DOI:
10.1039/D3RA06782C
(Paper)
RSC Adv., 2023,
13, 32039-32044
Formation of charge-transfer complexes in ionic crystals composed of 1,3-bis(dicyanomethylidene)indan anion and viologens bearing alkyl chains†
Received
6th October 2023
, Accepted 25th October 2023
First published on 1st November 2023
Abstract
The relationship between charge-transfer (CT) properties and the molecular arrangement formed from π-electronic ion pairs remains unclear because of the limited variety of π-electron anions. This study addressed this issue by synthesising a series of ion pair assemblies composed of viologen dications with diverse alkyl chains as π-electron cations and 1,3-bis(dicyanomethilidene)indan anion (CMI−) as a stable π-electron anion. We obtained seven ionic crystals and identified their assembled structures using single-crystal X-ray analysis. These structures are categorized into three types: “columnar”, “slipped columnar” and “independent”. The CT properties were characterised using UV-Vis absorption spectroscopy, which revealed that the CT absorption bands were dependent on the alkyl chain length. This intriguing variation in the CT transitions can be explained by the differences in the type of assembled structure.
Introduction
Organic functional cocrystals can modulate optical properties, electrical conductivity and other properties based on the combination of molecules.1 Electron-rich donors and electron-poor acceptors can be merged to obtain charge-transfer (CT) complex cocrystal materials, which are a common type of cocrystals. These materials have been reported to have potential applications in semiconductors2 and ferroelectric materials.3 It is widely acknowledged that the arrangement of π-electron-based assemblies has a significant impact on the functions of CT complexes. Specifically, CT complexes can exhibit conductivity or insulating behaviour depending on their arrangement.4 Thus, it is crucial to elucidate the molecular framework and arrangement for the development of organic functional cocrystals based on π-conjugated systems. The formation of CT complexes can be facilitated by utilising covalent and non-covalent bonds, such as hydrogen bonds.5 Furthermore, the formation of CT complexes using ionic interactions via the addition of a charge has also attracted attention. However, most previous studies have examined π-electron systems with ionic substituents, such as sulfonates and carboxylates, and only a few studies have investigated CT complexes using π-electron systems with delocalised charges. This is primarily due to the limited number of reports on π-electron anions, such as 7,7,8,8-tetracyanoquinodimethane (TCNQ) radical anions,6 pentacyanocyclopentadienide,7 and tris-(7H-dibenzo[c,g]fluorenylidenemethyl)methide ion (Kuhn's anion),8 compared to π-electron cations.9 Additionally, controlling the molecular arrangement in the crystal structure via π-electronic interactions is a challenging task. Meanwhile, peripheral modifications of ionic π-electron systems have led to the development of “charge-by-charge assemblies”, where anions and cations are stacked alternately, and “charge segregated assemblies”, where each charge species is stacked separately.10 Therefore, appropriate molecular modifications can be used to comprehend different interactions, including CT interactions, and construct various assembled forms consisting of ion pairs having the same main skeleton.
This study investigated using 1,3-bis(dicyanomethylidene)indan anion (CMI−), prepared via the self-deprotonation of 1,3-bis(dicyanomethylidene)indan (CMI), as a π-electron anion for the formation of ionic CT complexes.11 The fabrication of various ion-pair crystals using CMI− has been previously reported.12 Viologen, a well-known electron acceptor,13 was selected as the counter cation due to its molecular structure, which allows for facile modification and possesses high symmetry. Good packing and substantial CT properties can be achieved by selecting a combination of molecules with symmetric shapes and similar molecular sizes.14 In this study, new organic CT ion-pair crystals consisting of viologen dications with alkyl chains ranging from methyl to octyl (C1–C8) and CMI− were synthesised15 and their optical properties were characterised. The correlation between the crystal structure and the optical properties of the organic ion pairs was elucidated.
Results and discussion
Synthetic procedures
Salts of 1,3-bis(dicyanomethylidene)indan anions (CMI−) and N-alkyl viologen cations (C12+–C82+) were prepared via ion exchange using the sodium salt of CMI− (Na+-CMI−) and the chloride, bromide and iodide salts of the cations (Scheme 1). Their identifications were performed using 1H and 13C NMR spectroscopy (see ESI, Fig. S1–S12†).
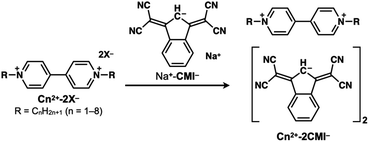 |
| | Scheme 1 Preparation of ion pairs comprising 1,1′-dialkyl-4,4′-bipyridinium cations (C12+–C82+) and 1,3-bis(dicyanomethylidene)indan anion (CMI−). | |
Single-crystal X-ray analysis
All synthesised compounds were screened using several crystallisation methods (e.g., vapour diffusion and vapour evaporation), and seven single crystals (C12+-2CMI−, C22+-2CMI−, C32+-2CMI−, C42+-2CMI−, C52+-2CMI−, C62+-2CMI− and C82+-2CMI−) appropriate for X-ray crystallographic analysis were obtained (Table S1†). All ionic crystals were obtained as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complexes of viologen cations and CMI− without other ions, and the packing structures are categorized into three types: “columnar structure”, “slipped columnar structure” and “independent structure” (Fig. 1–4).
:2 complexes of viologen cations and CMI− without other ions, and the packing structures are categorized into three types: “columnar structure”, “slipped columnar structure” and “independent structure” (Fig. 1–4).
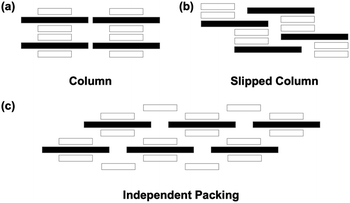 |
| | Fig. 1 Ion pair stacking motifs: (a) columnar structure; (b) slipped columnar structure; (c) independent packing structure. The black and white rectangles indicate cation and anion, respectively. | |
The ionic crystals of C12+-2CMI−, C22+-2CMI−, C32+-2CMI− and C42+-2CMI− were crystallised in the C2/c, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , P and P space groups, respectively. Columnar structures comprising alternating anion–cation–anion stacking were observed in the stacking structures of these four ion pairs (Fig. 1a, 2 and S13–16†). These columnar structures exhibited symmetric anion–cation–anion-type stacking for C12+-2CMI−, C22+-2CMI− and C42+-2CMI−, whereas asymmetric anion–cation–anion-type stacking was observed for C32+-2CMI−. The stacking distances between the average ring planes, calculated from the related sp2 atoms of CMI− (9 atoms) and bipyridinium moiety (12 atoms), were estimated as 3.75 Å, 3.31 Å, 3.37/3.35 Å and 3.26 Å in C12+-2CMI−, C22+-2CMI−, C32+-2CMI− and C42+-2CMI−, respectively. The rotations of the long axis of CMI− in relation to the long axis of the cation were 30.5°, 46.0° and 87.8° for C12+-2CMI−, C22+-2CMI− and C42+-2CMI−, respectively. In C32+-2CMI−, one CMI− was observed to be rotated 74.4° relative to the long axis of C32+, whereas the other CMI− was rotated 37.6° relative to the long axis of C32+ (Fig. 5 and S20†). Conversely, two CMI− were observed to exhibit face-to-face stacking within the column (Fig. 6). The anionic dimer was observed to exhibit anti-directional stacking, in which the dipole moments cancelled each other out, with intermolecular distances of 3.33 Å, 3.40 Å and 3.35 Å for C12+-2CMI−, C22+-2CMI− and C42+-2CMI−, respectively. Notably, the anion–anion stacking of C32+-2CMI− exhibited a rotational deviation of approximately 90°, resulting in an intermolecular distance of 3.43 Å.
, P and P space groups, respectively. Columnar structures comprising alternating anion–cation–anion stacking were observed in the stacking structures of these four ion pairs (Fig. 1a, 2 and S13–16†). These columnar structures exhibited symmetric anion–cation–anion-type stacking for C12+-2CMI−, C22+-2CMI− and C42+-2CMI−, whereas asymmetric anion–cation–anion-type stacking was observed for C32+-2CMI−. The stacking distances between the average ring planes, calculated from the related sp2 atoms of CMI− (9 atoms) and bipyridinium moiety (12 atoms), were estimated as 3.75 Å, 3.31 Å, 3.37/3.35 Å and 3.26 Å in C12+-2CMI−, C22+-2CMI−, C32+-2CMI− and C42+-2CMI−, respectively. The rotations of the long axis of CMI− in relation to the long axis of the cation were 30.5°, 46.0° and 87.8° for C12+-2CMI−, C22+-2CMI− and C42+-2CMI−, respectively. In C32+-2CMI−, one CMI− was observed to be rotated 74.4° relative to the long axis of C32+, whereas the other CMI− was rotated 37.6° relative to the long axis of C32+ (Fig. 5 and S20†). Conversely, two CMI− were observed to exhibit face-to-face stacking within the column (Fig. 6). The anionic dimer was observed to exhibit anti-directional stacking, in which the dipole moments cancelled each other out, with intermolecular distances of 3.33 Å, 3.40 Å and 3.35 Å for C12+-2CMI−, C22+-2CMI− and C42+-2CMI−, respectively. Notably, the anion–anion stacking of C32+-2CMI− exhibited a rotational deviation of approximately 90°, resulting in an intermolecular distance of 3.43 Å.
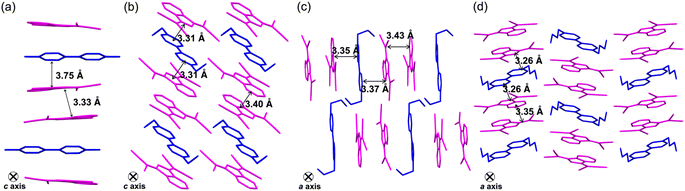 |
| | Fig. 2 Single-crystal X-ray structures of (a) C12+-2CMI−, (b) C22+-2CMI−, (c) C32+-2CMI− and (d) C42+-2CMI− wherein the magenta- and blue-coloured parts represent the anion and cation species, respectively. | |
The ionic crystal of C52+-2CMI− was crystallised in the triclinic P space group. The stacking structure of C52+-2CMI− revealed a slipped columnar structure consisting of alternating anion–cation–anion-type stacking along the a axis (Fig. 1b, 3 and S17†). The columnar structure exhibited asymmetrical anion–cation–anion stacking. The distances between the facing-conjugated planes of the adjacent cations and anions were measured to be 3.28/3.34 Å. Additionally, the orientation of the long axis of CMI− in relation to the long axis of the cation was calculated to be 74.2° (Fig. 5 and S20†). In contrast, the distance between the facing π-conjugated planes of the anions was recorded as 3.36 Å. Anion dimers exhibit anti-directional stacking, in which the dipole moments cancel each other (Fig. 6).
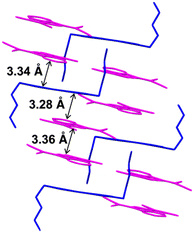 |
| | Fig. 3 Single-crystal X-ray structure of C52+-2CMI− wherein the magenta- and blue-coloured parts represent the anion and cation species, respectively. | |
The ionic crystals of C62+-2CMI− and C82+-2CMI− were crystallised in the space groups P21/c and P, respectively, and C82+-2CMI− contained isopropanol as the solvent. Despite the presence of anion–cation–anion-type stacking, the observed packing structures of both ion pairs lacked a columnar structure (independent packing; Fig. 1c, 4, S18 and S19†). The distances between the facing π-conjugated planes of the adjacent cation–anions were 3.31 Å and 3.32 Å for C62+-2CMI− and C82+-2CMI−, respectively. Furthermore, the orientations of the long axis of CMI− in relation to the long axis of the cation were calculated to be 25.7° and 76.4° for C62+-2CMI− and C82+-2CMI−, respectively (Fig. 5 and S20†).
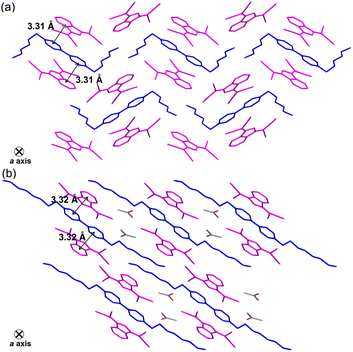 |
| | Fig. 4 Single-crystal X-ray structures of (a) C62+-2CMI− and (b) C82+-2CMI− wherein the magenta- and blue-coloured parts represent the anion and cation species, respectively. | |
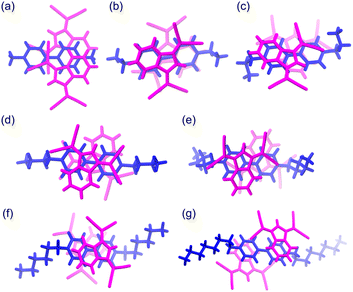 |
| | Fig. 5 The stacking structures of anion–cation–anion (top view) of (a) C12+-2CMI−, (b) C22+-2CMI−, (c) C32+-2CMI−, (d) C42+-2CMI−, (e) C52+-2CMI−, (f) C62+-2CMI− and (g) C82+-2CMI− wherein the magenta- and blue-coloured parts represent the anion and cation species, respectively. | |
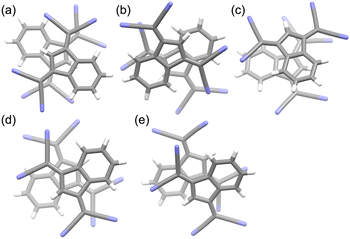 |
| | Fig. 6 The stacking structures of anion–anion (top view) of (a) C12+-2CMI−, (b) C22+-2CMI−, (c) C32+-2CMI−, (d) C42+-2CMI− and (e) C52+-2CMI−. Atom colour code: grey and blue refer to carbon and oxygen, respectively. | |
Thus, ion-pair crystals comprising Cn2+ and CMI− formed aggregates based on the anion–cation–anion structure. Moreover, it was established that the ion-pair crystals exhibited three distinct modes of assembly, determined by the length of the alkyl chain in Cn2+. In C12+-2CMI−, C22+-2CMI−, C32+-2CMI−, and C42+-2CMI−, characterised by short alkyl chains (n = 1–4), a highly ordered columnar structure was formed. Upon increasing the alkyl chain length (n = 5), the regularity of the structure was slightly impaired, resulting in the formation of a slipped columnar structure. This observation implies that the packing of the alkyl chains inhibits the electrostatic interactions between the ion pairs. In C62+-2CMI− and C82+-2CMI−, which supported longer alkyl chains (n = 6 and 8), the packing of the alkyl chains fully suppressed the formation of a columnar structure, and the anion–cation–anion structure existed independently. Interestingly, attention to the dihedral angles between the pyridinium moieties in Cn2+ tended towards 0° when the alkyl chain lengths were even, whereas they exhibited a range of 2.45–4.45° when the alkyl chain lengths were odd. Further investigation of this observable even–odd effect is imperative.
Optical properties
UV-Vis absorption spectrophotometry was performed by dissolving the sample in methanol to examine the interaction between Cn2+ and CMI− in a polar solvent (Fig S21†). The analysis of Na+-CMI− revealed two distinctive absorption bands. In the long-wavelength region, the initial absorption was observed at 440 nm, and the cut-off wavelength was determined to be approximately 670 nm in the visible range. This complicated absorption band with a maximum at 582 nm indicates that CMI− exists as the anion form without protonation in methanol. In contrast, the absorption spectra of Cn2+-2CMI− in methanol exhibited a similar absorption band with a maximum at 582 nm, indicating a substantial two-fold enhancement in its molar absorption coefficient. This result indicates the absence of an interaction between CMI− and Cn2+ in methanol.
Diffuse reflectance spectra were measured using powder samples to reveal the interactions between ion pairs (Fig. S22†). In Cn2+-2CMI− (n = 1–4), a significant redshift of the absorption edge compared to that of Na+-CMI− was observed (Fig. 7a). The presence of an absorption edge in the near-infrared (NIR) region suggests the formation of CT complexes with Cn2+ as the acceptor and CMI− as the donor. In contrast, in Cn2+-2CMI− (n = 5–8), only a small redshift of the absorption edge compared to that of Na+-CMI− was observed (Fig. 7b). These results correlated with the packing arrangement of the crystal structures. In particular, Cn2+-2CMI− (n = 1–4), which forms a columnar structure, demonstrated a remarkable CT absorption band because of the formation of a proficient stacking structure of anions and cations. In contrast, CT absorption was hardly observed in the slipped columnar structure with limited overlap between the anions and cations, as well as in the independent packing structures of the anion–cation–anion arrangements. Thus, the packing structure and the associated optical characteristics can be regulated by manipulating the length of the alkyl chain.
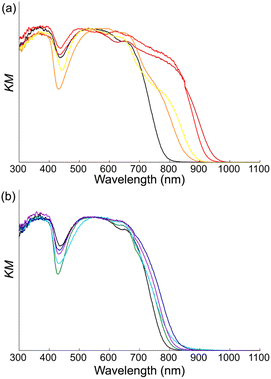 |
| | Fig. 7 Comparison of diffuse reflectance spectra of Cn2+-2CMI− (a) n = 1–4 and (b) n = 5–8 with Na-CMI−. Spectra line colour code: black, dark red, red, orange, yellow, green, light blue, blue and purple refer to Na-CMI−, C12+-2CMI−, C22+-2CMI−, C32+-2CMI−, C42+-2CMI−, C52+-2CMI−, C62+-2CMI−, C72+-2CMI− and C82+-2CMI−, respectively. | |
The optical energy bandgap (Eg) of optical functional materials is a critical parameter for producing optoelectronic application of these materials. Therefore, the optical bandgaps of the ionic crystals were calculated using the Tauc plots (Table 1). Based on the data of diffuse reflection spectra of powder sample, the Tauc plots were prepared by the following eqn (1).
Table 1 The optical bandgap (Eg) of Cn2+-2CMI− calculation by the Tauc plots
| Compound |
Bandgap Eg [eV] |
Wavelength of absorption edge [nm] |
| C12+-2CMI− |
1.27 |
974 |
| C22+-2CMI− |
1.30 |
957 |
| C32+-2CMI− |
1.37 |
904 |
| C42+-2CMI− |
1.35 |
916 |
| C52+-2CMI− |
1.47 |
842 |
| C62+-2CMI− |
1.34 |
927 |
| C72+-2CMI− |
1.41 |
878 |
| C82+-2CMI− |
1.44 |
864 |
α is UV-Vis absorption coefficient (=2.303logA/t, t: thickness: 1), h is Planck constant, ν is frequency, m is direct transitions (using for direct allowed transition, m = 1/2), A is absorbance and Eg is optical energy bandgap. (αhν)1/2 was plotted as a function of the hν, and hν is also photon energy, can calculate as 1240/λ. Calculated Eg by the Tauc plots of Cn2+-2CMI− were consistent with the photon energy at the absorption edge. Among Cn2+-2CMI−, the order of the bandgap from the regression curve of the Tauc plot is as follow; C12+-2CMI− < C22+-2CMI− < C62+-2CMI− < C42+-2CMI− < C32+-2CMI− < C72+-2CMI− < C82+-2CMI− < C52+-2CMI−. The bandgap of C62+-2CMI− was estimated to be low due to the presence of an extended absorption tail in the NIR range. However, the bandgap obtained from the Tauc plots clearly indicates a distinct boundary between C42+-2CMI− and C52+-2CMI−. Cn2+-2CMI− (n = 1–4) exhibit low bandgap values, approximately ranging from 1.30 to 1.45, suggesting the formation of a CT band. Thus, the presence of an elaborate columnar structure, consisting of the donor and acceptor, promotes the formation of the CT band. On the other hand, for Cn2+-2CMI− (n = 5–8, excepted for C62+-2CMI−), larger bandgap values were calculated compared to Cn2+-2CMI− (n = 1–4), approximately ranging from 1.47 to 1.54. In C62+-2CMI−, the transition derived from CT was observed even without the columnar structure, which was probably due to the small angle between the long axes of the cation and anion resulting in the substantial cation–anion overlap. However, it is noteworthy that the absorption derived from the CT transition was noticeably smaller compared to those of Cn2+-2CMI− (n = 1–4), which form columnar structures.
Thermal properties
The thermal properties of the ion pairs in their bulk states were examined using differential scanning calorimetry (DSC). The melting points were observed at 189, 235, 230, 221, 184, 146 and 157 °C for C22+-2CMI−, C32+-2CMI−, C42+-2CMI−, C52+-2CMI−, C62+-2CMI−, C72+-2CMI− and C82+-2CMI−, respectively, and C12+-2CMI− decomposed under its melting point (Fig. 8 and S23†). The melting point order was approximately dependent on the length of the alkyl chain.
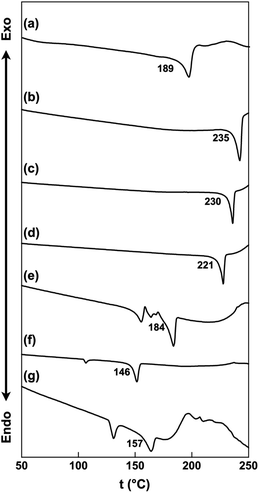 |
| | Fig. 8 DSC thermograms of (a) C22+-2CMI−, (b) C32+-2CMI−, (c) C42+-2CMI−, (d) C52+-2CMI−, (e) C62+-2CMI−, (f) C72+-2CMI− and (g) C82+-2CMI−. Onset temperatures (°C) of melting points are labeled. | |
Conclusions
Ion pairs consisting of CMI− and viologen dications with alkyl chains ranging from methyl to octyl groups were prepared via ion exchange using the sodium salt of CMI− as well as the chloride, bromide, and iodide salts of the cations. Seven ionic crystals were obtained, and their assembled structures were revealed via single-crystal X-ray analysis. In all the ionic crystals, anion–cation–anion-type stacking formations were observed. The assembled structures were classified into three types: “columnar”, “slipped columnar”, and “independent”, as shown in Fig. 1. The Cn2+-2CMI− crystals (n = 1–4) were sorted into columnar structures, whereas the C52+-2CMI− crystal was sorted into slipped columnar structures. The crystals of Cn2+-2CMI− (n = 6 and 8), including the long alkyl chains, were sorted into independent structures. These results indicate a correlation between the crystal structure and the optical properties. Ion pairs forming columnar and slipped columnar structures exhibited absorption bands derived from the CT transition, whereas the CT absorption band was not observed for the independent structure. Therefore, we successfully generated numerous assemblies by altering only the alkyl chain length while keeping the primary framework of the π-electron system unchanged. Moreover, we elucidated the correlation between the arrangement of anions and cations and the development of charge-transfer complexes. Systematic studies on the crystal structures of organic ionic species are just beginning to emerge, and further detailed analyses are ongoing and worth investigating.
Author contributions
E. Saito, T. Yoshida and R. Yamakado conceived and designed the experiments. T. Yasuhara and H. Yamaguchi conducted preliminary examinations. S. Okada assisted the measurement for the optical properties. E. Saito and R. Yamakado wrote the manuscript. All the authors have read and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A part of this work was supported by Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers, “Advanced Next Generation Energy Leadership (R2601, FY2014-2016)” JSPS KAKENHI Grant-in-Aid for Scientific Research (B) (JP 18H02068), JSPS Fellows (JP 21J22405 and 22KJ0332), Japan-Austria Bilateral Joint Research Project and Japan-Ukraine Bilateral Joint Research Project from Japan Society for the Promotion of Science (JSPS). The synchrotron radiation experiments were performed at the BL02B1 (2022B1944) of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) with the supports by Dr Y. Nakamura, JASRI/SPring-8 and Dr A. Matsunaga, Yamagata University. The NMR analysis was performed using a JEOL JNM-EC500, JEOL Ltd introduced by the subsidy program for development of advanced research infrastructure. We thank Mr Junya Kimura Yamagata University, for preliminary examinations.
Notes and references
-
(a) Y. Huang, Z. Wang, Z. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2019, 58, 9696–9711 CrossRef CAS PubMed
 ;
(b) M. Jiang, C. Zhen, S. Li, X. Zhang and W. Hu, Front. Chem., 2021, 9, 764628 CrossRef CAS PubMed .
;
(b) M. Jiang, C. Zhen, S. Li, X. Zhang and W. Hu, Front. Chem., 2021, 9, 764628 CrossRef CAS PubMed . - D. Jerome, Chem. Rev., 2004, 104, 5565–5592 CrossRef CAS PubMed .
-
(a) F. Kagawa, S. Horiuchi, H. Matsui, R. Kumai, Y. Onose, T. Hasegawa and Y. Tokura, Phys. Rev. Lett., 2010, 104, 227602 CrossRef CAS PubMed ;
(b) A. S. Tayi, A. K. Shveyd, A. C. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485–489 CrossRef CAS PubMed .
- H. Alves, A. S. Molinari, H. Xie and A. F. Morpurgo, Nat. Mater., 2008, 7, 574–580 CrossRef CAS PubMed .
- A. Das and S. Ghosh, Angew Chem. Int. Ed. Engl., 2014, 53, 2038–2054 CrossRef CAS PubMed .
-
(a) A. L. Sutton, B. F. Abrahams, D. M. D'Alessandro, R. W. Elliott, T. A. Hudson, R. Robson and P. M. Usov, CrystEngComm, 2014, 16, 5234–5243 RSC ;
(b) A. L. Sutton, B. F. Abrahams, D. M. D'Alessandro, T. A. Hudson, R. Robson and P. M. Usov, CrystEngComm, 2016, 18, 8906–8914 RSC ;
(c) K. Sambe, N. Hoshino, T. Takeda, T. Nakamura and T. Akutagawa, Cryst. Growth Des., 2020, 20, 3625–3634 CrossRef CAS .
-
(a) E. L. Goff and R. B. LaCount, J. Am. Chem. Soc., 1963, 85, 1354–1355 CrossRef ;
(b) O. W. Webster, J. Am. Chem. Soc., 1965, 87, 1820–1821 CrossRef CAS ;
(c) C. Richardson and C. A. Reed, Chem. Commun., 2004, 706–707 RSC ;
(d) R. J. Less, M. McPartlin, J. M. Rawson, P. T. Wood and D. S. Wright, Chem. –Eur. J., 2010, 16, 13723–13728 CrossRef CAS PubMed ;
(e) Y. Bando, Y. Haketa, T. Sakurai, W. Matsuda, S. Seki, H. Takaya and H. Maeda, Chem. –Eur. J., 2016, 22, 7843–7850 CrossRef CAS PubMed .
-
(a) R. Kuhn and D. Rewicki, Angew. Chem., Int. Ed., 1967, 6, 635–636 CrossRef CAS ;
(b) K. Okamoto, T. Kitagawa, K. Takeuchi, K. Komatsu, T. Kinoshita, S. Aonuma, M. Nagai and A. Miyabo, J. Org. Chem., 1990, 55, 996–1002 CrossRef CAS .
- J.-B. Ming, X. Li, J. Li, W. Yu and W. Wang, Cryst. Growth Des., 2022, 22, 1212–1220 CrossRef CAS .
-
(a) Y. Haketa and H. Maeda, Chem. Commun., 2017, 53, 2894–2909 RSC ;
(b) Y. Haketa and H. Maeda, Bull. Chem. Soc. Jpn., 2018, 91, 420–436 CrossRef CAS ;
(c) Y. Haketa, K. Urakawa and H. Maeda, Mol. Syst. Des. Eng., 2020, 5, 757–771 RSC .
-
(a) Y. Son and S. Kim, Dyes Pigm., 2005, 64, 153–155 CrossRef CAS ;
(b) J. Fujisawa, Chem. Phys. Lett., 2014, 608, 355–359 CrossRef CAS .
- Y. Tanaka, K. Ichijo, S. Kodama, S. Aoyama, T. Yoshida, R. Yamakado and S. Okada, Cryst. Growth Des., 2019, 19, 5811–5818 CrossRef CAS .
-
(a) R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147–156 RSC ;
(b) H. Chen, V. Brasiliense, J. Mo, L. Zhang, Y. Jiao, Z. Chen, L. O. Jones, G. He, Q. H. Guo, X. Y. Chen, B. Song, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 2886–2895 CrossRef CAS PubMed .
- P. Huo, Y.-H. Li, L.-J. Xue, T. Chen, L. Yu, Q.-Y. Zhu and J. Dai, CrystEngComm, 2016, 18, 1904–1910 RSC .
- E. Saito, T. Yasuhara, Y. Tanaka, R. Yamakado, S. Okada, T. Nohara, A. Masuhara and T. Yoshida, ECS Trans., 2019, 88, 301–311 CrossRef .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Taichi Yasuhara,
Hiroto Yamaguchi,
Shuji Okada
*,
Taichi Yasuhara,
Hiroto Yamaguchi,
Shuji Okada







