggested that the C-3/C-13 single bond and the C-7/C-12 double bond are approximately perpendicular to the average plane of the ten-membered ring in the same direction, whereas the C-3/C-4 double bond and the C-8/C-10 single bond preferentially face each other in parallel across the ten-membered ring. That is, the C-3/C-4 and C-7/C-12 double bonds are clearly distinguished inside and outside the ten-membered ring, suggesting the feasibility of a π-facial selective approach to alkene moieties.
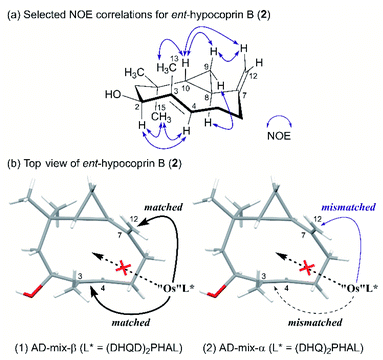 |
| | Fig. 2 Plausible mechanism of osmium-mediated π-facial selective dihydroxylation. (a) Selected NOE correlations for ent-hypocoprin B (2). (b) Top view of ent-hypocoprin B (2). | |
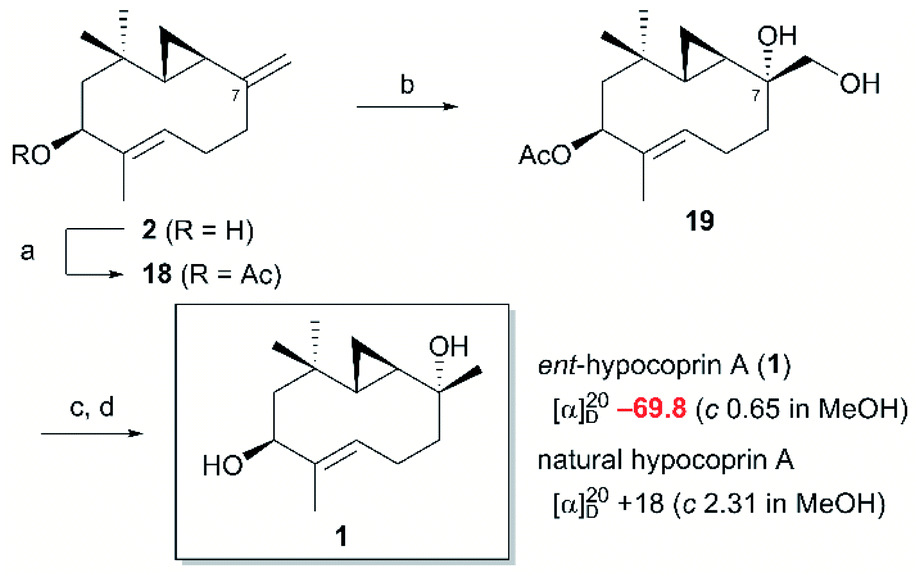
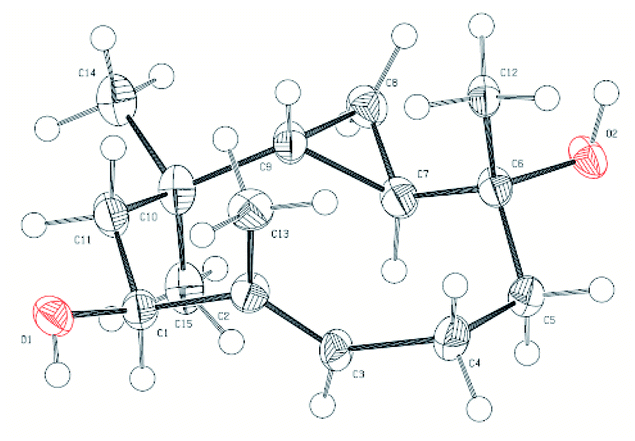
Diene 18 obtained by acetylation of ent-hypocoprin B (2) was subjected to osmium-mediated dihydroxylation using AD-mix-α or AD-mix-β (Scheme 5).12,13 In dihydroxylation using AD-mix-β, diene 18 was converted to completely unrecoverable high-polarity compounds within six hours. However, when AD-mix-α was attempted, the starting material 18 disappeared after around 24 hours, yielding the desired stereoisomer 19 with complete selectivity for C-7 as a mixture with MeSO2NH2. The stereochemistry of C-7 after derivatization to ent-hypocoprin A (1) was determined by X-ray crystallography. Interestingly, the C-3/C-4 trisubstituted alkene moiety was unaffected during this reaction. To understand this ideal π-facial selectivity, we employed a mnemonic device for predicting the stereoselectivity of dihydroxylation using AD-mix. The two alkene moieties of 18 were considered individually because they belong to different alkene-substitution classes. Considering the trisubstituted alkene (C-3/C-4) moiety, AD-mix-β and AD-mix-α are expected to preferentially proceed from outside and inside the ten-membered ring, respectively (Fig. 2b). As mentioned in Scheme 4, the inside is sterically congested, so dihydroxylation was unlikely to proceed in the presence of AD-mix-α. In contrast, 1,1-disubstituted alkene (C-7/C-12) moiety and AD-mix-β are undoubtedly a “matched” pair for dihydroxylation proceeding from outside the ring, as evidenced by the short reaction time. Nevertheless, AD-mix-α satisfactorily mediated the stereospecific dihydroxylation despite being mismatched for dihydroxylation proceeding from the outside. As is well known, the stereoselectivity of dihydroxylation is lower for 1,1-disubstituted alkenes than for trisubstituted alkenes.14 We thus hypothesized that the present regio- and diastereoselective dihydroxylations resulted from a combination of strict recognition of trisubstituted alkenes and permissive recognition of 1,1-disubstituted alkenes. Subsequently, mono-tosylation of diol 19, followed by reductive hydrogenation of the resulting tosylate with lithium triethylborohydride,15 led to ent-hypocoprin A (1). Compound 1 presented the same 1H NMR, 13C NMR, and high-resolution mass spectra as natural hypocoprin A but its specific rotation was opposite in sign [α]20D −69.8 (c 0.65 in MeOH) to that of natural hypocoprin A (ref. 1 [α]20D +18 (c 2.31 in MeOH). Furthermore, the relative configuration of synthetic compound 1 was confirmed by single-crystal X-ray diffraction (Fig. 3).16
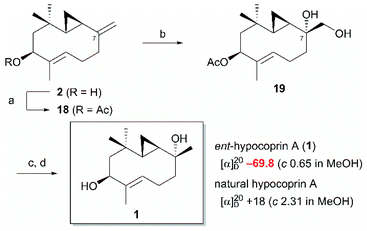 |
| | Scheme 5 Completion of the total synthesis of ent-hypocoprin A (1). Reagents and conditions (a) Ac2O, pyridine, r.t.; (b) AD-mix-α, MeSO2NH2, tBuOH/H2O, 0 °C; (c) pTsCl, pyridine, 30 °C; (d) LiEt3BH, THF, r.t., 94% (4 steps). | |
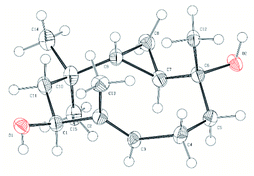 |
| | Fig. 3 Oak Ridge Thermal Ellipsoid Plot (ORTEP) of ent-hypocoprin A (1). | |
As expected, the specific rotation of the synthesized ent-hypocoprin A (1) was opposite in sign to that of natural hypocoprin A but the values of the two compounds were quite different. Many signals in the reported NMR spectrum of natural hypocoprin A were derived from impurities (mainly hypocoprin B), so the accuracy of the reported specific rotation was doubtful. In addition, as the sign of the synthesized ent-hypocoprin B (2) matched that of the reported specific rotation of hypocoprin B, one or both of the reported specific rotations might be incorrect. Therefore, we should not conclude that the synthesized ent-hypocoprin A (1) is a natural antipode only because the sign of its specific rotation is reversed. To confirm the correctness of the reported absolute configuration of hypocoprin A, we compared the NMR spectra of α-methoxy-α-trifluoromethylphenylacetic acid (MTPA) ester derivatives. In a previous study, the absolute configuration of natural hypocoprin A was determined by assigning the stereocenter of C-2 by the Mosher method.1,17,18 Therefore, ent-hypocoprin A (1) was converted into (R)-MTPA and (S)-MTPA esters and their NMR spectra were obtained. The spectra were consistent with the (S)-MTPA and (R)-MTPA esters of natural hypocoprin A described in the isolation paper. Therefore, one may reasonably conclude that the absolute configuration of the natural product reported in the isolation paper is correct.
Conclusions
In summary, we have accomplished a novel asymmetric total synthesis of ent-hypocoprin A (1) from optically active ketone 4 (19 steps, 13% overall yield). The key steps of our synthesis were TMSCl-accelerated conjugate addition, DBU-promoted cyclization to form the ten-membered ring, and osmium-mediated dihydroxylation under “mismatched” conditions to furnish the antipodes of the unique bicyclic sesquiterpenoids hypocoprin A and B. The biological activity of the two synthesized antipodes is currently under investigation. Moreover, we are applying our synthetic strategy to other unexplored 3/10 bicyclic sesquiterpenoids and diterpenoids.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- D. R. Jayanetti, Q. Yue, G. F. Bills and J. B. Gloer, J. Nat. Prod., 2015, 78, 396 CrossRef CAS.
- E. Zubia, A. Spinella, G. B. Guisto, A. Crispino and G. Cimino, Tetrahedron Lett., 1994, 35, 7069 CrossRef CAS.
- A. A. H. El-Gamal, S.-K. Wang, C.-F. Dai, I.-G. Chen and C.-Y. Duh, J. Nat. Prod., 2005, 68, 74 CrossRef CAS PubMed.
- A. A. H. El-Gamal, S.-K. Wang and C.-Y. Duh, Chem. Pharm. Bull., 2007, 55, 890 CrossRef CAS PubMed.
- The specific rotation of acetonide 3 was [α]27D –7.7 (c 1.07 in CHCl3) (ref. 6 [α]27D –7.9 (c 1.15 in CHCl3)).
- T. Morikawa, H. Sasaki, R. Hanai, A. Shibuya and T. Taguchi, J. Org. Chem., 1994, 59, 97 CrossRef CAS.
- J. B. Son, S. N. Kim, N. Y. Kim, M.-H. Hwang, W. Lee and D. H. Lee, Bull. Korean Chem. Soc., 2010, 31, 653 CrossRef CAS.
- Y. Kazuta, H. Abe, T. Yamamoto, A. Matsuda and S. Shuto, J. Org. Chem., 2003, 68, 3511 CrossRef CAS.
- F. Romanov-Michailidis, M. Pupier, L. Guénée and A. Alexakis, Chem. Commun., 2014, 50, 13461 RSC.
- K. Komine, Y. Nomura, J. Ishihara and S. Hatakeyama, Org. Lett., 2015, 17, 3918 CrossRef CAS PubMed.
- G. A. Molander and G. Hahn, J. Org. Chem., 1986, 51, 1135 CrossRef CAS.
- K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K.-S. Jeong, H.-L. Kwong, K. Morikawa, Z.-M. Wang, D. Xu and X.-L. Zhang, J. Org. Chem., 1992, 57, 2768 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS.
- D. Xu, G. A. Crispino and K. B. Sharpless, J. Am. Chem. Soc., 1992, 114, 7570 CrossRef CAS.
- H. Suemune, Y. Miyao and K. Sakai, Chem. Pharm. Bull., 1989, 37, 2523 CrossRef CAS.
- CCDC 2150062 contains the supplementary crystallographic data for ent-hypocoprin A (1).
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092 CrossRef CAS.
- J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Taiki Watanabe,
Shuntaro Igarashi,
Shinnosuke Okazaki,
Kazuo Kamaike and
Hiroaki Miyaoka
*,
Taiki Watanabe,
Shuntaro Igarashi,
Shinnosuke Okazaki,
Kazuo Kamaike and
Hiroaki Miyaoka